

Abstract
Background and purpose:
There are two important properties of receptor–ligand interactions: affinity (the ability of the ligand to bind to the receptor) and efficacy (the ability of the receptor–ligand complex to induce a response). Ligands are classified as agonists or antagonists depending on whether or not they have efficacy. In theory, it is possible to develop selective agonists based on selective affinity, selective intrinsic efficacy or both. This study examined the affinity and intrinsic efficacy of 31 β-adrenoceptor agonists at the three human β-adrenoceptors to determine whether the current agonists are subtype selective because of affinity or intrinsic efficacy.
Experimental approach:
Stable clonal CHO-K1 cell lines, transfected with either the human β1, β2 or β3-adrenoceptor, were used, and whole-cell [3H]-CGP 12177 radioligand binding and [3H]-cAMP accumulation were measured.
Key results:
Several agonists were found to be highly subtype selective because of selective affinity (e.g. salmeterol and formoterol, for the β2-adrenoceptor over the β1 or β3), while others (e.g. isoprenaline) had little affinity–selectivity. However, the intrinsic efficacy of salmeterol, formoterol and isoprenaline was similar across all three receptor subtypes. Other ligands (e.g. denopamine for β1; clenbuterol, AZ 40140d, salbutamol for β2) were found to have subtype-selective intrinsic efficacy. Several ligands appeared to activate two agonist conformations of the β1- and β3-adrenoceptors.
Conclusions and implications:
There are agonists with subtype selectivity based upon both selective affinity and selective intrinsic efficacy. Therefore, there is scope to develop better selective agonists based upon both selective affinity and selective intrinsic efficacy.
This article is commented on by Kenakin, pp. 1045–1047 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2010.00764.x
Keywords: β-adrenoceptor, β-blocker, β-agonist, β-antagonist, cAMP, drug selectivity, whole-cell binding, subtype selectivity, efficacy, intrinsic efficacy, GPCR
Introduction
There are two important properties of receptor–ligand interactions: affinity (the ability of the ligand to bind to the receptor) and efficacy (the ability of the receptor–ligand complex to induce a response; Strange, 2008). Ligands are classified as agonists or antagonists depending on whether or not they have efficacy (Clarke and Bond, 1998; Strange, 2008). Thus, a neutral antagonist binds to a given receptor but does not stimulate a response (i.e. it has affinity, but no efficacy). The affinity of antagonist ligands can be measured directly from radioligand binding or from antagonism of agonist responses in functional assays (Strange, 2008). Thus, the selectivity of antagonist drugs, either between receptors or between receptor subtypes, is relatively easily determined (e.g. Büscher et al., 1996; Smith and Teitler, 1999; Hoffmann et al., 2004; Baker, 2005a). Subtype-selective antagonist drugs have been sought for many clinical areas with the expected benefit that a high target receptor selectivity increases clinical effectiveness while reducing clinical side effects (Clarke and Bond, 1998).
In reality, many ligands considered as antagonists actually do stimulate responses, either by reducing the basal response (inverse agonist in systems with constitutive activity) or by stimulating a small response on their own (partial agonist). Truly neutral antagonists are few and far between, and a great many ‘antagonist’ drugs in clinical use are either inverse agonists or very weak partial agonists (e.g. Baker et al., 2003a,b; Kenakin, 2004 and examples in this study).
Measurements of agonist selectivity are more complex because agonists bind (and so have affinity), as well as being able to induce a response (efficacy; Kenakin, 1999a). Thus, the selectivity for agonist ligands between receptors or receptor subtypes depends upon these two variables, and it is theoretically possible to have subtype-selective agonists by different mechanisms. A ligand with higher affinity for a given receptor subtype would appear selective even if its efficacy was similar across different subtypes. Similarly, a ligand would appear as a subtype-selective agonist if its efficacy at a certain receptor subtype was significantly greater even if its affinity were the same across the other subtypes.
Agonist affinity can be measured in the same manner as that for antagonist ligands (e.g. binding studies); however, the response an agonist induces depends upon at least six factors: affinity, efficacy, number of receptors present, efficiency of the receptor–effector coupling, effector response measured and any desensitization that occurs within the time-frame of the measurement (Kenakin, 1999a; 2002;). Many of these factors alter between different cells, tissues and organisms, and between responses measured within the same tissue. Ligand affinity and ligand efficacy are, however, considered to depend upon the chemical interaction between the ligand and receptor, and are thus considered to be independent of the cells/tissues/organisms under study. Therefore, this chemical ability of a certain ligand to induce a response has been termed ‘intrinsic efficacy’, and is a measure of molecular efficacy for a given ligand at a given receptor, rather than at a tissue or organism level (Furchgott, 1966; Clarke and Bond, 1998).
Intrinsic efficacy is therefore an important property, and although direct measurements are difficult, relative intrinsic efficacies can be compared across different agonists if the other variables are controlled for. For example, using a stable cell line or single tissue preparation, and measuring a single response remove any tissue variables (including receptor expression level), and thus the differences in the response seen are due to those of ligand affinity and intrinsic efficacy. Thus, in a single system, partial agonists can be ranked in order of efficacy by the proportion of the maximal response they stimulate (Kenakin, 1999a; Strange, 2008). For full agonists, the intrinsic efficacy of ligands in the same system can be compared by means of an ‘efficacy ratio’ (KD/EC50; Furchgott, 1966; Kenakin, 1982; 1999b; Strange, 2008). For example, a ligand with an affinity (KD) of 100 nM, but an agonist response EC50 value of 1 nM has an intrinsic efficacy ratio of 100. This ligand would have a higher intrinsic efficacy than a ligand with a KD of 100 nM and an EC50 value of 10 nM (ratio of 10), provided both ligands were measured in the same system.
This study examines these two variables (affinity and intrinsic efficacy) for a range of ligands to determine whether the agonist ligands currently available are selective because of selective affinity, selective intrinsic efficacy or both. The three human β-adrenoceptors (Alexander et al., 2008) have been chosen because they are all Gs-coupled GPCRs, and therefore identical responses (cAMP) can be measured, and there is a large range of β-adrenoceptor agonist ligands available.
Methods
Materials
Fetal calf serum was obtained from PAA Laboratories (Teddington, Middlesex, UK). White-sided view plates were from Matrix Laboratories (supplied by Thermo Fisher Scientific, Basingstoke, UK), and Microscint 20 scintillation fluid was from PerkinElmer (Shelton, CT, USA). [3H]-CGP 12177 was from Amersham International (Buckinghamshire, UK). BRL 37344, cimaterol, formoterol, isoprenaline, L 755507, procaterol, SDZ 21009, SR 59230A, ZD 2079 and ZD 7114 were from Tocris Life Sciences (Avonmouth, UK). Carazolol was a gift from Dr Chris Tate (LMB, Cambridge, UK); nebivolol and butoxamine were a gift from Stefano Evangelista (Menarini Ricerche Spa, Florence, Italy); zinterol was a gift from Dr Torsten Christ (Department of Pharmacology and Toxicology, Dresden University of Technology, Dresden, Germany.); L 748337, TAK677 and (R)-N-[5-[2-[2-(9H-carbazole-2-iloxy)ethylamino]-1-hydroxyethyl]-2-hydroxyphenyl]methanesulphonamide were gifts from Dr Kerry af Forselles, Pfizer, Sandwich, UK. (R)-N-[5-[2-[2-(9H-carbazole-2-iloxy)ethylamino]-1-hydroxyethyl]-2-hydroxyphenyl]methanesulphonamide is compound d from US2003/0018061A1, which is believed to be AZ40140 (SB418790) and shall thus be referred to as AZ40140d throughout this paper. Christophe Fromont (Centre for Biomolecular Sciences, University of Nottingham) kindly performed mass spectrometry, which confirmed that it was (R)-N-[5-[2-[2-(9H-carbazole-2-iloxy)ethylamino]-1-hydroxyethyl]-2-hydroxyphenyl]methanesulphonamide). All other reagents were from Sigma Chemicals (Poole, Dorset, UK). The source, catalogue numbers, CAS RN number and isomeric status of the agonists used are given in Supporting Information Table S1.
Cell culture
CHO-K1 stably expressing either the human β1 (1146 fmol·mg−1 protein), the human β2 (466 fmol·mg−1 protein) or the human β3-adrenoceptor (790 fmol·mg−1 protein) were used throughout this study (Baker, 2005a). Parent CHO cells were also used (i.e. CHO cells without the transfected receptor). Cells were grown in Dulbecco's modified Eagle's medium nutrient mix F12 (DMEM/F12) containing 10% fetal calf serum and 2 mM l-glutamine in a 37°C humidified 5% CO2 : 95% air atmosphere.
[3H]-CGP 12177 whole-cell binding
Cells were grown to confluence in white-sided, clear-bottomed 96-well view plates. [3H]-CGP 12177 whole-cell binding was carried out the following day in a manner identical to that described in Baker (2005a). Briefly, the medium was removed from each well, and 100 µL of serum-free medium containing the competing ligand at twice the final required concentration was added to each well followed immediately by a fixed concentration of [3H]-CGP 12177. The cells were incubated for 2 h at 37°C, 5% CO2. The cells were washed twice by the addition and removal of 200 µL 4°C phosphate-buffered saline; 100 µL Microscint 20 was added to each well, a white sticky base applied to the bottom of the plate and a sealant top applied to the top of the plate. The plates were left at room temperature overnight in the dark, and the plates were counted on a Topcount at 21°C for 2 min per well.
[3H]-cAMP accumulation
Cells were grown to confluence in 24-well plates. The medium was removed and the cells were pre-labelled with [3H]-adenine by incubation for 2 h with 2 µCi·mL−1[3H]-adenine in serum-free medium (0.5 mL per well). The [3H]-adenine was removed, and each well was washed by the addition and removal of 1 mL serum-free medium. Then, 1 mL serum-free medium containing 1 mM IBMX was added to each well, and the cells were incubated for 15 min. Agonist (in 10 µL serum-free medium) was added to each well, and the plates were incubated for 10 min–5 h. The reaction was terminated by the addition of 50 µL concentrated HCl per well. The plates were then frozen, thawed and [3H]-cAMP separated from other 3H-nucleotides by sequential Dowex and alumina column chromatography, as previously described (Donaldson et al., 1988).
Data analysis
Whole-cell binding
All data points on each binding curve were performed in triplicate, and each 96-well plate also contained three to six determinations of total and non-specific binding. Non-specific binding was determined in the presence of 1 µM CGP 20712A or 1–10 µM propranolol for the β1-adrenoceptor, 1 µM ICI 118551 or 1–10 µM propranolol for the β2-adrenoceptor and 10 µM propranolol for the β3-adrenoceptor. In all cases where a KD value is stated, the competing ligand completely inhibited the specific binding of [3H]-CGP 12177.
One-site competition binding
A one-site sigmoidal binding curve (Equation 1) was then fitted to the data using Graphpad Prism 2.01, and the IC50 was then determined as the concentration required to inhibit 50% of the specific binding.
