

Abstract
19F NMR has proven to be a powerful technique in the study of protein structure and dynamics because the 19F nucleus is easily incorporated at specific labeling sites, where it provides a relatively nonperturbing yet sensitive probe with no background signals. Recent applications of 19F NMR in mapping out structural and functional features of proteins, including the galactose-binding protein, the transmembrane aspartate receptor, the CheY protein, dihydrofolate reductase, elongation factor-Tu, and D-lactose dehydrogenase, illustrate the utility of 19F NMR in the analysis of protein conformational states even in molecules too large or unstable for full NMR structure determination. These studies rely on the fact that the chemical shift of 19F is extremely sensitive to changes in the local conformational environment, including van der Waals packing interactions and local electrostatic fields. Additional information is provided by solvent-induced isotope shifts or line broadening of the 19F resonance by aqueous and membrane-bound paramagnetic probes, which may reveal the proximity of a 19F label to bulk solvent or a biological membrane. Finally, the effect of exchanging conformations on the 19F resonance can directly determine the kinetic parameters of the conformational transition.
Keywords: fluorine, paramagnetic broadening, spin-label, dynamics, X-ray crystallography
PERSPECTIVES AND OVERVIEW
Since the initial application of solution 19F NMR to protein systems in the late 1960s (80), the technique has evolved to become a versatile tool in the study of protein structure and dynamics and, thus, has provided an approach that complements established structure determination techniques. When high-resolution structural methods cannot be used, 19F NMR generates valuable low-resolution structural information. Perhaps the most powerful application of 19F NMR, however, is to probe proteins of known structure in their native solution environment, where the extreme sensitivity of the 19F resonance to its molecular surroundings can reveal important structural and kinetic features of protein conformational changes.
The rapidly expanding number of high-resolution protein crystal structures has emphasized the need for alternative approaches that can be used to monitor specific locations within a known structural frame-work in solution. For smaller proteins (Mr < 30 kDa), whose solution structures can be probed effectively by multidimensional NMR, such techniques clearly provide the maximum obtainable information and represent the method of choice to complement crystallographic data. Many proteins of known crystallographic structure, however, are too large for multidimensional NMR but still fall within the range of molecular weights accessible to solution 19F NMR (Mr < 100 kDa). For such a protein, one or more fluorine labels can be introduced, either biosynthetically or chemically, into specific sites, thereby enabling the use of 19F NMR to map out the regions of the molecule involved in a conformational change, characterize the kinetics of the event, and, in some cases, measure the intramolecular distance changes that result.
In this review, we use recent 19F NMR studies to illustrate the analysis of both proteins of known crystal or NMR structure and proteins for which no high-resolution structural information exists. We focus primarily on studies published during 1993–1995, in which the fluorine probes are introduced covalently into the protein itself. For a comprehensive summary of earlier applications, including theoretical aspects and studies that introduce fluorine into a small molecule or peptide ligand, see the excellent reviews by Gerig (30, 31), de Dios et al (21), Ho et al (37), and Sykes & Hull (85).
BACKGROUND AND METHODS
Useful Properties of the 19F Nucleus
Several factors contribute to the power of 19F NMR (20, 30, 31, 51):
The spin ½ 19F nucleus occurs at 100% natural abundance and has 83% the sensitivity of 1H.
19F does not occur naturally in proteins; thus, there are no background signals with which to contend.
Fluorine incorporation is generally nonperturbing, particularly when substituted for hydrogen in an amino acid sidechain (see below).
Although the large anisotropy of the chemical shift tensor leads to broader linewidths at high field strengths, the 19F chemical shift range is 100-fold larger than that of 1H. This resolution, coupled with the high detection sensitivity and absence of background signals, generally yields well-resolved 19F resonances in one-dimensional spectra.
One-dimensional 19F NMR studies generally require lower protein concentrations and shorter spectral acquisition times than do multi-dimensional NMR techniques.
The 19F chemical shift is controlled primarily by the fluorine lone-pair electrons, which provide a large paramagnetic term in the shielding formula. The chemical shift, therefore, is exquisitely sensitive to changes in the local van der Waals environment, as well as to local electrostatic fields.
The exposure of specific fluorine labels to paramagnetic centers, such as a bound or aqueous metal ion, a spin-labeled analogue of a ligand, or a spin-labeled lipid probe, can be easily detected.
Incorporation of 19F Labels
A wide variety of fluorine labels, including fluorinated amino acids, fluorinated reagents that react covalently with specific sidechains in proteins, and fluorinated ligands, are available. The most commonly used fluorinated amino acids are analogues of the aromatic amino acids, all of which are available commercially as mixtures of the D and L enantiomers (30). These amino acids include the ortho, meta, and para derivatives of phenylalanine (2-, 3-, and 4-F-Phe), the meta derivative of tyrosine (3-F-Tyr), and tryptophan fluorinated at specific indole ring positions (4-, 5-, and 6-F-Trp).
Several methods exist for incorporating fluorinated amino acids into a selected protein. Although chemical synthesis is impractical for any protein of appreciable size, several proteins have been labeled using a “semi-synthetic” approach (11, 46, 88). In this method, a peptide that contains a fluorinated amino acid is synthesized and combined with the remainder of the protein that has been produced biosynthetically, thereby producing an active, labeled protein. Recent advances in protein splicing techniques may soon revive this approach, which has the advantage of labeling a unique region of the protein (98).
More commonly, fluorinated amino acids are incorporated biosynthetically by microbial protein expression in the presence of the desired amino acid analogue. A fluorinated aromatic amino acid can be incorporated into a bacterial expression system easily, for example, by including it in the growth medium and eliminating the ability of the cell to synthesize that amino acid endogenously. The latter is accomplished either by using a bacterial strain auxotrophic for the amino acid of interest (85) or by adding glyphosate, which inhibits the synthesis pathways of all three aromatic amino acids (44). Depending on the application, the level of incorporation can be adjusted from less than 5% (22) to greater than 90% (50). Similarly, biosynthetic incorporation can be carried out by using a yeast expression system (96) or by including the amino acid analogue in the diet of such vertebrates as rabbit (95) avians (51), or primates (28).
When biosynthetic incorporation of fluorinated amino acids is impractical, or when alternative probe locations are desired, a fluorinated modification reagent can be used to label specific protein sidechains covalently. The most popular of these reagents are the cysteine-reactive probes, which target the unique chemistry of the cysteine thiol. Conveniently, the low frequency of cysteine usage in natural protein sequences generally guarantees a manageably small number of labeling sites or enables the engineering of labeling sites into important, selected locations in the protein structure. Other amino acids that may be labeled with fluorinated reagents include lysine (as well as the N-terminus), serine, tyrosine, and histidine. These reagents have been reviewed recently (30).
Finally, a wide variety fluorinated analogues can be used to substitute for natural ligands, substrates, and intermediates. For example, several fluorinated compounds have been found to be effective enzyme inhibitors (reviewed in 1). Other sources provide further information regarding these small molecules, as well as nucleic acid applications (16–18, 23, 36, 43, 58, 73–75, 81, 86).
Effect of 19F Labels on Protein Structure and Activity
Fluorine incorporation, particularly at a ring position of an aromatic sidechain, generally has little effect on the structure and function of a protein. Escherichia coli, for example, has been labeled with 5-F-Trp to a level of 80% at every tryptophan position within the cell without affecting growth seriously (44, 54), whereas higher order animals can tolerate at least 25% incorporation of 4-F-Phe (95). Similarly, at least 15 proteins have been shown to be structurally or functionally unperturbed by incorporation of fluorine (2, 11, 19, 22, 29, 33, 42, 49, 53, 54, 64, 71, 72, 77, 87, 91, 97). In contrast, detectable perturbations have been observed for a pentafluoro analogue of phenylalanine (8), as well as for monofluoro derivatives of tyrosine (47) and histidine (89), in which the ring fluorine alters the sidechain pKA substantially. Such perturbations, however, appear to be the exception. More caution must be used when fluorine is introduced by chemical modification, because the relatively large size of the modification reagent itself can generate a significant structural perturbation.
Several factors likely contribute to the surprisingly nonperturbing nature of fluorine when it is substituted for hydrogen within amino acid sidechains. The fluorine atom is similar in size to hydrogen [covalent radii of 1.35 Å and 1.2 Å, respectively (68)], such that fluorine substitution has little steric effect. Incorporation of a single fluorine atom into phenylalanine, for example, increases the volume of the side-chain only 0.7%. Moreover, the aliphatic C-F bond is only moderately polar, such that the fluorine atom is a weak hydrogen bond acceptor, at best (66). Aromatic C-F bonds, in which the fluorine electron density is significantly reduced by backbonding to the ring (68), are even less polar and, therefore, are correspondingly less able to accept a hydrogen bond. In fact, no case of hydrogen bond formation that involves aromatic fluorine has been documented. Finally, extensive mutagenic studies have shown that protein structures are relatively plastic and can accommodate a variety of amino acid substitutions, except for those targeted to the active site or locations critical for folding. Substitution of fluorine into a sidechain for hydrogen represents perhaps the most subtle type of “single-atom mutagenesis” possible.
To draw conclusions regarding the native protein from 19F NMR studies, however, it remains essential to establish that the fluorine label is nonperturbing. Two classes of approach have been used to test for perturbations in different types of applications. For those cases in which the level of fluorine incorporation is high enough, the effect of fluorine on protein structure and activity has been assayed directly (2, 11, 29, 33, 42, 49, 53, 54, 64, 71, 72, 77, 87, 91, 97). When the level of incorporation is low, however, an alternative mutagenic approach has been used (19, 22). The latter approach substitutes a natural amino acid, rather than a fluorinated analogue, at a position to be probed by fluorine incorporation. For example, tyrosine has been substituted at phenylalanine positions targeted for labeling with 4-F-Phe, thereby enabling the effect of substituents at the para position of specific phenylalanine residues to be measured directly (19, 22). In this example, the substitution of tyrosine is expected to overestimate any perturbation caused by the incorporation of 4-F-Phe, because the hydroxyl group is considerably larger, more polar, and better able to form hydrogen bonds than is fluorine.
Assignment of Resonances
Several methods have been used to assign 19F NMR resonances. The most rigorous assignments have used site-directed mutagenesis either to replace or to “nudge” each specific fluorine label. In the first method, the residue targeted for assignment is substituted with the most similar sidechain available, thereby causing the corresponding resonance in the 19F NMR spectrum to disappear while the remaining resonances are unperturbed (19, 22, 39, 50, 54, 76, 77). The direct replacement method fails occasionally, because each attempted substitution has perturbed the nontarget resonances. In proteins of known structure, the nudge method can then be used (19, 22). This approach identifies a sidechain in van der Waals contact with the target residue for conservative replacement, thereby generating a local nudge. In favorable cases, this nudge is not transmitted to the remaining resonances. Figure 1 illustrates both types of assignments in the 4-F-Phe-labeled CheY protein (22). A nonmutagenic, but similar, method of assignment has been illustrated in the egg white lysozyme system, for which 19F NMR spectra were obtained with the use of nonidentical lysozymes from several avian species (51).
Figure 1.
