

Although many viruses are capable of subclinical infection, only a few are known to undergo true latency. In latent infection, the full viral genome is retained in the host cell, but its expression is dramatically restricted, such that few viral antigens and no viral particles are produced. To qualify as latency, this cryptic form of infection must display two additional properties: persistence and reversibility. Reversibility – i.e. the capacity of the genome to, under the appropriate circumstances, reactivate full viral gene expression, with production of infectious progeny (so-called productive or lytic replication) - is the key requirement of latency. Cryptic states that lack this characteristic are more properly characterized as abortive infections, which typically occur when viruses infect cells that are nonpermissive for viral replication. Nonpermissivity can occur due to events at the cell surface – e.g. the absence of receptors or other entry cofactors – or due to post-entry blocks to subsequent steps in viral growth. When post-entry blocks are present (either due to the absence of a permissive factor or the presence of an inhibitory one), abortive infection results.
Abortive infections are well-described in cell culture, but because they cannot lead to persistence or spread in vivo, they are much less commonly identified in the intact host and likely have little biological significance - in essence, they are a dead end. It is the property of reversibility that allows latent infections to avoid a similar fate, and instead become an effective mechanism of viral persistence. Efficient establishment of latency allows the viral genome to persist despite host immune responses to many viral antigens, and in the face of other potentially adverse signals in the microenvironment. When environmental conditions warrant, appropriate signals can trigger the full repertoire of viral gene expression, allowing virus production and spread to resume.
Only a few virus families are known to be capable of true latency, as strictly defined above. Chief among these are the herpesviruses, a huge and widely distributed family of DNA viruses that are important pathogens in their native vertebrate hosts. The capacity for latency is a defining feature of herpesviruses: all known herpesviral infections display latency in every infected individual. Indeed, the anatomic sites of latency, and the frequency with which latency is reversed to engender lytic infection, are important determinants of the clinical manifestations of infection. The other important virus family in which latency has been described are the retroviruses. These small, enveloped RNA viruses replicate via reverse transcription of their RNA genomes; the resulting DNA establishes persistence by integrating into the host genome, from which it generally cannot be dislodged. In most infected cells in culture, such integrants continue to express genomic RNA and viral proteins, leading to the production of progeny virions. However, in some cultured cells such integrants are completely transcriptionally silent, though they can be exogenously stimulated to express viral mRNAs and resume virus production. These cells meet the molecular definition of latency.
In the intact host, the best-characterized example of retroviral latency occurs in HIV infection of humans, in which a small subpopulation of long-lived memory T cells can undergo this form of latent infection (Chun et al 1997). This latency has enormous clinical importance, for “archived” copies of proviral DNA in this locale cannot be eliminated with conventional antiretroviral drugs, and can lead to relapse of infection when such drugs are discontinued (see Han et al 2007, Peterson et al 2007 for reviews). But it is open to question whether such latency is critical for HIV persistence in a natural infection – active viral replication, which is continuously present in many more cells than are latently infected, appears more than adequate to assure this outcome in the host. Human HTLV infection is another case in which true latency likely exists in vivo. In that case, many circulating cells bear proviral DNA in the absence of overt viral replication or gene expression, though the exact role of these cells in the natural history of infection and persistence is still being debated (Asquith and Bangham, 2008). In most other retroviral infections, the issue of latency in vivo has been little studied. So even though latency clearly exists in retroviral infection, its universality and biological importance in vivo is not as well established for these agents as it is for herpesviruses.
Accordingly, in this review we focus on herpesviral latency and its regulation, with particular reference to the lymphotropic herpesviruses, where molecular analysis of latency is most highly advanced.
Herpesviral infection: a primer
Herpesviruses are large, enveloped DNA viruses that can engender either a latent or a lytic infection at the level of the single cell. Three major subfamilies of herpesviruses – termed α, β and γ - are recognized based on sequence phylogeny. The alphaviruses establish latent infections in neurons, while the gammaherpesviruses are markedly lymphotropic (the tropism of the betaherpesviruses is more variable). Among the alpha- and beta-viruses, lytic infection is the default pathway in culture. Moreover, for none of these viruses has an efficient cell culture model of latency been developed that faithfully recapitulates the cardinal features of latent infection in vivo. As a result, our understanding of lytic replication is very advanced in these viruses, but much less is known about the molecular basis of their latent states. For most of these viruses, latency can be studied only in an intact human or animal host. This poses a formidable experimental barrier to mechanistic understanding of latency, since (i) relatively few latently infected cells exist in any given tissue, and (ii) the extent and level of viral gene expression in latency is generally low.
By contrast, among the gammaherpesviruses, latency is generally (though not invariably) the default pathway in cell culture. As a result, it is usually straightforward to obtain substantial populations of latently infected cells following in vitro infection, facilitating the analysis of both viral and host gene expression in such populations, as well as analyses of the details of viral genome and chromatin structure. In some cases, stably latent cell populations can be continuously cultured, allowing analysis of viral genes and cis-acting elements required for genomic persistence. For all of these reasons, more is known about latency among the lymphotropic subgroup than about latency in any other subfamily of herpesviruses. Accordingly, in this review, we focus on how latency is established and maintained in the gammaherpesviruses, and how these processes are regulated.
Common features of the gammaherpesviral replication cycle
All gammaherpesviruses encapsidate duplex linear DNA genomes in their particles. Upon infection of cells, this DNA is delivered to the nucleus, where it is circularized (largely by host enzymatic machinery) generating a closed-circular DNA form that can persist in the nucleus as a plasmid. Incoming virion DNA generally lacks histones, but the resulting plasmid is rapidly chromatinized in the nucleus (Tempera and Lieberman 2010). Since latency is generally the default pathway in this group, most viral gene expression from the plasmid is silenced, but a handful of genes are expressed from the latent genome. As a general rule, one of more of these genes directs replication of the viral genome, using a cis-acting sequence (termed oriP) as the replication origin of the plasmid. Several latency genes are involved in modulation of host signaling, most notably influencing the activation of NFκB, which turns out to be important in the maintenance of latency (see below). Interestingly, however, these common functions are often carried out by viral proteins that share little homology across the different gammaherpesviruses. For example, none of the latency proteins of EBV are conserved in KSHV or MHV68 (though the latter two viruses do share a number of latency genes).
As noted above, latency is reversible. Because the full complement of viral DNA is retained in the nucleus, under the appropriate circumstances the second program of viral gene expression, lytic replication, can be activated. In this program, expression of virtually all viral ORFs is activated, in a temporally regulated cascade . Three major waves of viral gene expression occur: immediate early (IE) genes, the first wave, are predominantly regulators of gene expression; they turn on delayed early (DE) genes, many of which are functions involved in DNA replication, signal transduction, shutoff of host macromolecular synthesis and immune evasion. DE expression triggers robust viral DNA synthesis - which is now delinked from that of the host, and proceeds by a rolling-circle mechanism. Following DNA replication, late (L) genes are activated; these are mostly structural components of the virion, and their accumulation leads to encapsidation of newly replicated viral genomes into progeny virions. In contrast to the latency genes, there is extensive conservation of lytic cycle genes across all herpesviruses. In all three viruses under review here, lytic reactivation is controlled by one or more (usually one) master regulators of transcription. Expression of these so-called lytic switch protein genes is silenced in latency, but is turned on by all signals that trigger lytic reactivation; this activator, in turn, triggers expression downstream genes to kick-start the lytic cycle.
Gammaherpesviruses thus face a number of common problems that must be solved in order for latency to be established and maintained. First, incoming viral genomes must be chromatinized, and epigenetic and genetic controls established that allow for the latent transcriptional program. Second, latency functions must provide for stable replication from oriP, and for mechanisms to ensure segregation of the viral genome to daughter cells if the latently infected cell should divide. Third, latent regulators of signaling must create an environment that fosters the stability of latency without rendering it irreversible. Part of this involves establishing genetic and epigenetic controls over the expression of the lytic switch protein, as well as biochemical controls over its function. Finally, in the intact host successful latency may also require modulation of host cell functions, particularly those affecting the lifespan and proliferative potential of the latently infected cell. Our molecular understanding of all of these issues is still very incomplete, but some unifying themes are starting to come into view. Even though the latency proteins are poorly conserved among the viruses, commonalities in the modes by which they promote episome maintenance and in the signaling pathways they engage are emerging. In what follows, we discuss in detail the latency strategy of three individual gammaherpesviruses, with particular attention to these themes - and their variations.
EBV Latency and its control
EBV: An overview
EBV was the first identified gammaherpesvirus and remains the best characterized member of this family. Initially recovered from cell lines derived from African Burkitt's lymphoma (BL) in the 1960s, EBV has since been associated with a variety of lymphoproliferative diseases, including infectious mononucleosis (Purtilo 1987), 30-40% of Hodgkin's disease (Hjalgrim and Engels, 2008), and the majority of cases of post-transplant lymphoproliferative disease (PTLD) (Gottschalk et al 2005). In addition, EBV infection is also strongly linked several nonlymphoid disorders, including nasopharyngeal carcinoma (Busson et al 2004) and AIDS-related oral hairy leukoplakia (Greenspan and Greenspan 1989). In vitro and in vivo, the primary target of EBV infection is the B cell; following de novo infection of B cells in culture, latency is the default pathway. In vitro infection of primary B cells often results in immortalization, and the resulting lymphoblastoid cell lines (LCLs) have been an important source of information about EBV latency (see Rickinson and Kieff, 1996 for review). As we shall see, however, EBV latency is a complex subject that can present several different faces, not all of which are captured by the study of LCLs. Latently infected cells can also be derived from EBV-associated tumors, which often display different patterns of latent gene expression.
In EBV, the first regulator of the latent-lytic switch to be discovered was the protein Zta (aka BZLF1), a bZIP transcription factor that is centrally involved in turning on the subsequent genes of the lytic cycle (Countryman and Miller, 1985). Subsequently, a second viral protein, Rta, was also found to be essential for induction of the lytic cascade (Zalani et al 1996; Ragoczy et al 1998). Both Zta and Rta are transcriptional activators; they can each upregulate the other's expression, and together act to turn on downstream lytic genes. Ectopic expression of either gene can induce latently infected cells to commence lytic EBV production, and inactivation of either gene blocks lytic reactivation (Feederle et al 2000). As will be discussed later, Rta is well conserved among all the known gammaherpesviruses, while Zta is less well conserved . Typically, lytic replication is induced by chemicals like phorbol esters or sodium butyrate, which are thought to act by activating Zta expression (see Speck et al, 1997 for review). Thus, regulation of Zta expression and function is a key determinant of the latent-lytic switch. Upon de novo infection, incoming virion DNA is largely unmethylated. This is important because Zta binds preferentially to methylated sites at many of its recognition elements (Bhende et al., 2004); as such, even though there is a burst of Zta synthesis following de novo infection of B cells (Kalla et al, 2010), this does not trigger immediate entry into the lytic cycle. As the EBV genomes becomes chromatinized, CpG methylation proceeds; however, by the time most Zta target sites are methylated, expression of Zta has been quelled by the formation of repressive complexes on the BZLF1 promoter - most notably through binding of phosphorylated MEF2D (which binds to 3 sites within the Zta promoter) and recruitment of histone deacetylases (Bryant and Farrell, 2002). Thus, the epigenetic marking of the EBV genome plays a central role in regulating virus replication during the establishment of latency. It should also be pointed out that the core promoter driving Zta expression is devoid of CpG dinucleotides and thus is never methylated. This may be a key feature that allows the initial expression of Zta following appropriate reactivation stimuli.
A number of factors can inhibit the latent-lytic switch and thereby stabilize latency. Chief among these is the activation of the transcription factor NFkB. The p65 subunit of this transcription factor can interfere with Zta activation of target promoters; this inhibition is mediated through a direction interaction of p65 with Zta (Gutsch et al., 1994). Similar antagonism of lytic reactivation by active NFkB is also seen in KSHV and MHV68 infection (see below).
What are the in vivo cues that signal EBV to reactivate from a latently infected B cell? Data from several groups has now shown that stimulation of plasma cell differentiation leads to virus reactivation (Bhende et al., 2007; Laichalk and Thorley-Lawson, 2005; Sun and Thorley-Lawson, 2007). The critical cellular factor involved in initiating EBV reactivation is XBP-1s, which normally plays a central role in the cellular unfolded protein response (Bhende et al., 2007; Sun and Thorley-Lawson, 2007). XBP-1s expression is induced by BLIMP-1, the master regulator of plasma cell differentiation. XBP-1s is a member of the CREB family of transcription factors and binds to a site in the EBV Zta promoter that has previously been shown to bind CREB and AP-1 proteins. Thus, EBV is apparently able to usurp plasma cell differentiation to turn an antibody producing cell into a virus factory. The big remaining question is: what regulates differentiation of latently infected B cells to plasma cells? At present, the answer to this is unknown. Although cognate antigen recognition is one such stimulus, in the case of MHV68 a viral antigen has been identified that can drive plasma cell differentiation (Liang et al., 2009). This suggests the tantalizing possibility that plasma cell differentiation of latently infected B cells may be regulated by the virus rather than by antigen stimulation. But many other possibilities remain, and it is conceivable that both viral and host factors modulate this decision.
The EBV Latency Program
(i) The Growth Program
Shortly after the identification of EBV in Burkitt's lymphoma (BL) tumors, it was shown that the virus could efficiently immortalize primary B cells to LCLs (Henle et al., 1967). This discovery had a huge impact on the field, since it (i) provided a ready source of latently infected B cells for study; and (ii) appeared to cement the relationship between EBV infection and the genesis of BL. The next 20 years were largely spent identifying and characterizing viral genes expressed in such LCLs [reviewed in (Bornkamm and Hammerschmidt, 2001)]. This work led to the identification of 9 viral proteins, along with several non-coding RNAs, that were consistently detected. Because these gene products trigger B cell proliferation, they are sometimes referred to as the EBV “growth program.” The viral proteins expressed can be broken into 2 distinct groups - 6 nuclear antigens (Epstein-Barr nuclear antigens, EBNAs) and 3 membrane antigens (latency-associated membrane proteins, LMPs). The viral genes expressed during latency are not clustered in a single region, but rather are spread out over most of the viral genome. However, the promoters and cis-elements controlling these genes are in fact clustered in a relatively small region spanning the fused terminal repeats of the viral episome (see Fig. 1).
Figure 1.

Schematic illustration of EBV EBNA and LMP gene transcription, and auto-regulation of viral latency-associated gene expression by the EBNA gene products. An exploded view of Cp- and Wp –driven EBNA gene transcription is shown, depicting the organization of exons immediately downstream of each promoter. Cp-initiated transcript contain 2 unique exons, C1 and C2, which splice to a variable number of W1 and W2 exons encoded within the 3.0Kb internal repeats. Wp-initiated transcript contain a single unique exon, W0, which splices to a variable number of W1 and W2 repeat exons. See text for additional details regarding EBV gene expression during different stages of infection. Also shown in the inset is the predicted membrane topology of the LMP-1 and LMP-2a proteins, along with known cellular interacting partners and signaling pathways activated by these proteins.
An early indication of the complexity of EBV latency-associated gene expression came from the characterization of EBNA gene transcription. These studies revealed that the EBNA gene transcripts all share common 5′ exons that are alternatively spliced to coding exons located at the 3′ end of these transcripts (Bodescot et al., 1986; Bodescot and Perricaudet, 1986, 1987; Bodescot et al., 1987; Sample et al., 1986; Speck et al., 1986; Speck and Strominger, 1985, 1987). The exception is EBNA-LP, which is encoded by the common 5′ exons shared among all EBNA gene transcripts (Sample et al., 1986; Speck et al., 1986). The presence of the EBNA-LP translation initiation codon is determined by alternative splicing and thus both EBNA-LP coding and EBNA_LP- non-coding transcripts are generated - presumably ensuring efficient translation of downstream EBNA gene coding sequences present in the latter RNAs (Rogers et al., 1990).
During the initial phase of EBV infection, EBNA gene transcription is initiated from a promoter, Wp, located within the major internal repeat of the virus (IR1; 3Kb repeat length)(Woisetschlaeger et al., 1989; Woisetschlaeger et al., 1990). There are multiple copies of this promoter and these collectively appear to function as an “ignition switch” to launch EBNA gene transcription in the newly infected resting B cell. Expression of the most proximal EBNA genes, EBNA-LP and EBNA2, are the first viral proteins expressed (Allday et al., 1989; Rooney et al., 1989) and function together to drive a switch in promoter utilization to a second promoter, termed Cp, located just upstream of the IR1 repeat region (Jin and Speck, 1992; Woisetschlaeger et al., 1991; Woisetschlaeger et al., 1990). This promoter drives expression of the remaining EBNAs (SAM- is this an accurate statement?? Yes). This switch is driven by the interaction of EBNA2 with the cellular transcription factor RBP-Jk (also known as CBP-1 and CSL), which normally functions in the regulation of genes in the cellular Notch signaling pathway (Grossman et al., 1994; Henkel et al., 1994; Waltzer et al., 1995; Zimber-Strobl et al., 1994). Two other EBNAs, EBNA3A and EBNA3C, also interact with RBP-Jk and displace EBNA2 – thus serving as modulators of EBNA2's transcriptional regulatory activity (Bain et al., 1996; Johannsen et al., 1996; Radkov et al., 1997; Radkov et al., 1999; Robertson et al., 1995; Waltzer et al., 1996; Zhao et al., 1996). EBNA2 interaction with RBP-Jk also serves to turn on transcription of the LMP genes.
An important product of the Cp transcripts is EBNA-1, which plays a central role in latency by binding to sequences in ori P and promoting initiation of DNA replication of the viral episome, which is largely effected by cellular enzymes and proceeds in concert with host DNA replication (Yates et al. 1985; Reisman et al., 1985). In addition to this function, EBNA-1 also plays important roles in plasmid segregation in dividing cells, via its ability to affiliate with metaphase host chromosomes. As a result, the viral genome is tethered to host chromosomes and can therefore be passively distributed with them to daughter cells by the workings of the mitotic spindle. Mutational lesions in regions of EBNA-1 that are responsible for this tethering cannot establish stable episomes in dividing cells (Sears et al., 2004, Nayyar et al, 2009).
In addition to its replicative functions, EBNA-1 is also a transcriptional activator. When bound to oriP sequences, which are just upstream of Cp, it can upregulate transcription from this promoter (Puglielli et al., 1996; Reisman and Sugden, 1986; Sugden and Warren, 1989). Thus, the switch from Wp to Cp reflects a shift from utilizing a viral promoter regulated by cellular transcription factors to one which is tightly controlled by viral factors.
Which of the 9 latency-associated proteins are required for EBV immortalization of primary human B cells? Multiple studies have shown a requirement for EBNA1, EBNA2, EBNA3a, EBNA3c and LMP-1, while EBNA3b and LMP2a/b appear dispensable [reviewed in (Bornkamm and Hammerschmidt, 2001)]. No attempts to generate an EBNA-LP null virus have been reported, due to the difficulty of knocking out all possible alternatively spliced products that can/could encode a functional form of EBNA-LP. However, deletion of the unique C-terminal 45aa of EBNA-LP has been shown to significantly impair LCL formation (Mannick et al., 1991; Wang et al., 1985).
With respect to transforming activity, only one of the EBV latency-associated antigen, LMP-1, has been directly demonstrated to be an oncogene , as judged by standard growth transformation assays on fibroblasts. LMP-1 is a member of the TNF-receptor superfamily, and appears to function as a constitutively active CD40 receptor [for a details on LMP-1 structure and function see (Bornkamm and Hammerschmidt, 2001)]. LMP-1 is a ligandless receptor that constitutively trimerizes and recruits a number of cytoplasmic factors [e.g., TNF receptor associated factors (TRAFs)] involved in mediating receptor signaling. Most notably, LMP-1 expression in B lymphocytes strongly upregulates NF-κB activity (Izumi and Kieff, 1997). A LMP-1 null EBV is unable to immortalize primary human B cells (Izumi et al., 1997; Kilger et al., 1998), although it has been shown that LMP-1 function can be replaced by continued growth of LMP-1 null EBV infected B cells on fibroblasts feeder layers (Dirmeier et al., 2003).
