

Abstract
Phosphatidylinositol bisphosphate (PIP2) is an activator of mammalian inwardly rectifying potassium (Kir) channels. Multiscale simulations, via a sequential combination of coarse-grained and atomistic molecular dynamics, enabled exploration of the interactions of PIP2 molecules within the inner leaflet of a lipid bilayer membrane with possible binding sites on Kir channels. Three Kir channel structures were investigated: X-ray structures of KirBac1.1 and of a Kir3.1−KirBac1.3 chimera and a homology model of Kir6.2. Coarse-grained simulations of the Kir channels in PIP2-containing lipid bilayers identified the PIP2-binding site on each channel. These models of the PIP2−channel complexes were refined by conversion to an atomistic representation followed by molecular dynamics simulation in a lipid bilayer. All three channels were revealed to contain a conserved binding site at the N-terminal end of the slide (M0) helix, at the interface between adjacent subunits of the channel. This binding site agrees with mutagenesis data and is in the proximity of the site occupied by a detergent molecule in the Kir chimera channel crystal. Polar contacts in the coarse-grained simulations corresponded to long-lived electrostatic and H-bonding interactions between the channel and PIP2 in the atomistic simulations, enabling identification of key side chains.
Inwardly rectifying potassium (Kir)1 channels are intrinsic membrane proteins that control the selective permeation of K+ ions across cell membranes. Members of this family of K+ channels regulate membrane excitability and transmembrane K+ fluxes and play important roles in many cells, including secretion of insulin from pancreatic β-cells, repolarization of the cardiac action potential, and kidney function (1−3).
As revealed by X-ray diffraction (4,5), Kir channels are tetramers of four usually identical subunits. Each subunit has two transmembrane α-helices (M1 and M2) linked by a re-entrant pore loop that dips back into the membrane to form the K+ selectivity filter. The N-terminus is linked to M1 by an amphipathic slide helix (sometimes known as M0) that lies in the plane of the membrane. Like the N-terminus, the C-terminus is intracellular, and both N- and C-termini interact to form a tetrameric cytoplasmic domain (6) that regulates channel gating. Channel activity is modulated by pH, ATP binding, and/or interactions with G-proteins via this intracellular domain. While many of these modulators are subtype specific, the anionic lipid phosphatidylinositol 4,5-bisphosphate (PIP2) is thought to regulate all members of the Kir family (7,8). Intriguingly, although PIP2 activates mammalian Kir channels (9), it inhibits the bacterial channel KirBac1.1 (10,11).
Functional studies and mutagenesis data have implicated residues in both the N- and C-termini of the cytoplasmic domain in PIP2 binding (e.g., refs (12−17)). Many of these residues are basic and thus capable of interacting with the negatively charged inositol triphosphate (IP3) headgroup of PIP2. Recent crystallographic studies have enhanced our structural understanding of Kir channels (4−6,18), facilitating determination of the three-dimensional location of residues that functionally interact with the lipid. Unfortunately, it has not been possible to cocrystallize PIP2 bound to a Kir channel.
Two putative PIP2-binding sites for Kir channels have been described structurally. The first was derived from docking of IP3 to a homology model of Kir6.2 (19). The second was based on the location of a cocrystallized detergent molecule within the X-ray structure of a chimeric Kir3.1−KirBac1.3 channel [KirChim; PDB entry 2QKS, chain A (5)]. Although both models propose sites in the vicinity of the residues functionally implicated in PIP2 binding, they lie ∼14 Å apart from one another. Significantly, the KirChim structure revealed that the loop connecting M2 and the C-terminal domain contains a single-turn α-helix. This helix was not included in the earlier Kir6.2 homology model (19), as it is absent from the X-ray structures of both KirBac1.1 and the Kir3.1 C-terminal domain on which the Kir6.2 model was based.
Atomistic molecular dynamics (MD) simulations of membrane proteins have been used to identify lipid-binding sites for both KcsA (20) and the nicotinic acetylcholine receptor (21) and to characterize PIP2−PH domain interactions in a lipid bilayer (22). However, the computing power required means atomistic MD simulations are generally of limited duration. More recently, coarse-grained (CG) simulation approaches (23−28) that allow membrane protein simulations to be readily extended to a microsecond time scale have been used to predict nonspecific lipid−protein interactions for a wide range of membrane proteins (29). CG−MD simulations allow the dynamic self-assembly of lipids around a protein of interest, enabling prediction of its position and orientation within a membrane (29), and they have also been used to explore aspects of channel gating (30,31). It is therefore timely to address binding of PIP2 to Kir channels using improved computational approaches and a refined channel model that is based on the most recent structural data.
In this study, we use CG−MD simulations of PIP2-containing lipid (phosphatidylcholine) bilayers (Figure 1) to explore the binding of PIP2 to Kir channels. A multiscale simulation approach (32) is used in which the CG model of the Kir−PIP2 complex in a lipid bilayer is first generated and then subsequently converted to a full atomistic model, thus permitting detailed atomistic MD simulations and analysis of the protein−PIP2 interactions. We have applied this multiscale simulation approach to three Kir channels: a bacterial channel (KirBac1.1), a mammalian−bacterial hybrid channel (KirChim), and a mammalian channel (Kir6.2). X-ray structures are available for the former two; the latter is a novel homology model that incorporates the latest structural information and is supported by extensive functional data. The outcome of these simulations suggests a possible explanation for why PIP2 may activate mammalian Kir channels but not their bacterial homologues.