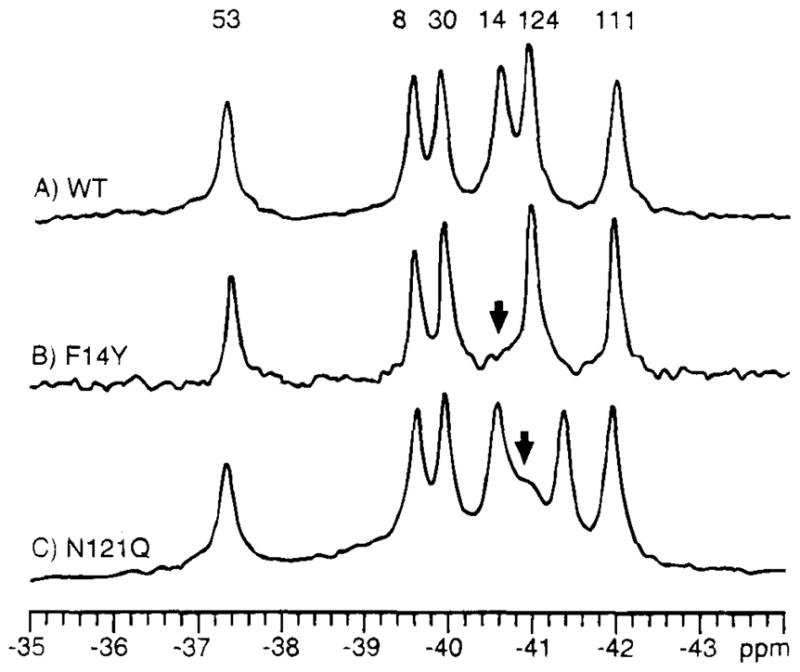
19F NMR spectra illustrating the two protein engineering approaches used to assign resonances of the phospho-signaling protein CheY (22). (A) The protein is labeled at its six phenylalanine positions with 4-F-Phe, thereby giving rise to a well-resolved 19F NMR spectrum at 470 MHz (B) The assignment of the Phe14 resonance by direct replacement with tyrosine. (C) The nudge assignment of Phe124 by replacement of the adjacent Asn121. The same two methods have also assigned the remaining resonances, as indicated.
Additional information, including solvent exposure (see below), perturbation of a resonance by a small molecule ligand or a substrate that binds to a specific site, or chemical modification of an amino acid near a fluorinated residue, can be used to confirm or tentatively assign resonances. These approaches can be complicated, however, by the ability of 19F NMR to detect the local structural and electrostatic effects triggered by a long-range conformational change that originates in a different region of the protein.
Detection of Solvent Exposure
Solvent exposure can be detected either by a solvent-induced isotopic shift (SIIS) or by paramagnetic broadening that stems from a phase-specific probe. The SIIS effect generates up to a 0.25-ppm chemical shift of a 19F resonance exposed to aqueous solution when the solvent is changed from H2O to D2O (30,42). In contrast, a 19F resonance that emanates from the protein interior does not experience solvation and the accompanying chemical shift change, unless the residue interacts with buried water molecules. The alternative method uses an aqueous or hydrophobic paramagnetic probe to broaden the resonances of water-exposed and lipid-proximal fluorine labels, respectively. The aqueous reagent Gd3+ EDTA4−, in which the metal ion possesses seven unpaired electrons, and the membrane probe 8-doxylpalmitic acid have proved useful in many studies (19, 20, 70, 77, 84, 90). Finally, a 19F NMR method has been developed recently to detect bound water molecules within a macromolecule (16).
Interpretation of Chemical Shifts
The large covalent chemical shift range of 19F, which approaches 1000 ppm, stems from the paramagnetic shielding generated by the fluorine lone-pair electrons. This paramagnetic term of the shielding equation also causes the chemical shift to be highly sensitive to the noncovalent tertiary interactions in a folded protein structure. In an unfolded protein that is labeled homogeneously with a given fluorine probe, the resonances from multiple probe positions collapse to give a single peak, because of the equivalent solvent environment experienced by each probe position. In a folded protein, however, the local packing and electrostatic fields perturb the lone-pair electrons of each probe differently, thereby providing a noncovalent chemical shift range as large as 17 ppm (69).
Several theoretical descriptions have been offered to explain observed range of environmental 19F chemical shifts in proteins. Millet & Raftery (61) divided fluorine shielding into contributions from (a) local magnetic fields that arise from electronically anisotropic groups, (b) hydrogen bond formation, (c) electrostatic fields from dipoles or formal charges, and (d) van der Waals interactions. Local magnetic fields from aromatic rings, carbonyls, and other electronically anisotropic groups generate chemical shift changes less than 2 ppm in magnitude and, therefore, cannot explain the observed protein chemical shift range (32). The contribution of hydrogen bonding is uncertain, because fluorine hydrogen bonds in proteins are weak or nonexistent. The two forces that dominate environmental 19F chemical shifts in proteins, therefore, are likely to be van der Waals interactions and electrostatic fields.
Considerable evidence suggests that van der Waals interactions play an important role. Hull & Sykes (42) observed a rough correlation between chemical shift and T1 relaxation for fluorines in alkaline phosphatase labeled with 3-F-Tyr. Because T1 relaxation and the van der Waals force have the same r−6 dependence on the distance to nearest neighbor protons, T1 relaxation was regarded as an indicator of the extent of van der Waals contacts or the extent of burial within the protein interior. The latter relationship was verified by solvent exposure measurements with the use of SIIS. These authors therefore concluded that van der Waals contacts were the primary factor influencing the chemical shift. Gregory & Gerig (32) reached a similar conclusion in a study comparing the results of molecular dynamics calculations with experimental data obtained for fluorinated ribonuclease-S (11). van der Waals interactions were proposed to be the dominant factor controlling 19F chemical shift, together with smaller contributions from local magnetic and electric fields. This study demonstrated the importance of molecular dynamics in chemical shift calculations, because calculations based on static structures yielded incorrect chemical shifts. More recently, van der Waals interactions within a molecular dynamics simulation have been used to explain the 19F chemical shifts of the E. coli galactose-binding protein in terms of its known high-resolution structure (12).
If van der Waals interactions were the sole effector of fluorine chemical shift changes, however, one would expect all 19F NMR resonances from buried probe positions to be shifted downfield from the denatured protein resonance. The fact that many 19F NMR resonances are observed upfield of the denatured protein resonance (30) indicates that another factor must also contribute to the observed chemical shifts. On the basis of electric field effects alone, Augspurger et al (3) could predict accurately chemical shift ranges in several nuclei, including 19F. Subsequently, a self-consistent field approach, denoted “charge field perturbation-gauge including atomic orbital,” was used to predict the chemical shifts of the E. coli galactose-binding protein solely on the basis of electric field effects (21, 69). A strong correlation was seen between the predicted and experimental chemical shifts, which led to the conclusion that weak electrical interactions dominate the fluorine chemical shielding term. Surface charge does not significantly contribute to the shielding (51, 69).
Overall, it appears likely that van der Waals and electrostatic interactions together control the environmental 19F chemical shifts observed in proteins. Significantly, such chemical shifts are regulated by local, rather than long-range, factors. Thus, although it remains difficult to convert either the magnitude or the sign of a chemical shift into a structural parameter, the observation of a chemical shift change indicates that the protein structure or electrostatics has been altered in the vicinity of that probe. In fact, the extreme sensitivity of the 19F chemical shift to its environment provides what is perhaps the most sensitive physical method available for the detection of local conformational changes.
PROBING CONFORMATIONAL CHANGES IN PROTEINS OF KNOWN STRUCTURE
X-ray crystallography and 19F NMR have proven to be splendidly complementary techniques. Starting with a high-resolution structure solved under relatively static conditions, 19F NMR allows us to observe the dynamics of the protein, as well as to map out areas where changes occur as the protein enters a conformation inaccessible to crystallography. Studies to date have primarily used proteins labeled with fluorinated aromatic amino acids.
Because of the importance of aromatic sidechains in many aspects of protein function, aromatic fluorine labels are often located in critical regions of the target protein, including most ligand and effector protein binding sites. For the most part, two 19F NMR approaches have been used to probe conformational changes. The first approach maps out the regions involved in the conformational change by observing which 19F NMR resonances undergo chemical shift changes. The second approach uses paramagnetic broadening to measure distances and changes in solvent exposure.
In addition to providing spatial maps of conformational changes in proteins, 19F NMR has been used to characterize the kinetic parameters of these conformational changes. The standard relationships of NMR exchange averaging places limits on the frequency of a conformational change, or, in favorable cases, can determine this rate directly (94):
| 1 |
| 2 |
| 3 |
When two conformations, A and B, which yield NMR frequencies vA and vB, interconvert over time at a frequency of vic two distinct resonances are observed if the interconversion is slow relative to the difference in their NMR frequencies (Equation 1). If, however, the interconversion rate is rapid relative to the NMR frequency difference, then a single exchange-averaged resonance is observed (Equation 3). In the intermediate exchange limit, where the interconversion rate approximately equals the NMR frequency difference, the two resonances become very broad and disappear into the baseline. Observation of the latter intermediate exchange limit enables direct determination of the interconversion rate, whereas the slow and rapid exchange cases provide upper and lower limits, respectively, on the interconversion rate. Spectral simulation can provide even more precise information regarding both interconversion kinetics and the relative populations of different conformational states.
The Proteins of Bacterial Chemotaxis
The chemotaxis pathway of E. coli and Salmonella typhimurium provides a well-suited system for the study of conformational changes involved in signal transduction (reviewed in 6, 35, 67, 83). The protein components of the pathway are defined fully, and several have been characterized structurally by crystallography or NMR. Such structural studies have helped define key mechanistic questions, the answers to which will provide a molecular understanding of signaling protein activation. To address these questions, 19F NMR has been utilized to augment high-resolution structures. Studies of the galactose-binding protein (54–56) represent the earliest application of 19F NMR to probe intrinsic sidechain labels in a protein of known crystal structure. The same approach has been extended to studies of the aspartate receptor (19, 26) and CheY (7, 22). Together, these three proteins illustrate how solution 19F NMR can complement the information provided by high-resolution structure determination. In some cases, the power and limitations of the 19F NMR method have been defined by direct comparison with results from independent approaches.
GALACTOSE-BINDING PROTEIN
Chemosensing in bacteria often begins with a soluble periplasmic receptor, also termed a binding protein, which engulfs a specific small molecule ligand, then docks to and activates one of several transmembrane receptors that span the cytoplasmic membrane. In general, the soluble receptor also transports its ligand by associating with a membrane-bound transport system distinct from the sensory pathway. As a representative member of the soluble receptor family, the galactose-binding protein plays both a sensory and transport role for its two ligands, D-galactose and D-glucose. The crystal structure of the monomeric 32-kDa protein has been solved to 1.9 Å resolution for the conformation that contains bound D-glucose, as illustrated in Figure 2 (93), but the apo-structure is undefined. Thus, little is known about the conformational change that the protein undergoes when it binds ligand. Overall, the structure consists of two homologous domains, each consisting of a β-sheet sandwiched between layers of α-helices. The two domains are connected by three polypeptide strands, and the sugar-binding site lies within the cleft between the domains. The protein also possesses an EF-hand-like calcium-binding site, which stabilizes the protein structure. Luck & Falke (54–56) have fluorine-labeled the protein with 5-F-Trp and 3-F-Phe to investigate the conformational changes induced by sugar and calcium binding.
Figure 2.