More recent studies have shown that the episomal maintenance protein EBNA1 is also dispensable for B cell immortalization, although EBNA1 null EBV was several thousand fold less efficient in generating LCLs (Humme et al., 2003). As expected, based on the known function of EBNA1 in maintaining the viral episome, all of the LCLs generated with EBNA1 null virus contained integrated EBV genomes. Integration of the viral genome would eliminate the requirement for EBNA1 in maintenance of the viral episome, but not its proposed role in enhancing LMP and EBNA gene transcription. Thus, it seems likely that in LCLs generated with EBNA1 null EBV there would be selection for integration events that compensate for the role of EBNA1 transcriptional activation of EBV latency-associated gene expression. The rarity of such events likely accounts for the sharp reduction in the frequency of LCL outgrowth with EBNA-1 null mutants.
(ii) Alternative latency programs
While the early studies on EBV immortalized lymphoblastoid cell lines and established BL tumor cell lines supported the notion of a direct role for EBV-driven B cell proliferation in the genesis of BL, a more careful analysis of fresh BL biopsies and early passage BL cell lines revealed a much more restricted pattern of EBV gene expression (Rowe et al., 1986; Rowe et al., 1987). In the vast majority of BL tumors the only viral antigen that can be detected is EBNA1 (this is now referred to as type I latency). Subsequent analyses of both nasopharyngeal carcinomas and EBV-positive Hodgkin's lymphomas revealed another restricted pattern of viral gene expression, in which LMP-1 and/or LMP-2 expression along with EBNA1 could be detected (now termed type II latency; the larger, growth-promoting program of LCLs is now designated type III latency) (Deacon et al., 1993; Herbst et al., 1991; Oudejans et al., 1996). In all of these tumors, the common theme was expression of EBNA1 in the absence of the other EBNA genes. Since all EBNA gene transcription in EBV LCLs is driven from a single transcriptional unit, this raised the question of how the EBNA1 only programs were regulated. This ultimately led to the identification of an alternative promoter, Qp, located downstream of the EBNA2 coding exons (Schaefer et al., 1995). Notably, Qp-initiated transcripts splice exclusively to the EBNA1 coding exon - bypassing the exons encoding the EBNA3 family of nuclear antigens (Figure 1). How this selectivity in splice site selection is achieved remains a mystery. While EBNA1 upregulates EBNA and LMP gene expression through binding to oriP, it potently inhibits transcription from Qp (Sample et al., 1992). A genome wide survey for EBNA1 binding sites in the EBV genome revealed only two sites outside oriP - both of which map just downstream of the Qp transcription initiation site (Jones et al., 1989). Unlike Cp and Wp, Qp is a TATA-less promoter that appears related to promoters found upstream of housekeeping genes - suggesting that it may function as the default promoter to ensure ongoing expression of EBNA1 in the absence of transcription initiation from Cp and/or Wp (Schaefer et al., 1997; Schaefer et al., 1995).
Given what we know about autoregulation of EBNA gene expression (see above), how is Cp/Wp-initiated EBNA gene transcription silenced in the EBV-associated tumors that arise in immunocompetent individuals? A large number of studies have shown that Cp and Wp are heavily methylated in these tumors, while Qp remains unmethylated and active [for review see (Tao and Robertson, 2003)]. That DNA methylation might be involved in modulating viral gene expression was not particularly surprising, since it had been shown that the EBV genome has undergone extensive CpG suppression during its evolution - presumably as a consequence of methylation of the viral genome. Furthermore, the importance of DNA methylation in the suppression of Cp/Wp transcriptional activity in BL cell lines is supported by three independent experimental observations: (i) the drift of some BL cell lines to expression of all the EBNA and LMP genes (type III latency) correlated with the loss of methylation around Cp and Wp; (ii) inhibition of DNA methyltransferase activity induces transcription from Cp/Wp in BL cell lines exhibiting the type I latency program; and (iii) reporter constructs containing extensive sequence upstream and downstream of Cp are active when transfected into BL cell lines exhibiting the type I latency program. The latter result argues that cells in latency type I contain the necessary transcription factors to drive Cp/Wp-initiated transcription, but are prevented from doing so because the viral genomes are methylated and in a transcriptionally inactive conformation.
Since the EBV growth/immortalization program isn't used in EBV-associated tumors, this raises the question of whether EBV is really causally associated with tumorigenesis in these cases – a concern hightened by the discovery that forms of BL exist that are unassociated with EBV infection. In fact, the common feature shared by all cases of BL is not the presence of EBV DNA but the translocation of the c-myc gene to one of the immunoglobulin loci, leading to dysregulation of c-myc expression. Although compelling data exist to link EBV infection with the development of BL, HD and NPC, it is now clear that the role for EBV in the generation of EBV-associated tumors is more complex than originally appreciated. For a more detailed treatment of these issues see the following reviews (Ambinder, 2007; Thorley-Lawson and Allday, 2008; Vereide and Sugden, 2009).
EBV latency in its natural context
All of the above studies reflect analyses of EBV latency in the highly abnormal context of EBV-induced tumors or LCLs. But what does latency look like in the majority case - i.e. persistent EBV infection of healthy seropositive individuals? Thorley-Lawson and colleagues have shown that in the peripheral blood of such individuals, EBV is found exclusively in resting memory B cells (CD20+, CD27+, CD5-, CD10-, IgD-), and not in activated lymphoblasts (Babcock et al., 1998). Analysis of viral gene expression in these resting memory B cells failed to detect expression of the viral antigens associated with the EBV growth program (type III latency), save for the sporadic detection of EBNA1 transcripts which are initiated from Qp [for review see (Thorley-Lawson, 2001; Thorley-Lawson et al., 2008)]. The latter appear to correspond to circulating memory B cells that have recently undergone a round of cell division. Whether these EBV infected memory B cells are antigen experienced remains unresolved - however, it has been determined that they have undergone both isotype switching and somatic hypermutation, suggesting antigen selection.
How does EBV get into the memory B cell pool? Examination of tonsil tissue has revealed the presence of EBV infection in: (i) naive B cells (CD19+, sIgD+) which exhibit a lymphoblastoid phenotype and display type III latency; (ii) germinal center B cells which exhibit a type II latency program; (iii) proliferating memory B cells (EBNA1 only; type I latency); and (iv) plasma cells, in which EBV enters the lytic cycle, as discussed earlier (Thorley-Lawson et al., 2008). Thus, EBV infection in germinal center B cells largely recapitulates the viral gene expression program in EBV-positive Hodgkin's lymphoma tumors, while EBV infection in cycling memory B cells recapitulates the latency program observed in endemic BL tumors. Why has EBV evolved this complicated set of latency programs? Based on these snap shots of EBV gene expression in distinct B cell populations, it has been proposed that EBV hijacks normal B cell differentiation to gain access to the memory B cell pool (see Fig. 2). This model postulates that virus infection of naive B cells results in the generation of activated lymphoblasts that appear phenotypically similar to antigen activated lymphoblasts. Some of these virus infected lymphoblasts then form and/or participate in germinal center reactions, in which LMP-1 expression provides signals that mimic CD4+ T cell help (CD40 signaling) while LMP-2a provides signals that mimic the essential survival signals normally provided by the B cell receptor - allowing EBV infected B cells to transit through the germinal center reaction. The only other viral antigen expressed in EBV infected germinal center B cells is EBNA1, which serves to maintain the viral episome in proliferating germinal center B cells. From these germinal centers, EBV infected memory B cells and plasma cells emerge. The memory B cells that exit the lymphoid tissue and enter the periphery then stop cycling and shut off EBNA1 gene transcription - presumably due to the accumulation of EBNA1 which negatively feedbacks on Qp, shutting down EBNA1 gene transcription. Analysis of viral genome methylation in B cells isolated from peripheral blood of healthy seropositive individuals revealed that, like the status of the viral genome in BL and Hodgkin's lymphoma tumors, Cp and Wp are methylated while Qp is protected from methylation (Paulson and Speck, 1999).
Figure 2.

Model depicting the different EBV latency programs expressed during the progression of infected naïve B cells through a germinal center reaction and establishment of latency in memory B cells. Also shown is virus reactivation linked to plasma cell differentiation. See text for additional details.
In summary, EBV has evolved a complex set of latency programs that allows the virus to navigate from infection of a naïve resting B cells (the dominant B cell population that the virus encounters during the initial stages of infection) to ultimately gain access to the memory B cell reservoir. While there might be a role for antigen in this process, the simplest model involves EBV driving the differentiation of B cells in the absence of any normal signals from the B cell receptor or CD4+ T cells. In this process, EBV takes advantage of an ancient host antimicrobial strategy – host cell-driven DNA methylation – to strategically silence viral gene expression as infection progresses from a naïve B cell to the germinal center reaction. In addition to methylation of the viral genome, modulation of NF-κB activity and manipulation of the Notch signaling pathway through interaction with the host DNA binding protein RBP-Jκ/CBF-1/CSL, represent two key pathways targeted by EBV. The end goal, establishing latency in memory B cells, clearly affords the virus a long lived cell type in which to persist and avoid immune detection.
KSHV Latency and its control
KSHV: an overview
KSHV (also called human herpesvirus 8) was discovered in 1994 in a search for viral agents in Kaposi's sarcoma, a neoplasm of endothelial cells. Subsequent epidemiologic research affirmed the etiologic link to KS (see Cohen et al 2005 for review), but also revealed that phylogenetically, KSHV belongs to the lymphotropic herpesviruses. This triggered a search for KSHV DNA in a variety of lymphoproliferative diseases, and led to its linkage to two disorders of B cells: primary effusion lymphoma (PEL) and multicentric Castlemen's disease (MCD) (Cesarman et al; 1995; Soulier et al 1995). Although rare sequelae of KSHV infection, they reflect the fact that in healthy seropositive hosts, CD19-positive B cells appear to be the primary target of KSHV infection (Ambroziak et al 1995).
KSHV can efficiently infect many adherent cell types in culture (endothelial cells, fibroblasts, epithelial cells), including many cells in which infection is never observed in vivo (Vieira et al 2001 et al; Bechtel et al 2003). As in EBV, the default pathway following these in vitro infections is latency. Although some cells infected in this fashion are lytically inducible, many are not - indicating that not all cultured cell lines that allow viral entry support true latency. Nonetheless, numerous lines are available in which the complete viral replicative cycle can be observed.
Paradoxically, however, KSHV stocks do not initiate infection of established B cell lines – e.g. BL lines or EBV-induced LCLs (Bechtel et al 2003, Renne et al 1998). Why this is so is unclear, but it has been a major impediment to the study of lymphoid infection by KSHV. However, recently two groups have been able to observe infection of primary B cells in vitro. If such B cells are derived from the peripheral blood, they must first be activated by treatment with IL4 and CD40 ligand (Rappocciolo et al 2008). However, B cells derived from tonsils can be infected without such pretreatments, probably reflecting the fact that tonsillar cells are already highly activated in vivo (Myoung and Ganem, 2010). In both situations, and in striking contrast to EBV infection of primary B cells in vitro, no immortalization of B cells follows KSHV infection. In fact, KSHV latency in all cell types lacks immortalizing activity (Bechtel et al 2003; Ciufo et al 2004; Grossmann and Ganem, 2006) – indicating that KSHV has no analog of the EBV growth program (latency III). Indeed, none of the known EBV latency genes have homologs in KSHV.
The KSHV genome, as extracted from the virion, is a linear duplex of 165kb (Renne et al 1996). Coding regions bearing at least 87 open reading frames (ORFs) comprise the central 140kb of the genome, and are flanked by extensive, noncoding, GC-rich repeats (terminal repeats, TRs) (Lagunoff and Ganem, 1997). Following infection, as in EBV, the genome circularizes in the nucleus (Renne et al 1996) and, in latently infected cells, is maintained as a chromatinized nuclear plasmid. Latent DNA replication proceeds from an origin (ori P) in the TRs (Ballastas et al 1999; 2001; Hu et al 2002; Grundhoff and Ganem 2003), and the genome is maintained at relatively low copy numbers.
In KSHV, a single viral gene, (RTA, for replication and transcription activator) controls the switch from latency to lytic growth (Sun et al 1998; Lukac et al 1998, 1999). RTA is a sequence-specific DNA binding protein that can function as a transcriptional activator, and null mutations engineered into full-length KSHV genomes result in viruses that cannot reactivate from latency (Xu et al 2005). RTA's transactivation function controls reactivation by turning on numerous IE and DE promoters,in a fashion formally analogous to the role of Zta in EBV. (KSHV does encode a distant homolog of Zta, but this protein, termed RAP or K8, functions primarily in lytic-cycle DNA replication in KSHV). High-affinity RTA binding sites exist in the promoters of several key lytic-cycle genes, and also near the origins of lytic-cycle DNA replication (Song et al 2003) (where it is presumed that RTA binding promotes an open chromatin configuation favorable to replication). However, many genes that are activated by RTA lack such sites. It is thought that for most such sites, RTA is recruited to the promoter via protein-protein interactions with other, predominantly host-encoded, transcription factors. Chief among these is RBP-Jκ or CSL-1, the main effector of the Notch signaling pathway; ablation of RBP-Jκ does not appear to affect establishment of latency, but efficiently blocks lytic reactivation (Liang et al 2002, 2003). [This is a very different role for RBP-Jk than is observed in EBV latency; however, at least one report suggests that a latent KSHV protein may also interact with RBP-Jk (Lan et al, 2005)]. RTA also interacts with C/EBPα, (Wang et al 2003a,b) Oct-1 (Sakakibara et al 2001; Carroll et al 2007), KRBP (Wang et al 2001) and other transcription factors to target the protein to additional promoters, as well as to components of the pol II-associated transcription machinery and to histone acetyl transferases (see (Deng, Liang and Sun, 2007) for review).
Many environmental signals can trigger the switch from KSHV latency to lytic growth in vitro – phorbol esters (Renne et al 1996b), histone deacetylase inhibitors (Miller et al 1996), interferon gamma and other cytokines (Chang et al 2000; Mercader et al 2000), proteasome inhibitors (Brown et al 2003), inhibitors of NFkB activation (Brown et al 2003; Grossmann and Ganem 2008), upregulation of protein kinase A (Chang et al 2005; Yu et al 2007a) or pim-1 and pim-3 kinases (Cheng et al 2009), expression of trancription factor XBP-1s (Yu et al 2007b; Wilson et al 2007); dopaminergic agonists (Lee et al 2008) and many others (Yu et al 2007a). It is striking that this list so extensively reduplicates the list of signals that trigger EBV reactivation – TPA, HDAc inhibitors, NFkB inhibition and XBP-1s induction all disrupt latency in both pathogens. Although the physiologic signals that trigger lytic KSHV reactivation in vivo are unknown, we do know that periodic “spontaneous” reactivation from latency occurs regularly, both in cell culture (Renne et al 1996) and in vivo (Vieira et al 1997; Pauk et al 2000; Casper et al 2007).
Presumably, all of the above inducers of lytic reactivation act by initiating biochemical events that ultimately converge on activation of the RTA promoter. (Don – Zta promoter lacks CpG and thus methylation doesn't appear to play a role - but EBV Rta promoter is methylated, so there is a parallel there), this promoter is methylated during latent infection, but undergoes demethylation in response to signaling events linked to induction (Chen et al 2001). It seems likely that other epigenetic marks (e.g. histone modifications) are also involved in regulation of RTA expression, but this is an area that has been relatively understudied. It may also be that reactivation is subject to additional controls beyond induction of RTA transcription. For example, RTA is heavily phosphorylated (Lukac et al 1999), and it is certainly possible that modulation of this or other post-translational modifications of RTA play adjunctive roles in regulating the efficiency with which latency is disrupted. RTA is also subject to regulation by viral proteins and miRNAs expressed during latency; these will be considered in the following section.
The Latency program of KSHV
Historically, definition of the latent KSHV transcriptional program has largely been carried out by studying B cell lines derived from primary effusion lymphomas (PEL). This led to recognition of a major latency locus that is abundantly and consistently transcribed in all latently infected cells (Figure 3). This region includes four open reading frames, encoding LANA (latency-associated nuclear antigen), v-cyclin, v-FLIP (Flice-inhibitory protein) and kaposins A, B and C (Figure 3). The first three genes are under the control of a single promoter, (the LANA promoter or LTc) which generates a series of coterminal mRNAs via differential splicing (Dittmer et al 1998; Sarid et al 1999; Talbot et al 1999). A second promoter (the kaposin promoter or LTd), located just downstream of LANA, encodes a spliced transcript encoding the kaposins (Li et al 2002; Pearce et al 2005; Cai & Cullen 2006), and can also generate a bicistronic RNA for v-cyclin and v-FLIP. This promoter also governs the expression of 12 pre-miRNAs (Fig 5), which can be processed to yield a total of 18 mature miRNAs (Cai et al 2005; Samols et al 2005; Cai et al 2006; Pfeffer et al 2005; Grundhoff et al 2008; Umbach and Cullen 2010). All of these latent products have been found to be expressed in KS spindle cells as well as PEL cells (Fakhari and Dittmer 2002; Dittmer 2003; Marshall et al 2007).
Figure 3.
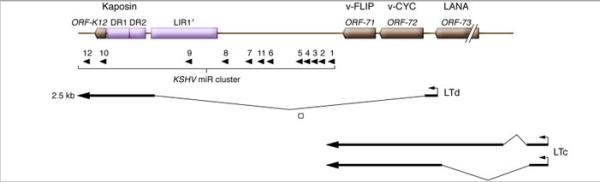
Major latency locus of KSHV. Top line: major ORFS of the locus. ORF-73 encodes LANA; ORF-72 encodes v-cyclin (v-CYC); ORF-71 encodes v-FLIP; ORF-K12 encodes kaposin A; DRs 1 and 2 encode direct repeats in which translation of kaposins B and C initiate. LIR, long interspersed repeats of unknown function. Middle panel: KSHV microRNA (miR) cluster, with pre-miRNAs indicated by arrowheads. Bottom panels: Structures of mRNAs directed by the kaposin (or LTd) promoter and by the LANA (or LTc) promoter. Figure modified with permission from Ganem (2010) J Clin Invest, in press.
A second, ulinked locus expressed in latent PEL cells encodes the v-IRF3 (or LANA-2) protein, a member of the IRF superfamily that dominantly inhibits the function of certain cellular IRFs and thereby blocks interferon induction (Rivas et al 2001). It has also been suggested that this protein can impair p53 function, but how this is achieved is not known. This gene has to date been found to be expressed only in PEL cells and not in KS cells –indicating that some latency genes may be lymphoid-specific.
Very recently (Chandriani and Ganem, 2010), a third latent locus has been identified – that encoding the K1 protein. This gene is on at exceedingly low levels in latency, and is upregulated during lytic growth. These properties made it difficult to identify as a latent gene, since in most latent cell lines the background level of spontaneous reactivation made it difficult to ascertain whether K1 mRNA was coming from latently- or lytically- infected cells. Limiting dilution RT-PCR experiments have resolved this controversy, and demonstrate that K1 is indeed expressed in latency. K1 is an interesting protein because it is a constitutively acting signaling molecule that mimics signaling via the B cell antigen receptor (Lee et al 1998; Lagunoff et al 1999; Lee et al 2002; Lee et al 2005). This is formally analogous to the output of EBV's LMP2, and raises the question of whether K1 and LMP2 play similar roles in the natural history of persistent B cell infection. At present, however, too little is known of KSHV latency in its human host to know if this analogy is accurate – we do not know, for example, whether KSHV positive B cells must traverse a germinal center, or whether they go on to establish residence in long-lived memory B cells.
In the following sections, we focus on the latent KSHV gene products whose functions have been most intensively investigated.
