

Abstract
An approach combining small-angle X-ray solution scattering (SAXS) data with coarse-grained (CG) simulations is developed to characterize the assembly states of Hck, a member of the Src-family kinases, under various conditions in solution. First, a basis set comprising a small number of assembly states is generated from extensive CG simulations. Second, a theoretical SAXS profile for each state in the basis set is computed by using the Fast-SAXS method. Finally, the relative population of the different assembly states is determined via a Bayesian-based Monte Carlo procedure seeking to optimize the theoretical scattering profiles against experimental SAXS data. The study establishes the concept of basis-set supported SAXS (BSS-SAXS) reconstruction combining computational and experimental techniques. Here, BSS-SAXS reconstruction is used to reveal the structural organization of Hck in solution and the different shifts in the equilibrium population of assembly states upon the binding of different signaling peptides.
Keywords: coarse-grained model, Bayesian analysis, folding, SAXS, simulation
Tyrosine kinases of the Src-family are large multidomain allosteric enzymes implicated in the signaling pathways regulating cell growth and proliferation (1). The key role that Src kinases play in the onset of many human diseases, particularly cancer, makes them important targets for therapeutic intervention (2).
All Src kinases share a common structural organization comprising the SH3 and SH2 binding domains followed by a highly conserved catalytic domain connected by flexible linkers (1). Crystal structures have offered a detailed view of the down-regulated forms of Hck and c-Src (3, 4), characterized by a compact assembled form stabilized by auto-inhibitory intramolecular interactions between the N and C terminus of the catalytic domain with the SH3 and SH2 modules, respectively. The SH2 and SH3 modules of the Src-family tyrosine kinases play dual roles: both are involved in the auto-inhibitory intramolecular interactions down-regulating kinase activity, but can also serve as possible binding receptors for cellular signals leading to kinase activation.
In a broadly accepted paradigm, Src inactivation/activation is pictured in terms of a simple two-state process involving an assembled (inactive) and a disassembled (active) conformation, i.e., once any of the intramolecular interaction is released, the kinase domain switches to a disassembled catalytically active state targeting down-stream cellular substrates (5–8). Crystal structures (3, 4) and mutational studies complemented by molecular dynamics (MD) simulations (9) are consistent with this view. However, one crystal structure of c-Src in a partially activated state in which the SH2-SH3 modules are reassembled in a different orientation with respect to the catalytic domain (10), suggests that the situation could be considerably more complex. In this regard, it seems likely that different signals could promote different assembly states, leading to differently configured activated kinases. While it is understood that Src increases its activity in response to molecular signals that interfere with the interactions stabilizing the assembled complex, little is known about the spatial organization of the up-regulated active form of Src. A quantitative characterization of the structural organization of the multidomain Src in solution under different conditions is critical for a better understanding of the molecular basis of the allosteric response.
Probing the spatial organization of a large and flexible multidomain macromolecular complex such as Src is challenging. The protein is expected to display substantial conformational dynamics, giving rise to a large number of possible conformations separated by small energy differences. Capturing any of those in a crystallographic state would be inherently difficult, and ideally one would like to observe the protein conformation in solution, where it is not affected by lattice packing. In principle, small-angle X-ray solution scattering (SAXS) is an experimental technique that can be used for mapping the three-dimensional organization of multidomain proteins in solution (11–15). Different conformations give rise to distinct scattering patterns containing information on the overall architecture and domain-domain separation within the complex. However, it is not straightforward to determine the three-dimensional shape directly from the low-resolution information provided by the SAXS data. More importantly, there are fundamental limitations associated with the naive notion of a unique molecular shape in the case of flexible multidomain complexes, which may display considerable conformational dynamics.
To address those issues, we present an integrative approach, the basis-set supported SAXS (BSS-SAXS) reconstruction, that aims at combining SAXS data with a simplified coarse-grained (CG) model to characterize the assembly states of the flexible multidomain Hck in solution. Simulations of the CG model are used to extensively explore and sample the accessible conformational space of the multidomain complex. The configurations are clustered into a small number of distinct putative assembly states, which are then used as a basis-set to analyze SAXS data. Comparison of the calculated SAXS patterns from all assembly states with experimental data makes it possible to determine the population fraction of each state of Hck under various conditions. The results indicate that multiple assembly states are generally present, and that the population equilibrium among these states responds to both mutations and the presence of signaling peptides binding to the SH2 or SH3 domains.