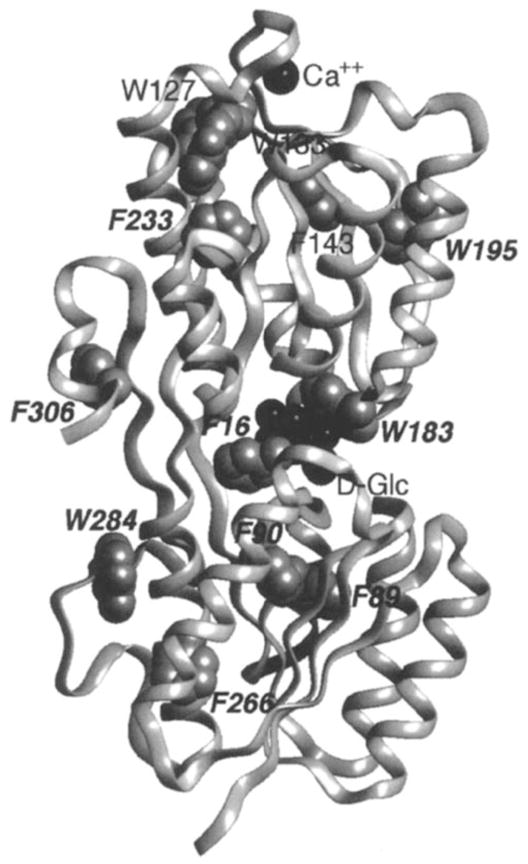
Backbone ribbon structure of the galactose-binding protein (93), which illustrates bound ligands (Ca2+ and D-glucose, both in black) and the 12 fluorine probe positions. The protein has been labeled at its five tryptophan positions with 5-F-Trp, or its seven phenylalanine positions with 3-F-Phe (gray VDW surfaces), thereby enabling the use of 19F NMR to probe ligand-induced conformational changes (54–56). The binding of D-galactose or D-glucose generates a long-range conformational change, thus yielding significant chemical shift changes for the nine probe positions indicated by italics (see Figure 5). In contrast, the three remaining probes, all in the vicinity of the Ca2+ binding site, are unperturbed. When Ca2+ binds, the converse pattern of frequency changes is observed, such that only the resonances of the latter three probes are observed to shift.
As seen in Figure 2, the targeted 12 probe locations (five tryptophans and seven phenylalanines) are well distributed throughout the molecule, including the sugar- and calcium-binding site regions. Biosynthetic incorporation of fluorinated amino acids into the appropriate E. coli auxotrophs yields 65–80% incorporation of 5-F-Trp or 20% incorporation of 3-F-Phe (54). The resonance that corresponds to each 5-F-Trp probe has been assigned by the direct-replacement mutagenesis method (see above).
The effect of fluorine labeling on the structure has been assayed directly, thereby revealing relatively minor effects caused by fluorine substitution (54). The estimated upper limits of these effects, calculated for the fully labeled protein, are as follows: (a) a 2.2- to 3-fold decrease in the D-galactose affinity caused by 5-F-Trp substitution, or a 1.5- to 4-fold affinity decrease caused by 3-F-Phe substitution, respectively; (b) a 1.5- to 2.5-fold increase in the calcium dissociation rate caused by 5-F-Trp substitution, whereas 3-F-Phe substitution has no detectable effect; and (c) a measurable decrease in protein stability, as revealed in urea denaturation curves yielding ΔΔGU(H2O) = −1.2 kcal mol −1 for 5-F-Trp, or −0.8 kcal mol −1 for 3-F-Phe. For this protein, then, the effects of fluorine incorporation on protein structure and function are quite small, especially considering that bound ligand is stabilized by direct van der Waals interactions with tryptophan and phenylalanine sidechains (one of each).
At the time the 19F NMR studies were carried out, it was proposed that a key feature of binding protein activation was closure of the ligand-binding cleft through bending of the interdomain linkage (57a). Such a hinged-cleft mechanism, however, had not been demonstrated experimentally for any member of the binding protein family. To test this mechanism, and to measure the minimum angle of cleft opening on removal of sugar, 19F NMR paramagnetic broadening measurements were carried out with the use of the aqueous paramagnet Gd3+ EDTA4−(56). With this approach, the inverse sixth power dependence of paramagnetic broadening is used to estimate the separation between the paramagnet and the 19F nucleus of a 5-F-Trp probe located at position 183 within the cleft. Addition of the paramagnet to the sugar-empty protein causes selective broadening of the probe resonance, as shown in Figure 3, whereas the paramagnet has no effect on the probe resonance when the cleft is closed with D-glucose trapped inside. The broadening of the probe resonance can be used to place an upper limit on the probe–paramagnet separation for the empty cleft. These results demonstrate that the cleft can open by at least 18 degrees in the absence of sugar, thereby enabling the paramagnetic probe to enter, as illustrated schematically in Figure 4 (56). Subsequent crystallographic studies have confirmed large, hinged-cleft motions in related binding proteins (27, 79).
Figure 3.
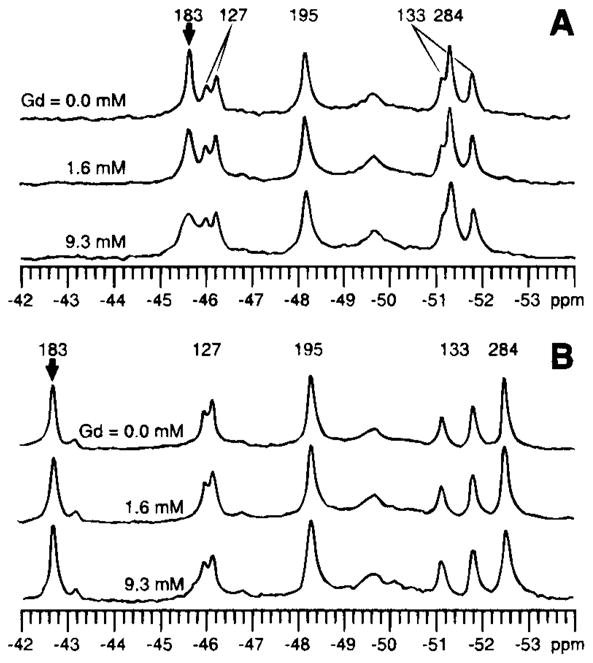
Effect of the aqueous paramagnetic probe Gd3+ EDTA on the 19F NMR spectrum of the galactose-binding protein (470 MHz) (56). The resonance of the 5-F-Trp probe at the Trp183 position within the sugar-binding cleft is broadened selectively by increasing concentrations of the paramagnet when the cleft is empty (A), whereas the same probe resonance in the closed cleft containing bound D-glucose is unaffected (B). The observed broadening of the Trp183 resonance in the empty cleft has been used to place an upper bound on the closest approach of the paramagnet (see Figure 4). The duplicity observed for the Trp127 and 133 resonances stems from two kinetically stable conformations of their environment.
Figure 4.
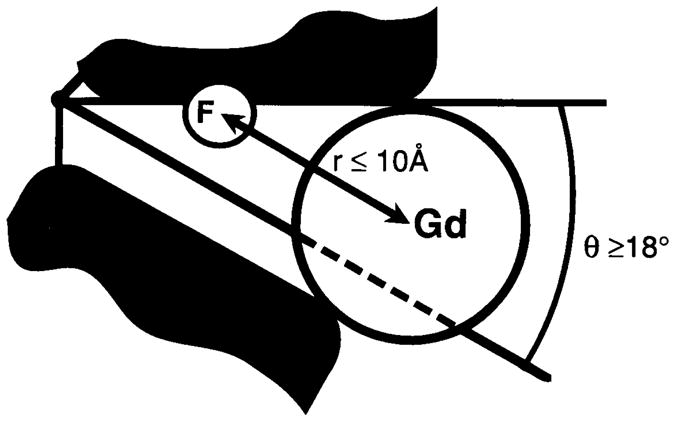
Schematic model for the empty sugar cleft of the galactose-binding protein (56). Addition of the aqueous paramagnet Gd3+ EDTA (Gd) to the empty cleft is observed to broaden the 19F NMR resonance of the 5-F-Trp probe (F) located at the Trp183 position within the cleft (see Figure 3). The resulting broadening indicates that the paramagnet can approach the fluorine probe at a distance of 10 Å or less, which places the paramagnet within the cleft. To accommodate the paramagnet, the known structure of the closed cleft must open by an angle of at least 18 degrees.
More subtle conformational changes are also revealed by 19F NMR chemical shift changes within the individual protein domains (Figure 5). Inside the sugar cleft itself, the binding of D-galactose causes a dramatic chemical shift change (+ 3.8 ppm) in the resonance that corresponds to Trp 183, which contacts the bound sugar. The binding of D-glucose, which differs from D-galactose by the stereochemistry about a single carbon atom, induces a slightly smaller chemical shift change (+ 2.8 ppm) in the same resonance. In contrast, at more distal 5-F-Trp or 3-F-Phe probe positions, the two sugars induce identical chemical shift changes, with the exception of resonances in the vicinity of the calcium-binding site. These resonances are unaffected by sugar binding (Figure 2). Conversely, calcium binding alters the chemical shifts of only those residues located in the vicinity of its binding site and does not affect the resonances coupled to the sugar site (55). Calcium binding, therefore, induces only a local structural change, whereas sugar binding generates a long-range conformational change; the two sites are allosterically independent of one another. The long-range nature of the sugar-induced conformational change is consistent with its function, which is to regulate protein-protein interactions that involve large regions of the binding protein surface.
Figure 5.

Effect of D-galactose and D-glucose on the 19F NMR spectrum of the 5-F-Trp labeled galactose-binding protein (470 MHz) (54). (A) The spectrum of the sugar-empty protein. (B) The protein saturated with D-galactose. (C) The protein saturated with D-glucose. (bold arrows) Resonances for which significant ligand-induced chemical shift changes are observed. The greatest changes are observed for the resonance from the Trp183 position, which lies in van der Waals contact with the bound sugar molecule.
Further analysis of 19F NMR spectra places a lower limit on the kinetics of sugar release. At substoichiometric concentrations of D-glucose and D-galactose, the spectrum of the 5-F-Trp-labeled protein shows distinct resonances that correspond to each apo- and sugar-bound state (54), thereby placing sugar release in the slow exchange limit. Equation 1 indicates that the lifetime of the D-galactose-bound state is τic ≫0.6 ms, whereas the lifetime of the D-glucose-bound state is τic≫ 0.8 ms, which is consistent with direct kinetic measurements that yield bound sugar lifetimes of 220 ms for D-galactose and 710 ms for D-glucose (60).
Altogether, the 19F NMR data indicate that the sugar-binding cleft does, indeed, close on sugar binding, thereby trapping the sugar inside until the complex can diffuse to the appropriate membrane receptor. Moreover, subtle, long-range conformational changes are induced in both domains by sugar, but not calcium, binding. These intradomain conformational changes, which are not easily detected by crystallography, likely stem from the conserved architecture of the binding protein family, in which most secondary structure elements originate or terminate in the substrate cleft. Such an architecture may transmit conformational information from the buried ligand-binding site to the protein surface, where it can be used to regulate the critical membrane docking sites (45).
ASPARTATE RECEPTOR
The transmembrane aspartate receptor is activated either directly, by the binding of the chemoattractant aspartate to its periplasmic ligand-binding domain, or indirectly, by association with the maltose binding protein. The resulting transmembrane signal regulates an associated cytoplasmic histidine kinase, which ultimately controls the swimming behavior of the cell. The receptor is a homodimer of identical 60-kDa subunits in both the presence and the absence of ligand (25, 62). The structure of the isolated periplasmic domain has been determined to 2.0 Å resolution by X-ray crystallography, as displayed in Figure 6 (59). This domain consists of a homodimer of four-helix bundles, with two symmetric, nonoverlapping attractant binding sites at the dimer interface. In the intact receptor, the structure of the transmembrane domain has been defined by disulfide mapping, which indicates that the N- and C-terminal α-helices of the ligand-binding domain continue uninterrupted through the membrane.