(i) LANA
The best understood of these functions is LANA (Latency-associated nuclear antigen), which clearly plays a role in the persistence and segregation of the latent viral episome that is formally analogous to that of EBNA-1 (Ballastas et al 1999). LANA is a large polypeptide with unique N- and C-terminal domains separated by central region composed of a series of acidic repeats. The C-terminal domain contains a sequence-specific DNA binding region that recognizes conserved sequences within the terminal repeats (TRs) of viral DNA (Ballestas & Kaye 2001; Garber et al 2001, 2002; Cotter et al 2001). This sequence represents the core of the replication origin of the latent viral plasmid (termed ori-P), and in transient assays, LANA can trigger rounds of DNA synthesis from oriP-bearing plasmids – in a fashion reminiscent of replication of the latent EBV origin by EBNA-1 (Hu et al 2002; Lim et al 2002; Grundhoff & Ganem 2003). Like EBNA-1, LANA also plays a role in the segregation of viral plasmids to daughter cells in proliferating cells (Ballestas et al 1999; Cotter et al 1999). The N-terminal domain of LANA contains motifs primarily responsible for its adherence to metaphase chromosomes (Piolot et al 2001), although a second chromosome binding region in LANA's C terminal domain has also been identified (Viejo-Borbolla et al 2005; Kelley-Clarke et al 2007a, 2007b, 2009). Recent studies indicate that much of the chromosome-binding activity is due to interactions of the N-terminus of LANA with histones H2A and H2B (Barbera et al 2006). But LANA also interacts with other chromatin-associated proteins like Brd2/RING3 and Brd4 (Platt et al (1999) ; Viejo-Borbolla et al (2005); Mattsson et al (2002); Ottinger et al 2006) and meCBP2 (Krivithas et al 2002), whose contributions to this activity are still under study. Brd2 and Brd4 interactions are mediated by the C-terminal region of LANA, and it has been proposed that this interaction may bind LANA to interphase chromosomes via interactions between Brd2/4 and acetylated histones H3 and H4. meCBP2 binds to the N-terminal chromosome-binding region, and expression of the human homolog in mouse cells renders LANA able to bind to mouse chromosomes.
By interacting with mitotic chromosomes on the one hand and viral episomes on the other, LANA can effectively tether the KSHV genome to host chromosomes and allow KSHV DNA to “hitch a ride” to daughter nuclei during mitosis. This function is critical to viral persistence in rapidly proliferating cells, like PEL cells in vivo or immortalized cells in culture. In evaluating the role of this activity in KSHV biology, it is important to note that while latency in PEL cells is very stable, the same may not be true of other latently infected cells. When most actively growing cell lines (endothelial, epithelial or fibroblastic) are infected in culture, KSHV genomes are rapidly lost unless a genetic selection is applied for their maintenance (Grundhoff & Ganem 2004). The same is true for KS cells cultured directly from KS biopsies (Flamand et al 1996; Aluigi et al 1996; Dictor et al 1996). Thus, the LANA/oriP system functions inefficiently in most cells; however, experiments in cultured cells suggest that cis-acting epigenetic modifications can stablize the episome (Grundhoff et al 2004; Skalsky et al 2007a). The exact nature of these modifications has not yet been determined. Presumably, PEL cells have undergone these adaptations in vivo, while most KS spindle cells have not.
These findings have important pathogenetic implications. Immunohistochemical studies of early and late KS lesions show that early in the evolution of KS, only ca 30% of spindle-like cells harbor latent KSHV (Dupin et al 1999), while in advanced lesions nearly all spindle cells are latently infected (Staskus et al 1997). Two inferences flow from this fact. First, latently infected KS spindle cells appear to have a growth or survival advantage in vivo, even though they are not transformed in vitro. That is, whatever advantage KSHV latency confers in vivo, it is much less dramatic than that which our current cell culture assays are geared to detect. It may be as subtle as a modest prolongation of lifespan, or an incremental increase in resistance to apoptosis induced by exposure to pro-apoptotic signaling molecules in the microenvironment. Second, if latency is unstable, and if it confers a survival or growth advantage, then either (i) a cell must undergo the epigenetic changes that stabilize the plasmid (as in PEL) or (ii) episomes lost during cell division must be replaced by exogenous reinfection. Interestingly, evidence consistent with the latter event has been accumulating in recent KS research (see Ganem (2010) for review).
What does all this mean for latency in its natural context- the healthy KSHV seropositive individual? We don't yet know, since the nature of the infected B cell in latency is still not established. If, as in EBV, latency primarily resides in infrequently dividing memory B cells, then there may have been no compelling evolutionary selection for a highly efficient system for plasmid maintenance. Interestingly, though the EBV EBNA-1/oriP system was initially believed to function very efficiently, it too displays remarkable instability in the absence of a genetic selection – suggesting that the EBV and KSHV latency maintenance machines may function at comparable efficiencies as well as by common mechanisms.
In addition to its function in viral plasmid maintenance, LANA has additional activities that may also more directly influence the behavior of latently infected cells. Isolated LANA expression outside the context of infection reveals that the protein inhibits p53-mediated transactivation–and therefore can block p53-mediated cell cycle arrest or apoptosis (Friborg et al 1999). Consistent with these activities, expression of LANA in primary endothelial cells has been shown to extend their survival in culture (Watanabe et al 2003) though it does not immortalize them. However, the finding that human PEL cells expressing LANA still respond to doxirubicin-induced DNA damage with p53 activation and growth arrest indicates that LANA does not ablate the p53 pathway entirely (Petre, Sin and Dittmer 2007). LANA also binds Rb and impairs Rb function (Radkov et al 2000), though evidence from PEL cells also suggests that this impairment is only partial (Platt et al 2002). Another LANA interaction partner is GSK-3β, a kinase that targets the cytosolic protein β-catenin for ubiquitination and proteasomal destruction (Fujimoro et al 2003). LANA relocates GSK-3β to the nucleus, thereby allowing cytosolic β-catenin to escape destruction; it can then oligomerize with the transcription factor LEF, and the resulting heterodimer activates a proliferative gene expression program that includes c-myc, c-jun and cyclin D. In accord with this, expression of LANA has been found to promote S phase entry.
LANA also has regulatory effects on transcription that potentially could influence the biology of latently infected cells. Stable expression of LANA is associated with numerous changes in host gene expression, as judged by array profiling (Renne et al 2001). When LANA is directly bound to DNA, it can repress transcription of adjacent reporter genes (Schwam et al 2000), an activity that has been linked to recruitment of mSin3 repressor complexes (Krivithas et al 2000) as well as to recruitment of epigenetic modifiers like meCBP2, Dnmt3a (Shamay et al 2006) and the histone methyltransferase SUV39H1 (Sakakibara et al 2004). Indeed, LANA expression has been shown to silence expression of TGF-β receptor II, impairing TGF signaling in latently infected cells, resulting in insensitivity to the growth-inhibitory effects of TGF-β (DiBartolo et al 2008).
LANA has also been proposed to negatively regulate viral lytic gene transcription (Li et al 2008), and thereby influence the control of latency. Potential mechanisms include (i) repression of the RTA promoter, to which it may be indirectly tethered via interactions with Sp1 and histones H2A/H2B (Lu, Day, Gao and Lieberman, 2006; Lan et al 2004) (ii) direct binding of LANA to RTA (Lan et al 2004); (iii) binding of LANA to RBPJk, an essential cofactor of RTA, with possible targeting of repressive LANA complexes to RBP-Jk sites in critical lytic promoters (Lan et al 2005) (Consistent with a role for LANA in suppression of lytic gene expression, LANA's repressive actions on the lytic program are also antagonized by its phosphorylation by the kinase pim-1, whose expression triggers and is required for lytic reactivation (Cheng et al 2009. Bajaj, B et al 2006). It is important to realize, however, that most cells infected by LANA deletion mutants of KSHV do not spontaneously enter the lytic program (Li et al 2008), indicating that there is considerably more to latency control than negative regulation of the lytic cycle by LANA.
V-cyclin
The major latency locus also encodes v-cyclin, the product of ORF72 and a viral homolog of cellular cyclin D. Like its cellular homolog, v-cyclin (Chang et al, 1996) activates cdk6, but unlike cyclin D it is poorly active on cdk4. Although both cyclins can trigger cdk6-mediated phosphorylation of Rb, v-cyclin expands the substrate specificity of the enzyme, leading to phosphrylation of p27, histone H1, Id-2 and cdc25a (Gooden-Kent et al 1997; Li et al 1997). V-cyclin expression can induce S-phase entry in quiescent 3T3 cells, and can overcome an Rb-mediated growth arrest induced by cdk-inhibitors (Swanton et al 1997). In fact, the v-cyclin/cdk6 complex is less sensitive to inhibition by cdk inhibitors like p27; moreover, phosphorylation of p27 by v-cyclin-cdk6 targets it for degradation in the proteosome, further releasing cdk6 from p27 control. (Ellis et al 1999, Mann et al 1999).
Because cellular cyclin D is so frequently involved in human cancer, many have presumed that v-cyclin expression is central to KS oncogenesis, and the term “oncogene” is often applied to this locus. However, there is very little evidence supporting this claim. Certainly, the in vitro activities cited above are consistent with cellular growth promotion. But direct experimental observations on human KSHV-associated tumors fails to confirm expectations derived from such in vitro expression studies. For example, despite the fact that v-cyclin overexpression destabilizes p27 in cultured cells, PEL tumors expressing v-cyclin often display abundant p27 expression (Carbone et al 2000). And the fact that many PEL tumors delete p16INK4a suggests that despite the action of v-cyclin, Rb function is not fully abrogated in latently infected B cells; further mutational lesions must accumulate in this pathway for full transformation. What v-cyclin does in the economy of the cell prior to the advent of such mutations remains unclear, but recent studies of v-cyclin expression in primary endothelial cells are instructive. While v-cyclin expression triggered enhanced S phase entry (and centrosome amplification), this was followed by activation of a DNA damage response characterized by induction of antiproliferative checkpoint mechanisms (phosphorylation of ATM and Chk2 kinases), p53 stabilization and potent growth arrest, with later induction of cellular senescence. Many of these features were reproduced by infection of primary ECs with KSHV itself, indicating that this was not the result of v-cyclin over-expression. Moreover, consistent with these data, Vershuren et al (2002) showed that in cultured cells, loss of p53 allowed cells to survive in the presence of ongoing v-cyclin expression. Taken together, the findings indicate that the primary effects of v-cyclin exposure are to induce replicative stress. Why such a function was selected in viral evolution is unclear, as it would appear to limit proliferation of latently infected cells. It seems safe to say that we are deeply in the dark about the true role of this enigmatic protein in KSHV infection in vivo. MHV68 encodes a homolog of this protein, and examination of that system (see below) has associated some phenotypes with mutational loss v-cyclin expression. At the very least, that system may provide a way to generate some new hypotheses about how v-cyclin may function in KSHV.
v-FLIP
The third gene in the major latency cluster encodes v-FLIP, a small polypeptide composed of two tandem death effector domains (DEDs). The acronym stands for FLICE inhibitory protein, with FLICE being an early name for Fas-activated caspase 8. The name v-FLIP derives from the fact that the protein is homologous to cellular FLIP proteins, which block caspase 8 activation by the Fas-FasL system by interacting with adaptor proteins like FADD via their DEDs (Thome et al, 1997). Early studies suggested that KSHV v-FLIP also shares this activity (Djerbi et al 1999, Belanger et al 2001), but many groups have failed to affirm this result (cf. Chugh et al 2005). Instead, there is general consensus that KSHV v-FLIP is a potent activator of NFκB (Chaudhary et al 1999). This is achieved via binding of the protein to the NEMO (or γ) subunit of the IkB kinase (IKK) (Liu et al 2002; Field et al 2003; Bagneris et al 2008). This binding is an activating event; the resulting phosphorylation of IkB triggers its ubiquitin-dependent proteolysis in the cytoplasm, releasing its bound NFκB subunits, which can then migrate into the nucleus to activate gene expression. The alternative pathway of NFκB activation is also upregulated by v-FLIP (Matta and Chaudhary 2004).
In the context of latency, this activation of NFkB has a number of consequences. First, NFκB activation antagonizes entry into the lytic cycle in many (but not all) cells, thereby stabilizing latency. Chemical (Brown et al 2003) or genetic (Grossmann and Ganem, 2008) inhibition of NFκB activation enhances spontaneous production of lytic markers, as does siRNA-mediated knockdown or mutational ablation of v-FLIP expression (Zhao et al 2007; Ye et al 2008) – clearly establishing that FLIP-mediated NFκB activation antagonizes lytic gene regulation. Recent studies by Izumiya et al (2009) reveal that active NFκB directly inhibits RTA's ability to activate lytic genes, by (i) binding and sequestering the RTA cofactor RBP-Jκ, and (ii) blocking the ability of RBPJκ to bind DNA, an important mechanism by which RTA is targeted to many viral lytic promoters. While it is satisfying that a latent gene can stabilize latency in this fashion, a paradox remains: during the lytic cycle, there is dramatic activation of NFkB (Sgarbanti et al 2004; Sadagopan et al 2007; Grossmann and Ganem 2008), probably due to the expression of several viral genes, including ORF 75, ORF K15 (Konrad et al 2009) and perhaps the GPCR encoded by ORF74 (Schwarz and Murphy 2001). Why this induction does not interfere with the lytic cycle remains unclear - perhaps NFkB activation occurs after the bulk of RTA-mediated gene induction is complete; alternatively, other mechanisms may be induced that nullify or mitigate NFkB's inhibitory effects on RTA.
It's noteworthy that activation of NFκB figures prominently in the latency programs of all three gammaherpesviruses under consideration in this review. In EBV, LMP-1 is a strong constitutive activator of NFκB, and LMP-1 signaling inhibits lytic reactivation from latency (Adler et al 2002). Similarly, evidence from MHV68 suggests that, in that system as well, NFκB activation is necessary to stabilize latency (see below, and Krug et al 2007).
In addition to its stabilizing effects on latency, NFκB is known to activate a pro-inflammatory and anti-apoptotic program in many cell types. The inflammatory activity is due to upregulation of numerous cytokines and chemokines, while the cell survival signals are thought to be mediated by upregulation of Bcl-xL, A1, and TNF receptor associated factors (TRAFs) 1 and 2, among other factors. Direct examination of chemokine production (Xu and Ganem, 2008; Punj et al 2009) and gene expression (Sakakibara et al 2009; Thurau et al 2009) in v-FLIP transduced HUVEC cells affirms the induction of a large array of proinflammatory and antiapoptotic factors. Clearly, v-FLIP expression in latently–infected KS spindle cells is a major cause of the proinflammatory microenvironment that characterizes KS; a second viral latency protein, Kaposin B, is also thought to contribute importantly to this activity (see below). Since infected endothelial cells are not immortalized in vitro, it is difficult to assess the contribution of v-FLIP's antiapoptotic signaling to spindle cell lifespan, although it does make the cells more resistant to anoikis (Efklidou et al 2008) and to superoxide-induced killing (Thurau et al 2009). However, ongoing v-FLIP expression is critical for PEL cell survival – siRNA knockdown of vFLIP (or inhibition of NFκB activation) in otherwise immortal PEL cells triggers prompt B cell death (Guaspari et al 2004; Keller et al 2000).
Finally, NFκB activation by vFLIP has an additional consequence for infected endothelial cells: it causes them to undergo a rearrangement of the actin cytoskeleton that produces the elongated (“spindle”) shape so characteristic of infected endothelium in KS. (Grossmann et al 2006; Matta et al 2007). This shape change is not observed in latent infection of any other cell type, and presumably reflects induction of endothelial-specific gene products. Whether this is important to the phenotype of latency in endothelia, or is merely an epiphenomenon, is unclear.
The Kaposin locus
The other major transcription unit active in latency encodes the Kaposin family of proteins and a series of viral microRNAs. As summarized in Fig 3, the latent Kaposin promoter generates a major spliced mRNA whose body encompasses two sets of 23 nt. direct repeats (DRs) of GC-rich sequences followed by a short (60 codon) ORF called ORF-K12. (A second, lytic promoter is located just 5’ to the DRs, allowing further upregulation of the kaposins during lytic growth (Sadler et al 1999)). Initially, only ORFK12 was recognized as having coding potential, generating a two-transmembrane domain polypeptide now called Kaposin A. Subsequently, however, it was recognized that the DRs can also engender protein products via translational initiation at CUG codons in their midst. One of these CUG inititators generates Kaposin B, a small polypeptide derived largely by translation of DR1 and DR2 sequences; a second CUG, located in a different reading frame, allows DR1 and DR2 to be translated in-frame with Kaposin A to generate a fusion protein called Kaposin C (Sadler et al 1999).
Kaposin A
Kaposin A is a small hydrophobic polypeptide found on intracellular and cell surface membranes (Tomkowicz et al 2002). It initially attracted attention because overexpression of this ORF in immortalized but untransformed rodent fibroblasts led to (inefficient) transformation in culture, as judged by growth in soft agar; the resulting clones are tumorigenic in nude mice (Muralidhar et al 1998). This makes kaposin A an obvious candidate for a role in growth deregulation in KS and PEL, though the fact that KSHV latent infection does not immortalize or transform cells means that the result cannot be directly transposed to explain KSHV tumorigenesis in vivo. How Kaposin A functions has remained something of an enigma. The best data indicate that Kaposin A binds cytohesin-1, a GEF for ARF family GTPases and a regulator of integrin-mediated cell adhesion (Kliche et al 2001). Genetic evidence suggests that this pathway is involved in rodent cell transformation, but the relationship of such transformation to the growth advantage of KSHV-infected cells in vivo is unknown. More recently, Kaposin A has been found to interact with Septin 4, a putative proapoptotic caspase 3 activator; kaposin A binding antagonizes septin 4's proapoptotic effect in transfected cells in culture, suggesting a potential role for kaposin A in lifespan prolongation (Lin et al 2007). However, such a role has yet to be demonstrated in vivo.
Kaposin B
This small protein consists largely of sets of 23 aa proline- and arginine-rich repeats derived from translation of DRs 1 and 2. Its simple amino acid composition and lack of known catalytic motifs suggested that it likely serves as a scaffold or adaptor protein, prompting searches for interacting proteins. The best understood of these is MAP kinase–associated protein kinase 2 (MAPKAPK2 or MK2), a target of the p38 signaling pathway (McCormick and Ganem, 2005). MK2 exists in the nucleus as an inactive enzyme; when the p38 MAP kinase pathway is activated (by inflammatory signals, oxidative stress or osmotic shock), p38 phosphorylates MK2, which activates its kinase activity and also causes it to translocate to the cytoplasm, where it phosphorylates many of its downstream targets (Duraisamy et al 2008). Some of these, like tristetraprolin (TTP), act to stabilize mRNAs containing AU-rich elements (AREs) (Sandler and Stoeklin 2008). AREs are found in many cytokine- and growth-factor mRNAs, and hence activation of MK2 is linked to upregulation of cytokine production. Importantly, binding of MK2 by kaposin B is an activating event, promoting its phosphorylation by p38 and thereby stimulating cytokine release during latency (McCormick and Ganem, 2005). As such, Kaposin B is the second latent KSHV gene that contributes to the proinflammatory microenvironment in KS (v-FLIP being the first). The two genes have complementary modes of action, with v-FLIP upregulating cytokine transcription while Kaposin B extends the half-life of the resulting transcripts. Together, these genes provide a satisfying explanation for the inflammatory nature of KS lesions.
However, they raise a deeper paradox. In most large DNA viruses, gene products have evolved to blunt rather than foster host inflammatory and immune responses (Johnston and McFadden 2003). Viral evolution is driven principally by factors influencing virus replication and spread; pathogenesis and disease is a sideshow, of relevance only in situations (e.g. respiratory tract infection or diarrhea) where disease promotes viral spread. The rarity of KS relative to infection by KSHV (see above) means that it can play no major role in the evolutionary shaping of the viral genome. Accordingly, we infer that something about an inflammatory microenvironment is advantageous to KSHV replication and spread. For example, the recruitment of suitable B cells or monocytes to sites of infection may facilitate more extensive lymphoid infection; additionally, local permeability changes may help assure egress and dissemination of infected cells. Sadly, the absence of a convenient animal model of KSHV infection has hampered the definitive examination of this interesting issue.
Kaposin C
To date, no published studies have examined kaposin C in detail. As such, we do not know if the signaling activities attributed to kaposins A or B are shared with their membrane-bound relative.
Viral miRNAs
As shown in Fig 3, the Kaposin transcription unit also encodes 12 pre-miRNAs, 10 of which emanate from the intron of the latent Kaposin mRNA (the remaining 2 are found in the body of the transcript). (Cai et al 2005; Samols et al 2005; Cai et al 2006; Pfeffer et al 2005). These pre-miRNAs are conserved in all isolates of KSHV (Marshall et al 2007), but are not conserved in other herpesviruses (Schafer et al 2007). The 12 KSHV pre-miRNAs actually engender 18 mature miRNAs (Umbach and Cullen 2010), primarily because a number of them can donate both strands of their hairpin precursors to RISC. (In addition, the RNA precursor to miR-K10 undergoes an RNA editing event in its seed sequence that generates related but distinct miRNAs; Gandy et al 2007). Because they are expressed in latency, they have the opportunity to target both cellular and viral mRNAs and could influence the phenotype of latently infected cells in an important way.