Results
Classes of Hck Assembly States.
The simplified CG model of Hck, though extremely simplified, incorporates the main generic features of the protein: (i) the native three-dimensional conformations of the SH3, SH2, and catalytic domains from the X-ray structures, (ii) the topological connectivity of the polypeptide chain via flexible linkers, and (iii) the excluded volume from the residue-residue core repulsion. The CG model also accounts for the intramolecular interactions involved in the stabilization of the down-regulated assembled form (3): the tyrosine-containing C-terminal “Ctail” of the kinase domain with the SH2 domain, and the polyproline SH2-kinase “linker” with the SH3 domain. As described in SI Appendix, both interactions were incorporated in the CG model, and adjusted to match the Kd from experiments for the SH2-binding Ctail peptide, and the SH3-binding polyproline peptide. The active and inactive conformations of the catalytic domain are also included in the multistate model as described previously (16, 17) to account for the ability of full-length Hck to adopt different conformations. To ensure proper sampling of various assembly conformations, simulations using different initial conditions were conducted, ranging from fully assembled to disassembled states, from increased to decreased binding interactions, and from destabilizing stable configurations to lowered transition barriers.
A large number of configurations were generated using MD simulations based on the CG model. A two-step clustering scheme was used to organize the large amount of information generated by the simulations of the CG model into a manageable form. Initially, 25 structural clusters were determined from the trajectories from a structural clustering based on residue-residue distances similarity criterion (16, 17). More detail on the clustering is given in SI Appendix: Fig. S1. Then, average scattering patterns were calculated for members of each cluster using the Fast-SAXS method (18), and the clusters were further regrouped into nine assembly states on the basis of a scattering pattern similarity criterion. The relationship between the nine states according to this metric is shown diagrammatically in SI Appendix: Fig. S1, and a representative conformation from each state is shown in Fig. 1. It should be noted that the configurations within those states are regrouped according to the similarity of their scattering patterns, and that they may display conformational differences. This set of nine assembly states, extracted from the simplified CG simulations, is then used as a basis-set for extracting the multidomain organization of Hck in solution from the experimentally observed SAXS patterns.
Fig. 1.

Multiple assembly conformational states adopted by the multidomain Hck from a CG model. Representatives of these nine scattering states, ranging in architecture from fully to partially assembled to disassembled states, in size from compact to extended forms, and in interdomain separation from assembled to fully or partially disassembled states. The catalytic domain is in blue, SH2 in green, and SH3 in yellow. States 1 and 8 are similar to the assembled Hck (pdb entry 1QCF) and the partially active c-Src (pdb entry 1Y57) structures, respectively. More information about the states is in SI Appendix: Figs. S4–S12.
Bayesian-Based Monte Carlo Analysis of SAXS Data.
The calculated profile Icalc is an average over the Ns = 9 states, weighted by the fractional population {Pi} ≡ P1,…,PNs of state i,
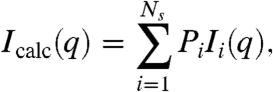 |
[1] |
where Ii(q) is the theoretical scattering profile of state i (see examples in SI Appendix: Fig. S2). The goal of the analysis is to determine the set of Ns fractional population {Pi} from the SAXS data. To this end, we define a dimensionless scoring function, χ2[{Pi}], measuring the agreement between the calculated and experimental SAXS profiles,
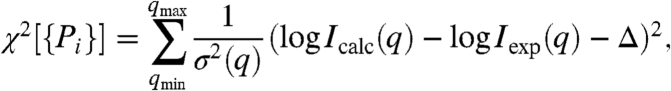 |
[2] |
where qmin and qmax are the lower and upper limit of q-range of the experimental profile of Iexp(q), respectively, and Δ is the offset between log Icalc and log Iexp(q) at q = qmin. The estimated experimental SAXS uncertainty, σ(q), is assumed to be [δ log Iexp(q)]3/2. Assuming independent experimental errors and a uniform prior, the probability distribution exp(-χ2) is sampled in the space of the model parameters, {Pi}, according to a Bayesian-Based Monte Carlo (BMC) algorithm. The procedure yields the fractional population Pi for the Ns states, along with their uncertainties extracted the fluctuations.