Figure 6.

Ribbon backbone structure of the ligand-binding domain from the aspartate receptor (59), which shows the residues (black) in one of the two symmetric ligand-binding sites, as well as the six fluorine probe positions in one of the two symmetric subunits. The isolated domain has been labeled with 4-F-Phe at all six phenylalanines (gray VDW surfaces), thereby enabling the use of 19F NMR to detect ligand-induced chemical shift changes (see Figure 7) (19). Aspartate is observed to induce a long-range conformational change, which yields significant changes in chemical shift for the three probe positions indicated by italics. In the intact receptor, two transmembrane helices from each subunit would extend downward into the bilayer located below the figure, thus carrying the ligand-induced signal across the membrane.
To map out the spatial and kinetic features of the aspartate-induced conformational change, both the intact receptor and the isolated ligand-binding domain have been probed by solution 19F NMR. The intact receptor has been labeled with 5-F-Trp in its native membrane (26). Because of the very slow tumbling of the protein-membrane complex, all but one of the receptor 5-F-Trp resonances are too broad to detect. The lone resonance observed lies near the mobile C-terminus of the receptor and is exposed to the cytoplasmic compartment, where it possesses considerable local motion. Aspartate binding to the periplasmic ligand-binding site induces a long-range, transmembrane conformational change that is transmitted to the C-terminal 5-F-Trp probe located more than 90 Å away, thereby perturbing the chemical shift of this resonance.
A higher resolution picture of the conformational change has been obtained with the use of the 36-kDa isolated ligand-binding domain, which has been cloned and shown to retain native aspartate binding (19, 63). The homodimeric domain has been biosynthetically labeled with 4-F-Phe, with the use of glyphosate to inhibit endogenous synthesis of phenylalanine (19). Each identical subunit possesses six phenylalanine labeling positions, which are distributed throughout the known structure (Figure 6). Significantly, two probe positions lie in or near the ligand-binding pocket, whereas the other four probes lie at positions distal to the bound aspartate, including distal sites on the N- and C-terminal α-helices. Assignment of the resulting resonances has been carried out with the use of a combination of the direct replacement and the nudge mutagenic techniques.
The chosen low level of fluorine incorporation (7%) ensures that the labeled domain typically possesses no more than one fluorine atom, thereby preventing pairwise or higher order interactions. To estimate the effects of such fluorine incorporation on domain structure and function, each of the six phenylalanine residues has been individually mutated to tyrosine (19). The resulting para-hydroxyl moiety is expected to be even more perturbing than the corresponding parafluorine (see above), yet the tyrosine substitutions yield little effect on the aspartate binding site, which retains glutamate affinities ranging from 4-fold to 0.6-fold of native. These effects are quite small on a free energy scale (≤0.8 kcal mol−1), which suggests that hydroxyl substitutions at the para position of each phenylalanine, as well as the corresponding fluorine substitutions, cause little or no perturbation of ligand binding and the ensuing conformational change.
The 19F NMR spectrum of the ligand-binding domain has been obtained for the apo-, aspartate-bound, and glutamate-bound states (19). Only six resonances are observed for the 12 4-F-Phe positions in the dimer, which indicates that the monomers are equivalent, on average, owing to rapid exchange of ligand between the two symmetric binding sites. As observed for the galactose-binding protein, different ligands elicit contrasting chemical shift changes for a resonance within their shared binding pocket (4-F-Phe150) but yield the same ligand-induced pattern of chemical shift changes at more distant probe positions. These ligands therefore generate similar or identical long-range conformational changes outside the binding site itself.
Figure 6 summarizes the probe positions for which significant ligand-induced chemical shift changes have been detected (19). Importantly, chemical shift changes are observed for probe positions lying on the C-terminal helix but not for those on the N-terminal helix, which implicates the former helix as the transmitter of the transmembrane signal. Subsequent analyses of data from X-ray crystallography and disulfide engineering have confirmed this identification of the C-terminal helix as the key transmembrane signaling element, whereas the N-terminal helix is proposed to play a structural role in stabilizing the dimer (13–15, 48).
Additional stoichiometric and kinetic information has been extracted from a titration of the 19F NMR spectrum with ligand (19), as illustrated in Figure 7. Note that the maximum effect of ligand on the resonances is attained at a ligand:dimer mole ratio of 1:1, although there are two symmetric binding sites in the apo-dimer. This result confirms the earlier conclusion of the crystallographic and direct binding measurements (5, 59), which indicated strong negative cooperativity between the two aspartate binding sites. The titration also reveals that one of the resonances, which arises from 4-F-Phe150 in the ligand-binding site, disappears because of exchange broadening at a nonsaturating aspartate concentration, thereby enabling the kinetics of aspartate binding and dissociation to be measured. The resulting calculation indicates that aspartate binding and release is rapid, approaching the diffusion controlled limit observed for the fastest enzymes (19). Ligand binding and release, therefore, are not rate limiting in the chemosensory pathway, because subsequent phosphorylation and/or protein diffusion events in the cytoplasm are significantly slower (78).
Figure 7.
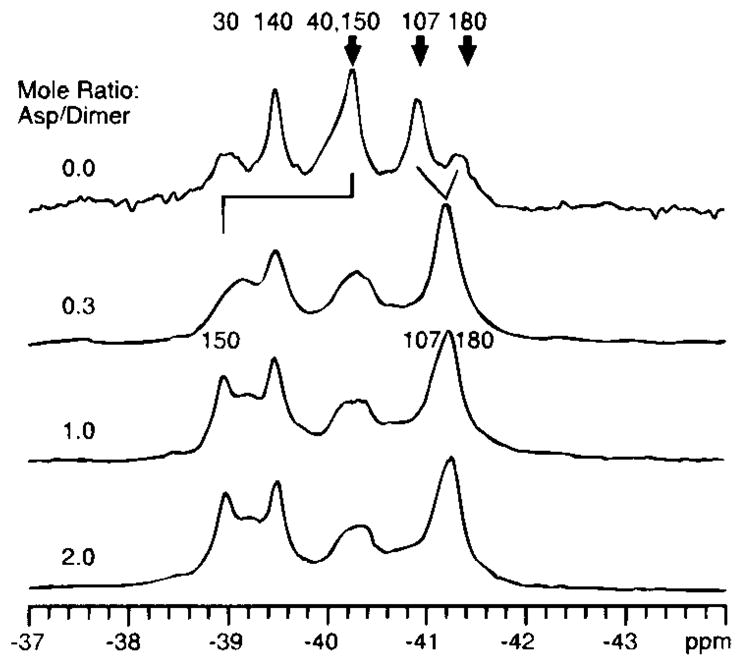
The ligand-binding domain of the aspartate receptor: titration of the 19F NMR spectrum with aspartate (470 MHz) (19). (upper) The spectrum of the 4-F-Phe-labeled apo-domain to which is added increasing concentrations of ligand aspartate. (bold arrows) The resonances that undergo chemical shift changes on ligand binding. The final chemical shifts are observed at a mole ratio of one aspartate molecule per dimer, which indicates half-of-sites occupancy. In addition, the Phe 150 resonance is observed to disappear at an intermediate loading (mole ratio = 0.3), then reappears at a new frequency on saturation (mole ratio = 1.0), thus demonstrating that this resonance passes through the intermediate exchange limit.
CHEY
CheY is the response regulator protein of the chemotaxis pathway. It receives a phosphate from the histidine kinase associated with the transmembrane receptors and subsequently docks to and regulates the behavior of the flagellar motor. The structure of the unphosphorylated 13-kDa protein has been determined crystallographically by several groups (4, 82, 92), and independent solution NMR structures have also been completed (9, 52, 65). CheY folds as a compact (α/β)5 motif illustrated in Figure 8, with a central antiparallel β-sheet sandwiched between layers of α-helices. The activation site, where phosphorylation occurs and activating mutantations are found, lies at one end of the β-sheet and includes four residues that exhibit 70–100% conservation across the response regulator family: Asp12, Asp13, Asp57, and Lys109. The Asp57 sidechain is the site of phosphorylation and provides one of the coordinating oxygens for a bound magnesium ion, which serves as a cofactor in phosphorylation and dephosphorylation of Asp57. A salt bridge has been proposed to exist between the highly conserved Asp57 and Lys109 residues (92). This salt bridge could provide a structural on-off switch in response regulator activation; alternatively, the Lys109 sidechain could play a role in the active site chemistry.
Figure 8.
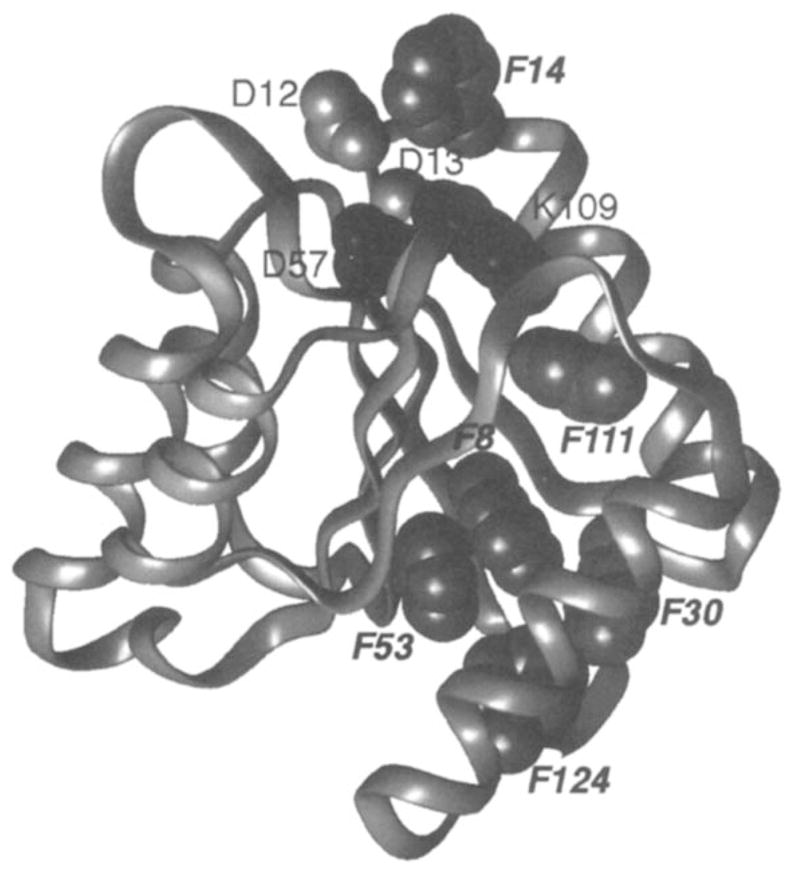
Ribbon backbone structure of the phospho-signaling protein CheY (92). Shown are Asp57, the site of phosphorylation, the adjacent highly conserved Lys109 (black), two other conserved active site residues (Asp12,13) and the fluorine probe positions. The protein has been labeled with 4-F-Phe at all six phenylalanines (gray VDW surfaces), thereby enabling the use of 19F NMR to detect activation-induced chemical shift changes (7,22). Phosphorylation of Asp57 is observed to generate significant chemical shift changes at all six probe positions, indicated by italics. In contrast, constitutive activation of the protein by the D13K mutation gives rise to large chemical shift changes for only the Phe14 and Phe111 resonances, both of which arise from the vicinity of the active site.