Studies with reporter genes and latently infected cell lines have suggested that several KSHV miRs may downregulate thrombospondin, a known antagonist of angiogenesis – as such, they could contribute to the neovascular phenotype of KS (Samols et al 2007). One of the viral miRNAs, miRK11, shares seed sequence identity with a lymphoid-specific host miRNA (miR155) whose targets affect B cell differentiation (Skalsky et al 2007b; Gottwein et al 2007); this miRNA may play important roles in B cell infection and possibly in PEL development. Similarly, several viral miRNAs have been shown to regulate the stability of latency. One, miRK9-3p (formerly K9*), targets a sequence in the 3’UTR of RTA; functional inactivation of this miRNA leads to a 2-3 fold upregulation of spontaneous lytic reactivation (Bellare and Ganem, 2009). By contrast, another viral microRNA, miRK5, targets a host function (BCLAF-1) that suppresses lytic reactivation 2 fold; as a result, this miRNA modestly enhances such reactivation (Ziegelbauer et al 2009). The latter phenotype, whose molecular basis is still not understood, probably contributes to maintaining the reversibility of latency. It's important to note that viral microRNAs are not the primary determinants of the regulation of latency – that role belongs to the transcriptional regulation of RTA. Rather, they appear to be ancillary effectors that allow for fine-tuning of the process. An attractive formulation envisions that miRK9-3p, for example, is designed to repress aberrant RTA transcripts arising from minor stochastic variations in the basal level of transcription (Bellare and Ganem, 2009). In this way, the miRNA prevents such “transcriptional noise” from triggering inappropriate entry into the lytic cycle.
MHV 68 Latency and its control
MHV68- an overview
Murine gammaherpesvirus 68 (MHV68, also referred to as γHV68 and MuHV4) was first isolated 30 years ago from bank voles and field mice in Eastern Europe (Blaskovic et al., 1980). Subsequent work indicates that wood mice are the likely principal reservoir for the virus (Blasdell et al., 2003). The MHV genome is estimated to have diverged from its primate counterparts ca 60 million years ago (McGeogh et al 2005). Its sequence reveals that it is 118Kb in length and is predicted to encode 79 ORFs, (Virgin et al 1997) (see Fig 4, in which the MHV68 genome is aligned with that of KSHV). In vitro, MHV68 infection grows readily in fibroblasts, and unlike EBV or KSHV, its default pathway in vitro is lytic replication. Viral growth in vitro is rapid and efficient, generating high-titer viral stocks. In addition, genetic systems based on homologous recombination allow facile genetic engineering of MHV68, making possible the construction of mutant viruses for virtually any viral gene. Indeed, transposon mutagenesis analyses have already pinpointed most of the genes that are dispensable for lytic growth in vitro, and identified genes that selectively affect replication and pathogenesis in the intact host. (Moorman et al., 2004; Song et al., 2005).
Figure 4.

Alignment of the MHV68 and KSHV genomes, depicting the large blocks of conserved genes interspersed with genes unique to each virus (K genes for KSHV - exception, K3 and K5 which are homologous the K3 gene in MHV68 - and M gene for MHV68). Shown below is data compiled from two independent transposon mutagenesis screens of the MHV68 genome, which identified genes involved in virus replication in permissive fibroblasts. Red arrow depict genes essential for replication, while orange arrows depict genes the play an important role in replication but are not essential. Green arrows depict genes that are dispensable for virus replication in tissue culture.
Both primary B cells, as well as established murine B cell lines, are difficult to efficiently infect with MHV68 and are not generally permissive for virus replication. However, several established B cell lines have been shown to support latent infection upon infection MHV68 (Sunil-Chandra et al., 1993; Forrest and Speck, 2008; Liang et al., 2009) , and a single MHV68-positive cell line (S11) has been established from a MHV68 infected mouse (Usherwood et al., 1996). However, the major asset of the MHV68 system is the availability of a robust and genetically manipulable animal model in which both latency and lytic reactivation reproducibly occur. MHV68 readily infects laboratory mice (Mus musculus), in which it establishes a chronic infection that is harbored for life. Most experimental infections are initiated by intranasal or intraperitoneal inoculation. Following intranasal infection, lytic replication occurs transiently in the respiratory tract and spleen, but is generally cleared by about 2 weeks post-infection, leading to latency establishment in B cells (predominantly in the spleen), macrophages (especially in the peritoneum) and splenic dendritic cells (Flano et al., 2000; Weck et al., 1999b). Following peritoneal infection, lytic replication also transiently occurs in the spleen, followed by establishment of latency in the same sites as the respiratory infection. Early in latency (e.g. by day 16-18), large numbers of latently infected cells are generated, with up to 1 in 100 splenocytes showing evidence of latency. At this time, up to 10% of these cells are capable of spontaneous re-entry into the lytic cycle, as judged by their ability to generate infectious centers when cultured on permissive mouse embryo fibroblast (MEF) monolayers (Sunil-Chandra et al., 1992; Weck et al., 1999a, b).. However, with the passage of time (and the development of antiviral immune responses) the profile of latently infected cells changes.
Early in latency, virus infection is found in naive (sIgD+), germinal center (GL7+/CD95+) and isotype switched memory B cells (Flano et al., 2002; Willer and Speck, 2003), as well as macrophages and dendritic cells. However, as infection progresses, infection of naive B cells rapidly wanes and is largely absent by 3 months post-infection (Willer and Speck, 2003). Concomitant with the loss of MHV68 infected naive B cells, is a significant overall contraction of the pool of latently infected splenocytes - from a peak of ca. 1 in 100 cells to a steady state level of ca. 1 in 10,000 cells by 3 months post-infection. The contribution of non-B cell reservoirs to the pool of latently infected splenocytes also rapidly diminishes, and is nearly non-existent by 3 months post-infection (Willer and Speck, 2003). Thus, like EBV, MHV68 ultimately is found nearly exclusively in isotype switched B cells that have undergone a germinal center reaction and appear to reflect memory B cells. [Caveat: the absence of definitive markers for murine memory B cells makes this assignment more provisional in MHV68 than in EBV]. Interestingly, as latency evolves, the frequency with which latently infected cells support spontaneous lytic reactivation also declines (Weck et al 1999a), suggesting that changes in latency are qualitative as well as quantitative. These changes occur in both B cells and peritoneal macrophages, though they are much more profound in B cells. However, the molecular nature of the changes affecting spontaneous induction are unknown.
Host immune function and MHV 68 persistence
Early studies on MHV68, utilizing mice deficient for specific aspects of the host immune response, revealed that many aspects of the host response (e.g., CD4+ or CD8+ T cells, interferon gamma, or β2-microglobulin) are individually dispensable for resolution of acute virus replication [reviewed in (Virgin and Speck, 1999)]. However, most of these responses play an important role in controlling chronic MHV68 infection. For example, interferon gamma receptor null (IFNγR-/-) mice clear acute MHV68 replication in the lungs following intranasal challenge with similar kinetics to wild type mice. However, unlike wild type mice, IFNγR-/- mice go on to develop a severe arteritis that affects the great elastic arteries, as well as developing fibrosis in multiple organs including the spleen and lungs, eventually succumbing to virus infection (Weck et al., 1997). This argues that the detente reached between MHV68 and the host in healthy immunocompetent individuals is an uneasy truce that requires constant vigilance by many components of the host immune system.
Even though B cells are the primary site of long-term latency in normal mice, persistent infections can be established in B cell-deficient (MuMT) mice. Infection of such mice via intraperitoneal inoculation results in a robust infection that persists for the life of these animals, with many latently infected peritoneal macrophages (Weck et al., 1996). Interestingly, in B cell deficient mice, latently infected cells display high rates of lytic reactivation, even many months into the infection, suggesting a major role for B cells (and/or antiviral antibody) in the long-term control of reactivation. A significant percentage of infected MuMT mice succumb to infection by day 100, likely due to the inability of these mice to adequately control persistent virus replication (Weck et al., 1999a). Many of these problems can be controlled by administration of anti-MHV serum, affirming the importance of humoral immunity in the control of chronic MHV68 infection (Gangappa et al 2002a).
Similarly, the absence of either CD4+ or CD8+ T cells, leads to chronic MHV68 infections that involve low level persistent virus replication (Virgin and Speck, 1999). Analyses of CD8+ or CD4+ T cell deficiency points toward production of IFNγ by these cells as an important effector that limits virus reactivation and/or persistent replication, especially in infected macrophages (Tibbetts et al., 2002). The absence of CD8+ T cells leads to prolonged virus replication in peritoneal macrophages compared to wild type mice (Dutia et al., 1997; Ehtisham et al., 1993; Tibbetts et al., 2002). This phenotype is even more pronounced in IFNγ null mice, and subsequent studies have shown that IFNγ can limit virus reactivation from latently infected peritoneal macrophages upon co-cultivation with MEFs (Tibbetts et al., 2002). Interestingly, there is evidence that a viral protein, M1, can elicit a selective expansion and activation of CD8+ T cells bearing the Vβ4 element (Tripp et al., 1997). Recombinant M1 protein induces Vβ4+ CD8+ T cells to express high levels of IFNγ (Evans et al., 2008). Thus, this population of CD8+ T cells may contribute significantly to the levels of IFNγ in vivo.
Similarly, analyses of the role of CD4+ T cells in controlling persistent virus replication shows that IFNγ is required (Sparks-Thissen et al., 2005). As for CD8 cells, inhibition by CD4 cell-generated IFNγ is targeted to latently infected macrophages (Steed et al., 2007; Steed et al., 2006); one of the targets of IFNγ is the MHV68 lytic switch protein Rta encoded by gene 50 (Goodwin et al.). The consequences of loss of IFNγ control of MHV68 infection, as briefly discussed above, are quite severe. IFNγR-/- mice develop a severe vasculitis upon MHV68 infection, along with fibrosis of multiple organs (Dutia et al., 1997; Ebrahimi et al., 2001; Evans et al., 2008; Weck et al., 1997). Thus, this is a clear demonstration of the importance of a host mechanism that plays a critical role in keeping a chronic gammaherpesvirus infection in check.
Role of NF-κB in MHV68 latency
Normal B cell responses are dependent on the activation of NF-κB for the initiation of a germinal center reaction, where both antigen stimulation through the B cell receptor and CD4+ T cell help mediated through CD40 are required. As discussed above, the EBV LMP-1 and KSHV vFLIP genes both act to upregulate NFkB activity, and this upregulation is important in the stabilization of the latent state. The importance of NF-κB activation in the establishment of chronic MHV68 infection is inferred from studies using a recombinant MHV68 expressing a mutant form of IκBα (IκBαM) that lacks two key serine residues. These serines serve as phosphorylation targets for IkB kinase (IKK), and upon phosphorylation signal the proteasomal degradation of IκBα and release of cytoplasmic NF-κB to the nucleus, where it can activate gene expression. Expression of IκBαM from the aforementioned virus prevents activation of NF-κB, and severely inhibits establishment of MHV68 latency in the spleen (Krug et al., 2007). Consistent with a role for NF-κB in establishment of MHV68 latency, it has been shown that inhibiting NF-κB activation triggers MHV68 reactivation from latency (Brown et al., 2003) - suggesting that NF-κB plays an important role in the decision between replication and latency. In addition, over-expression of the NF-κB subunit p65 inhibits MHV68 lytic replication in permissive cells, further arguing for an active role of NF-κB in promoting latency by suppressing virus replication (Brown et al., 2003).
A shortcoming of many of the above studies has been the reliance on over-expression of NF-κB subunits. However, a recent study (Krug et al., 2009) examining the role of the NF-κB p50 subunit, using knockout and mixed bone marrow chimeric mice infected with MHV68, provides further support for a role of NF-κB in suppressing reactivation of MHV68. In that study, the absence of p50 resulted in ongoing persistent virus replication in the lungs of infected mice at times when no virus replication could be detected in wild type infected mice. This was not due to immune defects in p50 null mice, since it was also observed in mixed bone marrow chimeric mice reconstituted with both p50-sufficient and p50-deficient bone marrow. The latter result argues for a cell intrinsic role of p50 in suppressing MHV68 reactivation from latency. A role of NF-κB in suppressing MHV68 reactivation is further supported by analyses of chronic MHV68 infection in CD40-/- mice, where significantly higher levels of persistent virus replication in the lungs can be detected at 2-3 months post-infection (Willer and Speck, 2005). Furthermore, in mixed bone marrow chimeric mice reconstituted with wild type and CD40-/- bone marrow, MHV68 infection of CD40-deficient B cells is lost over time - arguing for an active role of CD40 signaling in maintaining latency (Kim et al., 2003). Thus, just as in EBV and KSHV infection, NF-κB activation plays a critical role in latency in MHV68 infection. What is missing in the MHV68 case is an understanding of exactly how the virus manipulates the NF-κB pathway – i.e. which viral gene products are involved, and how they act.
However, there are some discordant notes in this symphony. It has been reported that the MHV68 LANA protein may down-regulate NF-κB activation, a function mediated through an unconventional SOCS-box motif in LANA (Rodrigues et al., 2009). This motif can target ubiquitination and proteosomal degradation of p65/RelA through assembly of an ElonginC/Cullin5/SOCS-like complex. Notably, mutations in the MHV68 LANA protein that disrupt this function prevent efficient establishment of MHV68 latency – this is counter to the above data that upregulation of NFkB promotes latency, and suggests that there is more to understand about the role of NFkB in the modulation of latency. Similarly, in KSHV, Grossmann and Ganem (2008) found that not all cell lines display enhanced lytic reactivation following inhibition of NFkB activation. At present, no explanation exists to reconcile these discrepancies.
The latency program of MHV68
Because (until rather recently) only a single B cell line harboring MHV68 has been widely available for study, there has been a paucity of in vitro latency systems that would allow rigorous, high-resolution characterization of the transcriptional program, as has been done for EBV and KSHV. As such, it has been harder to reach agreement on the details of the latent transcriptional program(s) in MHV68, which are dependent upon RT-PCR analyses of latently infected splenocytes and peritoneal macrophages. Spontaneous lytic replication and/or abortive infection in minor subpopulations of such cells, however, complicate interpretation of such analyses. For this reason, and for brevity, in this review we will not survey the complete roster of MHV68 gene products that have been posited to play roles in the establishment or disruption of latency. Some of these genes – for example those encoding homologs of GPCRs or Bcl2 – have interesting or subtle effects on reactivation from latency, but their homologs in EBV or KSHV are clearly lytic-cycle genes, raising questions as to how to interpret their functions in MHV68. Rather than confront this interpretive dilemma, we will focus instead on the small number of MHV68 genes that (i) are expressed by RT-PCR in latently infected tissues; and (ii) have homologs in other gammaherpesviruses that are clearly assigned to the latency program. Chief among these are LANA and v-cyclin (see below). However, it bears mentioning that MHV68 encodes a number of unique genes, not conserved in other gammaherpesviruses, that can have pronounced effects on latency and reactivation (e.g. M1 and M2). M1's role in eliciting IFN-γ has already been mentioned. M2 is a putative scaffolding protein with likely roles in modulation of signal transduction (Herskowitz et al 2008; Pires de Miranda et al 2008; Rodrigues et al 2006) and suppression of DNA-damage-induced apoptosis (Liang et al 2006). Additionally, M2 is a potent inducer of host IL-10 production, an activity that leads to enhanced survival and proliferation of primary B cells as well as suppression of host immune responses. M2 expression is also linked to the promotion of plasma cell differentiation (Liang et al 2009), which in MHV68, as in EBV and KSHV, can trigger lytic reactivation. The fact that M2 null mutants can establish latency but have a profound block to reactivation (Jacoby et al 2002; Herskowitz et al 2005) is likely linked to this fact.
LANA
The MHV68 LANA homolog (referred to as mLANA) is significant shorter than the KSHV LANA, being largely composed the C-terminal domain that is conserved among the other characterized LANA homologs and lacking the long internal repeat and amino-terminal domains present in the KSHV LANA. MHV68 LANA null viruses are severely compromised in establishing latency in the spleen following intranasal inoculation - consistent with the proposed role of LANA homologs in maintenance of the viral episome during latency (Fowler et al., 2003; Moorman et al., 2003b). It is possible to force the establishment of a chronic infection with the mLANA null MHV68 by either altering the route of inoculation of virus, or infecting immunocompromised mice (Paden, Forrest & Speck, unpublished results). However, under these experimental conditions the viral genome appears to be integrated and the infected B cells in the spleen are unable to reactivate virus. Thus, this is probably best described as an abortive infection - although the presence of splenocytes harboring viral genome persists for many months following infection.
The mLANA null MHV68 is also exhibits alterations in acute virus replication in the lungs following intranasal inoculation (Moorman et al., 2003b). This defect in virus replication can be partially mimicked under some infection conditions in tissue culture (Forrest et al., 2007), and correlates with significant dysregulation in viral lytic gene expression. The mLANA null MHV68 exhibits a substantially more rapid and higher level induction of early viral gene expression, consistent with a role for LANA in repression of lytic genes posited in KSHV (Li et al., 2008). However, LANA-null MHV68 infection also triggers a more rapid induction of cell death, likely due to the loss of LANA's ability to antagonize p53. The net result is that early cell death diminishes the ultimate virus yield in LANA-null MHV68 infection, despite the enhanced replication kinetics of the virus. Thus, in vivo studies with MHV68 affirm biologic roles for most of the biochemical activities previously ascribed to KSHV LANA on the basis of studies of the recombinant protein, including (i) episome maintenance; (ii) repression of at least some lytic genes; and (iii) inhibition of p53-mediated apoptosis.
vCyclin
Given the confusion about the biological role of KSHV v-cyclin (see above), it is hoped that further clues to its function can be gleaned from examination of its MHV68 homolog. MHV68 v-cyclin, however, displays some important biochemical differences from its KSHV counterpart. Both are fully functional as CDK activators, but the MHV68 protein preferentially binds and activates CDK2 (and can interact in vitro with CDK1), while KSHV v-cyclin preferentially binds CDK6, but can also bind CDK2 and CDK4. The crystal structure of the MHV68 v-cyclin in complex with CDK2 has been determined, and revealed that the MHV68 v-cyclin makes several novel contacts with CDK2 compared to CDK2 interaction with cyclin A, but overall its activation of CDK2 is likely similar to cyclin A (Card et al., 2000). In addition, the crystal structure provided critical insights into the resistance of gammaherpesvirus v-cyclins to inhibition by the CKI p27Kip1 (Card et al., 2000; Swanton et al., 1999).
The MHV68 v-cyclin is abundantly expressed during viral replication (SAM: lytic or latent replication?? Don – it is clearly abundantly expressed during virus replication, but appears to also be expressed during latency in some cases) from a promoter mapping just upstream of the v-cyclin coding exon (orf72). In addition, spliced v-cyclin transcripts have been identified in the MHV68 latently infected S11 B lymphoma cell line - driven by two distinct promoters, one mapping within the terminal repeats (P1) and another mapping just outside the terminal repeats (P2) (Allen et al., 2006). The is reminiscent of the EBV where the EBNA gene promoter Wp is encoded with the major internal repeat, and thus present in many copies like the P1 promoter, while Cp like P2 is present in a single copy being encoded outside of the repeat. Notably, like KSHV, transcripts initiating from P1 or P2 are alternatively splice to either the LANA or v-cyclin coding exons. Importantly, spliced LANA and v-cyclin transcripts could also detected using RNA prepared from splenocytes at day 16 post-infection - indicating that these transcripts are expressed in the context of MHV68 infection of mice (Allen et al., 2006).