Tests with the BSS-SAXS method indicate that the scattering pattern in the low-q regions, up to about 0.15 Å-1 contains most of the information that controls the structural differences in Hck (see SI Appendix: Fig. S3A,C). Including higher-q SAXS data has little effect on the average values of Pi, although it reduces the uncertainty of the Pi (see SI Appendix: Fig. S3B, D). Additional tests with the BSS-SAXS/BMC simulations show that the procedure yields the correct population Pi (see SI Appendix: Fig. S3E, F), although large increases in the estimated SAXS uncertainty can lead to inaccuracies (see SI Appendix: Fig. S3G, H). The present approach has some similarities with other methods, such as EOM (19) and BILBOMD (20), which were also designed to go beyond standard strategies based on a single conformation to fit SAXS data. For example, all three methods primarily rely on a large set of random conformers. Both EOM and BILBOMD use a Monte Carlo genetic algorithm to select a small number of specific conformers out of the large set to best-fit the SAXS data, which are attributed equal statistical weight, whereas BSS-SAXS uses a BMC algorithm to determine the fractional population Pi for the states in a scattering basis-set. Overall, the tests show that the BSS-SAXS analysis can provide quantitative estimates of the fractional population Pi of Hck in solution.
BSS-SAXS Characterization of Hck Assembly.
SAXS data were collected for the wild-type Hck, and for the high-affinity Ctail mutant (Hck-YEEI) in the presence of two types of external peptide ligands: (p2) a high-affinity phosphorylated tyrosine-containing SH2-binding peptide (6); (p3) a high-affinity SH3-binding PPII peptide (21 and 22). All the experimental scattering profiles are shown in Figs. 2, 3. For the sake of completeness, a simple Guinier analysis was performed for all the SAXS data shown in Figs. 2 and 3, and the radii of gyration Rg were extracted by fitting the scattering patterns in the range up to 0.05 Å-1. The results are reported in the Figure captions.
Fig. 2.

Analysis of wild-type Hck and high-affinity Ctail mutant (Hck-YEEI) in solution. The scattering pattern (left), the assembly state population (center), and the relative χ2 scores for different combinations (right) are shown. (A) SAXS data for the wild-type Hck (black) and BMC simulated scattering profile (red) are shown. Rg is 28.1 ± 0.2 Å. All the SAXS profiles of I(q) are shown in a logarithmic scale, where q = 2π/d and d is the Bragg spacing in scattering. Relative χ2 scores show that the inclusion of states 6 and 8 has improved the fit to the SAXS data. The relative χ2 score for the crystal structure is 7.6 × 107. (B) SAXS data for the Hck-YEEI mutant (black) and BMC simulated scattering (red) are shown. Rg is 27.1 ± 0.2 Å. Inclusion of state 6 greatly improves agreement with the SAXS data (right).
Fig. 3.

The wild-type and mutant Hck in solution in the absence and presence of SH2-binding (p2) and SH3-binding (p3) peptides. In all cases, the scattering pattern (left), the assembly state population (center), and the relative χ2 scores for different combinations (right) are shown. (A) SAXS data of Hck in the presence of p2 at a concentration of 250 μM (black) and the BMC simulated profile (red) are shown. Addition of p2 causes a shift in the equilibrium toward a coexistence of assembled and various disassembled states. Rg is 29.3 ± 0.2 Å. Relative χ2 scores for different solutions of the state combination. (B) SAXS data of Hck-YEEI in the presence of p2 at a concentration of 250 μM (black) and the BMC simulated profile (red) are shown. Rg is 28.1 ± 0.1 Å. Furthermore, in contrast to the Hck-YEEI scattering in the absence of p2 (Fig. 2B), these results demonstrate that the addition of p2 to a Hck-YEEI solution causes an increase in population of the disassembled and compact state 6. (C) SAXS data of Hck-YEEI in the presence of p3 peptide at a concentration of 1 mM (black) and the BMC simulated profile (red) are shown. Rg is 29.0 ± 0.3 Å. (D) Addition of both p2 and p3 peptides shifts the equilibrium of Hck conformations toward disassembled states. SAXS data of the wild-type Hck in the presence of both p2 at a concentration of 250 μM and p3 at a concentration of 1 mM (black) and the BMC simulated profile (red). Rg is 31.7 ± 0.2 Å.