The short lifetime of the phospho-Asp57 linkage (τ1/2 ~ 10 s) has prevented the crystallization of the protein in the phosphorylated state. Instead, 19F NMR has been used to probe CheY labeled with 4-F-Phe, thereby enabling the conformational changes triggered by both phosphorylation and activating mutations to be mapped out (22). Using an E. coli strain auxotrophic for phenylalanine, 4-F-Phe has been incorporated to a level of 5%. The resulting six fluorine probe positions (Figure 8) are found at interesting locations, including the active site (Phe14), the loop containing the putative “switch” residue Lys109 (Phe111), and a phenylalanine cluster on the opposite side of the molecule from the active site, which can serve as an antenna for long-range conformational changes. The six resonances have been assigned with the use of direct replacement and the nudge method (Figure 1), and the maximal effect of fluorine incorporation at each phenylalanine position has been estimated by the corresponding tyrosine substitutions (see above). Each of these tyrosine substitutions has a negligible effect on the chemotaxis pathway in vivo, which indicates that perturbations of CheY caused by para substituents on its phenylalanine rings are minimal or nonexistent (22).
To probe the conformational change induced by phosphorylation, the short lifetime of the phosphorylated state has been overcome by chemically phosphorylating Asp57 with the small molecule phosphodonor, acetyl phosphate (57). Excess acetyl phosphate can maintain a high steady-state level of phosphorylation for 10 min—sufficient time to obtain a one-dimensional 19F NMR spectrum. Phosphorylation induces a global conformational change in the CheY molecule, which extends from the active site to the distant phenylalanine cluster (Figure 8). Subsequent multidimensional solution NMR studies have confirmed this long-range conformational change and have provided a higher resolution map of its extent (52). The long-range nature of the phosphorylation-induced structural change may play an important role in the large family of two-domain response regulator proteins (67), in which phosphorylation of the regulatory domain could transmit an allosteric signal to the effector domain.
In contrast to the long-range structural change triggered by phosphorylation, activation of CheY by specific “lock-on” mutations generates a much more localized conformational perturbation (7). Six single or double mutations that activate CheY constitutively all generate an upfield chemical shift change for the resonance corresponding to Phe111, which lies in the same loop as Lys109 (Figure 8). The structural change detected for this loop could represent a key component of the on-off “switch.” On the basis of circumstantial evidence, it has been proposed (7) that the Asp57-Lys109 salt bridge is formed in the off state and broken in the on state; this conclusion remains controversial, however, and must be tested by other approaches. The localized nature of the conformational change triggered by one of the lock-on mutations has recently been confirmed by multidimensional NMR (FW Dahlquist, personal communication).
Nuclear magnetic resonance methods, including 19F NMR, have begun to reveal key aspects of the protein–protein interaction between CheY and its kinase protein, CheA. Interestingly, the high-affinity kinase binding site on CheY appears to be located at a surface distal from the phosphorylated residue Asp57. Thus, when the CheA fragment responsible for CheY docking is added to the labeled protein, large chemical shift changes at positions distant from the active site have been observed in both 19F (20; TB Morrison, M Welch, Y Blat, SL Butler, JJ Falke, M Eisenbach, and JS Parkinson, submitted for publication) and multidimensional (84a) NMR studies. The phosphorylation domain of the kinase, therefore, must be large enough to bridge this distance.
Other Examples
DIHYDROFOLATE REDUCTASE
The dihydrofolate reductase (DHFR) enzyme of E. coli is an 18-kDa monomeric protein that catalyzes the NADPH-dependent reduction of 7,8-dihydrofolate to 5,6,7,8-tetrahydrofolate, an important cofactor for several biosynthetic pathways. Because of its small size, well-characterized enzymatic mechanism, well-refined structure, and the reversibility of its folding reaction in the presence of chemical denaturants, Hoeltzi & Frieden (39) have deemed this a good model for studies of protein folding. The use of 19F NMR provides an advantage over fluorescence, absorbance, or circular diochroism in that it can provide information regarding specific sites within the protein. The five tryptophan residues, which have been labeled with 6-F-Trp, are distributed throughout the molecule illustrated in Figure 9 and give rise to well-resolved resonances that have been assigned by direct replacement mutagenesis (39).
Figure 9.
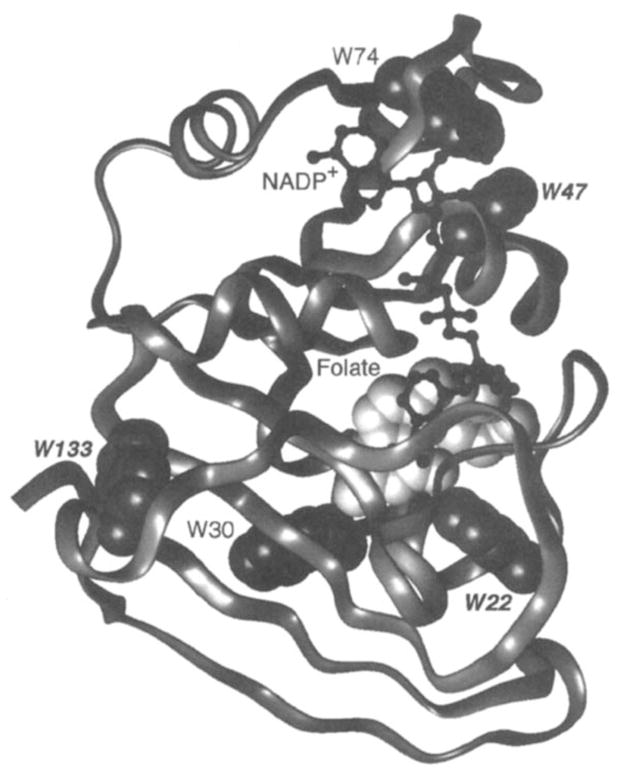
Ribbon backbone structure of the biosynthetic enzyme dihydrofolate reductase (10). Shown are the bound ligands NADP+ (black ball and stick) and folate (light VDW surface), as well as the fluorine probe positions. The protein has been labeled with 6-F-Trp at its five tryptophan positions (gray VDW surfaces), thereby enabling 19F NMR studies of ligand-induced chemical shift changes (40). Binding of methotrexate to the site that contains folate triggers a long-range conformational change, thereby yielding chemical shift changes at the probe positions indicated by italics and increasing the structural homogeneity of the protein (see text). In contrast, the binding of NADPH generates a more localized conformational change, detected by chemical shifts changes of the Trp22 and Trp74 resonances that arise from the NADPH binding pocket itself.
Dihydrofolate reductase possesses two ligand-binding sites, one for NADPH and the other for 7,8-dihydrofolate. The latter site also binds methotrexate, a clinically important anticancer drug. A comparison of the spectra observed for the apo, NADPH-bound, methotrexate-bound, and doubly liganded (NADPH and methotrexate) forms of the protein reveal that NADPH significantly shifts two of the five probe resonances in or near its binding site (Trp74 and Trp22), whereas methotrexate triggers a longer range conformational change revealed by large chemical shift changes of three probe resonances (Trp22, Trp133, and Trp47; see Figure 9). Moreover, methotrexate sharpens the resonances of two probe resonances (Trp22, Trp30) considerably, whereas NADPH broadens two resonances (Trp22, Trp74) relative to the apo state. The doubly liganded protein exhibits the maximum chemical shift dispersion and sharpest resonances, which suggests that this conformation is the most stable or least flexible and possesses less structural heterogeneity than the lower ligation states. Of the two ligands, the methotrexate appears to generate the greater increase in structural homogeneity (39). Crystallographic evidence also supports the ordering effect of this ligand, which is observed to order the loop containing one of the fluorine probe positions, Trp22 (10).
To study the unfolding process of this protein further, Hoeltzi et al (40, 41) have constructed a stopped-flow 19F NMR device to monitor unfolding in real time with a resolution of 1.5 s. They observe that immediately after the addition of urea, the resonances from the native protein disappear, but the denatured resonances exhibit only 20% of their final intensity. Together with fluorescence and circular dichroism data, these stopped-flow 19F NMR results indicate that the protein unfolds by two pathways. Approximately 20% of the population denatures rapidly, whereas 80% of the protein forms a molten globule intermediate that retains native-like secondary structure but loses tertiary contacts, thereby causing the tryptophan sidechains to exhibit considerable structural heterogeneity (40). Interestingly, when the protein is titrated with increasing concentrations of urea in a separate equilibrium experiment, the resonance corresponding to Trp22 narrows and moves toward its denatured chemical shift at urea concentrations well below the denaturation midpoint (39). Thus, as suggested by the 19F NMR ligand-binding and crystallographic results, the region of the methotrexate binding site, including the Trp22 residue, appears to be less folded than the rest of the molecule in the absence of its ligand.
ELONGATION FACTOR TU
In the GTP-bound form, elongation factor Tu (EF-Tu) delivers aminoacyl-transfer RNA to the ribosome. After hydrolysis of the GTP to GDP, EF-Tu dissociates, and the GTP-bound form is regenerated by a different elongation factor. The structure of each of the three domains of trypsin-treated EF-Tu have been determined crystallographically. Subsequently, 19F NMR has been used to monitor the effects of various ligands on the conformation of full-length EF-Tu and to examine the individual domains for perturbations that arise from their isolation through trypsin cleavage (24). Elongation factor Tu has been labeled with 3-F-Tyr at its ten tyrosine positions, and tentative assignments are postulated for two of the resulting resonances that exhibit the largest upfield shifts for the GDP-bound state relative to other conformations. One of the assigned resonances stems from a buried tyrosine in domain III of the known structure, which suggests that this domain interacts with domain I containing the GDP/GTP binding site. No major chemical shift changes are seen between the native and trypsin-treated forms of the protein, which indicates that, in all likelihood, the crystal structures represent native domain conformations.
INVESTIGATING A PROTEIN OF UNKNOWN STRUCTURE
We have seen how 19F NMR can complement a high-resolution protein structure in probing conformational changes. Even for proteins that have not been characterized structurally, however, 19F NMR can be used to define the solvent exposure or separations of specific probe positions. The best example of such an application is E. coli D-lactate dehydrogenase (D-LDH), a peripheral membrane protein for which Ho and coworkers have developed a low-resolution structural model on the basis of extensive 19F NMR data (38, 70, 77, 84, 90).
Rule et al (77) have incorporated 4-, 5-, and 6-fluorotryptophan into the protein to levels reaching 90% and assigned the 19F NMR resonances by direct replacement mutagenesis. None of the substitutions alter the kinetic parameters of the protein substantially, and stability studies at 55°C have demonstrated that both the 4- and 6-fluoro-Trp labels retain the native activity decay constant of 10 s. Moreover, 5-F-Trp labeling actually increases this decay constant to 40 s (77). The secondary structure of the 4- and 5-F-Trp-labeled D-LDH appears, by circular dichroism measurements, to be unaltered from native, although the 6-F-Trp-labeled D-LDH shows a minor perturbation (77). Because the 5-F-Trp resonances exhibit wider dispersion than the 4- or 6-F-Trp resonances (7 ppm vs 6 or 3 ppm, respectively), studies have focused on this analogue. To increase the molecular tumbling rate and thereby minimize the linewidths of the probe resonances, the protein was solubilized from its native membrane to yield a protein–detergent micellar complex, in which the resonances were resolved easily. Although this solubilization narrowed the linewidths significantly, the chemical shifts of the resonances were unperturbed, which indicated that detergent micelles did not induce a conformational change (77). This finding was verified later by repeating the studies in a small unilamellar vesicle system, which led to the same conclusions drawn in the detergent-solubilized system (90).