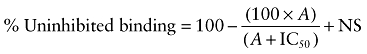 |
(1) |
where A is the concentration of the competing ligand, IC50 is the concentration at which half of the specific binding of [3H]-CGP 12177 has been inhibited and NS is the non-specific binding.
From the IC50 value and the known concentration of radioligand ([3H]-CGP 12177 conc), a KD (concentration at which half the receptors are bound by the competing ligand) value was calculated using the equation:
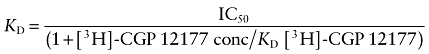 |
(2) |
Two-site competition binding
Several of the responses were best fitted to a two-site competition curve (e.g. Figure 2C). In these cases, Equation 3 was used:
Figure 2.
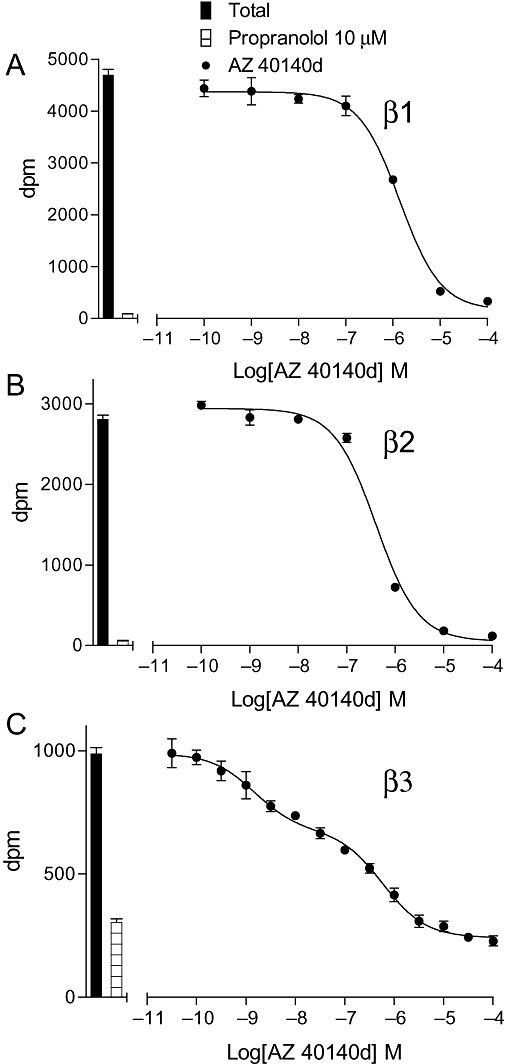
Inhibition of [3H]-CGP 12177 binding to whole cells by AZ 40140d in (A) CHO β1 cells, (B) CHO β2 cells and (C) CHO β3 cells. The columns represent total [3H]-CGP 12177 binding and non-specific binding determined in the presence of 10 µM propranolol. The concentrations of [3H]-CGP 12177 present in each case are (A) 1.41 nM, (B) 1.29 nM and (C) 19.31 nM. Data points are mean ± SEM of triplicate determinations. These single experiments are representative of (A) 8, (B) 10 and (C) 7 separate experiments.
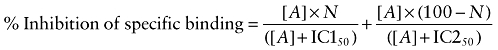 |
(3) |
where N is the percentage of site 1, [A] is the concentration of agonist and IC150 and IC250 are the respective IC50 values for the two agonist sites.
[3H]-cAMP accumulation
Most agonist responses were best described by a one-site sigmoidal concentration–response curve (Equation 4):
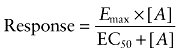 |
(4) |
where Emax is the maximum response, [A] is the agonist concentration and EC50 is the concentration of agonist that produces 50% of the maximal response.
Several of the responses were, however, best fitted to a two-site concentration response – Equation 5 (e.g. Figure 5A):
Figure 5.
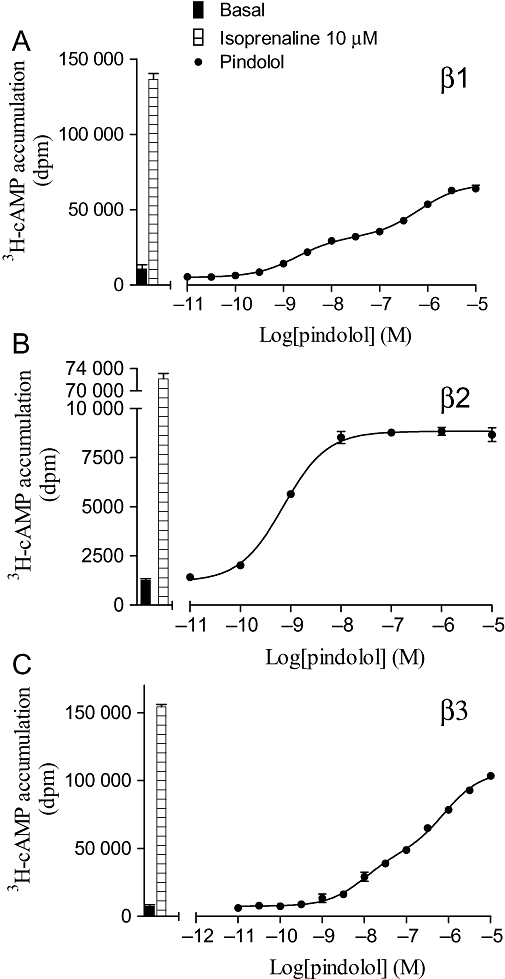
[3H]-cAMP accumulation in response to pindolol after 5 h incubation in (A) CHO β1 cells, (B) CHO β2 cells and (C) CHO β3 cells. The columns represent basal [3H]-cAMP accumulation and that in response to 10 µM isoprenaline. Data points are mean ± SEM of triplicate determinations. These single experiments are representative of (A) six, (B) nine and (C) six separate experiments.
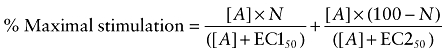 |
(5) |
where N is the percentage of site 1, [A] is the concentration of agonist and EC150 and EC250 are the respective EC50 values for the two agonist sites.
Isoprenaline (10 µM) was included in all experiments, and therefore all maximal responses are expressed as a percentage of this maximum.
Efficacy ratios
For comparative purposes, efficacy ratios were calculated by dividing the KD value by the EC50 value for each ligand as per method of Furchgott (1966).
Results
[3H]-CGP 12177 whole-cell binding
The KD values for [3H]-CGP 12177 have previously been determined in these cell lines as 0.42 nM for the β1-adrenoceptor, 0.17 nM for the β2-adrenoceptor and 109.2 nM for the β3-adrenoceptor (Baker, 2005a).
3H-CGP 12177 at concentrations from 0.34 to 4.65 nM was used for experiments involving the β1- and β2-adrenoceptors, and from 5.18 to 32.53 nM for the β3-adrenoceptor.
The affinity of ligands for the human β-adrenoceptors is presented in Table 1, arranged in order of selectivity for the β1- over the β2-adrenoceptor (Tables 1 and 2). Thus, several compounds originally considered as antagonists were found to have high affinity for the receptors, while as expected most ligands generally considered as agonists had relatively low affinity. Table 2 shows the selectivity of these ligands for the three adrenoceptors based on binding alone. Nebivolol was found to be the most selective ligand for the human β1-adrenoceptor in this current set of ligands (Table 2); however, the neutral antagonist CGP 20712A has previously been shown to have 501 times higher affinity for the β1- than for the β2-adrenoceptor in this system (Baker, 2005a). Salmeterol and formoterol were the most selective β2-compounds (Figure 1). There were several β3-selective compounds (e.g. AZ 40140d, L 755507, L 748337 and TAK 677); however, careful examination of their [3H]-CGP 12177 inhibitory responses yielded an inhibition that was best described by a two-component curve (Figure 2).
Table 1.