Several mutant versions of MHV68 cyclin have been constructed in the viral genome, and their phenotypes are somewhat variable, depending on the route of infection and MOI. Following high dose (106 pfu) intraperitoneal inoculation, v-cyclin null MHV68 mutants containing either a CMV-driven β-galactosidase expression cassette, or a translation termination codon and frame shift inserted into the v-cyclin orf, exhibited wild type levels of acute virus replication in the spleen and normal establishment of latency in both the spleen and peritoneum. However, both mutants exhibited a profound defect in virus reactivation from latently infected peritoneal macrophages at 6 weeks post-infection (van Dyk et al., 2000). A related study using a v-cyclin null MHV68, in which a β-galactosidase expression cassette was inserted in place of the v-cyclin orf, exhibited defects in both acute virus replication and reactivation from latently infected splencytes (Hoge et al., 2000). Further studies using both v-cyclin null virus and v-cyclin CDK binding mutants revealed acute replication defects in the lungs following low dose (1000 pfu) intranasal inoculation (Upton and Speck, 2006). The acute replication defects observed with the v-cyclin mutant viruses following low dose infection were significantly more severe when the viruses were administered intranasally vs. intraperitoneally (Upton and Speck, 2006). In addition, following intranasal inoculation, neither the vcyclin null mutant nor the CDK binding mutant viruses induced splenomegaly. Suprisingly, while the v-cyclin null mutant was highly attenuated for reactivation from macrophages, neither of the CDK binding mutants was impaired for reactivation from this latency reservoir. The latter argues that the v-cyclin has CDK-independent functions that play a role in virus reactivation from peritoneal macrophages.
While it's difficult to know how to translate these results to KSHV infection, it seems clear that v-cyclin consistently plays a role in reactivation of MHV68 from latency, and may affect other portions of the life cycle in a context-dependent way. The challenge for the future is to determine (i) how many of these activities relate to CDK activation, (ii) how CDK activation engenders these activities; and (iii) whether any v-cyclin actions are due to other biochemical activities not dependent upon CDKs.
Coda
The analysis of the latency programs of these three gammaherpesviruses reveals how superbly adapted they are to the lymphoid systems of their hosts. At the single cell level, they have evolved elegant mechanisms to assure extrachromosomal genomic persistence as autonomous replicons, and by using viral proteins to tether their genomes to host chromosomes during mitosis. Other viral proteins manipulate cellular signaling pathways, including those that activate NFkB and those that mimic BCR signaling, both of which are known to provide signals to promote prolonged survival of the latently infected cell. In the two cases where studies have been done on intact hosts, these activities are thought to allow the latent viral genome to access its ultimate reservoir, the long-lived memory B cell – an ideal locale from the point of view of assuring long-term persistence. In addition to extending host cell lifespan, latent gene products also act to repress lytic functions, activate latent promoters and (most likely) induce or regulate epigenetic marks that influence viral gene regulation. All of these activities are set up in a delicate balance, so as to maintain reasonable stability in latency without becoming locked into it irreversibly. As a result, spontaneous reactivation is frequent enough to promote efficient spread of infection between individuals, while ongoing latency allows the infection to persist lifelong in each newly infected subject. It's a winning formula, though not a simple one – and one we can expect to continue to be amazed by as we begin to understand it more fully in the years to come.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Allday MJ, Crawford DH, Griffin BE. Epstein-Barr virus latent gene expression during the initiation of B cell immortalization. J Gen Virol. 1989;70(Pt 7):1755–1764. doi: 10.1099/0022-1317-70-7-1755. [DOI] [PubMed] [Google Scholar]
- Allen RD, 3rd, Dickerson S, Speck SH. Identification of spliced gammaherpesvirus 68 LANA and v-cyclin transcripts and analysis of their expression in vivo during latent infection. J Virol. 2006;80:2055–2062. doi: 10.1128/JVI.80.4.2055-2062.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aluigi MG, Albini A, Carlone S, Repetto L, De Marchi R, Icardi A, Moro M, Noonan D, Benelli R. KSHV sequences in biopsies and cultured spindle cells of epidemic, iatrogenic and Mediterranean forms of Kaposi's sarcoma. Res Virol. 1996;147(5):267–75. doi: 10.1016/0923-2516(96)82285-0. 1996. [DOI] [PubMed] [Google Scholar]
- Ambinder RF. Epstein-barr virus and hodgkin lymphoma. Hematology Am Soc Hematol Educ Program. 2007:204–209. doi: 10.1182/asheducation-2007.1.204. [DOI] [PubMed] [Google Scholar]
- Ambroziak JA, Blackbourn DJ, Herndier BG, Glogau RG, Gullett JH, McDonald AR, Lennette ET, Levy JA. Herpes-like sequences in HIV-infected and uninfected Kaposi's sarcoma patients. Science. 1995;268(5210):582–3. doi: 10.1126/science.7725108. [DOI] [PubMed] [Google Scholar]
- Asquith B, Bangham CR. How does HTLV-I persist despite a strong cell-mediated immune response? Trends Immunol. 2008;29(1):4–11. doi: 10.1016/j.it.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock GJ, Decker LL, Volk M, Thorley-Lawson DA. EBV persistence in memory B cells in vivo. Immunity. 1998;9:395–404. doi: 10.1016/s1074-7613(00)80622-6. [DOI] [PubMed] [Google Scholar]
- Bagnéris C, Ageichik AV, Cronin N, Wallace B, Collins M, Boshoff C, Waksman G, Barrett T. Crystal structure of a vFlip-IKKgamma complex: insights into viral activation of the IKK signalosome. Mol Cell. 2008 Jun 6;30(5):620–3. doi: 10.1016/j.molcel.2008.04.029. 2008. [DOI] [PubMed] [Google Scholar]
- Bain M, Watson RJ, Farrell PJ, Allday MJ. Epstein-Barr virus nuclear antigen 3C is a powerful repressor of transcription when tethered to DNA. J Virol. 1996;70:2481–2489. doi: 10.1128/jvi.70.4.2481-2489.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj BG, Verma SC, Lan K, Cotter MA, Woodman ZL, Robertson ES. KSHV encoded LANA upregulates Pim-1 and is a substrate for its kinase activity. Virology. 2006;351(1):18–28. doi: 10.1016/j.virol.2006.03.037. [DOI] [PubMed] [Google Scholar]
- Ballestas ME, Chatis PA, Kaye KM. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science. 1999;284(5414):641–4. doi: 10.1126/science.284.5414.641. 1999. [DOI] [PubMed] [Google Scholar]
- Ballestas ME, Kaye KM. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (TR) sequence and specifically binds TR DNA. J Virol. 2001;75(7):3250–8. doi: 10.1128/JVI.75.7.3250-3258.2001. 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, Kaye KM. The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science. 2006 Feb 10;311(5762):856–61. doi: 10.1126/science.1120541. 2006. [DOI] [PubMed] [Google Scholar]
- Bechtel JT, Liang Y, Hvidding J, Ganem D. Host range of Kaposi's sarcoma-associated herpesvirus in cultured cells. J Virol. 2003;77:6474–6481. doi: 10.1128/JVI.77.11.6474-6481.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger C, Gravel A, Tomoiu A, Janelle ME, Gosselin J, Tremblay MJ, Flamand L. Human herpesvirus 8 viral FLICE-inhibitory protein inhibits Fas-mediated apoptosis through binding and prevention of procaspase-8 maturation. J. Hum. Virol. 2001;4:62–73. [PubMed] [Google Scholar]
- Bellare P, Ganem D. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe. 2009;6(6):570–5. doi: 10.1016/j.chom.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhende PM, Dickerson SJ, Sun X, Feng WH, Kenney SC. X-box-binding protein 1 activates lytic Epstein-Barr virus gene expression in combination with protein kinase D. J Virol. 2007;81:7363–7370. doi: 10.1128/JVI.00154-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhende PM, Seaman WT, Delecluse HJ, Kenney SC. The EBV lytic switch protein, Z, preferentially binds to and activates the methylated viral genome. Nat Genet. 2004;36:1099–104. doi: 10.1038/ng1424. [DOI] [PubMed] [Google Scholar]
- Blasdell K, McCracken C, Morris A, Nash AA, Begon M, Bennett M, Stewart JP. The wood mouse is a natural host for Murid herpesvirus 4. J Gen Virol. 2003;84:111–113. doi: 10.1099/vir.0.18731-0. [DOI] [PubMed] [Google Scholar]
- Blaskovic D, Stancekova M, Svobodova J, Mistrikova J. Isolation of five strains of herpesviruses from two species of free living small rodents. Acta Virol. 1980;24:468. [PubMed] [Google Scholar]
- Bodescot M, Brison O, Perricaudet M. An Epstein-Barr virus transcription unit is at least 84 kilobases long. Nucleic Acids Res. 1986;14:2611–2620. doi: 10.1093/nar/14.6.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodescot M, Perricaudet M. Epstein-Barr virus mRNAs produced by alternative splicing. Nucleic Acids Res. 1986;14:7103–7114. doi: 10.1093/nar/14.17.7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodescot M, Perricaudet M. Clustered alternative splice sites in Epstein-Barr virus RNAs. Nucleic Acids Res. 1987;15:5887. doi: 10.1093/nar/15.14.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodescot M, Perricaudet M, Farrell PJ. A promoter for the highly spliced EBNA family of RNAs of Epstein-Barr virus. J Virol. 1987;61:3424–3430. doi: 10.1128/jvi.61.11.3424-3430.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornkamm GW, Hammerschmidt W. Molecular virology of Epstein-Barr virus. Philos Trans R Soc Lond B Biol Sci. 2001;356:437–459. doi: 10.1098/rstb.2000.0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant H, Farrell PJ. Signal Transduction and Transcription Factor Modification during Reactivation of Epstein-Barr Virus from Latency. J Virol. 2002;76:10290–10298. doi: 10.1128/JVI.76.20.10290-10298.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busson P, Keryer C, Ooka T, Corbex M. EBV-associated nasopharyngeal carcinomas: from epidemiology to virus-targeting strategies. Trends Microbiol. 12:356–360. doi: 10.1016/j.tim.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Card GL, Knowles P, Laman H, Jones N, McDonald NQ. Crystal structure of a gamma-herpesvirus cyclin-cdk complex. EMBO J. 2000;19:2877–2888. doi: 10.1093/emboj/19.12.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown HJ, Song MJ, Deng H, Wu TT, Cheng G, Sun R. NF-kappaB inhibits gammaherpesvirus lytic replication. J Virol. 2003 Aug. 2003;77(15):8532–40. doi: 10.1128/JVI.77.15.8532-8540.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR. Kaposi's sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc Natl Acad Sci U S A. 2005;102(15):5570–5. doi: 10.1073/pnas.0408192102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Cullen BR. Transcriptional origin of Kaposi's sarcoma-associated herpesvirus microRNAs. J Virol. 2006;80(5):2234–42. doi: 10.1128/JVI.80.5.2234-2242.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone A, Gloghini A, Bontempo D, et al. Proliferation in HHV8-positive primary effusion lymphomas is associated with expression of HHV8 cyclin but independent of p27kip1. Am. J. Pathol. 2000;156:1209–1215. doi: 10.1016/S0002-9440(10)64991-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll KD, Khadim F, Spadavecchia S, Palmeri D, Lukac DM. Direct interactions of Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta protein with the cellular protein octamer-1 and DNA are critical for specifying transactivation of a delayed-early promoter and stimulating viral reactivation. J Virol. 2007 Aug. 2007;81(16):8451–67. doi: 10.1128/JVI.00265-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper C, Krantz E, Selke S, Kuntz SR, Wang J, Huang ML, Pauk JS, Corey L, Wald A. Frequent and asymptomatic oropharyngeal shedding of human herpesvirus 8 among immunocompetent men. J Infect Dis. 2007 Jan 1;195(1):30–6. doi: 10.1086/509621. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332:1186–91. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- Chang J, Renne R, Dittmer D, Ganem D. Induction of Kaposi's sarcoma-associated herpesvirus from latency by inflammatory cytokines. Virology. 2000;266:17–25. doi: 10.1006/viro.1999.0077. [DOI] [PubMed] [Google Scholar]
- Chang M, Brown HJ, Collado-Hidalgo A, Arevalo JM, Galic Z, Symensma TL, Tanaka L, Deng H, Zack JA, Sun R, Cole SW. beta-Adrenoreceptors reactivate Kaposi's sarcoma-associated herpesvirus lytic replication via PKA-dependent control of viral RTA. J Virol. 2005 Nov;79(21):13538–47. doi: 10.1128/JVI.79.21.13538-13547.2005. 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Moore PS, Talbot SJ, Boshoff CH, Zarkowska T, Godden K, Paterson H, Weiss RA, Mittnacht S. Cyclin encoded by KS herpesvirus. Nature. 1996;382(6590):410–11. doi: 10.1038/382410a0. [DOI] [PubMed] [Google Scholar]
- Chaudhary PM, Jasmin A, Eby MT, Hood L. Modulation of the NF-B pathway by virally encoded death effector domains-containing proteins. Oncogene. 1999;18:5738–5746. doi: 10.1038/sj.onc.1202976. [DOI] [PubMed] [Google Scholar]
- Chen J, Ueda K, Sakakibara S, Okuno T, Parravicini C, Corbellino M, Yamanishi K. Activation of latent Kaposi's sarcoma-associated herpesvirus by demethylation of the promoter of the lytic transactivator. Proc Natl Acad Sci U S A. 2001;98(7):4119–24. doi: 10.1073/pnas.051004198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F, Weidner-Glunde M, Varjosalo M, Rainio EM, Lehtonen A, Schulz TF, Koskinen PJ, Taipale J, Ojala PM. KSHV reactivation from latency requires Pim-1 and Pim-3 kinases to inactivate the latency-associated nuclear antigen LANA. PLoS Pathog. 2009;5(3):e1000324. doi: 10.1371/journal.ppat.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugh P, Matta H, Schamus S, Zachariah S, Kumar A, Richardson JA, Smith AL, Chaudhary PM. Constitutive NF-kappaB activation, normal Fas-induced apoptosis, and increased incidence of lymphoma in human herpes virus 8 K13 transgenic mice. Proc Natl Acad Sci U S A. 2005 Sep 6;102(36):12885–90. doi: 10.1073/pnas.0408577102. 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun T-W, Carruth L, Finzi D, Shen X, Digiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, Kuo Y-H, Brookmeyer R, Zeiger MA, Barditch-Crovo P, Siliciano RF. Quantitation of latent tissue reservoirs and total body load in HIV-1 infection. Nature. 1997;387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- Ciufo DM, Cannon JS, Poole LJ, Wu FY, Murray P, Ambinder RF, Hayward GS. Spindle cell conversion by Kaposi's sarcoma-associated herpesvirus: formation of colonies and plaques with mixed lytic and latent gene expression in infected primary dermal microvascular endothelial cell cultures. J Virol. 2001;75(12):5614–26. doi: 10.1128/JVI.75.12.5614-5626.2001. 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A, Wolf DG, Guttman-Yassky E, Sarid R. Kaposi's sarcoma-associated herpesvirus: clinical, diagnostic, and epidemiological aspects. Crit Rev Clin Lab Sci. 2005;42(2):101–53. doi: 10.1080/10408360590913524. 2005. [DOI] [PubMed] [Google Scholar]
- Cotter MA, II, Robertson ES. The latency-associated nuclear antigen tethers the Kaposi's sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology. 1999;264:254–264. doi: 10.1006/viro.1999.9999. [DOI] [PubMed] [Google Scholar]
- Cotter MA, 2nd, Subramanian C, Robertson ES. The Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen binds to specific sequences at the left end of the viral genome through its carboxy-terminus. Virology. 2001;291(2):241–59. doi: 10.1006/viro.2001.1202. 2001. [DOI] [PubMed] [Google Scholar]
- Countryman J, Miller G. Activation of expression of latent Epstein-Barr herpesvirus after gene transfer with a small cloned subfragment of heterogeneous viral DNA. Proc Natl Acad Sci U S A. 1985;82:4085–9. doi: 10.1073/pnas.82.12.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon EM, Pallesen G, Niedobitek G, Crocker J, Brooks L, Rickinson AB, Young LS. Epstein-Barr virus and Hodgkin's disease: transcriptional analysis of virus latency in the malignant cells. J Exp Med. 1993;177:339–349. doi: 10.1084/jem.177.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Liang Y, Sun R. Regulation of KSHV lytic gene expression. Curr Top Microbiol Immunol. 2007;312:157–83. doi: 10.1007/978-3-540-34344-8_6. 2007. [DOI] [PubMed] [Google Scholar]
- Di Bartolo DL, Cannon M, Liu YF, Renne R, Chadburn A, Boshoff C, Cesarman E. KSHV LANA inhibits TGF-beta signaling through epigenetic silencing of the TGF-beta type II receptor. Blood. 2008 May 1;111(9):4731–40. doi: 10.1182/blood-2007-09-110544. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dictor M, Rambech E, Way D, Witte M, Bendsoe N. Human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus) DNA in Kaposi's sarcoma lesions, AIDS Kaposi's sarcoma cell lines, endothelial Kaposi's sarcoma simulators, and the skin of immunosuppressed patients. Am J Pathol. 1996;148(6):2009–16. 1996. [PMC free article] [PubMed] [Google Scholar]
- Dirmeier U, Neuhierl B, Kilger E, Reisbach G, Sandberg ML, Hammerschmidt W. Latent membrane protein 1 is critical for efficient growth transformation of human B cells by epstein-barr virus. Cancer Res. 2003;63:2982–2989. [PubMed] [Google Scholar]
- Dittmer DP. Transcription profile of Kaposi's sarcoma-associated herpesvirus in primary Kaposi's sarcoma lesions as determined by real-time PCR arrays. Cancer Res. 2003 May 1;63(9):2010–5. 2003. [PubMed] [Google Scholar]
- Dittmer D, Lagunoff M, Renne R, Staskus K, Haase A, Ganem D. A cluster of latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J Virol. 1998;72:8309–15. doi: 10.1128/jvi.72.10.8309-8315.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djerbi M, Screpanti V, Catrina AI, Bogen B, Biberfeld P, Grandien A. The inhibitor of death receptor signaling, FLICE-inhibitory protein defines a new class of tumor progression factors. J. Exp. Med. 1999;190:1025–1032. doi: 10.1084/jem.190.7.1025. 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, van Marck E, Salmon D, Gorin I, Escande JP, Weiss RA, Alitalo K, Boshoff C. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc Natl Acad Sci U S A. 1999;96:4546–4551. doi: 10.1073/pnas.96.8.4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutia BM, Clarke CJ, Allen DJ, Nash AA. Pathological changes in the spleens of gamma interferon receptor-deficient mice infected with murine gammaherpesvirus: a role for CD8 T cells. J Virol. 1997;71:4278–4283. doi: 10.1128/jvi.71.6.4278-4283.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi B, Dutia BM, Brownstein DG, Nash AA. Murine gammaherpesvirus-68 infection causes multi-organ fibrosis and alters leukocyte trafficking in interferon-gamma receptor knockout mice. Am J Pathol. 2001;158:2117–2125. doi: 10.1016/s0002-9440(10)64683-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efklidou S, Bailey R, Field N, Noursadeghi M, Collins MK. vFLIP from KSHV inhibits anoikis of primary endothelial cells. J Cell Sci. 2008 Feb 15;121(Pt 4):450–7. doi: 10.1242/jcs.022343. 2008. [DOI] [PubMed] [Google Scholar]