The experimental SAXS data from wild-type Hck in the absence of external peptides is shown in Fig. 2A. It is observed that the SAXS profile calculated from the population derived from the BMC analysis (red) accurately reproduces the experimental data (black). The fit is achieved with the population of assembly states determined via the BMC analysis. The latter indicates that the enzyme is predominantly in the assembled state 1, with the population of 82%. The more weakly populated states include the fully disassembled state 6 and the partially disassembled state 8 with SH2-dissociated but SH3-bound similar to the partially disassembled crystal structure of c-Src (10). As shown in Fig. 2A, this combination of states best fits the SAXS data. The experimental SAXS data from the high-affinity Ctail mutant YEEI is shown in Fig. 2B. In this case too, the assembled state 1 dominates the three-dimensional organization, with the population of 83%.
To investigate how ligand binding affects the equilibrium assembly of the multidomain complex, SAXS data were also collected from Hck with two types of peptide ligands. As shown in Fig. 3A, there is a substantial shift of wild-type Hck from a compact state to disassembled states in the presence of 250 μM of the high-affinity phosphorylated SH2-binding peptide p2. Comparison of Figs. 2A and 3A shows that the population of the compact state 1 decreases from 82% to 22%, while that of the disassembled state 6 increases from 8% to 29%. Repeating this experiment for the Hck-YEEI Ctail mutant yields a very different result. The BMC analysis given in Fig. 3B indicates that, for the Hck-YEEI mutant, the population of the assembled state is reduced only to 62% upon addition of the peptide p2. Here, the disassembled and compact state 6, where SH2 and SH3 are disassociated but the overall Hck architecture remains compact, is also populated.
The effect of the SH3-binding peptide p3 is shown in Fig. 3C. BMC analysis of the SAXS data indicates that p3 destabilizes the assembled down-regulated state 1 and promotes the disassembled compact state 6. Comparison of Figs. 2A and 3C shows that the population of state 1 decreases from 82% to 7% and that of state 6 increases from 8% to 50% in the presence of peptide p3. Also, the population of the SH3-displaced states 2 and 3 increases, reaching a total combined population of about 39%. Cross-correlation analysis of the population of states 2 and 3, however, suggests that they are highly correlated and that determining their relative weight is at the limit of resolution of the present BMC analysis.
Lastly, the spatial organization of Hck was examined in the presence of both the p2 and p3 peptides simultaneously. The results of the BMC analysis of SAXS data, shown in Fig. 3D, demonstrate that any remnant of the assembled down-regulated state is wiped out upon the binding of p2 and p3. Here, the fully disassembled states 5 and 6 are representative of the spatial organization. Both states are disassembled when Ctail is released from SH2 and SH3 is disassociated, but they differ in overall size: state 5 is extended (SI Appendix: Fig. S8) and state 6 exhibits a compact form (SI Appendix: Fig. S9). Consistent with the BSS-SAXS analysis, Rg reaches its largest value (31.7 Å) under these conditions.
Discussion
The SAXS data is consistent with an increasing disassembled state for Hck as it is perturbed by the signaling peptides. In particular, the radius of gyration Rg extracted from the SAXS data via a Guinier analysis is typically smaller in the absence of binding peptides (Fig. 2) than in their presence (Fig. 3). Also consistent with this result, the smallest value of 27.1 Å is observed for the EEI Ctail mutant of Hck (Fig. 2B), whereas the largest value of 31.7 Å is observed for the wild-type HcK in the presence of peptides p2 and p3 (Fig. 3D). Nevertheless, although a simple analysis of SAXS data based on the variations of Rg is indicative of broad structural changes, the scattering pattern does not, by itself, provide a straightforward characterization of the assembly of a complex multidomain protein such as Hck.
To determine quantitatively how various factors can affect the configurational landscape of Hck, we set out to estimate the relative population of a set of possible assembly states from the experimental SAXS data using a BMC analysis. This analysis leans on the configurations generated by simulating a simplified CG model. The set of nine assembly states extracted from those simulations serves as a basis-set covering the wide range of compact and loose configurations that Hck could possibly adopt. In the BSS-SAXS reconstruction, the set of nine assembly states constitutes a Bayesian prior in the analysis (23). One configuration from each of the assembly states is illustrated in Fig. 1. However, it is emphasized that each state actually comprises a large ensemble of configurations (SI Appendix: Figs. S4–S12). Such conformational heterogeneity is consistent with the existence of a broad continuum of accessible configurations, but it could also reflect our limited ability to resolve the conformation of a multidomain protein unambiguously using only the information provided by the scattering patterns. Without additional experimental data permitting a more detailed structural description, it is best to exert some caution when interpreting the results of the BSS-SAXS analysis in terms of “state” population.