Figure 1.

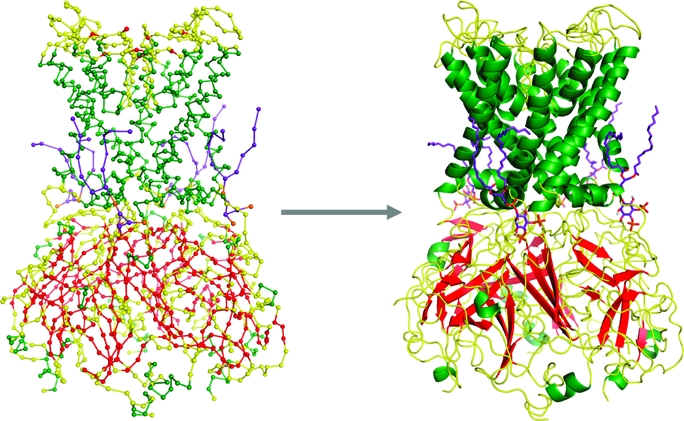
(A) Position of Kir6.2 in a POPC lipid bilayer, as predicted by the self-assembly CG simulations (see Figure S2 of the Supporting Information). PIP2 molecules are colored yellow-orange. (B) Atomistic (AT) and coarse-grained (CG) models of PIP2. The MARTINI particle types are indicated for the CG model. (C) Starting configurations used for the CG−MD simulation, as viewed from the extracellular side of the membrane. The PIP2 initial positions at the “sides of the box” are colored red, while those at the “corners of the box” are colored blue. In panels A and C, POPC lipids are colored green and the Cα trace of the Kir channel is colored gray.
Methods
Homology Modeling
Modeler 9v4 (33) was used to add missing residues to the crystal structures of KirBac1.1 [PDB entry 1P7B(4)] and the “dilated” (open cytosolic pore) conformation of the Kir3.1−KirBac1.3 chimera [KirChim; PDB entry 2QKS, chain A (5)]. Modeler was also used to create a homology model of Kir6.2 [SWISS-PROT (34) entry O35111]. This homology model was based on chain A of the KirChim structure, in which the loop connecting M2 to the cytoplasmic domain is complete. The sequence alignment is given in the Supporting Information (Figure S1). Each model was compared to its template to verify that the modeling step had not significantly altered backbone and side chain conformations.
Coarse-Grained PIP2
The CG model for POPC, from MARTINI version 2.1 (35,36), was used as a template for the 1′-phosphate, glycerol, and acyl tails of PIP2 (see Figure 1B for more details). The description of the ring of PIP2 was based on a similar three-particle CG representation used for the rings in cholesterol (35). All three particles of the ring were assigned the SP1 particle type. The description of the 1′-phosphate was used to generate the 4′- and 5′-phosphate particles, with both 4′- and 5′-phosphate particles assigned a charge of −2.
CG−MD Simulations
CG−MD simulations were performed using Gromacs (version 3.3.3) (37) (www.gromacs.org) with the MARTINI version 2.1 force field (36). An elastic network model was applied to the protein Cα particles, using a distance cutoff of 7 Å and a force constant of 10 kJ mol−1 Å−2(29).
A bilayer self-assembly protocol (29) was used to deduce the position of a Kir channel in a PIP2-containing lipid (POPC) bilayer. For each of these simulations, the Kir channel was positioned at the center of a 160 Å × 160 Å × 160 Å periodic box and randomly surrounded by 550 POPC, 8 PIP2, and ∼17500 water molecules. Each simulation was run for 50 ns, with the bilayer generally forming after 10 ns (see Figure S2 of the Supporting Information), with limited deformation around the protein. Each simulation was then extended by 0.5 μs to assess PIP2 protein contacts in either leaflet of the bilayer.
Although PIP2 molecules inserted into both leaflets of the bilayer during the self-assembly process, the majority of PIP2−Kir interactions occurred in the cytoplasmic leaflet (as defined by the channel orientation), close to the interfacial slide helix of the channel (Figure S3 of the Supporting Information). Thus, we conclude that, as might be anticipated from the restriction of PIP2 to the inner leaflet of cell membranes, PIP2 primarily interacts with the intracellular surface of Kir channels. In all subsequent simulations, we therefore restricted PIP2 to the inner leaflet of the bilayer.
To assess the interactions of PIP2 in the intracellular leaflet of the bilayer with the Kir channels, a preformed POPC bilayer was aligned with the self-assembled bilayer, with lipids overlapping the protein removed. Four PIP2 molecules were then positioned within the inner leaflet of the bilayer, at either the sides or the corners of the 140 Å × 140 Å × 160 Å simulation box. Each simulation was run for 0.5 μs, with a total of eight simulations performed for each Kir channel (i.e., four for each PIP2 starting position). In six cases, for each channel and PIP2 starting position, the simulations were extended to 1.5 μs.
CG to Atomistic Conversion
To convert the CG lipid to an atomistic model, energy-minimized atomistic lipid fragments were aligned with the CG particles, with the missing atoms between each particle extracted and appended to the CG lipids. The atomistic lipids were then energy minimized to refine bond lengths and angles and to remove any clashes that resulted from the conversion (see Figure S4 of the Supporting Information).