Log KD values obtained from [3H]-CGP 12177 whole-cell binding studies in CHO cells stably expressing either the human β1-, β2- or β3-adrenoceptors
| β1 | n | β2 | n | β3 | n | |
|---|---|---|---|---|---|---|
| Nebivolol | −9.06 ± 0.03 | 8 | −7.92 ± 0.04 | 8 | −7.04 ± 0.14 | 17 |
| Noradrenaline | −5.74 ± 0.03 | 5 | −5.41 ± 0.07 | 5 | −5.53 ± 0.10 | 8 |
| Denopamine | −6.12 ± 0.03 | 7 | −5.83 ± 0.04 | 7 | −5.30 ± 0.03 | 6 |
| ZD 7114 | −7.58 ± 0.09 | 8 | −7.31 ± 0.03 | 9 | −6.78 ± 0.07 | 12 |
| ZD 2079 | −4.21 ± 0.05 | 5 | >−4 | 9 | −4.59 ± 0.07 | 8 |
| TAK 677 | −7.44 ± 0.03 | 9 | −7.32 ± 0.05 | 9 | Site 1 −8.59 ± 0.07 | 10 |
| Site 2 −5.65 ± 0.14 | ||||||
| 57.4 ± 3.2% site 1 | ||||||
| Ractopamine | −6.97 ± 0.02 | 7 | −6.93 ± 0.02 | 7 | −5.82 ± 0.07 | 7 |
| Octopamine | −3.91 ± 0.03 | 6 | −4.03 ± 0.02 | 9 | IC50≥−3 | 6 |
| Dopamine | −3.57 ± 0.02 | 6 | −3.93 ± 0.11 | 6 | −3.10 ± 0.03 | 9 |
| Methoxyphenamine | −3.94 ± 0.02 | 11 | −4.32 ± 0.04 | 7 | −3.96 ± 0.03 | 7 |
| AZ 40140d | −6.69 ± 0.05 | 8 | −7.14 ± 0.09 | 10 | Site 1 −8.94 ± 0.23 | 7 |
| Site 2 −6.11 ± 0.23 | ||||||
| 47.4 ± 3.7% site 1 | ||||||
| Isoprenaline | −6.06 ± 0.08 | 9 | −6.64 ± 0.09 | 11 | −5.52 ± 0.08 | 9 |
| Metaproterenol | −4.71 ± 0.02 | 6 | −5.30 ± 0.02 | 7 | −4.06 ± 0.07 | 6 |
| L 755507 | −6.23 ± 0.03 | 20 | −6.83 ± 0.04 | 21 | Site 1 −8.60 ± 0.12 | 19 |
| Site 2 −6.20 ± 0.07 | ||||||
| 57.7 ± 2.4% site 1 | ||||||
| Bamethane | −4.30 ± 0.03 | 7 | −4.83 ± 0.03 | 7 | −4.42 ± 0.09 | 9 |
| Dobutamine | −5.23 ± 0.06 | 15 | −5.84 ± 0.05 | 14 | −5.09 ± 0.08 | 8 |
| Pindolol | −8.57 ± 0.03 | 8 | −9.23 ± 0.03 | 8 | −7.08 ± 0.08 | 7 |
| Bucindolol | −9.31 ± 0.03 | 6 | −9.99 ± 0.07 | 6 | −8.04 ± 0.08 | 5 |
| Cimaterol | −6.57 ± 0.06 | 11 | −7.26 ± 0.09 | 9 | −5.28 ± 0.06 | 6 |
| S-cyanopindolol | −10.39 ± 0.04 | 7 | −11.09 ± 0.08 | 7 | −8.36 ± 0.10 | 7 |
| Carazolol | −9.69 ± 0.06 | 7 | −10.49 ± 0.07 | 7 | −8.35 ± 0.10 | 7 |
| SR 59230A | −7.54 ± 0.05 | 9 | −8.45 ± 0.04 | 8 | −7.37 ± 0.09 | 8 |
| BAAM | −7.65 ± 0.07 | 7 | −8.57 ± 0.09 | 9 | −6.40 ± 0.09 | 7 |
| Adrenaline | −5.15 ± 0.06 | 9 | −6.13 ± 0.05 | 9 | −4.70 ± 0.08 | 8 |
| Oxprenolol | −7.96 ± 0.03 | 8 | −8.97 ± 0.04 | 8 | −6.25 ± 0.11 | 9 |
| L 748337 | −5.44 ± 0.06 | 16 | −6.47 ± 0.04 | 16 | Site 1 −8.04 ± 0.11 | 16 |
| Site 2 −5.46 ± 0.17 | ||||||
| 54.2 ± 3.7% site 1 | ||||||
| Isoxsuprine | −4.87 ± 0.06 | 7 | −5.93 ± 0.09 | 7 | −5.29 ± 0.08 | 7 |
| Tulobuterol | −5.62 ± 0.04 | 7 | −6.83 ± 0.09 | 7 | −4.72 ± 0.08 | 9 |
| Clenbuterol | −6.62 ± 0.03 | 5 | −7.90 ± 0.05 | 5 | −5.35 ± 0.07 | 7 |
| BRL 35135A | −6.08 ± 0.03 | 7 | −7.38 ± 0.04 | 7 | −6.73 ± 0.09 | 7 |
| BRL 37344 | −5.19 ± 0.07 | 7 | −6.51 ± 0.06 | 5 | −6.45 ± 0.06 | 7 |
| Ritodrine | −4.48 ± 0.02 | 7 | −5.81 ± 0.07 | 7 | −4.73 ± 0.11 | 7 |
| Salbutamol | −4.68 ± 0.03 | 12 | −6.01 ± 0.03 | 10 | −3.98 ± 0.06 | 9 |
| SDZ 21009 | −8.94 ± 0.05 | 11 | −10.28 ± 0.08 | 11 | −7.10 ± 0.10 | 9 |
| Butoxamine | −4.85 ± 0.02 | 10 | −6.23 ± 0.07 | 6 | >−4 | 6 |
| Terbutaline | −3.90 ± 0.03 | 12 | −5.51 ± 0.04 | 12 | −3.68 ± 0.06 | 12 |
| Fenoterol | −5.04 ± 0.03 | 5 | −7.03 ± 0.06 | 5 | −5.39 ± 0.07 | 8 |
| Zinterol | −5.96 ± 0.04 | 9 | −8.04 ± 0.10 | 9 | −6.27 ± 0.10 | 8 |
| Procaterol | −4.81 ± 0.02 | 8 | −7.11 ± 0.04 | 8 | −3.99 ± 0.11 | 7 |
| Formoterol | −6.11 ± 0.05 | 8 | −8.63 ± 0.02 | 8 | −5.82 ± 0.05 | 8 |
| Salmeterol | −5.73 ± 0.03 | 11 | −9.26 ± 0.06 | 10 | −6.33 ± 0.10 | 8 |
Values represent mean ± SEM of n separate experiments. The binding of four ligands to the human β3-adrenoceptor was best described by a two-component curve. In these instances, the log KD values are given for each component along with the percentage of the total inhibition that was represented by the first component.
Ligands are arranged in order of β1 versus β2 selectivity.
Table 2.
Selectivity ratios of the ligands for human β1-, β2- and β3-adrenoceptors, where a ratio of 1 demonstrates no selectivity (based on binding) for a given receptor subtype over another
|
Selectivity ratios (based on whole-cell binding) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| β1 | versus | β2 | β2 | versus | β3 | β1 | versus | β3 | ||
| Nebivolol | 13.8 | 7.6 | 104.7 | |||||||
| Noradrenaline | 2.1 | 1.3 | 1.6 | |||||||
| Denopamine | 1.9 | 3.4 | 6.6 | |||||||
| ZD 7114 | 1.9 | 3.4 | 6.3 | |||||||
| ZD 2079 | >1.6 | >3.9 | 2.4 | |||||||
| TAK 677 | Component 1 | 1.3 | 18.6 | 14.1 | ||||||
| Ractopamine | 1.1 | 12.9 | 14.1 | |||||||
| Octopamine | 1.3 | |||||||||
| Dopamine | 2.3 | 6.8 | 2.9 | |||||||
| Methoxyphenamine | 2.4 | 2.3 | 1.0 | |||||||
| AZ 40140d | Component 1 | 2.8 | 63.1 | 177.8 | ||||||
| Isoprenaline | 3.8 | 13.2 | 3.5 | |||||||
| Metaproterenol | 3.9 | 17.4 | 4.57 | |||||||
| L 755507 | Component 1 | 4.0 | 58.9 | 234.4 | ||||||
| Bamethane | 4.0 | 2.6 | 1.3 | |||||||
| Dobutamine | 4.1 | 5.6 | 1.4 | |||||||
| Pindolol | 4.6 | 141.3 | 30.9 | |||||||
| Bucindolol | 4.8 | 89.1 | 18.6 | |||||||
| Cimaterol | 4.9 | 95.5 | 19.5 | |||||||
| Cyanopindolol | 5.0 | 537.0 | 107.2 | |||||||
| Carazolol | 6.3 | 138.0 | 21.9 | |||||||
| SR 59230A | 8.1 | 12.0 | 1.5 | |||||||
| BAAM | 8.3 | 147.9 | 17.8 | |||||||
| Adrenaline | 9.6 | 26.9 | 2.8 | |||||||
| Oxprenolol | 10.2 | 524.8 | 51.3 | |||||||
| L 748337 | Component 1 | 10.7 | 37.2 | 398.1 | ||||||
| Isoxsuprine | 11.5 | 4.4 | 2.6 | |||||||
| Tulobuterol | 16.2 | 128.8 | 7.9 | |||||||
| Clenbuterol | 19.5 | 354.8 | 18.6 | |||||||
| BRL 35135A | 19.9 | 4.5 | 4.5 | |||||||
| BRL 37344 | 20.9 | 1.2 | 18.2 | |||||||
| Ritodrine | 21.4 | 12.0 | 1.8 | |||||||
| Salbutamol | 21.4 | 107.2 | 5.0 | |||||||
| SDZ 21009 | 21.9 | 1513.6 | 69.2 | |||||||
| Butoxamine | 24.0 | >169.8 | >7.1 | |||||||
| Terbutaline | 40.7 | 67.6 | 1.7 | |||||||
| Fenoterol | 97.7 | 43.7 | 2.2 | |||||||
| Zinterol | 120.2 | 58.9 | 2.0 | |||||||
| Procaterol | 199.5 | 1318.36 | 6.6 | |||||||
| Formoterol | 331.1 | 645.7 | 2.0 | |||||||
| Salmeterol | 3388.4 | 851.1 | 4.0 | |||||||
Where the inhibition of [3H]-CGP 12177 binding curve was best described by two components (Table 1), the selectivity ratio for the first component only is given.
Figure 1.
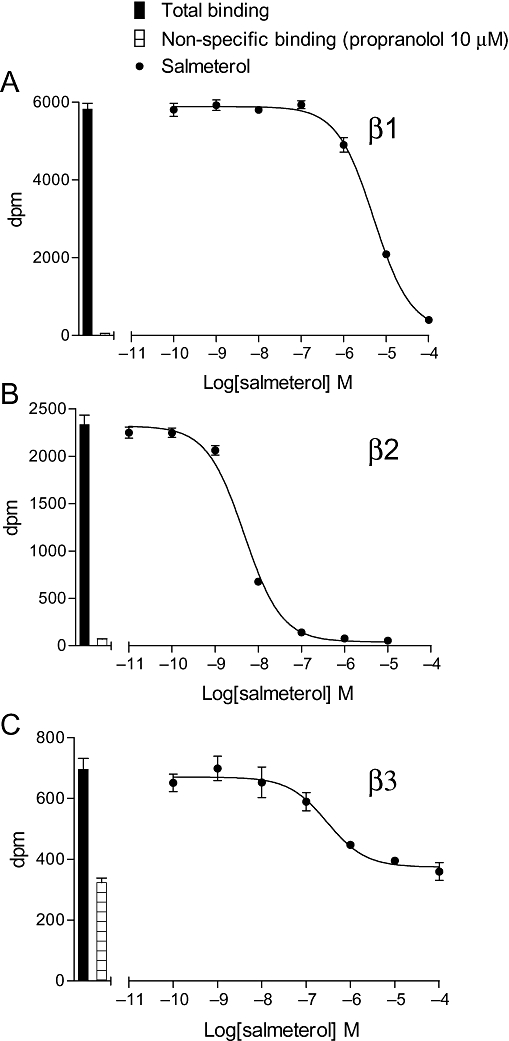
Inhibition of [3H]-CGP 12177 binding to whole cells by salmeterol in (A) CHO β1 cells, (B) CHO β2 cells and (C) CHO β3 cells. The columns represent total [3H]-CGP 12177 binding and non-specific binding determined in the presence of 10 µM propranolol. The concentrations of [3H]-CGP 12177 present in each case are (A) 0.90 nM, (B) 1.39 nM and (C) 17.9 nM. Data points are mean ± SEM of triplicate determinations. These single experiments are representative of (A) 11, (B) 10 and (C) 8 separate experiments.
[3H]-cAMP accumulation
To investigate whether the length of agonist incubation time had any effect on the log EC50 value or the percentage maximum response obtained, several ligands (full and partial) were incubated for 10 and 30 min, and 5 h before HCl was added to terminate the assay (Supporting Information Tables S2–S4). Isoprenaline stimulated [3H]-cAMP accumulation responses that were 13-, 27- and 12-fold over basal at 10 min; 45-, 69- and 28-fold at 30 min; and 92-, 154- and 34-fold at 5 h for the β1-, β2- and β3-adrenoceptors respectively. The log EC50 values and percent maximum responses remained constant over time for the great majority of ligands tested including full and partial agonists (Supporting Information Tables S2–S4; Figure 3) as found previously (Baker et al., 2004). However, for some ligands, the log EC50 values differed between 10 and 30 min (Figure 4). For some ligands [e.g. salmeterol, formoterol and CGP 12177 (β2)], this is most probably because of the slow onset of these ligands at this receptor (Lötvall, 2001; Baker et al., 2002), and indeed the degree of change reflects the known relative order of onset of action (Cote et al., 2009). The reason for the increase in potency for all three catecholamines between 10 and 30 min is not known; however, given this change for both the long acting ligands and the catecholamines, 30 min was chosen as the incubation time for investigation of majority of the other ligands (Tables 3–5).