- Ellis M, Chew YP, Fallis L, Freddersdorf S, Boshoff C, Weiss RA, Lu X, Mittnacht S. Degradation of p27(Kip) cdk inhibitor triggered by Kaposi's sarcoma virus cyclin-cdk6 complex. EMBO J. 1999;18:644–653. doi: 10.1093/emboj/18.3.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AG, Moser JM, Krug LT, Pozharskaya V, Mora AL, Speck SH. A gammaherpesvirus-secreted activator of Vbeta4+ CD8+ T cells regulates chronic infection and immunopathology. J Exp Med. 2008;205:669–684. doi: 10.1084/jem.20071135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhari FD, Dittmer DP. Charting latency transcripts in Kaposi's sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J Virol. 2002;76(12):6213–23. doi: 10.1128/JVI.76.12.6213-6223.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field N, Low W, Daniels M, Howell S, Daviet L, Boshoff C, Collins M. KSHV vFLIP binds to IKK-gamma to activate IKK. J Cell Sci. 2003 Sep 15;116(Pt 18):3721–8. doi: 10.1242/jcs.00691. 2003. [DOI] [PubMed] [Google Scholar]
- Feederle R, Kost M, Baumann M, Janz A, Drouet E, Hammerschmidt W, Delecluse HJ. The Epstein–Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 2000;19:3080–3089. doi: 10.1093/emboj/19.12.3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamand L, Zeman RA, Bryant JL, Lunardi-Iskandar Y, Gallo RC. Absence of human herpesvirus 8 DNA sequences in neoplastic Kaposi's sarcoma cell lines. J Acquir Immune Defic Syndr Hum Retrovirol. 1996;13(2):194–7. doi: 10.1097/00042560-199610010-00011. [DOI] [PubMed] [Google Scholar]
- Flano E, Husain SM, Sample JT, Woodland DL, Blackman MA. Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J Immunol. 2000;165:1074–1081. doi: 10.4049/jimmunol.165.2.1074. [DOI] [PubMed] [Google Scholar]
- Flano E, Kim IJ, Woodland DL, Blackman MA. Gamma-herpesvirus latency is preferentially maintained in splenic germinal center and memory B cells. J Exp Med. 2002;196:1363–1372. doi: 10.1084/jem.20020890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest JC, Paden CR, Allen RD, 3rd, Collins J, Speck SH. ORF73-null murine gammaherpesvirus 68 reveals roles for mLANA and p53 in virus replication. J Virol. 2007;81:11957–11971. doi: 10.1128/JVI.00111-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest JC, Speck SH. Establishment of B-cell lines latently infected with reactivation-competent murine gammaherpesvirus 68 provides evidence for viral alteration of a DNA damage-signaling cascade. J. Virol. 2008;82:7688–99. doi: 10.1128/JVI.02689-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler P, Marques S, Simas JP, Efstathiou S. ORF73 of murine herpesvirus-68 is critical for the establishment and maintenance of latency. J Gen Virol. 2003;84:3405–3416. doi: 10.1099/vir.0.19594-0. [DOI] [PubMed] [Google Scholar]
- Friborg J, Jr., Kong W, Hottiger MO, Nabel GJ. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature. 1999;402(6764):889–94. doi: 10.1038/47266. [DOI] [PubMed] [Google Scholar]
- Fujimuro M, Wu FY, ApRhys C, Kajumbula H, Young DB, Hayward GS, Hayward SD. A novel viral mechanism for dysregulation of beta-catenin in Kaposi's sarcoma-associated herpesvirus latency. Nat Med. 2003;9(3):300–6. doi: 10.1038/nm829. 2003. [DOI] [PubMed] [Google Scholar]
- Gandy SZ, Linnstaedt SD, Muralidhar S, Cashman KA, Rosenthal LJ, Casey JL. RNA editing of the human herpesvirus 8 kaposin transcript eliminates its transforming activity and is induced during lytic replication. Journal of Virology. 2007;81(24):13544–51. doi: 10.1128/JVI.01521-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem D. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Invest. 2010;120 doi: 10.1172/JCI40567. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangappa S, Kapadia SB, Speck SH, Virgin HW. Antibody to a lytic cycle viral protein decreases gammaherpesvirus latency in B-cell-deficient mice. J Virol. 2002a;76:11460–11468. doi: 10.1128/JVI.76.22.11460-11468.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber AC, Shu MA, Hu J, Renne R. DNA binding and modulation of gene expression by the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J Virol. 2001;75(17):7882–92. doi: 10.1128/JVI.75.17.7882-7892.2001. 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber AC, Hu J, Renne R. Latency-associated nuclear antigen (LANA) cooperatively binds to two sites within the terminal repeat, and both sites contribute to the ability of LANA to suppress transcription and to facilitate DNA replication. J Biol Chem. 2002;277(30):27401–11. doi: 10.1074/jbc.M203489200. 2002. [DOI] [PubMed] [Google Scholar]
- Godden-Kent D, Talbot S, Boshoff C, Chang Y, Moore P, Weiss R, Mittnacht S. The cyclin encoded by Kaposi's sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J. Virol. 1997;71:4193–4198. doi: 10.1128/jvi.71.6.4193-4198.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin MM, Canny S, Steed A, Virgin HW. Murine gammaherpesvirus 68 has evolved gamma interferon and stat1-repressible promoters for the lytic switch gene 50. J Virol. 84:3711–3717. doi: 10.1128/JVI.02099-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk S, Rooney CM, Heslop HE. Post-transplant lymphoproliferative disorders. Annu Rev Med. 2005;56:29–44. doi: 10.1146/annurev.med.56.082103.104727. [DOI] [PubMed] [Google Scholar]
- Gottwein E, Mukherjee N, Sachse C, Frenzel C, Majoros WH, Chi JT, Braich R, Manoharan M, Soutschek J, Ohler U, Cullen BR. A viral microRNA functions as an orthologue of cellular miR-155. Nature. 2007;450(7172):1096–9. doi: 10.1038/nature05992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenspan JS, Greenspan D. Oral hairy leukoplakia: diagnosis and management. Oral Surg Oral Med Oral Pathol. 1989;67(4):396–403. doi: 10.1016/0030-4220(89)90381-2. [DOI] [PubMed] [Google Scholar]
- Grossman C, Podogrobskina S, Skobe M, Ganem D. Activation of NF-κB by the latent v-FLIP gene of KSHV is required for the spindle shape of virus-infected endothelial cells and contributes to their pro-inflammatory phenotype. J Virol. 2006;80:7179–7185. doi: 10.1128/JVI.01603-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann C, Ganem D. Effects of NFκB activation on KSHV latency and lytic reactivation are complex and context-dependent. Virology. 2008;375(1):94–102. doi: 10.1016/j.virol.2007.12.044. 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SR, Johannsen E, Tong X, Yalamanchili R, Kieff E. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the J kappa recombination signal binding protein. Proc Natl Acad Sci U S A. 1994;91:7568–7572. doi: 10.1073/pnas.91.16.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundhoff A, Ganem D. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus (KSHV) permits replication of terminal repeat-containing plasmids. J Virol. 2003;77:2779–83. doi: 10.1128/JVI.77.4.2779-2783.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundhoff A, Ganem D. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J Clin Invest. 2004;113:124–136. doi: 10.1172/JCI200417803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundhoff A, Sullivan CS, Ganem D. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA. 2006 May;12(5):733–50. doi: 10.1261/rna.2326106. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasparri I, Keller SA, Cesarman E. KSHV vFLIP is essential for the survival of infected lymphoma cells. J Exp Med. 2004 Apr 5;199(7):993–1003. doi: 10.1084/jem.20031467. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gutsch DE, Holley-Guthrie EA, Zhang Q, Stein B, Blanar MA, Baldwin AS, Kenney SC. The bZIP transactivator of Epstein-Barr virus, BZLF1, functionally and physically interacts with the p65 subunit of NF-kappa B. Mol Cell Biol. 1994;14:1939–1948. doi: 10.1128/mcb.14.3.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Wind-Rotolo M, Yang HC, Siliciano JD, Siliciano RF. Experimental approaches to the study of HIV-1 latency. Nat Rev Microbiol. 2007;5(2):95–106. doi: 10.1038/nrmicro1580. [DOI] [PubMed] [Google Scholar]
- Henkel T, Ling PD, Hayward SD, Peterson MG. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein J kappa. Science. 1994;265:92–95. doi: 10.1126/science.8016657. [DOI] [PubMed] [Google Scholar]
- Henle W, Diehl V, Kohn G, Zur Hausen H, Henle G. Herpes-type virus and chromosome marker in normal leukocytes after growth with irradiated Burkitt cells. Science. 1967;157:1064–1065. doi: 10.1126/science.157.3792.1064. [DOI] [PubMed] [Google Scholar]
- Herbst H, Dallenbach F, Hummel M, Niedobitek G, Pileri S, Muller-Lantzsch N, Stein H. Epstein-Barr virus latent membrane protein expression in Hodgkin and Reed-Sternberg cells. Proc Natl Acad Sci U S A. 1991;88:4766–4770. doi: 10.1073/pnas.88.11.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjalgrim H, Engels EA. Infectious aetiology of Hodgkin and non-Hodgkin lymphomas: a review of the epidemiological evidence. J Intern Med. 2008;264(6):537–48. doi: 10.1111/j.1365-2796.2008.02031.x. [DOI] [PubMed] [Google Scholar]
- Hoge AT, Hendrickson SB, Burns WH. Murine gammaherpesvirus 68 cyclin D homologue is required for efficient reactivation from latency. J Virol. 2000;74:7016–7023. doi: 10.1128/jvi.74.15.7016-7023.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Garber AC, Renne R. The Latency-Associated Nuclear Antigen of Kaposi's Sarcoma-Associated Herpesvirus Supports Latent DNA Replication in Dividing Cells. J Virol. 2002;76(22):11677–87. doi: 10.1128/JVI.76.22.11677-11687.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humme S, Reisbach G, Feederle R, Delecluse HJ, Bousset K, Hammerschmidt W, Schepers A. The EBV nuclear antigen 1 (EBNA1) enhances B cell immortalization several thousandfold. Proc Natl Acad Sci U S A. 2003;100:10989–10994. doi: 10.1073/pnas.1832776100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi KM, Kaye KM, Kieff ED. The Epstein-Barr virus LMP1 amino acid sequence that engages tumor necrosis factor receptor associated factors is critical for primary B lymphocyte growth transformation. Proc Natl Acad Sci U S A. 1997;94:1447–1452. doi: 10.1073/pnas.94.4.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi KM, Kieff ED. The Epstein-Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB. Proc Natl Acad Sci U S A. 1997;94:12592–12597. doi: 10.1073/pnas.94.23.12592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumiya Y, Izumiya C, Hsia D, Ellison TJ, Luciw PA, Kung HJ. NF-kappaB serves as a cellular sensor of Kaposi's sarcoma-associated herpesvirus latency and negatively regulates K-Rta by antagonizing the RBP-Jkappa coactivator. J Virol. 2009 May;83(9):4435–46. doi: 10.1128/JVI.01999-08. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin XW, Speck SH. Identification of critical cis elements involved in mediating Epstein-Barr virus nuclear antigen 2-dependent activity of an enhancer located upstream of the viral BamHI C promoter. J Virol. 1992;66:2846–2852. doi: 10.1128/jvi.66.5.2846-2852.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannsen E, Miller CL, Grossman SR, Kieff E. EBNA-2 and EBNA-3C extensively and mutually exclusively associate with RBPJkappa in Epstein-Barr virus transformed B lymphocytes. J Virol. 1996;70:4179–4183. doi: 10.1128/jvi.70.6.4179-4183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JB, McFadden G. Poxvirus immunomodulatory strategies: current perspectives. J Virol. 2003 Jun;77(11):6093–100. doi: 10.1128/JVI.77.11.6093-6100.2003. 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CH, Hayward SD, Rawlins DR. Interaction of the lymphocyte-derived Epstein-Barr virus nuclear antigen EBNA-1 with its DNA-binding sites. J Virol. 1989;63:101–110. doi: 10.1128/jvi.63.1.101-110.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalla M, Schmeinck A, Bergbauer M, Pich D, Hammerschmidt W. AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc Natl Acad Sci U S A. 2010;107:850–855. doi: 10.1073/pnas.0911948107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller SA, Schattner EJ, Cesarman E. Inhibition of NF-B induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood. 2000;96:2537–2542. [PubMed] [Google Scholar]
- Kelley-Clarke B, De Leon-Vazquez E, Slain K, Barbera AJ, Kaye KM. Role of Kaposi's sarcoma-associated herpesvirus C-terminal LANA chromosome binding in episome persistence. J Virol. 2009 May;83(9):4326–37. doi: 10.1128/JVI.02395-08. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley-Clarke B, Ballestas ME, Srinivasan V, Barbera AJ, Komatsu T, Harris TA, Kazanjian M, Kaye KM. Determination of Kaposi's sarcoma-associated herpesvirus C-terminal latency-associated nuclear antigen residues mediating chromosome association and DNA binding. J Virol. 2007 Apr;81(8):4348–56. doi: 10.1128/JVI.01289-06. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley-Clarke B, Ballestas ME, Komatsu T, Kaye KM. Kaposi's sarcoma herpesvirus C-terminal LANA concentrates at pericentromeric and peri-telomeric regions of a subset of mitotic chromosomes. Virology. 2007 Jan 20;357(2):149–57. doi: 10.1016/j.virol.2006.07.052. 2007. [DOI] [PubMed] [Google Scholar]
- Kilger E, Kieser A, Baumann M, Hammerschmidt W. Epstein-Barr virus mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO J. 1998;17:1700–1709. doi: 10.1093/emboj/17.6.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IJ, Flano E, Woodland DL, Lund FE, Randall TD, Blackman MA. Maintenance of long term gamma-herpesvirus B cell latency is dependent on CD40-mediated development of memory B cells. J Immunol. 2003;171:886–892. doi: 10.4049/jimmunol.171.2.886. [DOI] [PubMed] [Google Scholar]
- Kliche S, Kremmer E, Hammerschmidt W, Koszinowski U, Haas J. Persistent infection of Epstein-Barr virus-positive B lymphocytes by human herpesvirus 8. J Virol. 1998 Oct;72(10):8143–9. doi: 10.1128/jvi.72.10.8143-8149.1998. 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliche S, Nagel W, Kremmer E, Atzler C, Ege A, Knorr T, Koszinowski U, Kolanus W, Haas J. Signaling by human herpesvirus 8 kaposin A through direct membrane recruitment of cytohesin-1. Mol Cell. 2001 Apr;7(4):833–43. doi: 10.1016/s1097-2765(01)00227-1. 2001. [DOI] [PubMed] [Google Scholar]
- Konrad A, Wies E, Thurau M, Marquardt G, Naschberger E, Hentschel S, Jochmann R, Schulz TF, Erfle H, Brors B, Lausen B, Neipel F, Stürzl M. A systems biology approach to identify the combination effects of human herpesvirus 8 genes on NF-kappaB activation. J Virol. 2009 Mar;83(6):2563–74. doi: 10.1128/JVI.01512-08. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopal S, Furuhjelm JH, Järviluoma A, Jäämaa S, Pyakurel P, Pussinen C, Wirzenius M, Biberfeld P, Alitalo K, Laiho M, Ojala PM. Viral oncogene-induced DNA damage response is activated in Kaposi sarcoma tumorigenesis. PLoS Pathog. 2007 Sep 7;3(9):1348–60. doi: 10.1371/journal.ppat.0030140. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krithivas A, Young DB, Liao G, Greene D, Hayward SD. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J Virol. 2000;74(20):9637–45. doi: 10.1128/jvi.74.20.9637-9645.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krithivas A, Fujimuro M, Weidner M, Young DB, Hayward SD. Protein interactions targeting the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus to cell chromosomes. J Virol. 2002;76(22):11596–604. doi: 10.1128/JVI.76.22.11596-11604.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug LT, Collins CM, Gargano LM, Speck SH. NF-kappaB p50 plays distinct roles in the establishment and control of murine gammaherpesvirus 68 latency. J Virol. 2009;83:4732–4748. doi: 10.1128/JVI.00111-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug LT, Moser JM, Dickerson SM, Speck SH. Inhibition of NF-kappaB activation in vivo impairs establishment of gammaherpesvirus latency. PLoS Pathog. 2007;3:e11. doi: 10.1371/journal.ppat.0030011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagunoff M, Ganem D. Organization of the termini of the genome of the Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8). Virology. 1997;236:147–154. doi: 10.1006/viro.1997.8713. [DOI] [PubMed] [Google Scholar]
- Lagunoff M, Majeti R, Weiss A, Ganem D. Deregulated signal transduction by the K1 gene product of Kaposi's sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A. 1999;96:5704–9. doi: 10.1073/pnas.96.10.5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laichalk LL, Thorley-Lawson DA. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J Virol. 2005;79:1296–1307. doi: 10.1128/JVI.79.2.1296-1307.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan K, Kuppers DA, Verma SC, Robertson ES. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus-mediated control of latency. J Virol. 2004;78(12):6585–94. doi: 10.1128/JVI.78.12.6585-6594.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan K, Kuppers DA, Robertson ES. Kaposi's sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J Virol. 2005;79(6):3468–78. doi: 10.1128/JVI.79.6.3468-3478.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BS, Lee SH, Feng P, Chang H, Cho NH, Jung JU. Characterization of the Kaposi's sarcoma-associated herpesvirus K1 signalosome. J Virol. 2005;79:12173–84. doi: 10.1128/JVI.79.19.12173-12184.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BS, Paulose-Murphy M, Chung YH, Connlole M, Zeichner S, Jung JU. Suppression of tetradecanoyl phorbol acetate-induced lytic reactivation of Kaposi's sarcoma-associated herpesvirus by K1 signal transduction. J Virol. 2002;76:12185–99. doi: 10.1128/JVI.76.23.12185-12199.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Guo J, Li M, Choi JK, DeMaria M, Rosenzweig M, Jung JU. Identification of an immunoreceptor tyrosine-based activation motif of K1 transforming protein of Kaposi's sarcoma-associated herpesvirus. Mol Cell Biol. 1998;18:5219–28. doi: 10.1128/mcb.18.9.5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Deng H, Yu F, Melega WP, Damoiseaux R, Bradley KA, Sun R. Regulation of Kaposi's sarcoma-associated herpesvirus reactivation by dopamine receptor-mediated signaling pathways. J Acquir Immune Defic Syndr. 2008 Aug 15;48(5):531–40. doi: 10.1097/QAI.0b013e31817fbdcf. 2008. [DOI] [PubMed] [Google Scholar]
- Li H, Komatsu T, Dezube BJ, Kaye KM. The Kaposi's sarcoma-associated herpesvirus K12 transcript from a primary effusion lymphoma contains complex repeat elements, is spliced, and initiates from a novel promoter. J Virol. 2002 Dec;76(23):11880–8. doi: 10.1128/JVI.76.23.11880-11888.2002. 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Lee H, Yoon DW, Albrecht JC, Fleckenstein B, Neipel F, Jung JU. Kaposi's sarcoma-associated herpesvirus encodes a functional cyclin. J Virol. 1997;71(3):1984–91. doi: 10.1128/jvi.71.3.1984-1991.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Zhou F, Ye F, Gao SJ. Genetic disruption of KSHV major latent nuclear antigen LANA enhances viral lytic transcriptional program. Virology. 2008;379:234–244. doi: 10.1016/j.virol.2008.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Collins CM, Mendel JB, Iwakoshi NN, Speck SH. Gammaherpesvirus-driven plasma cell differentiation regulates virus reactivation from latently infected B lymphocytes. PLoS Pathog. 2009;5:e1000677. doi: 10.1371/journal.ppat.1000677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Chang J, Lynch SJ, Lukac D, Ganem D. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jk, the target of the Notch signaling pathway. Genes Dev. 2002;16:1977–1989. doi: 10.1101/gad.996502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Ganem D. Lytic but not latent infection by Kaposi's sarcoma-associated herpesvirus requires host CSL protein, the mediator of Notch signaling. Proc Natl Acad Sci U S A. 2003;100:8490–5. doi: 10.1073/pnas.1432843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C, Sohn H, Lee D, Gwack Y, Choe J. Functional dissection of latency-associated nuclear antigen 1 of Kaposi's sarcoma-associated herpesvirus involved in latent DNA replication and transcription of terminal repeats of the viral genome. J Virol. 2002;76(20):10320–31. doi: 10.1128/JVI.76.20.10320-10331.2002. 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C, Lee D, Seo T, Choi C, Choe J. Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus functionally interacts with heterochromatin protein 1. J Biol Chem. 2003;278(9):7397–405. doi: 10.1074/jbc.M211912200. [DOI] [PubMed] [Google Scholar]