To convert the protein representation, atomistic protein side chains were aligned with and substituted for the CG particles used to model the side chain. Pulchra was used to grow the protein backbone from the Cα trace (38) before Modeler added any missing atoms. The protein structure was then energy minimized using the conjugant gradient approach (33).
Atomistic MD Simulations
Atomistic MD simulations were performed with Gromacs using the GROMOS96 43a1 force field (39). Simulations were performed using semi-isotropic pressure coupling with the Parrinello−Rahman barostat (40). The temperature of the system was coupled to an external bath held at 300 K, using the Berendsen themostat (41). The water model used was SPC (42). The LINCS algorithm was used to constrain bond lengths (43). Long-range electrostatics, up to 10 Å, were modeled using the particle mesh Ewald (PME) method, while a cutoff of 10 Å was also used for van der Waals interactions. Gromacs was used to add hydrogen atoms to the models. The system was then subjected to unrestrained MD for 20 ns, during which coordinates were saved every 10 ps for analysis. Analysis of the simulations was performed using Gromacs and locally written code (37). Visualization used VMD (44) and Pymol (version 1.0) (45).
Results
Coarse-Grained MD Simulations
To identify the PIP2-binding sites, Kir channels were positioned in a preformed bilayer in orientations defined by self-assembly simulations (29) (see the Supporting Information for details). In each case, four PIP2 molecules were placed in the inner leaflet of the bilayer, between 30 and 50 Å from the protein (Figure 1A). For each Kir channel, a series of eight CG simulations was performed, each with a duration of 0.5−1.5 μs, with the four PIP2 molecules (Figure 1B) positioned either at the corners or at the sides (Figure 1C) of a 140 Å × 140 Å × 160 Å simulation box.
In these simulations, the PIP2 molecules diffused from their initial positions in the lipid bilayer to form contacts with the channel protein that were initiated within ∼0.1 μs and became established within 0.5 μs (Figure 2A). A distance cutoff of 6 Å [as used in previous CG studies (46)] was used to define contacts made between the PIP2 headgroup and the Kir channels during the CG simulations. Analysis of the total number of contacts as a function of simulation time (Figure 3) suggests that the CG interactions between the protein and ligand have generally equilibrated within ∼0.25 μs. Once bound, the lipid remained attached to the protein even if the simulation was extended to ∼5 μs (data not shown).
Figure 2.
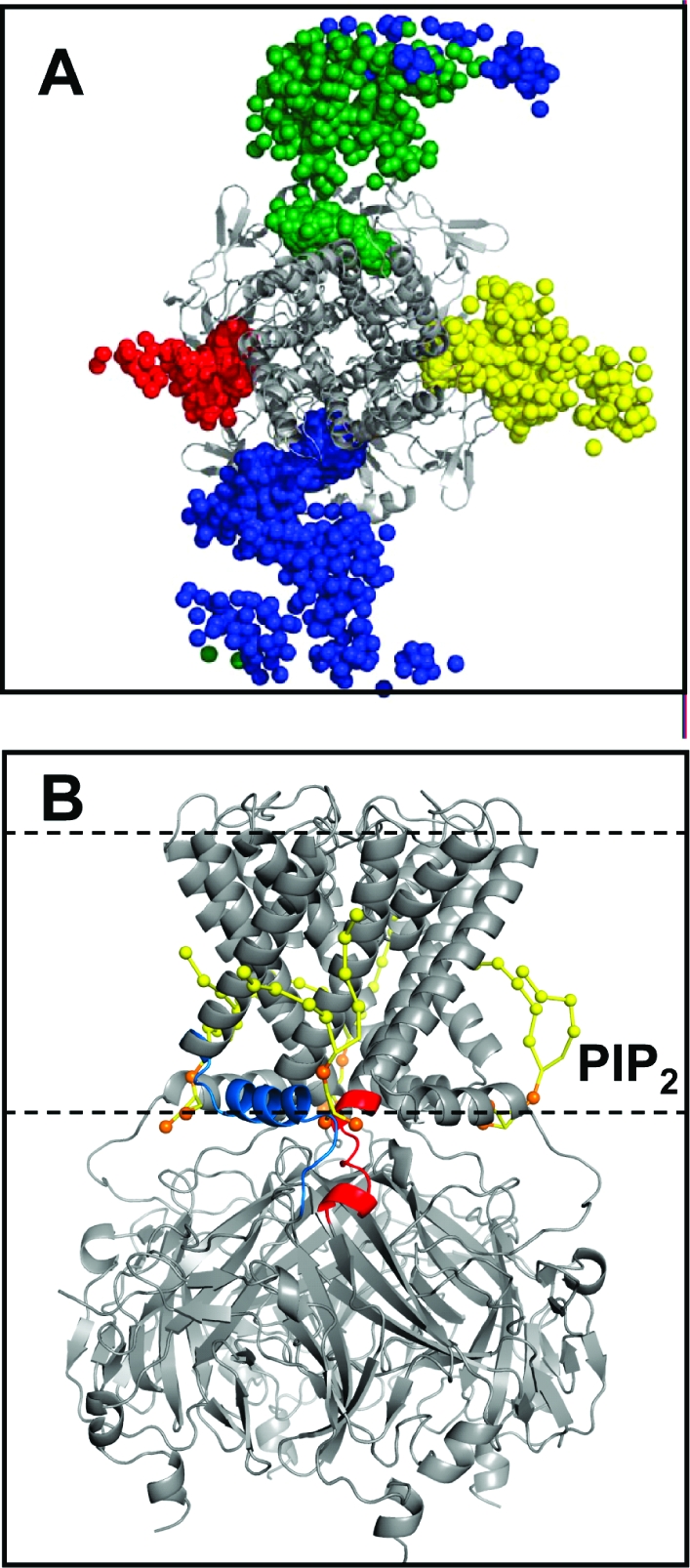
(A) Positions of the four PIP2 molecules (shown as their 1′-phosphate particles) over the course of a 0.5 μs CG simulation of KirBac1.1. In this example, the PIP2 molecules start at the sides of the box (see Figure 1). (B) Position of PIP2 molecules (yellow-orange) bound to KirChim after a 0.5 μs simulation. The pre-M0 loop [i.e., residues 50−54 (Kir6.2 numbering)] and slide helix are colored blue, while the post-M2 loop [residues 173−180 (Kir6.2)] is colored red. The horizontal dashed lines indicate the approximate position of the phosphate particles of the POPC lipid molecules. The Cα trace of the Kir channel is colored gray.