The nine states in Fig. 1 can be resolved from SAXS data, i.e., differences in scattering intensity are larger than experimental uncertainty. Distinguishability of the states included in the set is measured from the uncertainty-weighted difference in average SAXS patterns 〈I(q)〉 (see also SI Appendix: Table S1). The set includes a wide range of different configurations, varying in their multidomain organization and overall architecture. State 1 is similar to the X-ray structure of Hck corresponding to the fully assembled down-regulated kinase (3, 4), while state 8 is analogous to the partially activated state observed in one X-ray structure of c-Src (10). Fluctuations are important and there are subtle, though important, differences among some states. For example, states 5 and 6 are both disassembled, but the former is more extended than the latter. State 2 and 3 correspond to SH3-displaced configurations, lacking the direct interaction between the SH3 and the PPII linker. In some disassembled configurations, the SH3 and SH2 domains maintain the relative orientation observed in the assembled form (state 1), consistent with a previous simulation study (24).
The result of the BMC analysis of SAXS data for the wild-type Hck is shown in Fig. 2A. The analysis indicates that the enzyme is predominantly in the assembled state 1, with a probability of 82%. As shown in the figure (left), the calculated scattering profile with the combination of states determined from the BMC analysis (red curve) is in excellent agreement with the SAXS data (black curve). Although the assembly state 1 is similar to the X-ray structure of Hck (3, 4), the ensemble of configurations includes dynamical fluctuations with rmsd on the order of 2–3 Å (see SI Appendix: Fig. S4). The scattering pattern calculated from the static X-ray structure alone does not fit the SAXS data accurately. In fact, the assembly state 1 (with dynamical fluctuations) also yields a poor score (Fig. 2A, right). The contribution from assembly states 6 and 8 is critical and improves the goodness of fit. In a previous SAXS study of c-Src, it was concluded that about 83% of the enzymes were in a conformation similar to the assembled form (25). In those experiments, Tyr527 in the C terminus of the catalytic domain was phosphorylated by the kinase Csk, which is required for kinase inactivation. It is particularly noteworthy that the assembled form is predominant, even though Tyr527 is unphosphorylated in the present experiments. The interaction of the phosphorylated tyrosine with the SH2 module is critical to stabilize the assembled form of the kinase (3, 4). It is well known that dephophorylation of Tyr527 leads to kinase activation and truncation of the C terminus leads to a constitutively active kinase. Yet, the present results indicate that the assembled conformation is also predominant in solution (84%), even when the tyrosine in the Ctail is unphosphorylated. Thus, despite the expectation that Hck with an unphosphorylated Tyr527 in the Ctail might adopt a multiplicity of disassembled states, the assembled conformation typical of the inactive kinase is predominant. The present observations challenge the notion that dephophorylation of the Ctail immediately triggers a large conformational change toward a disassembled state leading to kinase activation.