Of the five labeled tryptophan positions, two (Trp384 and 567) are revealed by their solvent-induced isotopic shifts to be near the aqueous surface of the molecule. One position (Trp369) displays an altered chemical shift and line broadening on substrate binding and the accompanying reduction of the bound flavin cofactor (FAD), which suggests that this position may lie in or near the lactate-binding site (77). The observed line broadening is attributed to exchange of the probe between multiple environments, presumably owing to fluctuations of the local structure (38). In further studies, the proximity of the fluorine positions to the detergent micelle or membrane has been examined with the use of the nitroxide-containing fatty acid 8-doxylpalmitic acid, a paramagnetjc probe that will broaden any resonance within approximatelyo15 Å. Because the nitroxide lies in the middle of the fatty acid, 7 Å from either end, any resonance broadened would lie in or near the hydrocarbon phase. None of the intrinsic tryptophan positions are broadened by this probe, which suggests that they are not located near the membrane-binding surface (77).
To elaborate on these results, the repertoire of labeling positions has been expanded by substituting tryptophan for other aromatic sidechains within D-LDH through site-directed mutagenesis (70, 84). Only those mutants that retain significant enzymatic activities were used in the subsequent structural analysis. Figure 10 summarizes the resulting engineered 5-F-Trp positions, as well as the intrinsic 5-F-Trp probes, located within in a region of D-LDH proposed to contain the membrane-binding site. Also summarized are three types of structural information obtained by 19F NMR: (a) chemical shift changes induced by substrate binding and accompanying FAD reduction, (b) line broadenings caused by the paramagnetic fatty acid probe placed in the associated micelle or bilayer, and (c) solvent-induced isotopic shifts. The identified membrane-binding region extends from Tyr226 to Trp384. Within this region, the 19F NMR data have allowed the development of a low-resolution structural model (84), as illustrated in Figure 10.
Figure 10.
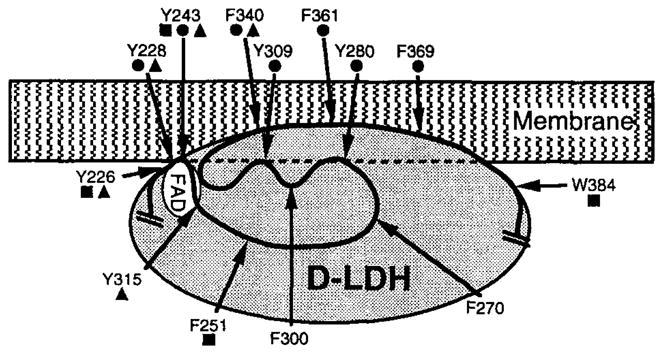
Schematic low-resolution structural model for the membrane-binding domain of D-LDH, developed by 19F NMR studies (84). Shown are the predicted locations of the FAD binding site and the locations of fluorine probes within the proposed membrane-binding domain, spanning residues 226–384. The protein has been labeled with 5-F-Trp at its native tryptophan sidechains. In addition, engineered tryptophans have been substituted for other aromatic sidechains and labeled with 5-F-Trp as indicated. (filled squares) Probe positions judged to be exposed to aqueous solvent by their solvent isotope-induced shifts; (filled triangles) positions yielding FAD-induced chemical shift changes; and (filled circles) positions for which paramagnetic line broadening is observed on addition of a spin-labeled fatty acid, which implies close proximity to the membrane-binding surface.
In regions of D-LDH lying outside the proposed membrane-binding domain, limited structural information has been obtained from the chemical shift and linewidth changes induced by sidechain substitutions. In general, the effects of such point mutations stem from local structural perturbations of the region surrounding the substitution, thereby identifying fluorine probes in the vicinity of the altered sidechain (70). Such identifications must be regarded as tentative, however, because of the ability of 19F NMR to detect long-range conformational changes triggered at a distant site.
CONCLUSIONS AND FUTURE DIRECTIONS
In summary, the systematic use of solution 19F NMR has shed light on the kinetics and spatial ranges of conformational changes in several proteins of known structure and has been used to develop a low-resolution conformational map for one protein of unknown structure. In cases where the conclusions of 19F NMR studies can be checked against newer information from crystallographic or multidimensional NMR structures, it generally has been observed that 19F NMR provides an accurate and powerful method for probing protein conformations. As with any method, the main caution is to avoid the overinterpretation of a limited data set.
Challenges for the future include the use of 19F NMR to probe specifically labeled proteins in vivo. One example of such an application has already been described in living yeast cells (96). Further, it should be possible to use paramagnetic probes that are attached covalently to a protein functionality, or paramagnetic metal ions bound in an intrinsic site, to map out multiple fluorine-paramagnet distances, thereby facilitating the development of a low-resolution structure for macromolecules inaccessible to other structural techniques. Such an approach has been used successfully in 1H NMR studies (34) but still needs to be tested for 19F NMR in a protein of known structure. Finally, the assignment of 19F NMR resonances in proteins of known structure could be facilitated by 19F-1H nuclear Overhauser effect experiments. Such approaches could, in favorable cases, allow full assignment of the 19F NMR spectrum without the aid of protein engineering. Overall, as additional high-resolution protein structures become available, and as the techniques of molecular cloning make new proteins of unknown structure accessible, 19F NMR will continue to be a useful technique in the toolbox of the protein chemist.
Acknowledgments
The authors thank a large number of colleagues in the field for sending reprints and preprints. We apologize to those whose work was, unfortunately, outside the scope of the present review. Special thanks are due to Steven Drake, Dr. Linda Luck, and Dr. Olve Peersen for helpful discussions and important experimental contributions, and to the National Institutes of Health for funding (GM40731 and GM48203 to JJF).
Literature Cited
- 1.Abeles RH, Alston TA. Enzyme inhibition by fluoro compounds. J Biol Chem. 1990;265:16705–8. [PubMed] [Google Scholar]
- 2.Arseniev AS, Kuryatov AB, Tsetlin VI, Bystrov VF, Ivanov VT, Orchinnikov YA. 19F NMR study of 5-fluorotrytophan-labeled bacteriorhodopsin. FEBS Lett. 1987;213:283–88. [Google Scholar]
- 3.Augspurger J, Pearson JG, Oldfield E, Dykstra CE, Park KD, Schwartz D. Chemical-shift ranges in proteins. J Magn Reson. 1992;100:342–57. [Google Scholar]
- 4.Bellsolell L, Prieto J, Serrano L, Coll M. Magnesium binding to the bacterial chemotaxis protein CheY results in large conformational changes involving its functional surface. J Mol Biol. 1994;238:489–95. doi: 10.1006/jmbi.1994.1308. [DOI] [PubMed] [Google Scholar]
- 5.Biemann H-P, Koshland DE., Jr Aspartate receptors of Escherichia coli and Salmonella typhimurium bind ligand with negative and half-of-sites cooperativity. Biochemistry. 1994;33:629–34. doi: 10.1021/bi00169a002. [DOI] [PubMed] [Google Scholar]
- 6.Bourret RB, Brokovich KA, Simon MI. Signal transduction pathways involving protein phosphorylation in prokaryotes. Annu Rev Biochem. 1991;60:401–41. doi: 10.1146/annurev.bi.60.070191.002153. [DOI] [PubMed] [Google Scholar]
- 7.Bourret RB, Drake SK, Chervitz SA, Simon MI, Falke JJ. Activation of the phosphosignaling protein CheY II. Analysis of activated 19F NMR and protein engineering. J Biol Chem. 1993;268:13089–96. [PMC free article] [PubMed] [Google Scholar]
- 8.Bovy PR, Getman DP, Matsoukas JM, Moore GJ. Influence of polyfluorination of the phenylalanine ring of angiotensin II on conformation and biological activity. Biochim Biophys Acta. 1991;1079:23–28. doi: 10.1016/0167-4838(91)90019-v. [DOI] [PubMed] [Google Scholar]
- 9.Bruix M, Pascual J, Santoro J, Prieto J, Serrano L, Rico M. 1H and 15N-NMR assignment and solution structure of the chemotactic Escherichia coli Che Y Protein. Eur J Biochem. 1993;215:573–85. doi: 10.1111/j.1432-1033.1993.tb18068.x. [DOI] [PubMed] [Google Scholar]
- 10.Bystroff C, Oatley SJ, Kraut J. Crystal strucutres of Escherichia coli dihydrofolate recuctase: the NADP+ holoenzyme and the folate NADP+ ternary complex. Substrate binding and a model for the transition state. Biochemistry. 1990;29:3263–77. doi: 10.1021/bi00465a018. [DOI] [PubMed] [Google Scholar]
- 11.Chaiken IM, Freedman MH, Lyerla JR, Jr, Cohen JS. Preparation and studies of 19F-labeled and enriched 13C-labeled semisynthetic ribonuclease-S′ analogues. J Biol Chem. 1973;248:884–91. [PubMed] [Google Scholar]
- 12.Chambers SE, Lau EY, Gerig JT. Origins of fluorine chemical shifts in proteins. J Am Chem Soc. 1994;116:3603–4. [Google Scholar]
- 13.Chervitz SA, Falke JJ. Molecular mechanism of transmembrane signaling by the aspartate receptor. Proc Natl Acad Sci USA. 1996 doi: 10.1073/pnas.93.6.2545. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chervitz SA, Falke JJ. Lock on/off disulfides identify the transemembrane signaling helix of the aspartate receptor. J Biol Chem. 1995;41:24043–53. doi: 10.1074/jbc.270.41.24043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chervitz SA, Lin CM, Falke JJ. Transmembrane signaling by the aspartate receceptor: engineered disulfides reveal static regions of the subunit interface. Biochemistry. 1995;34:9722–33. doi: 10.1021/bi00030a010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cistola DP, Hall KB. Probing internal water molecules in proteins using two-dimensional 19F-1H NMR. J Biomol Nucl Magn Reson. 1995;5:415–19. doi: 10.1007/BF00182285. [DOI] [PubMed] [Google Scholar]
- 17.Colmenares LU, Liu RSH. Fluorinated phenylrhodopsin analogs-binding selectivity, restricted rotation, and 19F NMR studies. Tetrahedron. 1996 In press. [Google Scholar]