Figure 3.
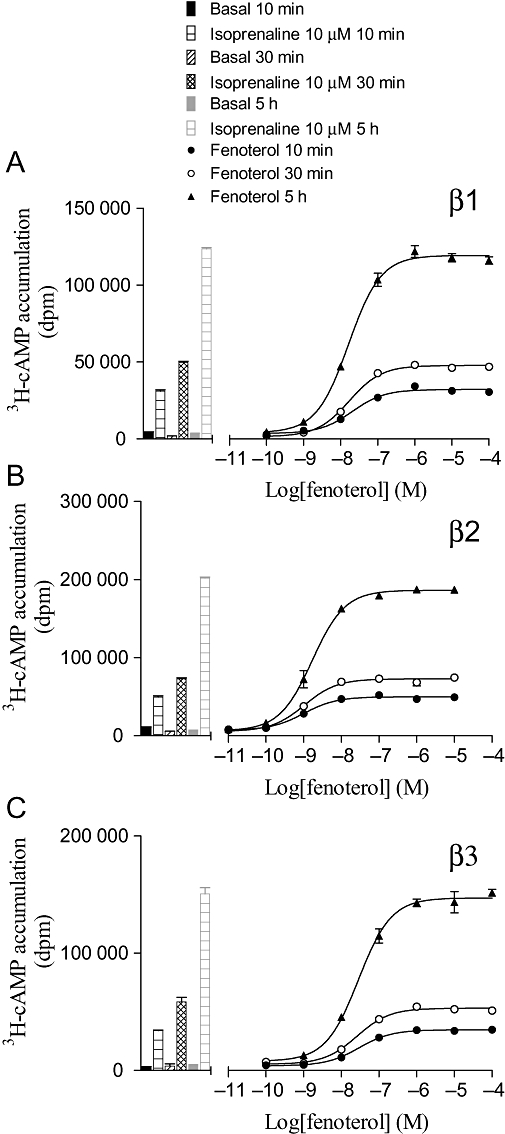
[3H]-cAMP accumulation in response to fenoterol after 10 and 30 min, and 5 h incubation in (A) CHO β1 cells, (B) CHO β2 cells and (C) CHO β3 cells. The columns represent basal [3H]-cAMP accumulation and that in response to 10 µM isoprenaline at each time-point. Data points are mean ± SEM of triplicate determinations. These single experiments are representative of (A) four, (B) four and (C) three separate experiments.
Figure 4.
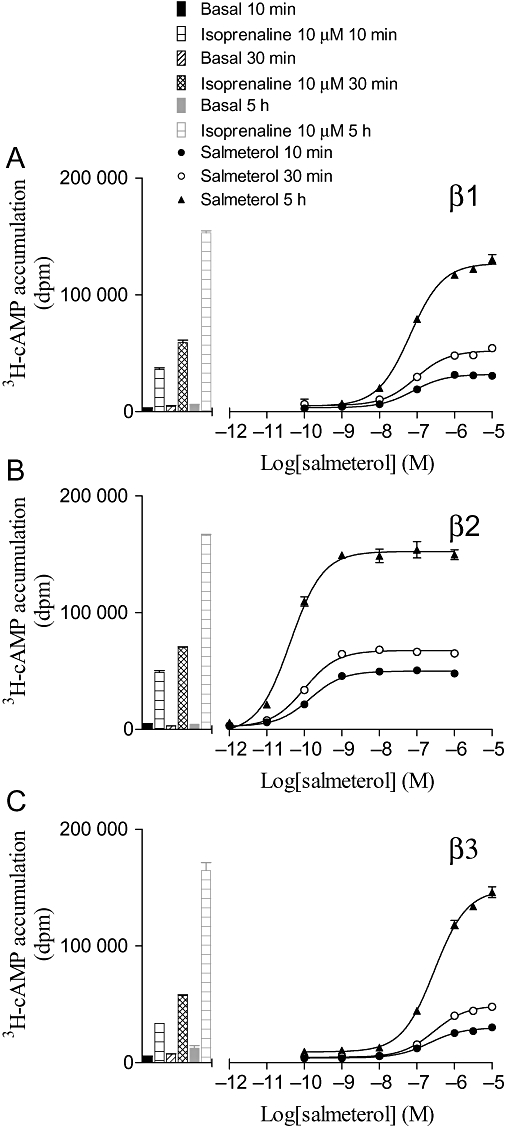
[3H]-cAMP accumulation in response to salmeterol after 10 and 30 min, and 5 h incubation in (A) CHO β1 cells, (B) CHO β2 cells and (C) CHO β3 cells. The columns represent basal [3H]-cAMP accumulation and that in response to 10 µM isoprenaline at each time-point. Data points are mean ± SEM of triplicate determinations. These single experiments are representative of (A) four, (B) four and (C) three separate experiments.
Table 3.
Log KD values from [3H]-CGP 12177 whole-cell binding (from Table 1), log EC50 values and % isoprenaline maximal responses obtained from [3H]-cAMP accumulation and intrinsic efficacy ratios (KD/EC50) for cells expressing the human β1-adrenoceptor
| β1 | Log KD from binding (β1) | Log EC50 from cAMP (β1) | % Isoprenaline maximal response | n | Efficacy ratio (log) |
|---|---|---|---|---|---|
| Isoprenaline | −6.06 | −8.59 ± 0.10 | 100 | 11 | 2.53 |
| Metaproterenol | −4.71 | −7.21 ± 0.07 | 113.0 ± 4.9 | 4 | 2.50 |
| Fenoterol | −5.04 | −7.53 ± 0.07 | 106.0 ± 1.7 | 4 | 2.49 |
| Adrenaline | −5.15 | −7.61 ± 0.10 | 101.7 ± 2.1 | 8 | 2.46 |
| Noradrenaline | −5.74 | −7.94 ± 0.13 | 102.4 ± 3.6 | 10 | 2.20 |
| Formoterol | −6.11 | −8.29 ± 0.08 | 111.4 ± 3.3 | 4 | 2.18 |
| Terbutaline | −3.90 | −5.76 ± 0.09 | 110.0 ± 2.2 | 4 | 1.86 |
| Cimaterol | −6.57 | −8.42 ± 0.08 | 109.7 ± 3.6 | 4 | 1.85 |
| Octopamine | −3.91 | −5.73 ± 0.03 | 107.2 ± 2.3 | 4 | 1.82 |
| Ractopamine | −6.97 | −8.74 ± 0.09 | 103.2 ± 1.5 | 5 | 1.77 |
| Ritodrine | −4.48 | −6.20 ± 0.06 | 103.9 ± 2.0 | 5 | 1.72 |
| BRL 35135A | −6.08 | −7.79 ± 0.10 | 104.0 ± 5.0 | 6 | 1.71 |
| Isoxsuprine | −4.87 | −6.54 ± 0.10 | 99.4 ± 4.5 | 5 | 1.67 |
| Denopamine | −6.12 | −7.73 ± 0.06 | 121.2 ± 4.1 | 5 | 1.61 |
| Dobutamine | −5.23 | −6.81 ± 0.08 | 97.4 ± 3.6 | 4 | 1.58 |
| Salbutamol | −4.68 | −6.21 ± 0.05 | 103.7 ± 3.8 | 4 | 1.53 |
| Dopamine | −3.57 | −5.02 ± 0.03 | 112.5 ± 4.9 | 5 | 1.45 |
| TAK 677 | −7.44 | −8.86 ± 0.08 | 108.4 ± 1.6 | 8 | 1.42 |
| L 755507 | −6.23 | −7.63 ± 0.05 | 101.6 ± 3.0 | 5 | 1.40 |
| BRL 37344 | −5.19 | −6.54 ± 0.02 | 100.7 ± 8.6 | 4 | 1.35 |
| Bamethane | −4.30 | −5.63 ± 0.04 | 108.5 ± 5.3 | 4 | 1.33 |
| Salmeterol | −5.73 | −7.03 ± 0.09 | 98.1 ± 1.7 | 5 | 1.30 |
| Zinterol | −5.96 | −7.23 ± 0.10 | 113.5 ± 3.8 | 5 | 1.27 |
| Procaterol | −4.81 | −5.80 ± 0.09 | 106.7 ± 4.6 | 5 | 0.99 |
| Tulobuterol | −5.62 | −6.59 ± 0.09 | 97.8 ± 2.7 | 5 | 0.97 |
| L 748337 | −5.44 | −6.23 ± 0.03 | 43.4 ± 1.7 | 5 | 0.79 |
| Butoxamine | −4.85 | −5.56 ± 0.12 | 11.3 ± 0.7 | 10 | 0.71 |
| Clenbuterol | −6.62 | −7.29 ± 0.04 | 99.9 ± 5.1 | 4 | 0.67 |
| Methoxyphenamine | −3.94 | −4.59 ± 0.04 | 9.6 ± 0.8 | 7 | 0.65 |
| ZD 2079 | −4.21 | −4.79 ± 0.03 | 81.7 ± 4.9 | 4 | 0.58 |
| AZ 40140d | −6.69 | −7.23 ± 0.08 | 98.3 ± 5.6 | 6 | 0.54 |
| Oxprenolol | −7.96 | Site 1 −8.46 ± 0.03 | 37.1 ± 3.4 | 6 | 0.50 |
| Site 2 −6.03 ± 0.12 | |||||
| Site 1 65.4 ± 0.8% | |||||
| ZD 7114 | −7.58 | −8.05 ± 0.08 | 72.0 ± 2.9 | 4 | 0.47 |
| SR 59230A | −7.54 | −7.96 ± 0.06 | 52.8 ± 1.9 | 4 | 0.42 |
| Pindolol | −8.57 | Site 1 −8.84 ± 0.08 | 51.2 ± 4.1 | 6 | 0.27 |
| Site 2 −6.12 ± 0.11 | |||||
| Site 1 38.4 ± 1.4% | |||||
| Nebivolol | −9.06 | −8.97 ± 0.09 | 2.8 ± 0.3 | 5 | −0.09 |
| BAAM | −7.65 | −7.51 ± 0.07 | 34.4 ± 3.1 | 5 | −0.14 |
| Bucindolol | −9.31 | Site 1 −9.11 ± 0.07 | 73.9 ± 1.3 | 6 | −0.20 |
| Site 2 −7.46 ± 0.07 | |||||
| Site 1 65.3 ± 4.9% | |||||
| SDZ 21009 | −8.94 | Site 1 −8.50 ± 0.11 | 67.8 ± 2.5 | 12 | −0.44 |
| Site 2 −6.79 ± 0.10 | |||||
| Site 1 48.9 ± 3.8% | |||||
| Carazolol | −9.69 | Site 1 −9.15 ± 0.09 | 38.1 ± 4.9 | 8 | −0.54 |
| Site 2 −7.39 ± 0.06 | |||||
| Site 1 21.9 ± 2.4% | |||||
| Cyanopindolol | −10.39 | Site 1 −9.63 ± 0.11 | 62.7 ± 6.3 | 7 | −0.76 |
| Site 2 −8.43 ± 0.13 | |||||
| Site 1 42.4 ± 7.5% |
Values represent mean ± SEM of n separate experiments. For several ligands, the concentration–response curve was best described by a two-component curve. In these cases, the log EC50 values are given for each component, the percentage of the response represented by the first component and the percentage of the total response in relation to the isoprenaline stimulation.