Comparison of the results obtained on the wild-type and mutant kinase sheds an interesting light on the competition between the binding of the Ctail and an external p2 peptide, corresponding intramolecular and bimolecular association processes, respectively. The experimental SAXS data from the high-affinity Ctail mutant Hck-YEEI is shown in Fig. 2B. Here as well, the assembled state 1 dominates the three-dimensional organization. The lack of increase in the population of assembly state 1 is somewhat surprising because the high-affinity Ctail mutant is known to down-regulate the kinase (6), although a Guinier analysis indicates that Rg shifts from 28.1 Å to 27.1 Å (Fig. 2 A and B). As shown in Fig. 3 A and B, the presence of the SH2-binding peptide p2 destabilizes the assembled down-regulated state, the effects being more important for the wild-type Hck than for the Hck-YEEI Ctail mutant. The population of assembly state 1 is reduced to 22% for the wild-type Hck, while it is reduced only to 62% for the Hck-YEEI mutant. This destabilization is also reflected in the Rg of 29.3 Å and 28.1 Å, for the wild-type and Hck-YEEI mutant, respectively. The shift toward disassembled configurations is consistent with the increase in kinase activity observed in the presence of SH2-binding ligands (7). Nevertheless, the nonnegligible population of assembly state 1 remaining in the presence of high-affinity SH2-binding peptide is somewhat surprising. The peptide comprises a phosphorylated tyrosine and is present at a nearly saturating concentration of 250 μM. In spite of this saturation, the intramolecular SH2-Ctail interactions for both the Hck-YEEI mutant and, to a lesser extent, wild-type Hck compete against the binding of a very high-affinity peptide and protect the assembly state 1. The observation can be rationalized in terms of a simple equilibrium model between an assembled state A and a disassembled state D, with the assumption that only the latter can bind the external ligand. The equilibrium for the three state process, A⇌D + L⇌DL, is governed by [D]/[A] = p, and [DL]/[D] = [L]/Kd, where p is the natural propensity of occurrence of the D state, and Kd is the dissociation constant, respectively. The probability of the assembled state is then equal to, PA = (1 + p + p[L]/Kd)-1. Because p is smaller for the high-affinity Ctail mutant Hck- YEEI than for the wild-type Hck, the intramolecular interaction is able to override the strong bimolecular association (pYEEI[L]/Kd ≪ pwt[L]/Kd), thus protecting the assembled down-regulated conformation.
The action of the SH3-binding peptide p3 is meant to mimic a signal such as produced by the Nef PxxP motif, which was previously used to induce kinase activation by releasing the SH3 module from the PPII linker (26). The analysis of the SAXS data for the Hck-YEEI mutant in the presence of p3 shown in Fig. 3C indicates that the binding of p3 increases the population of assembly states 2 and 3, corresponding to two possible SH3-displaced orientations (see also SI Appendix: Fig. S5 and Fig. S6). A number of in vitro and in vivo data including cell based assays and mutagenesis have also supported the concept of such an SH3-displaced model in kinase activation (5, 27). While the present results are clearly at the limit of resolution available from the analysis of SAXS data, they support the concept of SH3-displaced states.
The classic view of Src activation has typically invoked some loose disassembled conformation, in which all autoinhibitory intramolecular interactions are released. The analysis of the SAXS data for the wild-type Hck mutant in the presence of both the p2 and p3 peptides shown in Fig. 3D yields a representation of such a state of Hck in solution. Of particular interest, the assembled state 1 is wiped out while the most populated states, 5 and 6, differ from those detected under the previous conditions. This result demonstrates that the multidomain protein has access to a wide basin of different conformations and that different conditions can shift the population of the states in various directions.
Conclusion
An integrated framework combining SAXS experiments and computational modeling was introduced to quantitatively characterize the assembly of Hck kinase in solution. The BSS-SAXS analysis provides a view of the rich possibilities of three-dimensional conformational organizations accessible to Src in solution, going beyond the down-regulated assembled or fully disassembled states, which may be associated with kinase activation. The method offers a complementary and powerful approach to characterize multidomain molecular assemblies in solution, especially when multiple conformations can coexist. An important finding from the analysis is that multiple assembly states are required to construct a proper basis-set covering the conformational landscape of Hck. Available crystal structures are not enough to match the experimental SAXS data. Of particular interest, a large fraction of the kinase conformations exist in the assembled down-regulated state, even when the Tyr527 in the C terminus of the catalytic domain is not phosphorylated. Furthermore, different external signals lead to different distributions among the possible assembly states. These observations point to a molecular interpretation of the allosteric response of Hck to various biochemical signals that is in accord with the general concept of a shift in equilibrium populations (28, 29).
Methods
Experiments.
Wild-type HCK3D or the HCK3D.YEEI mutant were coexpressed in bacteria with YopH phosphatase and purified as described by Seeliger et al. (30). The SH2 binding peptide (referred to as p2; sequence EPQ[pY]EEIPIKQ, where pY is phosphotyrosine) and the SH3 binding peptide (referred to as p3; sequence VSLARRPLPPLP) were synthesized in house. SAXS data were collected at the BioCAT-18ID from the Advanced Photon Source. Data were collected using a flow-cell kept at 10 °C with a series of 1 s exposures for the Hck protein sample, the buffer, and the empty capillary. SAXS profiles were determined by the subtraction of the buffer and the empty capillary scattering from the protein sample scattering. See more details in SI Appendix.