- 18.Connick TJ, Reilly RT, Dunlap RB, Ellis PD. Fluorine-19 nuclear magnetic resonance studies of binary and ternary nuclear magnetic resonance studies of binary and ternary complexes of thymidilate synthase utilizing a fluorine-labeled folate analogue. Biochemistry. 1993;32:9888–95. doi: 10.1021/bi00089a003. [DOI] [PubMed] [Google Scholar]
- 19.Danielson MA, Biemann H-P, Koshland DE, Jr, Falke JJ. Attractant-and disulfide-induced conformational changes in the ligand binding domain of the chemotaxis aspartate receptor:a 19F NMR study. Biochemistry. 1994;33:6100–9. doi: 10.1021/bi00186a009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Danielson MA, Falke JJ. Fluorine NMR of proteins involved in chemotaxis. In: Grant DM, Harris RK, editors. Encyclopedia of Nuclear Magnetic Resonance. Chichester: Wiley; 1996. pp. 855–61. [Google Scholar]
- 21.de Dios AC, Pearson JG, Oldfield E. Secondary and tertiary structural effects on protein NMR chemical shifts: an ab initio approach. Science. 1993;260:1491–96. doi: 10.1126/science.8502992. [DOI] [PubMed] [Google Scholar]
- 22.Drake SK, Bourret RB, Luck LA, Simon MI, Falke JJ. Activation of the phosphosignaling protein CheY I. Analysis of the phosphorylated conformation by 19F NMR and protein engineering. J Biol Chem. 1993;268:13081–88. [PMC free article] [PubMed] [Google Scholar]
- 23.Dubois BW, Cherian SF, Evers AS. Volatile anesthetics compete for common binding sites on bovine serum albumin: a 19F NMR study. Proc Natl Acad Sci USA. 1993;90:6478–82. doi: 10.1073/pnas.90.14.6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eccleston JF, Molloy DP, Hinds MG, King RW, Feeney J. Conformational differences between complexes of elongation factor Tu studied by 19F-NMR spectroscopy. Eur J Biochem. 1993;218:1041–47. doi: 10.1111/j.1432-1033.1993.tb18463.x. [DOI] [PubMed] [Google Scholar]
- 25.Falke JJ, Koshland DE., Jr Global flexibility in a sensory receptor: a site-directed cross-linking approach. Science. 1987;237:1596–600. doi: 10.1126/science.2820061. [DOI] [PubMed] [Google Scholar]
- 26.Falke JJ, Luck LA, Scherrer J. 19F nuclear magnetic resonance studies of aqueous and transmembrane receptors. Examples from the Escherichia coli chemosensory pathway. Biophys J. 1992;62:82–86. doi: 10.1016/S0006-3495(92)81787-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flocco MM, Mobray SL. The 1.9 Å X-ray structure of a closed unliganded form of the glucose/galactose receptor from Salmonella typhimurium. J Biol Chem. 1994;269:8931–36. [PubMed] [Google Scholar]
- 28.Gamcsik MP, Gerig JT, Swenson RB. Fluorine-NMR studies of chimpanzee hemoglobin. Biochim Biophys Acta. 1986;874:372–74. doi: 10.1016/0167-4838(86)90038-5. [DOI] [PubMed] [Google Scholar]
- 29.Gammon KL, Smallcombe SH, Richards JH. Magnetic resonance studies of protein-small molecule interactions. Binding of N-trifluoroacetyl-D-(and L-)-p-fluorophenylalanine to chymotrypsin. J Am Chem Soc. 1972;94:4573–80. doi: 10.1021/ja00768a027. [DOI] [PubMed] [Google Scholar]
- 30.Gerig JT. Fluorine NMR of proteins. Prog Nucl Magn Reson Spectrosc. 1994;26:293–370. [Google Scholar]
- 31.Gerig JT. Fluorine nuclear magnetic resonance of fluorinated ligands. Methods Enzymol. 1989;177:3–23. doi: 10.1016/0076-6879(89)77003-8. [DOI] [PubMed] [Google Scholar]
- 32.Gregory DH, Gerig JT. Prediction of fluorine chemical shifts in proteins. Biopolymers. 1991;31:845–58. doi: 10.1002/bip.360310705. [DOI] [PubMed] [Google Scholar]
- 33.Gregory DH, Gerig JT. Structural effects of fluorine substitution in proteins. J Comp Chem. 1991;12:180–85. [Google Scholar]
- 34.Guiles RD, Basus VJ, Sarma S, Malpure S, Fox KM, et al. Novel heteronuclear methods of assignment transfer from a diamagnetic to a paramagnetic protein: application to rat cytochrome b5. Biochemistry. 1993;32:8329–40. doi: 10.1021/bi00083a037. [DOI] [PubMed] [Google Scholar]
- 35.Hazelbauer GL, Berg HC, Matsumura P. Bacterial motility and signal transduciton. Cell. 1993;73:15–22. doi: 10.1016/0092-8674(93)90156-k. [DOI] [PubMed] [Google Scholar]
- 36.Henry GD, Maruta S, Ikebe M, Sykes BD. Observation of multiple myosin subfragment 1-ADP-fluoroberyllate complexes by 19F NMR spectroscopy. Biochemistry. 1993;32:10451–56. doi: 10.1021/bi00090a022. [DOI] [PubMed] [Google Scholar]
- 37.Ho C, Dowd SR, Post JFM. 19F NMR investigations of membranes. Curr Top Bioenerg. 1985;14:53–95. [Google Scholar]
- 38.Ho C, Pratt EA, Wu C-S, Yang JT. Membrane-bound D-lactate dehydrogenase of Escherichia coli: a model for protein interactions in membranes. Biochim Biophys Acta. 1989;988:173–84. doi: 10.1016/0304-4157(89)90018-x. [DOI] [PubMed] [Google Scholar]
- 39.Hoeltzi SD, Frieden C. 19F NMR spectroscopy of [6–19F]tryptophan-labeled Escherichia coli dihydrofolate reductase: equilibrium folding and ligand binding studies. Biochemistry. 1994;33:5502–9. doi: 10.1021/bi00184a019. [DOI] [PubMed] [Google Scholar]
- 40.Hoeltzi SD, Frieden C. Stopped-flow NMR spectroscopy: real-time unfolding studies of 6–19F-tryptophan labeled E. coli dihydrofolate reductase. Proc Natl Acad Sci USA. 1995;92:9318–22. doi: 10.1073/pnas.92.20.9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoeltzi SD, Ropson IJ, Freiden C. Application of equilibrium and stopped-flow 19F NMR spectroscopy to protein folding: studies of E. coli dihydrofolate reductase. Tech Protein Chem. 1994;5:455–65. [Google Scholar]
- 42.Hull WE, Sykes BD. Fluorine-19 nuclear magnetic resonance study of fluorotyrosine alkaline phosphatase: the influence of zinc on protein structure and a conformational change induced by phoshate binding. Biochemistry. 1976;15:1535–46. doi: 10.1021/bi00652a027. [DOI] [PubMed] [Google Scholar]
- 43.Johnson CD, Gerig JT. Dynamics at the active site of N-(4-flurophenyl), N-(2,6-difluorophenyl)carbamoyl-α-chymotrypsin. Magn Res Chem. 1995 In press. [Google Scholar]
- 44.Kim H-W, Perez JA, Ferguson SJ, Campbell ID. The specific incorporation of labelled aromatic amino acids into proteins through growth of bacteria in the presence of glyphosate. FEBS Lett. 1990;272:34–36. doi: 10.1016/0014-5793(90)80442-l. [DOI] [PubMed] [Google Scholar]
- 45.Kossman M, Wolff C, Manson MD. Maltose chemoreceptor of Escherichia coli interaction of maltose-binding protein and the tar signal transducer. J Bacteriol. 1988;170:4516–21. doi: 10.1128/jb.170.10.4516-4521.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koul AK, Wasserman GF, Warme PK. Semi-synthetic analogs of cytochrome C at positions 67 and 74. Biochem Biophys Res Commun. 1979;89:1253–59. doi: 10.1016/0006-291x(79)92143-0. [DOI] [PubMed] [Google Scholar]
- 47.Labroo VM, Hebel D, Krik KL, Cohen LA, Lemieux C, Schiller PW. Direct electrophilic fluorination of tyrosine in dermorphin analogs and its effect on biological activity, receptor affinity, and selectivity. Int J pept Protein Res. 1991;37:440–49. doi: 10.1111/j.1399-3011.1991.tb00758.x. [DOI] [PubMed] [Google Scholar]
- 48.Lee GF, Dutton DP, Hazelbauer GL. Identification of, functionally important helical faces in transmembrane segments by scanning mutagenesis. Proc Natl Acad Sci USA. 1995;92:5416–20. doi: 10.1073/pnas.92.12.5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li E, Qian S-J, Nader L, Young N-CC, d’Avignon A, et al. Nuclear magnetic resonance studies of 6-fluorotryptophan-substituted rat cellular retinol-binding protein II produced in Escherichia coli. J Biol Chem. 1989;264:17041–48. [PubMed] [Google Scholar]
- 50.Li E, Quian S-J, Yan N-CC, d’Avignon A, Gordon JI. Nuclear magnetic resonance studies of 6-fluorotryptophan-substituted rat cellular retinol binding protein II produced in Escherichia coli. J Biol Chem. 1990;265:11549–54. [PubMed] [Google Scholar]
- 51.Lian C, Le H, Montez B, Patterson J, Harrell S, et al. Fluoine-19 nuclear magnetic resonance spectroscopic study of fluorophenylalanine-and flurortryptophan-labeled avian egg white lysozymes. Biochemistry. 1994;33:5238–45. doi: 10.1021/bi00183a029. [DOI] [PubMed] [Google Scholar]
- 52.Lowry DF, Roth AF, Rupert AF, Rupert PB, Dahlquist FW, et al. Signal transduction in chemotaxis. A propogating conformation change upon phosphorylation of cheY. J Biol Chem. 1994;269:26358–62. [PubMed] [Google Scholar]
- 53.Lu P, Jarema M, Mosser K, Daniel WE. Lac repressor: 3-fluorotyrosine substitution for nuclear magnetic resonance studies. Proc Natl Acad Sci USA. 1976;73:3471–75. doi: 10.1073/pnas.73.10.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luck LA, Falke JJ. 19F NMR studies of the D-galactose chemosensory receptor. 1 Sugar binding yields a global structural change. Biochemistry. 1991;30:4248–56. doi: 10.1021/bi00231a021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luck LA, Falke JJ. 19F NMR studies of the D-galactose chemosensory receptor. 2 Ca(II) Binding yields a local structural change. Biochemistry. 1991;30:4257–61. doi: 10.1021/bi00231a022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luck LA, Falke JJ. Open conformation of a substrate-binding cleft: 19F NMR studies of cleft angle in the D-galactose chemosensory receptor. Biochemistry. 1991;30:6484–90. doi: 10.1021/bi00240a019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lukat GS, McCleary WR, Stock AM, Stock JB. Phosphorylation of bacterial response regulator proteins by low molecular weight phospho-donors. Proc Natl Acat Sci USA. 1992;89:718–22. doi: 10.1073/pnas.89.2.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57a.Mao B, Pear MR, McCammon JA, Quiocho FA. Hinge-bending in L-arabinose binding protein: the “Venus-flytrap” model. J Biol Chem. 1982;257:1131–33. [PubMed] [Google Scholar]
- 58.Maruta S, Henry GD, Sykes BD, Ikebe M. Conformation of the stable myosin-ADP-aluminum fluoride and myosin-ADP-beryllium fluoride complexes and their analysis using 19F NMR. J Biol Chem. 1993;268:7093–100. [PubMed] [Google Scholar]