- Lin CW, Tu PF, Hsiao NW, Chang CY, Wan L, Lin YT, Chang HW. Identification of a novel septin 4 protein binding to human herpesvirus 8 kaposin A protein using a phage display cDNA library. J Virol Methods. 2007;143(1):65–72. doi: 10.1016/j.jviromet.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Liu C, Okruzhnov Y, Li H, Nicholas J. Human herpesvirus 8 (HHV-8)-encoded cytokines induce expression of and autocrine signaling by vascular endothelial growth factor (VEGF) in HHV-8-infected primary-effusion lymphoma cell lines and mediate VEGF-independent antiapoptotic effects. J. Virol. 2001;75:10933–10940. doi: 10.1128/JVI.75.22.10933-10940.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Eby MT, Rathore N, Sinha SK, Kumar A, Chaudhary PM. The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the IB kinase complex. J. Biol. Chem. 2002;277:13745–13751. doi: 10.1074/jbc.M110480200. [DOI] [PubMed] [Google Scholar]
- Liu J, Martin H, Shamay M, Woodard C, Tang QQ, Hayward SD. Kaposi's sarcoma-associated herpesvirus LANA protein downregulates nuclear glycogen synthase kinase 3 activity and consequently blocks differentiation. J Virol. 2007;81(9):4722–31. doi: 10.1128/JVI.02548-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Martin HJ, Liao G, Hayward SD. The Kaposi's sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc. J Virol. 2007;81(19):10451–9. doi: 10.1128/JVI.00804-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Day L, Gao SJ, Lieberman PM. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi's sarcoma-associated herpesvirus lytic transcription. J Virol. 2006;80(11):5273–82. doi: 10.1128/JVI.02541-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukac DM, Renne R, Kirshner JR, Ganem D. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology. 1998;252(2):304–12. doi: 10.1006/viro.1998.9486. [DOI] [PubMed] [Google Scholar]
- Lukac DM, Kirshner JR, Ganem D. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J Virol. 1999;73:9348–9361. doi: 10.1128/jvi.73.11.9348-9361.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DJ, Child ES, Swanton C, Laman H, Jones N. Modulation of p27(Kip1) levels by the cyclin encoded by Kaposi's sarcoma-associated herpesvirus. EMBO J. 1999;18:654–663. doi: 10.1093/emboj/18.3.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannick JB, Cohen JI, Birkenbach M, Marchini A, Kieff E. The Epstein-Barr virus nuclear protein encoded by the leader of the EBNA RNAs is important in B-lymphocyte transformation. J Virol. 1991;65:6826–6837. doi: 10.1128/jvi.65.12.6826-6837.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall V, Parks T, Bagni R, Wang CD, Samols MA, Hu J, Wyvil KM, Aleman K, Little RF, Yarchoan R, Renne R, Whitby D. Conservation of virally encoded microRNAs in Kaposi sarcoma--associated herpesvirus in primary effusion lymphoma cell lines and in patients with Kaposi sarcoma or multicentric Castleman disease. J Infect Dis. 2007;195(5):645–59. doi: 10.1086/511434. [DOI] [PubMed] [Google Scholar]
- Matta H, Chaudhary PM. Activation of alternative NF-kappa B pathway by human herpes virus 8-encoded Fas-associated death domain-like IL-1 beta-converting enzyme inhibitory protein (vFLIP). Proc Natl Acad Sci U S A. 2004 Jun 22;101(25):9399–404. doi: 10.1073/pnas.0308016101. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson K, Kiss C, Platt GM, Simpson GR, Kashuba E, Klein G, Schulz TF, Szekely L. Latent nuclear antigen of Kaposi's sarcoma herpesvirus/human herpesvirus-8 induces and relocates RING3 to nuclear heterochromatin regions. J Gen Virol. 2002 Jan;83(Pt 1):179–88. doi: 10.1099/0022-1317-83-1-179. 2002. [DOI] [PubMed] [Google Scholar]
- McCormick C, Ganem D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science. 2005;307:739–41. doi: 10.1126/science.1105779. [DOI] [PubMed] [Google Scholar]
- McGeoch DJ, Gatherer D, Dolan A. On phylogenetic relationships among major lineages of the Gammaherpesvirinae. J Gen Virol. 2005;86:307–316. doi: 10.1099/vir.0.80588-0. [DOI] [PubMed] [Google Scholar]
- Mercader M, Taddeo B, Panella JR, Chandran B, Nickoloff BJ, Foreman KE. Induction of HHV-8 lytic cycle replication by inflammatory cytokines produced by HIV-1-infected T cells. Am J Pathol. 2000 Jun;156(6):1961–71. doi: 10.1016/S0002-9440(10)65069-9. 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G, Rigsby MO, Heston L, Grogan E, Sun R, Metroka C, Levy JA, Gao SJ, Chang Y, Moore P. Antibodies to butyrate-inducible antigens of Kaposi's sarcoma-associated herpesvirus in patients with HIV-1 infection. N. Engl. J. Med. 1996;334:1292–1297. doi: 10.1056/NEJM199605163342003. [DOI] [PubMed] [Google Scholar]
- Moorman NJ, Lin CY, Speck SH. Identification of candidate gammaherpesvirus 68 genes required for virus replication by signature-tagged transposon mutagenesis. J Virol. 2004;78:10282–10290. doi: 10.1128/JVI.78.19.10282-10290.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman NJ, Willer DO, Speck SH. The gammaherpesvirus 68 latency associated nuclear antigen homolog is critical for the establishment of splenic latency. J Virol. 2003b;77:10295–10303. doi: 10.1128/JVI.77.19.10295-10303.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidhar S, Pumfery AM, Hassani M, Sadaie MR, Azumi N, Kishishita M, Brady JN, Doniger J, Medveczky P, Rosenthal LJ. Identification of kaposin (ORF K12) as a human herpesvirus 8 (Kaposi's sarcoma associated herpesvirus) transforming gene. J. Virol. 1998;72:4980–4988. doi: 10.1128/jvi.72.6.4980-4988.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myoung J, Ganem D. KSHV Infection of primary tonsillar lymphoid cells (submitted) 2010. [DOI] [PMC free article] [PubMed]
- Nayyar VK, Shire K, Frappier L. Mitotic chromosome interactions of Epstein-Barr nuclear antigen 1 (EBNA1) and human EBNA1-binding protein 2 (EBP2). J Cell Sci. 2009;122:4341–50. doi: 10.1242/jcs.060913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottinger M, Christalla T, Nathan K, Brinkmann MM, Viejo-Borbolla A, Schulz TF. Kaposi's sarcoma-associated herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J Virol. 2006;80(21):10772–86. doi: 10.1128/JVI.00804-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudejans JJ, Dukers DF, Jiwa NM, van den Brule AJ, Grasser FA, de Bruin PC, Horstman A, Vos W, van Gorp J, Middeldorp JM, et al. Expression of epstein-barr virus encoded nuclear antigen 1 in benign and malignant tissues harbouring EBV. J Clin Pathol. 1996;49:897–902. doi: 10.1136/jcp.49.11.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauk J, Huang ML, Brodie SJ, Wald A, Koelle DM, et al. Mucosal Shedding of Human Herpesvirus 8 in Men. N Engl J Med. 2000;343:1369–1377. doi: 10.1056/NEJM200011093431904. [DOI] [PubMed] [Google Scholar]
- Paulson EJ, Speck SH. Differential methylation of Epstein-Barr virus latency promoters facilitates viral persistence in healthy seropositive individuals. J Virol. 1999;73:9959–9968. doi: 10.1128/jvi.73.12.9959-9968.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce M, Matsumura S, Wilson AC. Transcripts encoding K12, v-FLIP, v-cyclin, and the microRNA cluster of Kaposi's sarcoma-associated herpesvirus originate from a common promoter. J Virol. 2005 Nov;79(22):14457–64. doi: 10.1128/JVI.79.22.14457-14464.2005. 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson S, Reid AP, Kim S, Siliciano RF. Treatment implications of the latent reservoir for HIV-1. Adv Pharmacol. 2007;55:411–25. doi: 10.1016/S1054-3589(07)55012-X. 2007. [DOI] [PubMed] [Google Scholar]
- Petre CE, Sin SH, Dittmer DP. Functional p53 signaling in Kaposi's sarcoma-associated herpesvirus lymphomas: implications for therapy. J Virol. 2007;81(4):1912–22. doi: 10.1128/JVI.01757-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer S, Sewer A, Lagos-Quintana M, Sheridan R, Sander C, Grässer FA, van Dyk LF, Ho CK, Shuman S, Chien M, Russo JJ, Ju J, Randall G, Lindenbach BD, Rice CM, Simon V, Ho DD, Zavolan M, Tuschl T. Identification of microRNAs of the herpesvirus family. Nat Methods. 2005 Apr;2(4):269–76. doi: 10.1038/nmeth746. 2005. [DOI] [PubMed] [Google Scholar]
- Piolot T, Tramier M, Coppey M, Nicolas JC, Marechal V. Close but distinct regions of human herpesvirus 8 latency-associated nuclear antigen 1 are responsible for nuclear targeting and binding to human mitotic chromosomes. J Virol. 2001;75(8):3948–59. doi: 10.1128/JVI.75.8.3948-3959.2001. 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt GM, Simpson GR, Mittnacht S, Schulz TF. Latent nuclear antigen of Kaposi's sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J Virol. 1999;73(12):9789–95. doi: 10.1128/jvi.73.12.9789-9795.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt G, Carbone A, Mittnacht S. p16INK4a loss and sensitivity in KSHV associated primary effusion lymphoma. Oncogene. 2002 Mar 14;21(12):1823–31. doi: 10.1038/sj.onc.1205360. 2002. [DOI] [PubMed] [Google Scholar]
- Platt GM, Simpson GR, Mittnacht S, Schulz TF. Latent nuclear antigen of Kaposi's sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J Virol. 1999;73(12):9789–95. doi: 10.1128/jvi.73.12.9789-9795.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puglielli MT, Woisetschlaeger M, Speck SH. oriP is essential for EBNA gene promoter activity in Epstein-Barr virus-immortalized lymphoblastoid cell lines. J Virol. 1996;70:5758–5768. doi: 10.1128/jvi.70.9.5758-5768.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punj V, Matta H, Schamus S, Yang T, Chang Y, Chaudhary PM. Induction of CCL20 production by Kaposi sarcoma-associated herpesvirus: role of viral FLICE inhibitory protein K13-induced NF-kappaB activation. Blood. 2009 May 28;113(22):5660–8. doi: 10.1182/blood-2008-10-186403. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purtilo DT. Epstein-Barr virus: the spectrum of its manifestations in human beings. South Med J. 1987;80(8):943–7. doi: 10.1097/00007611-198708000-00003. [DOI] [PubMed] [Google Scholar]
- Radkov SA, Bain M, Farrell PJ, West M, Rowe M, Allday MJ. Epstein-Barr virus EBNA3C represses Cp, the major promoter for EBNA expression, but has no effect on the promoter of the cell gene CD21. J Virol. 1997;71:8552–8562. doi: 10.1128/jvi.71.11.8552-8562.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radkov SA, Touitou R, Brehm A, Rowe M, West M, Kouzarides T, Allday MJ. Epstein-Barr virus nuclear antigen 3C interacts with histone deacetylase to repress transcription. J Virol. 1999;73:5688–5697. doi: 10.1128/jvi.73.7.5688-5697.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragoczy T, Heston L, Miller G. The Epstein–Barr virus Rta protein activates lytic cycle genes and can disrupt latency in B lymphocytes. J Virol. 1998;72:7978–7984. doi: 10.1128/jvi.72.10.7978-7984.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappocciolo G, Hensler HR, Jais M, Reinhart TA, Pegu A, Jenkins FJ, Rinaldo CR. Human herpesvirus 8 infects and replicates in primary cultures of activated B lymphocytes through DC-SIGN. J Virol. 2008;82(10):4793–806. doi: 10.1128/JVI.01587-07. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisman D, Sugden B. trans activation of an Epstein-Barr viral transcriptional enhancer by the Epstein-Barr viral nuclear antigen 1. Mol Cell Biol. 1986;6:3838–3846. doi: 10.1128/mcb.6.11.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisman D, Yates J, Sugden B. A putative origin of replication of plasmids derived from Epstein-Barr virus is composed of two cis-acting components. Mol Cell Biol. 1985;5:1822–1832. doi: 10.1128/mcb.5.8.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renne R, Barry C, Dittmer D, Compitello N, Brown PO, Ganem D. Modulation of cellular and viral gene expression by the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Virol. 2001;75:458–468. doi: 10.1128/JVI.75.1.458-468.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renne R, Lagunoff M, Zhong W, Ganem D. The size and conformation of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) DNA in infected cells and virions. J. Virol. 1996;70:8151–8154. doi: 10.1128/jvi.70.11.8151-8154.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med. 1996;2:342–6. doi: 10.1038/nm0396-342. [DOI] [PubMed] [Google Scholar]
- Renne R, Blackbourn D, Whitby D, Levy J, Ganem D. Limited transmission of Kaposi's sarcoma-associated herpesvirus in cultured cells. J.Virol. 1998;72:5182–5188. doi: 10.1128/jvi.72.6.5182-5188.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickinson A, Kieff E. Epstein-Barr virus. In: Fields BN, Knipe DM, Howley PM, Chanock RM, Melnick LL, Monath TP, Roizmann B, Strauss SE, editors. Fields’ Virology. 3rd ed. Lippincott-Raven; Philadephia, PA: 1996. pp. 2397–2446. [Google Scholar]
- Rivas C, Thlick AE, Parravicini C, Moore PS, Chang Y. Kaposi's sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J Virol. 2001;75(1):429–38. doi: 10.1128/JVI.75.1.429-438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson ES, Grossman S, Johannsen E, Miller C, Lin J, Tomkinson B, Kieff E. Epstein-Barr virus nuclear protein 3C modulates transcription through interaction with the sequence-specific DNA-binding protein J kappa. J Virol. 1995;69:3108–3116. doi: 10.1128/jvi.69.5.3108-3116.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues L, Filipe J, Seldon MP, Fonseca L, Anrather J, Soares MP, Simas JP. Termination of NF-kappaB activity through a gammaherpesvirus protein that assembles an EC5S ubiquitin-ligase. EMBO J. 2009;28:1283–1295. doi: 10.1038/emboj.2009.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RP, Woisetschlaeger M, Speck SH. Alternative splicing dictates translational start in Epstein-Barr virus transcripts. EMBO J. 1990;9:2273–2277. doi: 10.1002/j.1460-2075.1990.tb07398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney C, Howe JG, Speck SH, Miller G. Influence of Burkitt's lymphoma and primary B cells on latent gene expression by the nonimmortalizing P3J-HR-1 strain of Epstein-Barr virus. J Virol. 1989;63:1531–1539. doi: 10.1128/jvi.63.4.1531-1539.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe DT, Rowe M, Evan GI, Wallace LE, Farrell PJ, Rickinson AB. Restricted expression of EBV latent genes and T-lymphocyte-detected membrane antigen in Burkitt's lymphoma cells. EMBO J. 1986;5:2599–2607. doi: 10.1002/j.1460-2075.1986.tb04540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe M, Rowe DT, Gregory CD, Young LS, Farrell PJ, Rupani H, Rickinson AB. Differences in B cell growth phenotype reflect novel patterns of Epstein-Barr virus latent gene expression in Burkitt's lymphoma cells. EMBO J. 1987;6:2743–2751. doi: 10.1002/j.1460-2075.1987.tb02568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc Natl Acad Sci U S A. 1996;93:14862–7. doi: 10.1073/pnas.93.25.14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadagopan S, Sharma-Walia N, Veettil MV, Bottero V, Levine R, Vart RJ, Chandran B. Kaposi's sarcoma-associated herpesvirus upregulates angiogenin during infection of human dermal microvascular endothelial cells, which induces 45S rRNA synthesis, antiapoptosis, cell proliferation, migration, and angiogenesis. J Virol. 2009 Apr;83(7):3342–64. doi: 10.1128/JVI.02052-08. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler R, Wu L, Forghani B, Renne R, Zhong W, Herndier B, Ganem D. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi's sarcoma-associated herpesvirus. J Virol. 1999;73(7):5722–30. doi: 10.1128/jvi.73.7.5722-5730.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara S, Ueda K, Chen J, Okuno T, Yamanishi K. Octamer-binding sequence is a key element for the autoregulation of Kaposi's sarcoma-associated herpesvirus ORF50/Lyta gene expression. J Virol. 2001;75(15):6894–900. doi: 10.1128/JVI.75.15.6894-6900.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara S, Ueda K, Nishimura K, et al. Accumulation of heterochromatin components on the terminal repeat sequence of Kaposi's sarcoma-associated herpesvirus mediated by the latency-associated nuclear antigen. J Virol. 2004;78:7299–7310. doi: 10.1128/JVI.78.14.7299-7310.2004. 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakibara S, Pise-Masison CA, Brady JN, Tosato G. Gene regulation and functional alterations induced by Kaposi's sarcoma-associated herpesvirus-encoded ORFK13/vFLIP in endothelial cells. J Virol. 2009 Mar;83(5):2140–53. doi: 10.1128/JVI.01871-08. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samols MA, Hu J, Skalsky RL, Renne R. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi's sarcoma-associated herpesvirus. J Virol. 2005 Jul;79(14):9301–5. doi: 10.1128/JVI.79.14.9301-9305.2005. 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samols MA, Skalsky RL, Maldonado AM, Riva A, Lopez MC, et al. Identification of Cellular Genes Targeted by KSHV-Encoded MicroRNAs. PLoS Pathog. 2007;3(5):e65. doi: 10.1371/journal.ppat.0030065. doi:10.1371/journal.ppat.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sample J, Henson EB, Sample C. The Epstein-Barr virus nuclear protein 1 promoter active in type I latency is autoregulated. J Virol. 1992;66:4654–4661. doi: 10.1128/jvi.66.8.4654-4661.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sample J, Hummel M, Braun D, Birkenbach M, Kieff E. Nucleotide sequences of mRNAs encoding Epstein-Barr virus nuclear proteins: a probable transcriptional initiation site. Proc Natl Acad Sci U S A. 1986;83:5096–5100. doi: 10.1073/pnas.83.14.5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler H, Stoecklin G. Control of mRNA decay by phosphorylation of tristetraprolin. Biochem Soc Trans. 2008 Jun;36(Pt 3):491–6. doi: 10.1042/BST0360491. 2008. [DOI] [PubMed] [Google Scholar]
- Sarid R, Wiezorek JS, Moore PS, Chang Y. Characterization and cell cycle regulation of the major Kaposi's sarcoma-associated herpesvirus (Human herpesvirus 8) latent genes and their promoter. J Virol. 1999;73:1438–46. doi: 10.1128/jvi.73.2.1438-1446.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer A, Cai X, Bilello JP, Desrosiers RC, Cullen BR. Cloning and analysis of microRNAs encoded by the primate gamma-herpesvirus rhesus monkey rhadinovirus. Virology. 2007;364(1):21–7. doi: 10.1016/j.virol.2007.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer BC, Paulson E, Strominger JL, Speck SH. Constitutive activation of Epstein-Barr virus (EBV) nuclear antigen 1 gene transcription by IRF1 and IRF2 during restricted EBV latency. Mol Cell Biol. 1997;17:873–886. doi: 10.1128/mcb.17.2.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer BC, Strominger JL, Speck SH. Redefining the Epstein-Barr virus encoded nuclear antigen EBNA-1 gene promoter and transcription initiation site in group I Burkitt lymphoma cell lines. Proc Natl Acad Sci U S A. 1995;92:10565–10569. doi: 10.1073/pnas.92.23.10565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwam DR, Luciano RL, Mahajan SS, Wong L, Wilson AC. Carboxy terminus of human herpesvirus 8 latency-associated nuclear antigen mediates dimerization, transcriptional repression, and targeting to nuclear bodies. J Virol. 2000 Sep;74(18):8532–40. doi: 10.1128/jvi.74.18.8532-8540.2000. 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz M, Murphy PM. Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor constitutively activates NF-kappa B and induces proinflammatory cytokine and chemokine production via a C-terminal signaling determinant. J Immunol. 2001 Jul 1;167(1):505–13. doi: 10.4049/jimmunol.167.1.505. 2001. [DOI] [PubMed] [Google Scholar]