Figure 3.

Average (over eight simulations) number of PIP2 headgroup contacts (6 Å cutoff) with each Kir channel as a function of time during the CG−MD simulations, for Kir6.2 (red), KirChim (black), and KirBac1.1 (blue).
The PIP2 molecules generally converge upon a single site that is common to all four subunits (Figure 4). As frequently found, this site is located at the interface between subunits, with residues in and around the slide (i.e., M0) helix and within the immediate post-M2 region [post-M2 loop, residues 173−180 (Kir6.2 numbering)] making the primary interactions with the PIP2 headgroup (Figure 2B and Figure S5 of the Supporting Information).
Figure 4.

Contacts between PIP2 and the three Kir channels during the 24 CG simulations. The left-hand panel for each channel shows the initial (○) and final (●) positions for the 1′-phosphate of PIP2. The right-hand panel for each channel shows the distribution (color-coded from blue to dark red) of positions of the 1′-phosphate of PIP2 over the final 250 ns of the simulation. A final snapshot of the protein (TM domain) Cα trace is colored green: (A) Kir6.2, (B) KirChim, and (C) KirBac1.1.
In the simplest scenario, all four sites on the channel (i.e., one per subunit) became occupied by PIP2 molecules. However, this only occurred in ∼25% of simulations. In ∼50% of cases, competition between two PIP2 molecules for a single site resulted in one or two sites being left unoccupied. In only ∼15% of the simulations did a PIP2 molecule not make contact with the protein within 0.5 μs. This may be summarized by calculating the distribution, in the bilayer plane, of the 1′-phosphate of the PIP2 headgroup relative to the protein over the latter 0.25 μs of each simulation (Figure 4).
Although PIP2 binds to the same region in all three channels, the amino acid compositions of these sites are different for each channel. The most divergent is KirBac1.1, which shares the lowest degree of sequence homology with the other two channels. As might be expected for a binding site for an anionic lipid like PIP2, basic side chains and amphipathic aromatic side chains predominate in the region occupied by the PIP2 headgroup. Indeed, basic residues alone are predicted to account for 40−50% of the contacts of the PIP2 headgroup with the proteins. In KirBac1.1, seven Arg, two Lys, three Trp, and three Tyr residues interact with PIP2, whereas in KirChim, the contacts are formed by five Arg, four Lys, and two Trp residues; in Kir6.2, the contacts are formed by six Arg, five Lys, and three His residues and one Trp residue (Figure 5). The interactions of the lipid tail are less specific than that of the PIP2 headgroup (reflecting the less directional nature of hydrophobic contacts), with contacts made primarily with hydrophobic residues in the slide, M1, and exterior face of the M2 helices. The primary role for the tails appears to be to anchor the PIP2 molecule in the bilayer, at a position that enables the IP3 headgroup to interact with the key basic residues. As with the headgroup contacts, these interactions generally involve two adjacent subunits.
Figure 5.

Contacts made by the headgroup PIP2 to protein side chains expressed as a percentage of the total contacts made during the coarse-grained simulations: (A) Kir6.2, (B) KirChim, and (C) KirBac1.1. For the sake of clarity, only residues that form more than 1% of the total contacts are shown. The asterisks indicate residues that form hydrogen bonds with PIP2 over the course of the subsequent atomistic simulations.
The predicted binding site is proximal to, but distinct from, that of the nonyl glucoside detergent molecule, which was cocrystallized in the KirChim structure (5). The detergent molecule is smaller than PIP2 and lacks the anionic headgroup, so it is not surprising that it binds to a slightly different site. Indeed, it is possible that the detergent inhibited binding of the PIP2 analogue that was included in the crystallization buffer (5).
Atomistic MD Simulations
To investigate the PIP2-binding site in more detail, the coarse-grained system was converted to an atomistic representation, including proteins, bound PIP2 molecules, and lipids (see Methods for details). For each channel, a CG system in which all four PIP2 molecules were bound to the channel at distinct sites was selected for conversion. This was used to initiate an atomistic MD simulation 20 ns in duration for each channel species. These simulations were conformationally stable, with an overall Cα rmsd of ∼4.5 Å over 20 ns for each Kir channel and of ∼3 Å for the core structural elements (i.e., excluding the loops) (Figure S6 of the Supporting Information). Furthermore, analysis of residue by residue fluctuations [root-mean-square fluctuations (rmsfs) (Figure S6 of the Supporting Information)] reveals that for nonloop regions the Cα rmsf values are <1.5 Å, again indicative of conformational stability on a 20 ns time scale [as seen in earlier atomistic Kir simulations (47)].