The ligands are arranged in order of intrinsic efficacy.
Table 5.
Log KD values from [3H]-CGP 12177 whole-cell binding (from Table 1), log EC50 values and % isoprenaline maximal responses obtained from [3H]-cAMP accumulation and intrinsic efficacy ratios (KD/EC50) for cells expressing the human β3-adrenoceptor
| β3 | Log KD from binding (β3) | Log EC50 from cAMP (β3) | % Isoprenaline maximal response | n | Efficacy ratio (log) |
|---|---|---|---|---|---|
| Fenoterol | −5.39 | −7.63 ± 0.07 | 100.5 ± 4.0 | 4 | 2.24 |
| Terbutaline | −3.68 | −5.85 ± 0.08 | 101.4 ± 2.7 | 4 | 2.17 |
| Salbutamol | −3.98 | −6.01 ± 0.06 | 97.2 ± 3.4 | 4 | 2.03 |
| Metaproterenol | −4.06 | −6.08 ± 0.07 | 95.8 ± 6.2 | 6 | 2.02 |
| Formoterol | −5.82 | −7.82 ± 0.05 | 99.0 ± 3.2 | 4 | 2.00 |
| Isoprenaline | −5.52 | −7.41 ± 0.08 | 100 | 10 | 1.89 |
| Zinterol | −6.27 | −8.09 ± 0.10 | 101.2 ± 3.4 | 5 | 1.82 |
| Ritodrine | −4.73 | −6.54 ± 0.08 | 99.1 ± 3.1 | 6 | 1.81 |
| Adrenaline | −4.70 | −6.48 ± 0.07 | 102.1 ± 2.0 | 8 | 1.78 |
| Cimaterol | −5.28 | −6.96 ± 0.06 | 98.2 ± 3.4 | 4 | 1.68 |
| Dopamine | −3.10 | −4.76 ± 0.07 | 90.5 ± 2.5 | 6 | 1.66 |
| Noradrenaline | −5.53 | −7.17 ± 0.09 | 104.1 ± 1.8 | 6 | 1.64 |
| L 755507 | Site 1 −8.60 | −10.10 ± 0.05 | 101.1 ± 3.4 | 8 | 1.50 |
| Site 2 −6.20 | |||||
| 57.7% site 1 | |||||
| ZD 2079 | −4.59 | −6.00 ± 0.04 | 97.9 ± 1.4 | 4 | 1.41 |
| Dobutamine | −5.09 | −6.37 ± 0.06 | 87.3 ± 3.7 | 4 | 1.28 |
| Procaterol | −3.99 | −5.22 ± 0.04 | 91.6 ± 5.3 | 4 | 1.23 |
| Bamethane | −4.42 | −5.61 ± 0.08 | 98.3 ± 5.0 | 5 | 1.19 |
| Ractopamine | −5.82 | −7.00 ± 0.07 | 88.8 ± 2.1 | 5 | 1.18 |
| AZ 40140d | Site 1 −8.94 | −9.99 ± 0.09 | 104.4 ± 2.4 | 9 | 1.05 |
| Site 2 −6.11 | Site 2 max not reached | ||||
| 47.4% Site 1 | |||||
| Oxprenolol | −6.25 | Site 1 −7.30 ± 0.04 | 47.7 ± 8.3 | 6 | 1.05 |
| Site 2 −5.62 ± 0.13 | |||||
| Site 1 56.6 ± 1.9% | |||||
| BRL 37344 | −6.45 | −7.47 ± 0.09 | 79.7 ± 3.6 | 6 | 1.02 |
| Denopamine | −5.30 | −6.27 ± 0.08 | 92.4 ± 3.4 | 5 | 0.97 |
| TAK 677 | Site 1 −8.59 | −9.54 ± 0.06 | 96.0 ± 2.2 | 7 | 0.95 |
| Site 2 −5.65 | |||||
| 57.4% Site 1 | |||||
| Tulobuterol | −4.72 | −5.65 ± 0.06 | 83.0 ± 3.6 | 6 | 0.93 |
| BRL 35135A | −6.73 | −7.61 ± 0.03 | 77.5 ± 2.5 | 6 | 0.88 |
| Clenbuterol | −5.35 | −6.19 ± 0.04 | 86.1 ± 2.6 | 4 | 0.84 |
| Isoxsuprine | −5.29 | −6.08 ± 0.06 | 71.4 ± 1.9 | 5 | 0.79 |
| ZD 7114 | −6.78 | Site 1 −7.55 ± 0.06 | 58.2 ± 2.0 | 10 | 0.77 |
| Site 2 −5.64 ± 0.20 | |||||
| Site 1 74.8 ± 3.8% | |||||
| Carazolol | −8.35 | Site 1 −8.84 ± 0.07 | 75.7 ± 1.1 | 8 | 0.49 |
| Site 2 −6.96 ± 0.08 | |||||
| Site 1 30.6 ± 3.3% | |||||
| Cyanopindolol | −8.36 | Site 1 −8.84 ± 0.12 | 57.0 ± 2.1 | 8 | 0.48 |
| Site 2 −7.01 ± 0.05 | |||||
| Site 1 27.6 ± 2.7% | |||||
| L 748337 | Site 1 −8.04 | Site 1 −8.43 ± 0.09 | 42.6 ± 0.9 | 8 | 0.39 |
| Site 2 −5.46 | Site 2 −5.54 ± 0.12 | ||||
| 54.2% Site 1 | Site 1 60.9 ± 4.4% | ||||
| Pindolol | −7.08 | Site 1 −7.43 ± 0.12 | 67.2 ± 3.6 | 6 | 0.35 |
| Site 2 −5.63 ± 0.11 | |||||
| Site 1 44.5 ± 2.9% | |||||
| Salmeterol | −6.33 | −6.60 ± 0.05 | 85.5 ± 1.4 | 5 | 0.27 |
| SDZ 21009 | −7.10 | Site 1 −7.37 ± 0.04 | 87.3 ± 3.7 | 12 | 0.27 |
| Site 2 −5.75 ± 0.11 | |||||
| Site 1 52.4 ± 4.2% | |||||
| SR 59230A | −7.37 | −7.59 ± 0.07 | 35.5 ± 1.9 | 4 | 0.22 |
| BAAM | −6.40 | −6.58 ± 0.07 | 33.5 ± 2.0 | 4 | 0.18 |
| Bucindolol | −8.04 | Site 1 −8.19 ± 0.08 | 80.4 ± 3.7 | 6 | 0.15 |
| Site 2 −6.43 ± 0.17 | |||||
| Site 1 61.4 ± 2.4% | |||||
| Octopamine | IC50 =−3 | −5.38 ± 0.07 | 91.7 ± 2.0 | 4 | |
| Butoxamine | >−4 | No response | 11 | ||
| Methoxyphenamine | −3.96 | No response | 8 | ||
| Nebivolol | −7.04 | No response | 8 |
Values represent mean ± SEM of n separate experiments. For several ligands, the concentration–response curve was best described by a two-component curve. In these cases, the log EC50 values are given for each component, the percentage of the response represented by the first component and the percentage of the total response in relation to the isoprenaline stimulation.
The ligands are arranged in order of intrinsic efficacy.
The percentage maximum response obtained was the same for the ligands at each receptor at each time-point. However, the counts collected after 5 h agonist incubation were greater than those after 30 min incubation, thus allowing a bigger window to examine the small responses. Weak partial agonists and those with a two-component agonist response curves were therefore incubated for 5 h to allow more accurate measurement (with no change in either log EC50 values or percentage maximum response obtained).
Many of the agonists stimulated full (as compared to isoprenaline) concentration–response curves. However, many ligands often considered as ‘antagonists’ stimulated partial agonist responses (e.g. nebivolol, cyanopindolol, carazolol, oxprenolol and bucindolol) (Tables 3–5, arranged in order of ‘efficacy ratio’ for each receptor). For the human β1- and β3-adrenoceptors, several of these partial agonist responses were found to best fit a two-component concentration–response curve (e.g. Figures 5 and 6). This is similar to previous findings which suggest that the first high-affinity component is acting via the catecholamine conformation of the receptor, and the second low-affinity component via a secondary agonist conformation (Baker et al., 2003a; Baker, 2005b).
Figure 6.
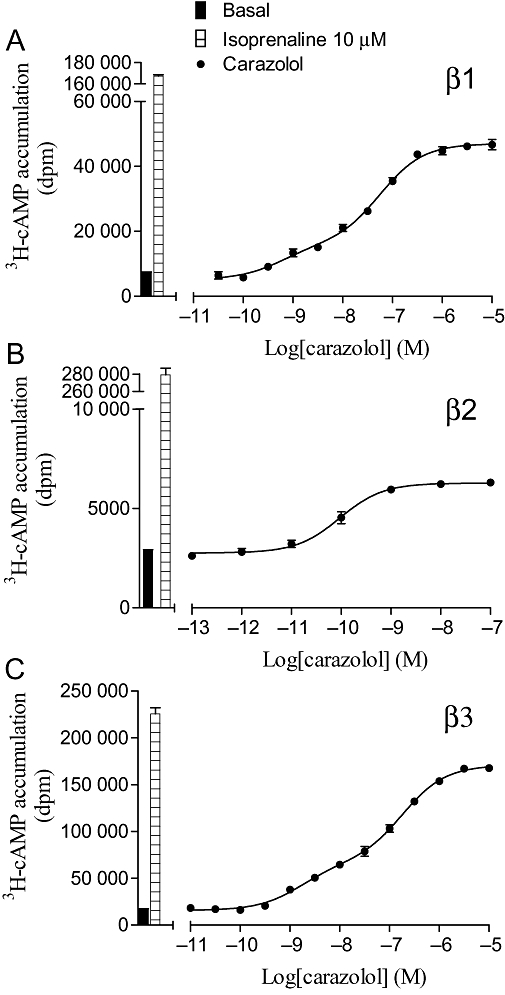
[3H]-cAMP accumulation in response to carazolol after 5 h incubation in (A) CHO β1 cells, (B) CHO β2 cells and (C) CHO β3 cells. The columns represent basal [3H]-cAMP accumulation and that in response to 10 µM isoprenaline. Data points are mean ± SEM of triplicate determinations. These single experiments are representative of (A) eight, (B) four and (C) eight separate experiments.