Multidomain Modeling of Hck.
Extensive configurational sampling was carried out using a multistate CG model of Hck following methods similar to previous studies (16, 31, 32). Models of inactive and active states of the catalytic domain were built from the crystal structures of Hck (PDB entry: 1QCF) and c-Src (PDB entry: 1Y57), respectively (4, 10), and models of the SH3 and SH2 were taken from the inactive state of the down-regulated Hck (4). The models are built at the residue level, where each residue is represented by its Cα atom. For postdata analysis, a clustering and mapping method as described previously was applied to simulation data (17). The resulting configurations were clustered into 25 structural clusters, where a list of 89 interdomain pair wise residue distances were used in a K-means clustering calculation. For each cluster, a number of 100 randomly selected configurations was used to compute the theoretical scattering profiles using the Fast-SAXS method (18), yielding 25 profiles (and their standard deviations) log Ii(q) ± δ log Ii(q) where i = 1,⋯,25. A second level K-means clustering procedure was performed using a metric based on the similarity between calculated scattering patterns. This second step assigns the 25 structural clusters to Ns = 9 scattering classes or states (see SI Appendix: Fig. S1). See the SI Appendix for more details.
Supplementary Material
Acknowledgments.
We thank Markus Seeliger and John Kuriyan for providing the pHCK3D and YopH Duet plasmids and Richard Jones for synthesizing p2 and p3 peptides. SAXS experiments were carried out at the BioCAT of the Argonne National Laboratory with the help of Dr. Liang Guo. We thank Alper Dagcan for helping with SAXS experiments. This work was supported by the National Institutes of Health via Grant CA-093577 from the National Cancer Institute. The computations were supported by Teragrid under project MCA01S018 and by the San Diego Supercomputer Center.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1004569107/-/DCSupplemental.
References
- 1.Martin GS. The hunting of the Src. Nat Rev Mol Cell Biol. 2001;2:467–475. doi: 10.1038/35073094. [DOI] [PubMed] [Google Scholar]
- 2.Noble MEM, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 3.Sicheri F, Moarefi I, Kuriyan J. Crystal structure of the Src family tyrosine kinase Hck. Nature. 1997;385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- 4.Schindler T, et al. Crystal structure of Hck in complex with a Src family-selective tyrosine kinase inhibitor. Mol Cell. 1999;3:639–648. doi: 10.1016/s1097-2765(00)80357-3. [DOI] [PubMed] [Google Scholar]
- 5.Moarefi I, et al. Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature. 1997;385:650–653. doi: 10.1038/385650a0. [DOI] [PubMed] [Google Scholar]
- 6.Porter M, Schindler T, Kuriyan J, Miller WT. Reciprocal regulation of Hck activity by phosphorylation of tyr527 and tyr416. effect of introducing a high affinity intramolecular SH2 ligand. J Biol Chem. 2000;275:2721–2726. doi: 10.1074/jbc.275.4.2721. [DOI] [PubMed] [Google Scholar]
- 7.Goldberg GS, et al. Src phosphorylates cas on tyrosine 253 to promote migration of transformed cells. J Biol Chem. 2003;278:46533–46540. doi: 10.1074/jbc.M307526200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller W. Determinants of substrate recognition in nonreceptor tyrosine kinases. Acc Chem Res. 2003;36:393–400. doi: 10.1021/ar020116v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Young MA, Gonfloni S, Superti-Furga G, Roux B, Kuriyan J. Dynamic coupling between the SH2 and SH3 domains of c-Src and Hck underlies their inactivation by C-terminal tyrosine phosphorylation. Cell. 2001;105:115–126. doi: 10.1016/s0092-8674(01)00301-4. [DOI] [PubMed] [Google Scholar]
- 10.Cowan-Jacob SW, et al. The crystal structure of a c-Src complex in an active conformation suggests possible steps in c-Src activation. Structure. 2005;13:861–871. doi: 10.1016/j.str.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 11.Koch MHJ, Vachette P, Svergun DI. Small-angle scattering: a view on the properties, structures, and structural changes of biological macromolecules in solution. Q Rev Biophys. 2003;36:147–227. doi: 10.1017/s0033583503003871. [DOI] [PubMed] [Google Scholar]