To evaluate the specific binding of PIP2, the hydrogen bonds formed between PIP2 headgroups and the protein side chains during the course of the 20 ns of simulation were calculated. These correlated well with the close contacts identified by the CG simulations (Figure 5). In particular, most of the PIP2−basic side chain interactions were retained. Thus, from the atomistic simulations, the interactions with basic residues are as follows: four Arg and two Lys residues for KirBac1.1 and for KirChim and three Arg and four Lys residues and one Trp residue for Kir6.2. The interactions of the lipid tails are similar to those observed in the CG simulations.
A more detailed comparison of the H-bonding interactions between the headgroup of PIP2 and the three Kir proteins, and a sequence alignment of the regions involved, is presented in Figure 6. Residues implicated in simulations are in good agreement with the available functional data (which is limited to KirBac and Kir6.2). Thus, for Kir6.2, the residues that form the majority of H-bonds in simulations [R50, R54, K67, R176, and R206 (Figure 5)] are also implicated in PIP2 binding and channel modulation (7,8,15,19). Residues that interact in the KirChim simulation include R28 (54), K41 (67), R43 (69), K130 (170), K135 (175), K136 (176), and R166 (206) (numbers in parentheses indicate the corresponding residues in Kir6.2). All of these residues are provided by the mammalian Kir3.1 segment of the KirChim construct and have been implicated in interactions of PIP2 with Kir2.1 (48−50).
Figure 6.

(A) Interactions of PIP2 (purple) with Kir6.2 (gray) after a 5 ns atomistic simulation. Key residues are shown as yellow sticks. (B) Percentage of hydrogen bonds made between the PIP2 molecules and the side chains of the three different Kir channels. The numbering for Kir6.2 is shown along the y-axis. For the sake of clarity, only residues that form more than 5% of the total hydrogen bonds over the course of the simulations are shown. (C) Sequence alignment of the three Kir channels investigated. Residues that form greater than 5% of the total hydrogen bonds with PIP2 are highlighted in bold. The three residues absent from the post-M2 loop of KirBac1.1 are shown as hyphens.
Thus, we may be reasonably confident that the PIP2 sites identified by our multiscale simulations are in good agreement with the available functional data. It is notable that (using the Kir6.2 numbering) residues equivalent to R50, R54, K67, R69, K170, H175, R176, and R206 are predicted to contribute to the PIP2-binding site in both the Kir6.2 model and the (semiopen) KirChim X-ray structure. Significantly, the bacterial KirBac1.1 channel forms different interactions, with the primary H-bonds to KirBac1.1 including R36 (44), S46 (54), R49 (57), K57 (65), R148 (170), R151 (173), and K155 (177) (numbers in parentheses indicate the corresponding residues in Kir6.2). Residue 49 has been previously suggested to interact with membrane phospholipids on the basis of mutagenesis studies (51).
Discussion
Biological Implications of Results
Multiscale simulations have enabled us to predict PIP2-binding sites for three different Kir channels: Kir6.2, KirBac1.1, and a chimeric channel, KirChim, that contains elements of both pro- and eukaryotic channels. For the KirChim channel, it is possible to compare the PIP2 site predicted by multiscale simulations with that based on the observation of a bound nonylglucoside detergent molecule in one conformation (the dilated state) of the X-ray structure of the channel (21). The two sites are close, but the headgroup of the simulated PIP2 molecule is displaced by ∼5 Å laterally, relative to the glucosyl group of the detergent. There are also differences in contacts between the two sites, such that the site predicted by the simulation optimizes the number of phosphate−basic side chain contacts between the PIP2 headgroup and the protein. As discussed above, there are some detailed differences between the PIP2 headgroup−protein side chain contacts for the three different Kir channels, but the overall pattern of PIP2 interacting with a cluster of basic side chains [as is the case in other PIP2-binding proteins, e.g., PH domains (52)] is conserved.
We note that for Kir6.2, the simulated−predicted binding site for PIP2 differs somewhat from that proposed previously by Haider et al. (19). This is probably due to two factors. (i) The structure of the post-M2 loop in the current model of Kir6.2 differs from that in the earlier model. (ii) In the earlier study, the IP3 headgroup of PIP2 was docked to the Kir6.2 model to identify the binding site, followed by addition of lipid acyl tails, while in our study, the initial protein−lipid contact was simulated in the presence of a bilayer. The latter approach was possible because of the multiscale strategy adopted.
Thus, the combination of a multiscale method with improved structural data for modeling has enabled a refined prediction of the PIP2-binding site. We note that related multiscale approaches have been used, in a different context, to explore the interaction of antibiotic inhibitors with bacterial ribosomes (53). Our modeling studies, in combination with published functional data, enable the formulation of testable hypotheses concerning the relationship between the structure and mechanism of Kir channels.