S-pindolol
Racemic ligands were used in the majority of cases, but the different enantiomers of pindolol have previously been shown to have different properties in certain studies (Walter et al., 1984; Joseph et al., 2003). S-pindolol was therefore examined. This enantiomer was found to have [3H]-CGP 12177 binding affinities of −9.16 ± 0.09 (n = 5) and −9.55 ± 0.04 (n = 5) at the β1- and β2-adrenoceptors respectively. When the [3H]-cAMP accumulation response was examined, S-pindolol stimulated a biphasic concentration–response curve (log EC501 =−9.09 ± 0.09, log EC502 =−6.43 ± 0.09, 43.3% site 1, 53.5 ± 1.6% isoprenaline maximum) at the β1-adrenoceptor very similar to that of racemic pindolol (Table 3). Thus, as expected, S-pindolol appears to have slightly higher affinity than racemic pindolol, and the isomer S-pindolol appears to be able to activate both conformations of the β1-adrenoceptor.
Parent CHO cells
There was no specific binding of [3H]-CGP 12177 to the parent CHO cells (i.e. CHO cells without a transfected receptor). In the [3H]-cAMP accumulation assay, the parent CHO cells responded to forskolin (stimulation 21.5 ± 2.2-fold over basal, n = 10), but there was no response to any of the other ligands.
Discussion
Several recent studies have examined the selectivity (affinity) of β-antagonists for the β-adrenoceptors in different recombinant cell systems, and concluded that many clinically available β-blockers are not very selective (Smith and Teitler, 1999; Schnabel et al., 2000; Hoffmann et al., 2004; Baker, 2005a). This study set out to examine the binding and function of β-adrenoceptor agonists, measured under similar conditions, to determine if agonists are subtype selective and whether it was affinity or intrinsic efficacy that made them so. Whole-cell techniques were used, as this is how agonists would behave in the clinical situation (i.e. with GTP present in living cells). This probably means that measured KD values obtained from binding represent the binding affinity for the basal R state of the receptor (rather than the R* state; Leff, 1995; Leff et al., 1997).
The affinity measurements can be compared across the three cell lines, as affinity from ligand binding is a direct measure. From these studies, it can be seen that several very selective β2- and β3-agonists exist on the basis of affinity; however, there are few β1-selective agonists. Competition binding curves were all best described by a one-site sigmoidal response curve for the human β1- and β2-adrenoceptors; however, AZ 40140d, L 755507, L 748337 and TAK 677 were found to have a two-component displacement curve (Table 1; Figure 2). The human β3-receptor has previously been suggested to exist in at least two agonist conformations (Baker, 2005b), and it may be that this represents binding to two different agonist conformations of this receptor. The β1-adrenoceptor also exists in at least two conformations (Pak and Fishman, 1996; Granneman, 2001; Baker et al., 2003a; Molenaar, 2003; Arch, 2004; Baker, 2005c; Kaumann and Molenaar, 2008). However, the affinity between these conformations is greater for the β1- than the β3-adrenoceptor, and this probably explains why [3H]-CGP 12177 at the concentrations used only demonstrated binding to one conformation at the β1-adrenoceptor. As several GPCRs have allosteric functions, it is also possible that there is a degree of allosteric interference from the radioligand or the competing agonist affecting the binding of the other.
Recent evidence has also shown that the functional response measured from a given receptor–ligand interaction can vary depending on which cellular response is being measured. For example, propranolol acts as an inverse agonist at the human β2-adrenoceptor by decreasing cAMP production below basal, but at the same time stimulates the ERK pathway (e.g. Azzi et al., 2003; Baker et al., 2003b). Thus, the binding of a ligand (e.g. adrenaline) may generate a plethora of active ‘states’ of the receptor, some of which may be coupled to different G-proteins or intracellular effector mechanisms (e.g. β-arrestin), and these in turn may have different allosteric influences on the receptor itself. As the human β-adrenoceptors are all Gs-coupled receptors, the robust, Gs-coupled, rapid upstream cAMP response was used as a measure of functional efficacy in this study. As the whole-cell binding assay and the cAMP accumulation assay were both performed in the same confluent intact living cells, the ‘states’ of the receptor induced by the competing ligand (e.g. adrenaline) in the binding assay would be the same ‘states’ as those induced by the same ligand measured in the cAMP assay. However, the binding assay will only detect the state that [3H]-CGP 12177 is able to bind to. Thus, the [3H]-CGP 12177 binding assay might not detect all of the active states generated by the binding of the agonist (e.g. adrenaline). The cAMP assay measured the response generated from the plethora of states generated from, for example, adrenaline. So, although adrenaline will generate the same plethora of states in both assays, potentially, the group of states detected in the binding assay might not overlap completely with the states that induce the production of cAMP. There is however likely to be substantial overlap as [3H]-CGP 12177 is completely displaceable by virtually all the ligands (Table 1), and CGP 12177 itself stimulates a cAMP response (Supporting Information Tables S2–S4).
When examining the cAMP responses, several things must be considered before comparisons are made. Three different cell lines have been used (one for each receptor), and each has a different receptor expression level and, because they are different cell lines, will have different effector coupling efficiencies. Direct comparisons of functional responses across the three cell lines are therefore not possible. However, a rank order of intrinsic efficacy can be determined from a ratio between affinity (KD from binding) and the EC50 from the functional response (Furchgott, 1966; Kenakin, 1982; 1999b; Strange, 2008). Thus, at the β1-adrenoceptor, fenoterol (log KD−5.04, log EC50−7.53) has an ‘intrinsic efficacy ratio’ of 309 (log 2.49), and salbutamol has an intrinsic efficacy ratio of 33.9 (log 1.53). Therefore, fenoterol has c. 10-fold more intrinsic efficacy at the human β1-adrenoceptor than salbutamol. Thus, although intrinsic efficacy has not been measured directly, a meaningful measure has been obtained to allow comparison between ligands.
Tables 3–5 are arranged in order of intrinsic efficacy for each receptor. At the top of the table are full agonists with the highest intrinsic efficacy. As the intrinsic efficacy of the ligands decreases, there comes a stage when they are no longer efficacious enough to stimulate a maximum response. The maximum responses then decrease until there is no response at all (as for nebivolol at β2- and β3-receptors). Here, nebivolol is a neutral antagonist at these receptors in this assay. This pattern is as would be predicted (Kenakin, 1999b; Strange, 2008), but there are some notable exceptions (butoxamine and methoxyphenamine at β1, bucindolol at β2 and oxprenolol and bucindolol at β3).
In addition, the agonist concentration–response curves are not always best described by a one-site sigmoidal response curve. Several agonist concentration–response curves at both the human β1- and β3-adrenoceptors are best described by a two-component response. This is similar to previous findings (Walter et al., 1984; Baker et al., 2003a; Baker, 2005b) where both of these receptors (but as yet not the human β2-adrenoceptor) have been described as having at least two active agonist conformations (Pak and Fishman, 1996; Granneman, 2001; Arch, 2004; Kaumann and Molenaar, 2008). Interestingly, some compounds appear to bind to or induce two conformations of the receptor, while others stimulate a two-component response, and L 748337 is best described by a two-component response in both binding and [3H]-cAMP accumulation. Ligands are known to have different affinities and different efficacies for the two sites (agonist or antagonist; Baker et al., 2003a; Baker, 2005b,c;), so the differences probably represent ligands binding to the two components with similar affinity, but one conformation being more functionally active (efficacious) than the other and vice versa.
Finally, comparisons of the rank order of ligands for the three different receptors provide information about relative intrinsic efficacies. Fenoterol is a full and efficacious agonist at the β1-adrenoceptor, ranking third out of the agonists studied. It was also a full agonist at the β2- and β3-adrenoceptors with the highest intrinsic efficacy (i.e. top of Tables 4 and 5, rank 1). Thus, fenoterol is a full agonist with high intrinsic efficacy across all three β-adrenoceptors. Likewise, BAAM is a partial agonist at all three receptors, with rankings of 37, 34 and 36 at β1, β2 and β3, respectively, and can therefore be said to have low intrinsic efficacy at all three receptors.
Table 4.
Log KD values from [3H]-CGP 12177 whole-cell binding (from Table 1), log EC50 values and % isoprenaline maximal responses obtained from [3H]-cAMP accumulation and intrinsic efficacy ratios (KD/EC50) for cells expressing the human β2-adrenoceptor
| β2 | Log KD from binding (β2) | Log EC50 from cAMP (β2) | % Isoprenaline maximal response | n | Efficacy ratio (log) |
|---|---|---|---|---|---|
| Fenoterol | −7.03 | −8.90 ± 0.07 | 100.9 ± 2.7 | 4 | 1.87 |
| Adrenaline | −6.13 | −7.93 ± 0.07 | 101.9 ± 1.8 | 8 | 1.80 |
| Terbutaline | −5.51 | −7.29 ± 0.02 | 102.7 ± 3.7 | 4 | 1.78 |
| Metaproterenol | −5.30 | −7.07 ± 0.06 | 109.2 ± 3.7 | 5 | 1.77 |
| Salbutamol | −6.01 | −7.72 ± 0.07 | 95.8 ± 2.4 | 4 | 1.71 |
| Cimaterol | −7.26 | −8.94 ± 0.07 | 96.6 ± 3.5 | 4 | 1.68 |
| Isoprenaline | −6.64 | −8.22 ± 0.11 | 100 | 12 | 1.58 |
| Formoterol | −8.63 | −10.08 ± 0.02 | 104.3 ± 2.8 | 4 | 1.45 |
| Zinterol | −8.04 | −9.48 ± 0.07 | 105.3 ± 4.6 | 5 | 1.44 |
| Procaterol | −7.11 | −8.43 ± 0.06 | 107.9 ± 2.0 | 6 | 1.32 |
| BRL 35135A | −7.38 | −8.69 ± 0.05 | 86.3 ± 3.4 | 5 | 1.31 |
| Clenbuterol | −7.90 | −9.18 ± 0.04 | 95.3 ± 3.1 | 4 | 1.28 |
| AZ 40140d | −7.14 | −8.40 ± 0.09 | 95.6 ± 2.0 | 7 | 1.26 |
| Ritodrine | −5.81 | −6.82 ± 0.06 | 91.4 ± 2.9 | 6 | 1.01 |
| Noradrenaline | −5.41 | −6.36 ± 0.04 | 103.4 ± 2.1 | 11 | 0.95 |
| Tulobuterol | −6.83 | −7.60 ± 0.08 | 89.3 ± 1.8 | 7 | 0.77 |
| Isoxsuprine | −5.93 | −6.65 ± 0.06 | 68.1 ± 2.4 | 5 | 0.72 |
| Dopamine | −3.93 | −4.64 ± 0.03 | 89.8 ± 3.5 | 6 | 0.71 |
| TAK 677 | −7.32 | −8.03 ± 0.06 | 89.6 ± 1.9 | 8 | 0.71 |
| Ractopamine | −6.93 | −7.63 ± 0.05 | 83.3 ± 2.6 | 5 | 0.70 |
| Salmeterol | −9.26 | −9.89 ± 0.08 | 94.1 ± 2.3 | 6 | 0.63 |
| Bamethane | −4.83 | −5.38 ± 0.08 | 71.5 ± 2.3 | 5 | 0.55 |
| Dobutamine | −5.84 | −6.32 ± 0.05 | 64.8 ± 2.7 | 4 | 0.48 |
| Octopamine | −4.03 | −4.45 ± 0.08 | 32.0 ± 2.9 | 5 | 0.42 |
| BRL 37344 | −6.51 | −6.88 ± 0.03 | 80.1 ± 4.1 | 5 | 0.37 |
| ZD 7114 | −7.31 | −7.65 ± 0.12 | 2.2 ± 0.4 | 7 | 0.34 |
| Methoxyphenamine | −4.32 | −4.62 ± 0.07 | 10.0 ± 0.8 | 10 | 0.30 |
| L 755507 | −6.83 | −7.05 ± 0.07 | 3.0 ± 0.2 | 6 | 0.22 |
| Butoxamine | −6.23 | −6.41 ± 0.17 | 1.4 ± 0.4 | 5 | 0.18 |
| Oxprenolol | −8.97 | −9.10 ± 0.03 | 5.9 ± 0.3 | 4 | 0.13 |
| L 748337 | −6.47 | −6.44 ± 0.07 | 9.5 ± 2.2 | 6 | −0.03 |
| Pindolol | −9.23 | −9.12 ± 0.10 | 9.9 ± 0.9 | 9 | −0.11 |
| Denopamine | −5.83 | −5.65 ± 0.08 | 11.2 ± 0.7 | 5 | −0.18 |
| BAAM | −8.57 | −8.24 ± 0.08 | 6.9 ± 0.1 | 5 | −0.33 |
| SR 59230A | −8.45 | −8.10 ± 0.10 | 3.1 ± 0.2 | 4 | −0.35 |
| Bucindolol | −9.99 | −9.40 ± 0.06 | 16.2 ± 1.2 | 5 | −0.59 |
| Carazolol | −10.49 | −9.75 ± 0.07 | 1.9 ± 0.3 | 4 | −0.74 |
| SDZ 21009 | −10.28 | −9.39 ± 0.09 | 3.7 ± 0.4 | 13 | −0.89 |
| Cyanopindolol | −11.09 | −10.03 ± 0.03 | 9.9 ± 1.7 | 4 | −1.06 |
| ZD 2079 | >−4 | Stimulation at 100 µM | 16.1 ± 1.1 | 4 | |
| Nebivolol | −7.92 | No response | 7 |
Values represent mean ± SEM of n separate experiments.