- 12.Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations, and assemblies in solution. Q Rev Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 13.Lipfert J, Doniach S. Small-angle X-ray scattering from RNA, proteins, and protein complexes. Annu Rev Bioph Biom. 2007;36:307–327. doi: 10.1146/annurev.biophys.36.040306.132655. [DOI] [PubMed] [Google Scholar]
- 14.Forster F, et al. Integration of small-angle X-ray scattering data into structural modeling of proteins and their assemblies. J Mol Biol. 2008;382:1089–1106. doi: 10.1016/j.jmb.2008.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hura GL, et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang S, Roux B. Src kinase conformational activation: thermodynamics, pathways, and mechanisms. PLoS Comput Biol. 2008;4:e1000047. doi: 10.1371/journal.pcbi.1000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang S, Banavali NK, Roux B. Mapping the conformational transition in Src activation by cumulating the information from multiple molecular dynamics trajectories. Proc Natl Acad Sci USA. 2009;106:3776–3781. doi: 10.1073/pnas.0808261106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang S, Park S, Makowski L, Roux B. A rapid coarse residue-based computational method for X-ray solution scattering characterization of protein folds and multiple conformational states of large protein complexes. Biophys J. 2009;96:4449–4463. doi: 10.1016/j.bpj.2009.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bernadó P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. Structural characterization of flexible proteins using Small-Angle X-ray scattering. J Am Chem Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 20.Pelikan M, Hura GL, Hammel M. Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen Physiol Biophys. 2009;28:174–189. doi: 10.4149/gpb_2009_02_174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zarrinpar A, Bhattacharyya RP, Lim WA. The structure and function of proline recognition domains. Science's STKE. 2003;2003:1–10. doi: 10.1126/stke.2003.179.re8. [DOI] [PubMed] [Google Scholar]
- 22.Lerner EC, et al. Activation of the Src family kinase Hck without SH3-linker release. J Biol Chem. 2005;280:40832–40837. doi: 10.1074/jbc.M508782200. [DOI] [PubMed] [Google Scholar]
- 23.Sriraman S, Kevrekidis IG, Hummer G. Coarse master equation from bayesian analysis of replica molecular dynamics simulations. J Phys Chem B. 2005;109:6479–6484. doi: 10.1021/jp046448u. [DOI] [PubMed] [Google Scholar]
- 24.Faraldo-Gomez J, Roux B. On the importance of a funneled energy landscape for the assembly and regulation of multidomain Src tyrosine kinases. Proc Natl Acad Sci USA. 2007;104:13643–13648. doi: 10.1073/pnas.0704041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernadó P, Perez Y, Svergun DI, Pons M. Structural characterization of the active and inactive states of Src kinase in solution by small-angle X-ray scattering. J Mol Biol. 2008;376:492–505. doi: 10.1016/j.jmb.2007.11.066. [DOI] [PubMed] [Google Scholar]
- 26.Grzesiek S, et al. The solution structure of HIV-1 nef reveals an unexpected fold and permits delineation of the binding surface for the SH3 domain of Hck tyrosine protein kinase. Nat Struct Biol. 1996;3:340–345. doi: 10.1038/nsb0496-340. [DOI] [PubMed] [Google Scholar]
- 27.Lerner EC, Smithgall TE. SH3-dependent stimulation of Src-family kinase autophosphorylation without tail release from the SH2 domain in vivo. Nat Struct Biol. 2002;9:365–369. doi: 10.1038/nsb782. [DOI] [PubMed] [Google Scholar]
- 28.Vendruscolo M, Dobson CM. Structual biology: dynamic visions of enzymatic reactions. Science. 2006;313:1586–1587. doi: 10.1126/science.1132851. [DOI] [PubMed] [Google Scholar]
- 29.Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 30.Seeliger MA, et al. High yield bacterial expression of active c-Abl and c-Src tyrosine kinases. Protein Sci. 2005;14:3135–3139. doi: 10.1110/ps.051750905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang S, Onuchic JN, Levine H. Effective stochastic dynamics on a protein folding energy landscape. J Chem Phys. 2006;125:054910. doi: 10.1063/1.2229206. [DOI] [PubMed] [Google Scholar]
- 32.Best RB, Chen YG, Hummer G. Slow protein conformational dynamics from multiple experimental structures: the helix/sheet transition of arc repressor. Structure. 2005;13:1755–1763. doi: 10.1016/j.str.2005.08.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.