A mechanism for PIP2-driven gating of Kir channels remains somewhat elusive. However, the steps involved in Kir gating may be proposed on the basis of the two conformations of the cytoplasmic domains found in the KirChim crystal structure (while noting that detailed functional data are not available for KirChim). In the constricted pore conformation of the cytosolic domain, the C-terminal domain is positioned ∼6 Å lower (relative to the transmembrane domain) than in the alternative state in which the cytoplasmic pore is in a dilated conformation. The gate at the M2 helix bundle crossing is closed in both structures. The position of the PIP2 molecule in its binding site is in an ideal location to facilitate the upward motion of the C-terminal domain associated with a switch from the constricted to dilated conformation, through interactions with basic residues.
Among the differences between the sequences of mammalian Kir channels (which are activated by PIP2) and bacterial Kir channels (which are inhibited by PIP2), there is an insertion of three residues that form a single turn of an α-helix in mammalian channels (see Figure 6C). This alters the orientation of a number of residues within the PIP2-binding site, including R176 and R177 (using Kir6.2 numbering), and so is likely to modify the mechanism by which PIP2 molecules induce conformational change(s) in Kir channels. In the majority of Kir channels crystallized to date, a salt bridge is found between a glutamate (E292 in Kir6.2) in the cytoplasmic gating loop and a highly conserved arginine (R301 in Kir6.2). This interaction is disrupted in the more dilated conformation of the gating loop, being replaced by a possible interaction between E292 and the residue equivalent to R177. This latter interaction is too distant in the KirChim crystal structure (∼6 Å) but is frequently observed in MD simulations of the Kir channels. We suggest that the observed interactions of R176 with PIP2 could pull on the post-M2 loop, which includes the single-turn α-helix specific to the mammalian Kirs. In turn, this could draw upon the R177−E292 salt bridge and open the cytoplasmic gating loop.
The absence of the single-turn α-helix in the KirBac1.1 channel alters this mechanism. In this case, we suggest that K155, the residue equivalent to R177, forms a salt bridge with the gating loop glutamate (E262) in the open state of the channel. However, upon PIP2 binding, the absence of the single-turn helix (and hence of a residue equivalent to R176) allows K155 to alter its conformation and interact with PIP2. In turn, this breaks the salt bridge with the gating loop glutamate, resulting in collapse and closure of the cytoplasmic region of the channel.
The mechanism that leads to transmission of these changes to the transmembrane region is also poorly understood. In the mammalian channels, it seems likely that the binding of PIP2 modifies the position of the slide (M0) helix to release restraints upon the packing of the M2 helices. This, in addition to the contacts made with the post-M2 loop, would allow the M2 helices to kink and swivel about the glycine hinge (54−57) thus widening the pore in the region of the helix bundle crossing (58,59) of the channel.
Critical Evaluation of Methods
The primary aim of this study was to identify the binding site for PIP2 in both mammalian and bacterial Kir channels, and to compare the differences between these two classes of Kir channels. CG−MD simulations have provided a successful tool for predicting interactions of lipids with proteins (29), and therefore, this method was used to predict the interactions of the anionic lipid PIP2 with Kir channels. Unlike a previous computational study (19), our simulations take into account the whole of the PIP2 molecule, encompassing both the role of the tails in anchoring PIP2 to the membrane and specific contacts made by the IP3 headgroup. The simulations also allow for a degree of protein flexibility during the encounter of the protein with PIP2; i.e., this method allows “dynamic docking” of PIP2 to the Kir channels over an extended time scale, with enhanced sampling in comparison with previous atomistic simulations.
To test the specificity of the Kir6.2-binding site for PIP2, a set of five CG simulations were performed, in which a simpler anionic lipid [palmitoyloleoylphosphatidylglycerol (POPG)] was substituted for PIP2. In these instances, the interactions of POPG were far more transient (dwell time of <50 ns), with the POPG lipids binding and unbinding within the PIP2 site throughout the course of the simulations. There are also differences in the interactions of the POPG headgroup, with interactions with the N-terminal R50 and C-terminal R176 and R201 residues greatly weakened.
Our approach combined extended (0.5 μs) CG−MD simulations with relatively short (20 ns) atomistic MD simulations to evaluate and refine the CG models of the Kir−PIP2 complexes. Such a serial multiscale approach (32) differs from a parallel multiscale approach in which, for example, the protein and some lipids would be represented atomistically while the surrounding bilayer environment was represented in a more coarse-grained fashion. The serial multiscale approach is relatively straightforward to implement, avoiding issues of, for example, changes in granularity as a lipid molecule moves between outer and inner regions (60).
The behaviors of the PIP2 molecules in both the CG−MD and atomistic MD simulations are comparable to those observed recently (61), with the 1′-phosphate sitting within the average phosphate plane, ∼20 Å from the bilayer center, while the 4′-phosphate extends ∼5.2 Å into the solvent along the bilayer normal (z). Further more detailed comparisons with published atomistic MD simulations of PIP2-containing bilayers will be possible in the near future.
Multiscale simulations thus provide a novel approach to identifying lipid-binding sites in ion channel proteins. Previous computational approaches to interactions of lipids with KcsA (20), with Kv channel voltage sensors (62), and with the nicotinic acetylcholine receptor (21) have relied upon analysis of relatively short atomistic simulations guided by preexisting structural and/or modeling data. In this study, we were able to use multiscale simulations to both predict and subsequently refine and analyze channel−lipid interactions. The resultant models are likely to be suitable starting points for more extended (∼1 μs) atomistic simulations (63), which may reveal aspects of conformational changes induced by interactions with PIP2 and other ligands of Kir channels.