The ligands are arranged in order of intrinsic efficacy.
Salmeterol appears very selective for the human β2-adrenoceptor in the cAMP assay (log EC50 values of −7.03, −9.89 and −6.60 for β1, β2 and β3 respectively). This could be due to either selective affinity or selective intrinsic efficacy for the β2-adrenoceptor over the other subtypes, or a mixture of both. The affinity measurements (log KD values of −5.73, −9.26 and −6.33 for β1, β2 and β3, respectively), show that salmeterol has high affinity for the β2-adrenoceptor. The intrinsic efficacy of the compound is relatively poor in all cases (rank order 22 at β1, 21 at β2 and 33 at β3). Thus, overall, salmeterol is a highly selective β2-adrenoceptor agonist because of its higher β2-affinity and not because of higher β2-intrinsic efficacy. A similar reasoning can be applied to formoterol, although this agonist has higher intrinsic efficacy at all three receptors (rank 6, 8 and 5 at β1, β2 and β3). This is similar to the finding of Kenakin and Beek (1980) where prenaterol was found to have equal intrinsic efficacy at β1- and β2-adrenoceptors.
Of the agonists studied here, there was a general trend that those with highest intrinsic efficacy were so across all three receptor subtypes (i.e. at the top of Tables 3–5, e.g. fenoterol, terbutaline, metaproterenol and adrenaline), and those with lower intrinsic efficacy likewise tended to have it across all three receptors (bottom of the tables). Thus, the approximate rank order of ligands is similar across all three receptors. However, in order to examine this further, the efficacy ratios (log KD/log EC50) were plotted for the receptors. Figure 7A (affinity values) shows that salmeterol has high selectivity for β2-adrenoceptors based on affinity, whereas denopamine, AZ 40140d and isoprenaline have little affinity-based selectivity. Figure 7B (efficacy ratios) shows that isoprenaline and salmeterol are equally efficacious at the human β1- and β2-adrenoceptors. Denopamine is the most selective ligand for β1-receptors, with regard to intrinsic activity and efficacy, and clenbuterol, procaterol, zinterol, AZ 40140d and salbutamol are more selective for the β2-adrenoceptor than the β1-adrenoceptor based on intrinsic activity and efficacy. Although difficult to directly compare, the selectivity based on affinity appears to be greater than the selectivity based on efficacy.
Figure 7.
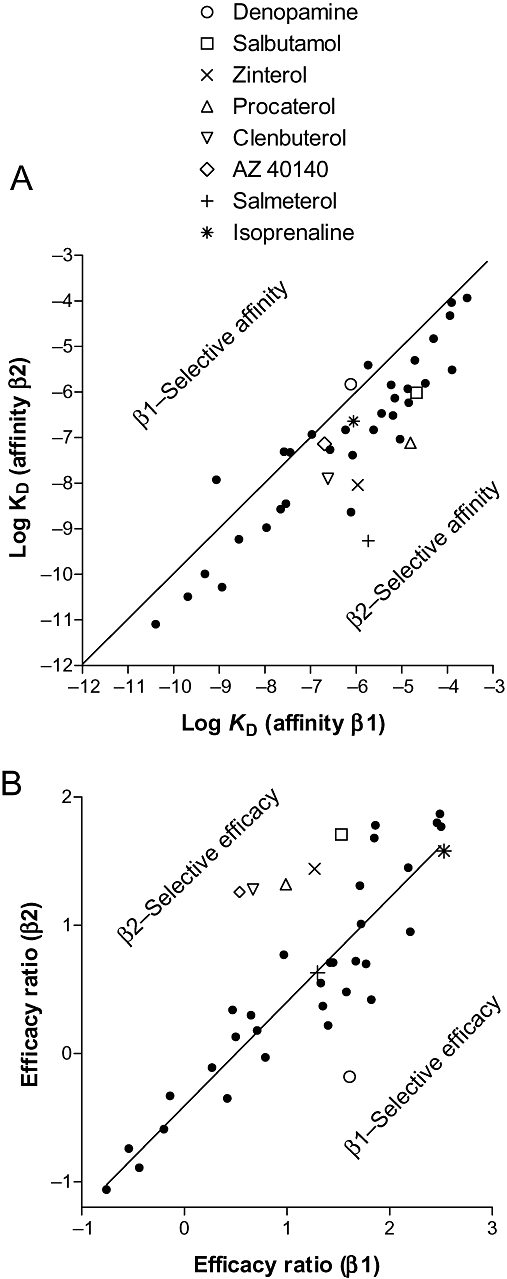
(A) Plot of log KD for compounds from Table 1 for human β1- versus human β2-adrenoceptors. The line is through the origin and represents equal affinity, so compounds occurring to the right and below are β2-selective. Thus, salmeterol can be seen to be highly β2-selective based on affinity, whereas isoprenaline, denopamine and AZ 40140d have little selectivity based on affinity. (B) Plot of efficacy values (log KD/log EC50) for the same compounds (taken from Tables 3 and 4). The line is that of best fit – the slope is not 1 nor does it go though the origin as this represents a function of efficacy (i.e. differences in cell line which include receptor number, receptor–effector coupling, etc.). Denopamine can be seen to be β1-selective based on efficacy, whereas AZ 40140d is β2-selective, and isoprenaline and salmeterol were non-selective based on efficacy.
There are two current theories on the matter of efficacy: conformational selection and conformational induction (Clarke and Bond, 1998; Hunyady et al., 2003).
Conformational selection suggests that receptors spontaneously exist in basal (R) and pre-determined active forms [R* in two-state (Leff, 1995) and R** in three-state models (Leff et al., 1997)] in the cell membranes that are in equilibrium with each other. Agonists bind with greater affinity to R* (or R**) over R, and therefore reset the equilibrium with more R* present, and hence a response is generated. Here, intrinsic efficacy is explained on the basis of selective affinity where ‘intrinsic efficacy’ is actually the ratio of affinity for R* over R. Conformational (or agonist) induction suggests that receptors are in a basal state in the cell membrane, and the binding of the ligand creates the agonist R* state of the receptor. Here, efficacy is the ability of the ligand to change the receptor conformation. This model allows for an infinite number of active receptor states, but suggests that there is something distinct about the chemical interaction that gives some molecules the receptor-activating property of intrinsic efficacy. However, both models suggest that it should be possible to develop agonist ligands that are selective because of selective intrinsic efficacy.
In conclusion, therefore, agonist ligands can, in theory, appear selective for different receptor subtypes for different reasons: selective binding affinity or selective intrinsic efficacy. This study shows that some β-adrenoceptor agonists (e.g. salmeterol) are highly selective purely because of selective binding affinity. Others have very similar affinity but appear subtype selective because of a degree of selective intrinsic efficacy. Thus, it is possible to develop selective ligands based on either affinity or intrinsic efficacy, and there is therefore scope for the development of better, more selective agonists with both of these properties.
Acknowledgments
J.G.B. is a Wellcome Trust clinician scientist and would like to thank June McCulloch, Marleen Groenen and Richard Proudman for technical assistance in running the cAMP chromatography columns; Steve Hill for comments on the manuscript; Christophe Fromont for confirming the structure of AZ 40140d; and Peter Fischer for assistance in constructing Supporting Information Table S1.