- Sears J, Ujihara M, Wong S, Ott C, Middeldorp J, Aiyar A. The amino terminus of Epstein-Barr Virus (EBV) nuclear antigen 1 contains AT hooks that facilitate the replication and partitioning of latent EBV genomes by tethering them to cellular chromosomes. J Virol. 2004;78:11487–505. doi: 10.1128/JVI.78.21.11487-11505.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgarbanti M, Arguello M, tenOever BR, Battistini A, Lin R, Hiscott J. A requirement for NF-kappaB induction in the production of replication-competent HHV-8 virions. Oncogene. 2004 Jul 29;23(34):5770–80. doi: 10.1038/sj.onc.1207707. 2004. [DOI] [PubMed] [Google Scholar]
- Shamay M, Krithivas A, Zhang J, Hayward SD. Recruitment of the de novo DNA methyltransferase Dnmt3a by Kaposi's sarcoma-associated herpesvirus LANA. Proc Natl Acad Sci U S A. 2006;103:14554–14559. doi: 10.1073/pnas.0604469103. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalsky RL, Hu J, Renne R. Analysis of viral cis elements conferring Kaposi's sarcoma-associated herpesvirus episome partitioning and maintenance. J Virol. 2007;81(18):9825–37. doi: 10.1128/JVI.00842-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalsky RL, Samols MA, Plaisance KB, Boss IW, Riva A, Lopez MC, Baker HV, Renne R. Kaposi's sarcoma-associated herpesvirus encodes an ortholog of miR-155. J Virol. 2007;81(23):12836–45. doi: 10.1128/JVI.01804-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MJ, Deng H, Sun R. Comparative study of regulation of RTA-responsive genes in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J Virol. 2003;77(17):9451–62. doi: 10.1128/JVI.77.17.9451-9462.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MJ, Hwang S, Wong WH, Wu TT, Lee S, Liao HI, Sun R. Identification of viral genes essential for replication of murine gamma-herpesvirus 68 using signature-tagged mutagenesis. Proc Natl Acad Sci U S A. 2005;102:3805–3810. doi: 10.1073/pnas.0404521102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, et al. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood. 1995;86:1276–80. [PubMed] [Google Scholar]
- Sparks-Thissen RL, Braaten DC, Hildner K, Murphy TL, Murphy KM, Virgin H.W.t. CD4 T cell control of acute and latent murine gammaherpesvirus infection requires IFNgamma. Virology. 2005;338:201–208. doi: 10.1016/j.virol.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Speck SH, Chatila T, Flemington E. Reactivation of Epstein-Barr virus: regulation and function of the BZLF1 gene. Trends Microbiol. 1997;5:399–405. doi: 10.1016/S0966-842X(97)01129-3. [DOI] [PubMed] [Google Scholar]
- Speck SH, Pfitzner A, Strominger JL. An Epstein-Barr virus transcript from a latently infected, growth-transformed B-cell line encodes a highly repetitive polypeptide. Proc Natl Acad Sci U S A. 1986;83:9298–9302. doi: 10.1073/pnas.83.24.9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck SH, Strominger JL. Analysis of the transcript encoding the latent Epstein-Barr virus nuclear antigen I: a potentially polycistronic message generated by long-range splicing of several exons. Proc Natl Acad Sci U S A. 1985;82:8305–8309. doi: 10.1073/pnas.82.24.8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck SH, Strominger JL. Epstein-Barr virus transformation. Prog Nucleic Acid Res Mol Biol. 1987;34:189–207. [PubMed] [Google Scholar]
- Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J Virol. 1997;71:715–719. doi: 10.1128/jvi.71.1.715-719.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steed A, Buch T, Waisman A, Virgin H.W.t. Gamma interferon blocks gammaherpesvirus reactivation from latency in a cell type-specific manner. J Virol. 2007;81:6134–6140. doi: 10.1128/JVI.00108-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steed AL, Barton ES, Tibbetts SA, Popkin DL, Lutzke ML, Rochford R, Virgin H.W.t. Gamma interferon blocks gammaherpesvirus reactivation from latency. J Virol. 2006;80:192–200. doi: 10.1128/JVI.80.1.192-200.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugaya M, Watanabe T, Yang A, Starost MF, Kobayashi H, Atkins AM, Borris DL, Hanan EA, Schimel D, Bryant MA, Roberts N, Skobe M, Staskus KA, Kaldis P, Blauvelt A. Lymphatic dysfunction in transgenic mice expressing KSHV k-cyclin under the control of the VEGFR-3 promoter. Blood. 2005;105(6):2356–63. doi: 10.1182/blood-2004-08-3364. [DOI] [PubMed] [Google Scholar]
- Sugden B, Warren N. A promoter of Epstein-Barr virus that can function during latent infection can be transactivated by EBNA-1, a viral protein required for viral DNA replication during latent infection. J Virol. 1989;63:2644–2649. doi: 10.1128/jvi.63.6.2644-2649.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun CC, Thorley-Lawson DA. Plasma cell-specific transcription factor XBP-1s binds to and transactivates the Epstein-Barr virus BZLF1 promoter. J Virol. 2007;81:13566–13577. doi: 10.1128/JVI.01055-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A. 1998;95(18):10866–71. doi: 10.1073/pnas.95.18.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunil-Chandra NP, Efstathiou S, Nash AA. Murine gammaherpesvirus 68 establishes a latent infection in mouse B lymphocytes in vivo. J Gen Virol. 1992;73(Pt 12):3275–3279. doi: 10.1099/0022-1317-73-12-3275. [DOI] [PubMed] [Google Scholar]
- Sunil-Chandra NP, Efstathiou S, Nash AA. Interactions of murine gammaherpesvirus 68 with B and T cell lines. Virology. 1993;193:825–833. doi: 10.1006/viro.1993.1191. [DOI] [PubMed] [Google Scholar]
- Swanton C, Card GL, Mann D, McDonald N, Jones N. Overcoming inhibitions: subversion of CKI function by viral cyclins. Trends Biochem Sci. 1999;24:116–120. doi: 10.1016/s0968-0004(99)01354-7. [DOI] [PubMed] [Google Scholar]
- Swanton C, Mann DJ, Fleckenstein B, Neipel F, Peters G, Jones N. Herpes viral cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature (Lond.) 1997;390:184–187. doi: 10.1038/36606. [DOI] [PubMed] [Google Scholar]
- Talbot SJ, Weiss RA, Kellam P, Boshoff C. Transcriptional analysis of human herpesvirus-8 open reading frames 71, 72, 73, K14, and 74 in a primary effusion lymphoma cell line. Virology. 1999;257(1):84–94. doi: 10.1006/viro.1999.9672. [DOI] [PubMed] [Google Scholar]
- Tao Q, Robertson KD. Stealth technology: how Epstein-Barr virus utilizes DNA methylation to cloak itself from immune detection. Clin Immunol. 2003;109:53–63. doi: 10.1016/s1521-6616(03)00198-0. [DOI] [PubMed] [Google Scholar]
- Tempera I, Lieberman PM. Chromatin organization of gammaherpesvirus latent genomes. Biochim Biophys Acta. 2010;1799(3-4):236–245. doi: 10.1016/j.bbagrm.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–21. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- Thorley-Lawson DA. Epstein-Barr virus: exploiting the immune system. Nat Rev Immunol. 2001;1:75–82. doi: 10.1038/35095584. [DOI] [PubMed] [Google Scholar]
- Thorley-Lawson DA, Allday MJ. The curious case of the tumour virus: 50 years of Burkitt's lymphoma. Nat Rev Microbiol. 2008;6:913–924. doi: 10.1038/nrmicro2015. [DOI] [PubMed] [Google Scholar]
- Thorley-Lawson DA, Duca KA, Shapiro M. Epstein-Barr virus: a paradigm for persistent infection - for real and in virtual reality. Trends Immunol. 2008;29:195–201. doi: 10.1016/j.it.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Thurau M, Marquardt G, Gonin-Laurent N, Weinländer K, Naschberger E, Jochmann R, Alkharsah KR, Schulz TF, Thome M, Neipel F, Stürzl M. Viral inhibitor of apoptosis vFLIP/K13 protects endothelial cells against superoxide-induced cell death. J Virol. 2009 Jan;83(2):598–611. doi: 10.1128/JVI.00629-08. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbetts SA, van Dyk LF, Speck SH, Virgin H.W.t. Immune control of the number and reactivation phenotype of cells latently infected with a gammaherpesvirus. J Virol. 2002;76:7125–7132. doi: 10.1128/JVI.76.14.7125-7132.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkowicz B, Singh SP, Cartas M, Srinivasan A. Human herpesvirus-8 encoded Kaposin: subcellular localization using immunofluorescence and biochemical approaches. DNA Cell Biol. 2002 Mar;21(3):151–62. doi: 10.1089/10445490252925413. 2002. [DOI] [PubMed] [Google Scholar]
- Tripp RA, Hamilton-Easton AM, Cardin RD, Nguyen P, Behm FG, Woodland DL, Doherty PC, Blackman MA. Pathogenesis of an infectious mononucleosis-like disease induced by a murine gamma-herpesvirus: role for a viral superantigen? J Exp Med. 1997;185:1641–1650. doi: 10.1084/jem.185.9.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JL, Cullen BR. In-depth analysis of Kaposi's sarcoma-associated herpesvirus microRNA expression provides insights into the mammalian microRNA-processing machinery. J Virol. 2010;84(2):695–703. doi: 10.1128/JVI.02013-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Speck SH. Evidence for CDK-dependent and CDK-independent functions of the murine gammaherpesvirus 68 v-cyclin. J Virol. 2006;80:11946–11959. doi: 10.1128/JVI.01722-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usherwood EJ, Stewart JP, Nash AA. Characterization of tumor cell lines derived from murine gammaherpesvirus-68-infected mice. J. Virol. 1996;70:6516–8. doi: 10.1128/jvi.70.9.6516-6518.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dyk LF, Virgin H.W.t., Speck SH. The murine gammaherpesvirus 68 vcyclin is a critical regulator of reactivation from latency. J Virol. 2000;74:7451–7461. doi: 10.1128/jvi.74.16.7451-7461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vereide D, Sugden B. Proof for EBV's sustaining role in Burkitt's lymphomas. Semin Cancer Biol. 2009;19:389–393. doi: 10.1016/j.semcancer.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschuren EW, Klefstrom J, Evan GI, Jones N. The oncogenic potential of Kaposi's sarcoma-associated herpesvirus cyclin is exposed by p53 loss in vitro and in vivo. Cancer Cell. 2002;2(3):229–41. doi: 10.1016/s1535-6108(02)00123-x. [DOI] [PubMed] [Google Scholar]
- Vieira J, Huang ML, Koelle DM, Corey L. Transmissible Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in saliva of men with a history of Kaposi's sarcoma. J. Virol. 1997;71:7083–7087. doi: 10.1128/jvi.71.9.7083-7087.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira J, O'Hearn P, Kimball L, Chandran B, Corey L. Activation of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) lytic replication by human cytomegalovirus. J. Virol. 2001;75:1378–1386. doi: 10.1128/JVI.75.3.1378-1386.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viejo-Borbolla A, Ottinger M, Brüning E, Bürger A, König R, Kati E, Sheldon JA, Schulz TF. Brd2/RING3 interacts with a chromatin-binding domain in the Kaposi's Sarcoma-associated herpesvirus latency-associated nuclear antigen 1 (LANA-1) that is required for multiple functions of LANA-1. J Virol. 2005 Nov;79(21):13618–29. doi: 10.1128/JVI.79.21.13618-13629.2005. 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin HW, Speck SH. Unraveling immunity to gamma-herpesviruses: a new model for understanding the role of immunity in chronic virus infection. Curr Opin Immunol. 1999;11:371–379. doi: 10.1016/s0952-7915(99)80063-6. [DOI] [PubMed] [Google Scholar]
- Virgin H.W.t., Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol. 1997;71:5894–5904. doi: 10.1128/jvi.71.8.5894-5904.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin HW, Presti RM, Li XY, Liu C, Speck SH. Three distinct regions of the murine gammaherpesvirus 68 genome are transcriptionally active in latently infected mice. J Virol. 1999;73:2321–2332. doi: 10.1128/jvi.73.3.2321-2332.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltzer L, Bourillot PY, Sergeant A, Manet E. RBP-J kappa repression activity is mediated by a co-repressor and antagonized by the Epstein-Barr virus transcription factor EBNA2. Nucleic Acids Res. 1995;23:4939–4945. doi: 10.1093/nar/23.24.4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltzer L, Perricaudet M, Sergeant A, Manet E. Epstein-Barr virus EBNA3A and EBNA3C proteins both repress RBP-J kappa-EBNA2-activated transcription by inhibiting the binding of RBP-J kappa to DNA. J Virol. 1996;70:5909–5915. doi: 10.1128/jvi.70.9.5909-5915.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Liebowitz D, Kieff E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell. 1985;43:831–840. doi: 10.1016/0092-8674(85)90256-9. [DOI] [PubMed] [Google Scholar]
- Wang S, Liu S, Wu MH, Geng Y, Wood C. Identification of a cellular protein that interacts and synergizes with the RTA(ORF50) protein of Kaposi's sarcoma-associated herpesvirus in transcriptional activation. J. Virol. 2001;75:11961–11973. doi: 10.1128/JVI.75.24.11961-11973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SE, Wu FY, Fujimuro M, Zong J-C, Hayward SD, Hayward GS. Role of the CCAAT/enhancer-binding protein alpha (C/EBP) in activation of the Kaposi's sarcoma-associated herpesvirus (KSHV) lytic-cycle replication-associated protein (RAP) promoter in cooperation with the KSHV replication and transcription activator (RTA) and RAP. J. Virol. 2003;77:600–623. doi: 10.1128/JVI.77.1.600-623.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SE, Wu FY, Yu Y, Hayward GS. CCAAT/enhancer-binding protein- is induced during the early stages of Kaposi's sarcoma-associated herpesvirus (KSHV) lytic cycle reactivation and together with the KSHV replication and transcription activator (RTA) cooperatively stimulates the viral RTA, MTA, and PAN promoters. J. Virol. 2003;77:9590–9612. doi: 10.1128/JVI.77.17.9590-9612.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Sugaya M, Atkins AM, Aquilino EA, Yang A, Borris DL, Brady J, Blauvelt A. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen prolongs the life span of primary human umbilical vein endothelial cells. J Virol. 2003;77:6188–6196. doi: 10.1128/JVI.77.11.6188-6196.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weck KE, Barkon ML, Yoo LI, Speck SH, Virgin HI. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J Virol. 1996;70:6775–6780. doi: 10.1128/jvi.70.10.6775-6780.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weck KE, Dal Canto AJ, Gould JD, O'Guin AK, Roth KA, Saffitz JE, Speck SH, Virgin HW. Murine gamma-herpesvirus 68 causes severe large-vessel arteritis in mice lacking interferon-gamma responsiveness: a new model for virus-induced vascular disease. Nat Med. 1997;3:1346–1353. doi: 10.1038/nm1297-1346. [DOI] [PubMed] [Google Scholar]
- Weck KE, Kim SS, Virgin HI, Speck SH. B cells regulate murine gammaherpesvirus 68 latency. J Virol. 1999a;73:4651–4661. doi: 10.1128/jvi.73.6.4651-4661.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weck KE, Kim SS, Virgin HI, Speck SH. Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J Virol. 1999b;73:3273–3283. doi: 10.1128/jvi.73.4.3273-3283.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer DO, Speck SH. Long-term latent murine Gammaherpesvirus 68 infection is preferentially found within the surface immunoglobulin D-negative subset of splenic B cells in vivo. J Virol. 2003;77:8310–8321. doi: 10.1128/JVI.77.15.8310-8321.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer DO, Speck SH. Establishment and maintenance of long-term murine gammaherpesvirus 68 latency in B cells in the absence of CD40. J Virol. 2005;79:2891–2899. doi: 10.1128/JVI.79.5.2891-2899.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SJ, Tsao EH, Webb BL, Ye H, Dalton-Griffin L, Tsantoulas C, Gale CV, Du MQ, Whitehouse A, Kellam P. X box binding protein XBP-1s transactivates the Kaposi's sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency. J Virol. 2007 Dec. 2007;81(24):13578–86. doi: 10.1128/JVI.01663-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woisetschlaeger M, Jin XW, Yandava CN, Furmanski LA, Strominger JL, Speck SH. Role for the Epstein-Barr virus nuclear antigen 2 in viral promoter switching during initial stages of infection. Proc Natl Acad Sci U S A. 1991;88:3942–3946. doi: 10.1073/pnas.88.9.3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woisetschlaeger M, Strominger JL, Speck SH. Mutually exclusive use of viral promoters in Epstein-Barr virus latently infected lymphocytes. Proc Natl Acad Sci U S A. 1989;86:6498–6502. doi: 10.1073/pnas.86.17.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woisetschlaeger M, Yandava CN, Furmanski LA, Strominger JL, Speck SH. Promoter switching in Epstein-Barr virus during the initial stages of infection of B lymphocytes. Proc Natl Acad Sci U S A. 1990;87:1725–1729. doi: 10.1073/pnas.87.5.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, AuCoin DP, Huete AR, Cei SA, Hanson LJ, Pari GS. A Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J Virol. 2005;79(6):3479–87. doi: 10.1128/JVI.79.6.3479-3487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Ganem D. Induction of chemokine production by latent Kaposi's sarcoma-associated herpesvirus infection of endothelial cells. J Gen Virol. 2007 Jan;88(Pt 1):46–50. doi: 10.1099/vir.0.82375-0. 2007. [DOI] [PubMed] [Google Scholar]
- Yates J, Warren N, Reisman D, Sugden B. A cis-acting element from the Epstein-Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proc Natl Acad Sci U S A. 1984;81:3806–3810. doi: 10.1073/pnas.81.12.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates JL, Warren N, Sugden B. Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature. 1985;313:812–5. doi: 10.1038/313812a0. [DOI] [PubMed] [Google Scholar]
- Ye FC, Zhou FC, Xie JP, Kang T, Greene W, Kuhne K, Lei XF, Li QH, Gao SJ. Kaposi's sarcoma-associated herpesvirus latent gene vFLIP inhibits viral lytic replication through NF-kappaB-mediated suppression of the AP-1 pathway: a novel mechanism of virus control of latency. J Virol. 2008 May. 2008;82(9):4235–49. doi: 10.1128/JVI.02370-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Harada JN, Brown HJ, Deng H, Song MJ, Wu TT, Kato-Stankiewicz J, Nelson CG, Vieira J, Tamanoi F, Chanda SK, Sun R. Systematic identification of cellular signals reactivating Kaposi sarcoma-associated herpesvirus. PLoS Pathog. 2007 Mar;3(3):e44. doi: 10.1371/journal.ppat.0030044. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Feng J, Harada JN, Chanda SK, Kenney SC, Sun R. B cell terminal differentiation factor XBP-1 induces reactivation of Kaposi's sarcoma-associated herpesvirus. FEBS Lett. 2007 Jul 24;581(18):3485–8. doi: 10.1016/j.febslet.2007.06.056. 2007. [DOI] [PubMed] [Google Scholar]
- Zalani S, Holley-Guthrie E, Kenney S. Epstein–Barr viral latency is disrupted by the immediate-early BRLF1 protein through a cell-specific mechanism. Proc Natl Acad Sci U S A. 1996;93:9194–9199. doi: 10.1073/pnas.93.17.9194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Punj V, Matta H, Mazzacurati L, Schamus S, Yang Y, Yang T, Hong Y, Chaudhary PM. K13 blocks KSHV lytic replication and deregulates vIL6 and hIL6 expression: a model of lytic replication induced clonal selection in viral oncogenesis. PLoS One. 2007;2(10):e1067. doi: 10.1371/journal.pone.0001067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Marshall DR, Sample CE. A conserved domain of the Epstein-Barr virus nuclear antigens 3A and 3C binds to a discrete domain of Jkappa. J Virol. 1996;70:4228–4236. doi: 10.1128/jvi.70.7.4228-4236.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegelbauer J, Sullivan C, Ganem D. Serial array-based expression screens identify BCLAF/Btf as a target of multiple KSHV-encoded miRNAs. Nature Genetics. 2009;41:130–134. doi: 10.1038/ng.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimber-Strobl U, Strobl LJ, Meitinger C, Hinrichs R, Sakai T, Furukawa T, Honjo T, Bornkamm GW. Epstein-Barr virus nuclear antigen 2 exerts its transactivating function through interaction with recombination signal binding protein RBP-J kappa, the homologue of Drosophila Suppressor of Hairless. EMBO J. 1994;13:4973–4982. doi: 10.1002/j.1460-2075.1994.tb06824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]