In summary, our studies indicate that multiscale simulations enable us to predict lipid interaction sites on membrane proteins, starting from either X-ray structures or homology models. Given the growing body of evidence for the structural (64) and functional importance of protein−lipid interactions (65,66), this is a valuable contribution of simulation-based approaches.
Acknowledgments
We thank members of the Sansom and Ashcroft laboratories for helpful discussions, especially Kathryn Scott, Timothy Carpenter, Robert D’Rozario, and Philip Fowler.
Supporting Information Available
Further details of simulation and analytical methods. This material is available free of charge via the Internet at http://pubs.acs.org.
Research in the laboratories of M.S.P.S. and F.M.A. is supported by the Wellcome Trust.
Footnotes
Abbreviations: Kir, inwardly rectifying potassium; MD, molecular dynamics; PDB, Protein Data Bank; PIP2, phosphatidylinositol bisphosphate; POPC, palmitoyloleoylphosphatidylcholine; POPG, palmitoyloleoylphosphatidylglycerol; rmsd, root-mean-square deviation; TM, transmembrane.
Supplementary Material
References
- Doupnik C. A.; Davidson N.; Lester H. A. (1995) The inward rectifier potassium channel family. Curr. Opin. Neurobiol. 5, 268–277. [DOI] [PubMed] [Google Scholar]
- Nichols C. G.; Lopatin A. N. (1997) Inward rectifier potassium channels. Annu. Rev. Physiol. 59, 171–191. [DOI] [PubMed] [Google Scholar]
- Reimann F.; Ashcroft F. M. (1999) Inwardly rectifying potassium channels. Curr. Opin. Cell Biol. 11, 503–508. [DOI] [PubMed] [Google Scholar]
- Kuo A.; Gulbis J. M.; Antcliff J. F.; Rahman T.; Lowe E. D.; Zimmer J.; Cuthbertson J.; Ashcroft F. M.; Ezaki T.; Doyle D. A. (2003) Crystal structure of the potassium channel KirBac1.1 in the closed state. Science 300, 1922–1926. [DOI] [PubMed] [Google Scholar]
- Nishida M.; Cadene M.; Chait B. T.; MacKinnon R. (2007) Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J. 26, 4005–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida M.; MacKinnon R. (2002) Structural basis of inward rectification: Cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 Å resolution. Cell 111, 957–965. [DOI] [PubMed] [Google Scholar]
- Fan Z.; Makielski J. C. (1997) Anionic phospholipids activate ATP-sensitive potassium channels. J. Biol. Chem. 272, 5388–5395. [DOI] [PubMed] [Google Scholar]
- Logothetis D. E.; Jin T.; Lupyan D.; Rosenhouse-Dantsker A. (2007) Phosphoinositide-mediated gating of inwardly rectifying K+ channels. Pfluegers Arch. 455, 83–95. [DOI] [PubMed] [Google Scholar]
- Fan Z.; Makielski J. C. (1999) Phosphoinositides decrease ATP sensitivity of the cardiac ATP-sensitive K+ channel. A molecular probe for the mechanism of ATP-sensitive inhibition. J. Gen. Physiol. 114, 251–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkvetchakul D.; Bhattacharyya J.; Jeliazkova I.; Groesbeck D. K.; Cukras C. A.; Nichols C. G. (2004) Functional characterization of a prokaryotic Kir channel. J. Biol. Chem. 279, 47076–47080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W. W.; Enkvetchakul D.; Nichols C. G. (2009) KirBac1.1: It's an inward rectifying potassium channel. J. Gen. Physiol. 133, 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng S. L.; Cukras C. A.; Harwood J.; Nichols C. G. (2000) Structural determinants of PIP2 regulation of inward rectifier K(ATP) channels. J. Gen. Physiol. 116, 599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukras C. A.; Jeliazkova I.; Nichols C. G. (2002) The role of NH2-terminal positive charges in the activity of inward rectifier K-ATP channels. J. Gen. Physiol. 120, 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukras C. A.; Jeliazkova I.; Nichols C. G. (2002) Structural and functional determinants of conserved lipid interaction domains of inward rectifying Kir6.2 channels. J. Gen. Physiol. 119, 581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze D.; Krauter T.; Fritzenschaft H.; Soom M.; Baukrowitz T. (2003) Phosphatidylinositol 4,5-bisphosphate (PIP2) modulation of ATP and pH sensitivity in Kir channels. A tale of an active and a silent PIP2 site in the N terminus. J. Biol. Chem. 278, 10500–10505. [DOI] [PubMed] [Google Scholar]
- John S. A.; Weiss J. N.; Ribalet B. (2005) ATP sensitivity of ATP-sensitive K+ channels: Role of the γ-phosphate group of ATP and the R50 residue of mouse Kir6.2. J. Physiol. 568, 931–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribalet B.; John S. A.; Xie L. H.; Weiss J. N. (2006) ATP-sensitive K+ channels: Regulation of bursting by the sulphonylurea receptor, PIP2 and regions of Kir6.2. J. Physiol. 571, 303–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegan S.; Arrabit C.; Zhou W.; Kwiatkowski W.; Collins A.; Slesinger P. A.; Choe S. (2005) Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat. Neurosci. 8, 279–287. [DOI] [PubMed] [Google Scholar]