Glossary
Abbreviations:
- AZ 40140d
(R)-N-[5-[2-[2-(9H-carbazole-2-iloxy)ethylamino]-1-hydroxyethyl]-2-hydroxyphenyl]methanesulphonamide
- BAAM
bromoacetylalprenololmenthane
- BRL 35135A
(R*,R*)-[4-2-[[2-(3-chlorophenyl)-2-hydroxyethyl]amino]propyl]phenoxy]-acetic acid methyl ester
- BRL 37344
(R*,R*)- (±)-4-[2-[(2-(3-chlorophenyl)-2-hydroxyethyl)amino]propyl]phenoxyacetic acid
- CGP 12177
(–)-4-(3-tert-butylamino-2-hydroxypropoxy)-benzimidazol-2-one
- CGP 20712A
2-hydroxy-5-(2-[{hydroxy-3-(4-[1-methyl-4-trifluoromethyl-2-imidazolyl]phenoxy)propyl}amino]ethoxy)benzamide
- CHO
Chinese hamster ovary
- COPD
chronic obstructive pulmonary disease
- ICI 118551
(–)-1-(2,3-[dihydro-7-methyl-1H-inden-4-yl]oxy)-3-([1-methylethyl]-amino)-2-butanol
- L 748337
N-[[3-[(2S)-2-Hydroxy-3-[[2-[4-[(phenylsulphonyl)amino]phenyl]ethyl]amino]propoxy]phenyl]methyl]-acetamide
- L 755507
4-[[(hexylamino)carbonyl]amino]-N-[4-[2-[[(2S)-2-hydroxy-3-(4-hydroxyphenoxy)propyl]amino]ethyl]phenyl]-benzenesulphonamide
- SDZ 21009
4-[3-[(1,1-dimethylethyl)amino]-2-hydroxypropoxy]-1H-indole-2-carboxylic acid, 1-methylethyl ester
- SR59230A
1-(2-ethylphenoxy)-3-[[(1S)-1,2,3,4-tetrahydro-1-naphthalenyl]amino]-(2S)-2-propanol
- TAK 677
[3-[(2R)-[[(2R)-(3-chlorophenyl)-2-hydroxyethyl]amino]propyl]-1H-indol-7-yloxy]-acetic acid
- ZD 2079
4-[2-[[(2R)-2-hydroxy-2-phenylethyl]amino]ethoxy]-benzeneacetic acid hydrochloride
- ZD 7114
(S)-4-[2-hydroxy-3-phenoxypropylaminoethoxy]-N-(2-methoxyethyl)phenoxyacetamide hydrochloride
Conflicts of interest
The author has no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Table S1 Agonist, source, catalogue number, CAS RN number and isomeric status for each of the agonists used.
Table S2 Log EC50 values and % maximal isoprenaline responses measured from [3H]-cAMP accumulation in CHO β1 cells at 10 min, 30 min and 5 h.
Table S3 Log EC50 values and % maximal isoprenaline responses measured from [3H]-cAMP accumulation in CHO β2 cells at 10 min, 30 min and 5 h.
Table S4 Log EC50 values and % maximal isoprenaline responses measured from [3H]-cAMP accumulation in CHO β3 cells at 10 min, 30 min and 5 h.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arch JR. Do low-affinity states of beta-adrenoceptors have roles in physiology and medicine? Br J Pharmacol. 2004;143:517–518. doi: 10.1038/sj.bjp.0705991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzi M, Charest PG, Angers S, Rosseau G, Kohout T, Bouvier M, et al. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci USA. 2003;100:11406–11411. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG. The selectivity of β-adrenoceptor antagonists at the human β1, β2 and β3 adrenoceptors. Br J Pharmacol. 2005a;144:317–322. doi: 10.1038/sj.bjp.0706048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG. Evidence for a secondary state of the human β3-adrenoceptor. Mol Pharmacol. 2005b;68:1645–1655. doi: 10.1124/mol.105.015461. [DOI] [PubMed] [Google Scholar]
- Baker JG. Sites of action of β-ligands at the human β1-adrenoceptor. J Pharmacol Exp Ther. 2005c;313:1163–1171. doi: 10.1124/jpet.104.082875. [DOI] [PubMed] [Google Scholar]
- Baker JG, Hall IP, Hill SJ. Pharmacological characterization of CGP 12177 at the human β2-adrenoceptor. Br J Pharmacol. 2002;137:400–408. doi: 10.1038/sj.bjp.0704855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG, Hall IP, Hill SJ. Agonist actions of ‘β-blockers’ provide evidence for two agonist activation sites or conformations of the human β1-adrenoceptor. Mol Pharmacol. 2003a;63:1312–1321. doi: 10.1124/mol.63.6.1312. [DOI] [PubMed] [Google Scholar]
- Baker JG, Hall IP, Hill SJ. Agonist and inverse agonist actions of ‘β-blockers’ at the human β2-adrenoceptor provide evidence for agonist-directed signalling. Mol Pharmacol. 2003b;64:1357–1369. doi: 10.1124/mol.64.6.1357. [DOI] [PubMed] [Google Scholar]
- Baker JG, Hall IP, Hill SJ. Temporal characteristics of CRE-mediated gene transcription: requirement for sustained cAMP production. Mol Pharmacol. 2004;65:986–998. doi: 10.1124/mol.65.4.986. [DOI] [PubMed] [Google Scholar]
- Büscher R, Heeks C, Taguchi K, Michel MC. Comparison of guinea-pig, bovine and rat alpha 1-adrenoceptor subtypes. Br J Pharmacol. 1996;117:703–711. doi: 10.1111/j.1476-5381.1996.tb15247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke WP, Bond RA. The elusive nature of intrinsic efficacy. Trends Pharmacol Sci. 1998;19:270–276. doi: 10.1016/s0165-6147(97)01138-3. [DOI] [PubMed] [Google Scholar]
- Cote C, Pearle JL, Sharafkhaneh A, Spangenthal S. Faster onset of action of formoterol versus salmeterol in patients with chronic obstructive pulmonary disease: a multicenter, randomized study. Pulm Pharmacol Ther. 2009;22:44–49. doi: 10.1016/j.pupt.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Donaldson J, Brown AM, Hill SJ. Influence of rolipram on the cyclic-3′,5′-adenosine monophosphate response to histamine and adenosine in slices of guinea-pig cerebral cortex. Biochem Pharmacol. 1988;37:715–723. doi: 10.1016/0006-2952(88)90146-3. [DOI] [PubMed] [Google Scholar]
- Furchgott RF. The use of β-haloalkylamines in the differentiation of receptors and in the determination of dissociation constants of receptor-agonist complexes. In: Harper NJ, Simmonds AB, editors. Advances in Drug Research. New York: Academic Press; 1966. pp. 21–55. Vol. 3. [Google Scholar]
- Granneman JG. The putative beta4-adrenergic receptor is a novel state of the beta1-adrenergic receptor. Am J Physiol Endocrinol Metab. 2001;280:E199–E202. doi: 10.1152/ajpendo.2001.280.2.E199. [DOI] [PubMed] [Google Scholar]
- Hoffmann C, Leitz MR, Oberdorf-Maass S, Lohse MJ, Klotz KN. Comparative pharmacology of human β-adrenergic receptor subtypes – characterization of stably transfected receptor in CHO cells. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:151–159. doi: 10.1007/s00210-003-0860-y. [DOI] [PubMed] [Google Scholar]
- Hunyady L, Vauquelin G, Vanderheyden P. Agonist induction and conformational selection during activation of a G-protein-coupled receptor. Trends Pharmacol Sci. 2003;24:81–86. doi: 10.1016/S0165-6147(02)00050-0. [DOI] [PubMed] [Google Scholar]
- Joseph SS, Lynham JA, Molenaar P, Grace AA, Colledge WH, Kaumann AJ. Intrinsic sympathomimetic activity of (–)-pindolol mediated through a (–)-propranolol-resistant site of the beta1-adrenoceptor in human atrium and recombinant receptors. Naunyn Schmiedebergs Arch Pharmacol. 2003;368:496–503. doi: 10.1007/s00210-003-0835-z. [DOI] [PubMed] [Google Scholar]
- Kaumann AJ, Molenaar P. The low-affinity site of the beta1-adrenoceptor and its relevance to cardiovascular pharmacology. Pharmacol Ther. 2008;118:303–336. doi: 10.1016/j.pharmthera.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Kenakin TP. Theoretical and practical problems with the assessment of intrinsic efficacy of agonists: efficacy of reputed beta-1 selective adrenoceptor agonists for beta-2 adrenoceptors. J Pharmacol Exp Ther. 1982;223:416–423. [PubMed] [Google Scholar]
- Kenakin T. Efficacy in drug receptor theory: outdated concept or under-valued tool? Trends Pharmacol Sci. 1999a;20:400–405. doi: 10.1016/s0165-6147(99)01361-9. [DOI] [PubMed] [Google Scholar]
- Kenakin T. The measurement of efficacy in the drug discovery agonist selection process. J Pharmacol Toxicol Methods. 1999b;42:177–187. doi: 10.1016/s1056-8719(00)00070-8. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Drug efficacy at G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2002;42:349–379. doi: 10.1146/annurev.pharmtox.42.091401.113012. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Efficacy as a vector: the relative prevalence and paucity of inverse agonism. Mol Pharmacol. 2004;65:2–11. doi: 10.1124/mol.65.1.2. [DOI] [PubMed] [Google Scholar]
- Kenakin TP, Beek D. Is prenalterol (H133/80) really a selective beta 1 adrenoceptor agonist? Tissue selectivity resulting from differences in stimulus–response relationships. J Pharmacol Exp Ther. 1980;213:406–413. [PubMed] [Google Scholar]
- Leff P. The two-state model of receptor activation. Trends Pharmacol Sci. 1995;16:89–97. doi: 10.1016/s0165-6147(00)88989-0. [DOI] [PubMed] [Google Scholar]
- Leff P, Scaramellini C, Law C, McKechnie K. The three-state model of agonist action. Trends Pharmacol Sci. 1997;18:355–362. doi: 10.1016/s0165-6147(97)01105-x. [DOI] [PubMed] [Google Scholar]
- Lötvall J. Pharmacological similarities and differences between beta2-agonists. Respir Med. 2001;95:S7–11. doi: 10.1053/rmed.2001.1139. [DOI] [PubMed] [Google Scholar]
- Molenaar P. The ‘state’ of beta-adrenoceptors. Br J Pharmacol. 2003;140:1–2. doi: 10.1038/sj.bjp.0705420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak MD, Fishman PH. Anomalous behaviour of CGP 12177A on beta 1-adrenergic receptors. J Recept Signal Transduct Res. 1996;16:1–23. doi: 10.3109/10799899609039938. [DOI] [PubMed] [Google Scholar]
- Schnabel P, Maack C, Mies F, Tyroller S, Scheer A, Bohm M. Binding properties of β-blockers at recombinant β1, β2 and β3-adrenoceptors. J Cardiovasc Pharmacol. 2000;36:466–471. doi: 10.1097/00005344-200010000-00008. [DOI] [PubMed] [Google Scholar]
- Smith C, Teitler M. Beta-blocker selectivity at cloned human beta1- and beta2-adrenoceptors. Cardiovasc Drugs Ther. 1999;13:123–126. doi: 10.1023/a:1007784109255. [DOI] [PubMed] [Google Scholar]
- Strange PG. Agonist binding, agonist affinity and agonist efficacy at G protein-coupled receptors. Br J Pharmacol. 2008;153:1353–1363. doi: 10.1038/sj.bjp.0707672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter M, Lemoine H, Kaumann AJ. Stimulant and blocking effects of optical isomers of pindolol on the sinoatrial node and trachea of guinea pig. Role of beta-adrenoceptor subtypes in the dissociation between blockade and stimulation. Naunyn Schmiedebergs Arch Pharmacol. 1984;327:159–175. doi: 10.1007/BF00500912. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.