- 59.Milburn MV, Prive GG, Milligan DL, Scott WG, Yeh J, et al. Three-dimensional structures of the ligand-binding domain of the bacterial aspartate receptor with and without a ligand. Science. 1991;254:1342–47. doi: 10.1126/science.1660187. [DOI] [PubMed] [Google Scholar]
- 60.Miller DM, Olson JS, Quiocho FA. The mechanism of sugar binding to the periplasmic receptor for galactose chemotaxis and transport in Escherichia coli. J Biol Chem. 1980;255:2465–71. [PubMed] [Google Scholar]
- 61.Millet F, Raftery MA. An NMR method for characterizing conformation changes in proteins. Biochem Biophys Res Commun. 1972;47:625–32. doi: 10.1016/0006-291x(72)90924-2. [DOI] [PubMed] [Google Scholar]
- 62.Milligan DL, Koshland DE., Jr Site-directed crosslinking. Establishing the dimeric structure of the aspartate receptor of bacterial chemotaxis. J Biol Chem. 1988;263:6268–75. [PubMed] [Google Scholar]
- 63.Milligan DL, Koshland DE., Jr Purification and characterization of hte periplasmic domain of the aspartate chemoreceptor. J Biol Chem. 1993;268:19991–97. [PubMed] [Google Scholar]
- 64.Mirmira RG, Tager HS. Disposition of the phenylalanine-B25 sidechain during insulin-receptor and insulin-insulin interactions. Biochemistry. 1991;30:8222–29. doi: 10.1021/bi00247a019. [DOI] [PubMed] [Google Scholar]
- 65.Moy FJ, Low DF, Matsumura P, Dahlquist FW, Krywko JE, Domaille PJ. Assignments, secondary structure, global fold, and dynamics of chemotaxis Y protein using three- and four-dimensional heteronuclear (13C, 15N) NMR spectroscopy. Biochemistry. 1994;33:10731–42. doi: 10.1021/bi00201a022. [DOI] [PubMed] [Google Scholar]
- 66.Murray-Rust P, Stallings WC, Monti CT, Preston RK, Glusker JP. Intermolecular interactions of the C-F bond: the crystallographic environment of fluorinated carboxylic acids and related structures. J Am Chem Soc. 1983;105:3206–14. [Google Scholar]
- 67.Parkinson JS, Kofoid EC. Communication modules in bacterial signaling proteins. Annu Rev Genet. 1992;26:71–112. doi: 10.1146/annurev.ge.26.120192.000443. [DOI] [PubMed] [Google Scholar]
- 68.Pauling L. The Nature of the Chemical Bond. 3 Ithaca: Cornell Univ. Press; 1960. p. 644. [Google Scholar]
- 69.Pearson JG, Oldfield E, Lee FS, Warshell A. Chemical shifts in proteins: a shielding trajectory analysis of the fluorine nuclear magnetic resonance spectrum of the Escherichia coli galactose binding protein using a multipole shielding polarizability-local reaction field-molecular dynamics approach. J Am Chem Soc. 1993;115:685–62. [Google Scholar]
- 70.Peersen OB, Pratt EA, Truong H-TN, Ho C, Rule GS. Site-specific incoporation of 5-fluorotryptophan as a probe of the structure and function of the membrane-bound D-lactate dehydrogenase of Escherichia coli: a 19F nuclear magnetic resonance study. Biochemistry. 1990;29:3256–62. doi: 10.1021/bi00465a017. [DOI] [PubMed] [Google Scholar]
- 71.Post JF, Cottam PF, Simplaceanu V, Ho C. Fluorine-19 nuclear magnetic resonance study of 5-fluorotryptophan-labeled histidine-binding protein J of Salmonella typhimurium. J Mol Biol. 1984;179:729–43. doi: 10.1016/0022-2836(84)90164-5. [DOI] [PubMed] [Google Scholar]
- 72.Pratt EA, Ho C. Incorporation of fluorotryptophans into proteins of Escherichia coli. Biochemistry. 1975;14:3035–40. doi: 10.1021/bi00684a037. [DOI] [PubMed] [Google Scholar]
- 73.Rastinejad F, Artz P, Lu P. Origin of the asymmetrical contact between lac repressor and lac operator DNA. J Mol Biol. 1993;233:389–99. doi: 10.1006/jmbi.1993.1519. [DOI] [PubMed] [Google Scholar]
- 74.Rastinejad F, Evilia C, Lu P. Studies of nucleic acids and their protein interactions by 19F NMR. Methods Enzymol. 1995;261:560–75. doi: 10.1016/s0076-6879(95)61025-1. [DOI] [PubMed] [Google Scholar]
- 75.Rastinejad F, Lu P. Bacteriophage T7 RNA polymerase: 19F nuclear magnetic resonance observations at 5-fluorouracil-substituted promotor DNA and RNA transcript. J Mol Biol. 1993;232:105–22. doi: 10.1006/jmbi.1993.1373. [DOI] [PubMed] [Google Scholar]
- 76.Ropson IJ, Frieden C. Dynamic NMR spectral analysis and protein folding: identification of a highly populated folding intermediate of rat intestinal fatty acid-binding protein by 19F NMR. Proc Natl Acad Sci USA. 1991;89:7222–26. doi: 10.1073/pnas.89.15.7222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rule GS, Pratt EA, Simplaceanu V, Ho C. Nuclear magnetic resonance and molecular genetic studies of the membrane-bound D-lactate dehydrogenase of Escherichia coli. Biochemistry. 1987;26:549–56. doi: 10.1021/bi00376a029. [DOI] [PubMed] [Google Scholar]
- 78.Segall JE, Manson MD, Berg HC. Signal processing times in bacterial chemotaxis. Nature. 1982;296:855–57. doi: 10.1038/296855a0. [DOI] [PubMed] [Google Scholar]
- 79.Sharff AJ, Rodseth LE, Spurlino JC, Quiocho FA. Crystallographic evidence of a large ligand-induced hinge-twist motion between the two domains of the maltodextrin binding protein involved in active transport and chemotaxis. Biochemistry. 1992;31:10657–63. doi: 10.1021/bi00159a003. [DOI] [PubMed] [Google Scholar]
- 80.Spotswood TM, Evans JM, Richards JH. Enzyme-substrate interaction by nuclear magnetic resonance. J Am Chem Soc. 1967;89:5052–54. doi: 10.1021/ja00995a047. [DOI] [PubMed] [Google Scholar]
- 81.Stirtan W, Withers SG. Direct 19F NMR titration of phosphorylase molecules binding to fluorine-labeled glycogen particles. Carbohydr Res. 1993;249:253–58. [Google Scholar]
- 82.Stock AM, Martinez-Hackert E, Rasmussen BF, West AH, Stock JB, et al. Structure of the Mg2+-bound form of CheY and mechanism of phosphoryl transfer in bacterial chemotaxis. Biochemistry. 1993;32:13375–80. doi: 10.1021/bi00212a001. [DOI] [PubMed] [Google Scholar]
- 83.Stock JB, Lukat GS. Bacterial chemotaxis and the molecular logic of intracellular signal transduction networks. Annu Rev Biophys Biophys Chem. 1991;20:109–36. doi: 10.1146/annurev.bb.20.060191.000545. [DOI] [PubMed] [Google Scholar]
- 84.Sun Z-Y, Truong H-TN, Pratt EA, Sutherland DC, Kulig CE, et al. A 19F-NMR study of the membrane-binding region of D-lactate dehydrogenase of Escherichia coli. Protein Sci. 1993;2:1938–47. doi: 10.1002/pro.5560021115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84a.Swanson RV, Lowry DF, Matsumura P, McEvoy MM, Simon MI, Dahlquist FW. Localized perturbations in CheY structure monitored by NMR identify a CheA binding interface. Nat Struct Biol. 1995;2:906–10. doi: 10.1038/nsb1095-906. [DOI] [PubMed] [Google Scholar]
- 85.Sykes BD, Hull WE. Fluorine nuclear magnetic resonance studies of proteins. Methods Enzymol. 1978;49:270–95. doi: 10.1016/s0076-6879(78)49015-9. [DOI] [PubMed] [Google Scholar]
- 86.Sylvia LA, Gerig JT. NMR studies of the a-chymotrypsin-〉-1-acetamido -2-(4-fluorophenyl)ethane–1-boronic acid complex. Biochim Biophys Acta. 1993;1163:321–34. doi: 10.1016/0167-4838(93)90169-r. [DOI] [PubMed] [Google Scholar]
- 87.Tanaka H, Osaka F, Ohashi M, Shiraki M, Munekata E. Substance-P analogues containing para-fluoro-L-phenylalanine. Chem Lett. 1986:391–94. [Google Scholar]
- 88.Taylor HC, Komoriya A, Chaiken IM. Crystallographic structure of an active, sequence-engineered ribonuclease. Proc Natl Acad Sci USA. 1985;82:6423–26. doi: 10.1073/pnas.82.19.6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Taylor HC, Richardson DC, Richardson JS, Wlodawer A, Komoriya A, Chaiken IM. Active conformation of an inactive semi-synthetic ribonuclease-S. J Mol Biol. 1981;149:313–17. doi: 10.1016/0022-2836(81)90305-3. [DOI] [PubMed] [Google Scholar]
- 90.Truong T-TN, Pratt EA, Ho C. Interaction of the membrane-bound D-lactate dehydrogenase of Escherichia coli with phospholipid vesicles and reconstitution of activity using a spin-labeled fatty acid as an electron acceptor: a magnetic resonance and biochemical study. Biochemistry. 1991;30:3893–98. doi: 10.1021/bi00230a013. [DOI] [PubMed] [Google Scholar]
- 91.Vine WH, Brueckner DA, Needleman P, Marshall GR. Synthesis, biological activity, and 19F nuclear magnetic resonance spectra of angiotensin II analogs containing fluorine. Biochemistry. 1973;12:1630–37. doi: 10.1021/bi00732a026. [DOI] [PubMed] [Google Scholar]
- 92.Volz K, Matsumura P. Crystal structure of Escherichia coli CheY refined at 1.7 Å resolution. J Biol Chem. 1991;266:15511–19. doi: 10.2210/pdb3chy/pdb. [DOI] [PubMed] [Google Scholar]
- 93.Vyas NK, Vyas MN, Quiocho FA. A novel calcium binding site in the galactose-binding protein of bacterial transport and chemotaxis. Nature. 1987;327:635–38. doi: 10.1038/327635a0. [DOI] [PubMed] [Google Scholar]
- 94.Wagner G, Wütrich K. Observation of internal motility of proteins by nuclear magnetic resonance in solution. Methods Enzymol. 1986;131:307–26. doi: 10.1016/0076-6879(86)31047-4. [DOI] [PubMed] [Google Scholar]
- 95.Westhead EW, Boyer PD. The incorporation of p-fluorophenylalanine into some rabbit enzymes and other proteins. Biochim Biophys Acta. 1961;54:145–50. doi: 10.1016/0006-3002(61)90947-7. [DOI] [PubMed] [Google Scholar]
- 96.Williams S-P, Fulton AM, Brindle KM. Estimation of the intracellular free ADP concentration by 19F NMR studies of fluorine-labeled yeast phosphoglycerate kinase in vivo. Biochemistry. 1993;32:4895–902. doi: 10.1021/bi00069a026. [DOI] [PubMed] [Google Scholar]
- 97.Wilson ML, Dahlquist FW. Membrane protein conformational change dependent on the hydrophobic environment. Biochemistry. 1985;24:1920–28. doi: 10.1021/bi00329a018. [DOI] [PubMed] [Google Scholar]
- 98.Wuttke DS, Gray HB, Fisher SL, Imperaili B. Semisynthesis of bipyridyl-alanine cytochrome c mutants: novel proteins with enhanced electron-transfer properties. J Am Chem Soc. 1993;115:8455–56. [Google Scholar]