- Haider S.; Tarasov A.; Craig T. J.; Sansom M. S. P.; Ashcroft F. M. (2007) Identification of the PIP2-binding site on Kir6.2 by molecular modelling and functional analysis. EMBO J. 26, 3749–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deol S. S.; Domene C.; Bond P. J.; Sansom M. S. P. (2006) Anionic phospholipids interactions with the potassium channel KcsA: Simulation studies. Biophys. J. 90, 822–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannigan G.; Hénin J.; Law R.; Eckenhoff R.; Klein M. L. (2008) Embedded cholesterol in the nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. U.S.A. 105, 14418–14423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psachoulia E.; Bond P. J.; Fowler P. W.; Sansom M. S. P. (2008) Helix-helix interactions in membrane proteins: Coarse grained simulations of glycophorin helix dimerization. Biochemistry 47, 10503–10512. [DOI] [PubMed] [Google Scholar]
- Shelley J. C.; Shelley M. Y.; Reeder R. C.; Bandyopadhyay S.; Klein M. L. (2001) A coarse grain model for phospholipid simulations. J. Phys. Chem. B 105, 4464–4470. [Google Scholar]
- Marrink S. J.; de Vries A. H.; Mark A. E. (2004) Coarse grained model for semiquantitative lipid simulations. J. Phys. Chem. B 108, 750–760. [Google Scholar]
- Nielsen S. O.; Lopez C. F.; Srinivas G.; Klein M. L. (2004) Coarse grain models and the computer simulation of soft materials. J. Phys.: Condens. Matter 16, R481–R512. [Google Scholar]
- Izvekov S.; Voth G. A. (2005) A multiscale coarse-graining method for biomolecular systems. J. Phys. Chem. B 109, 2469–2473. [DOI] [PubMed] [Google Scholar]
- Bond P. J.; Holyoake J.; Ivetac A.; Khalid S.; Sansom M. S. P. (2007) Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J. Struct. Biol. 157, 593–605. [DOI] [PubMed] [Google Scholar]
- Voth G. A. (2008) Coarse-Graining of Condensed Phase and Biomolecular Systems, CRC Press, Boca Raton, FL. [Google Scholar]
- Scott K. A.; Bond P. J.; Ivetac A.; Chetwynd A. P.; Khalid S.; Sansom M. S. P. (2008) Coarse-grained MD simulations of membrane protein-bilayer self-assembly. Structure 16, 621–630. [DOI] [PubMed] [Google Scholar]
- Yefimov S.; van der Giessen E.; Onck P. R.; Marrink S. J. (2008) Mechanosensitive membrane channels in action. Biophys. J. 94, 2994–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treptow W.; Marrink S.-J.; Tarek M. (2008) Gating motions in voltage-gated potassium channels revealed by coarse-grained molecular dynamics simulations. J. Phys. Chem. B 112, 3277–3282. [DOI] [PubMed] [Google Scholar]
- Ayton G. A.; Noid W. G.; Voth G. A. (2007) Multiscale modeling of biomolecular systems: In serial and in parallel. Curr. Opin. Struct. Biol. 17, 192–198. [DOI] [PubMed] [Google Scholar]
- Sali A.; Blundell T. L. (1993) Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815. [DOI] [PubMed] [Google Scholar]
- Boeckmann B.; Bairoch A.; Apweiler R.; Blatter M. C.; Estreicher A.; Gasteiger E.; Martin M. J.; Michoud K.; O'Donovan C.; Phan I.; Pilbout S.; Schneider M. (2003) The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink S. J.; Risselada J.; Yefimov S.; Tieleman D. P.; de Vries A. H. (2007) The MARTINI forcefield: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824. [DOI] [PubMed] [Google Scholar]
- Monticelli L.; Kandasamy S. K.; Periole X.; Larson R. G.; Tieleman D. P.; Marrink S. J. (2008) The MARTINI coarse grained force field: Extension to proteins. J. Chem. Theory Comput. 4, 819–834. [DOI] [PubMed] [Google Scholar]
- van der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. (2005) GROMACS: Fast, flexible, and free. J. Comput. Chem. 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- Rotkiewicz P.; Skolnick J. (2008) Fast procedure for reconstruction of full-atom protein models from reduced representations. J. Comput. Chem. 29, 1460–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott W. R. P.; Hunenberger P. H.; Tironi I. G.; Mark A. E.; Billeter S. R.; Fennen J.; Torda A. E.; Huber T.; Kruger P.; van Gunsteren W. F. (1999) The GROMOS biomolecular simulation program package. J. Phys. Chem. A 103, 3596–3607. [Google Scholar]
- Parrinello M.; Rahman A. (1981) Polymorphic transitions in single-crystals: A new molecular-dynamics method. J. Appl. Phys. 52, 7182–7190. [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. [Google Scholar]
- Hermans J.; Berendsen H. J. C.; van Gunsteren W. F.; Postma J. P. M. (1984) A consistent empirical potential for water-protein interactions. Biopolymers 23, 1513–1518. [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J. C.; Fraaije J. G. E. M. (1997) LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472. [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. (1996) VMD: Visual Molecular Dynamics. J. Mol. Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
- DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific, San Carlos, CA. [Google Scholar]
- Hall B. A.; Sansom M. S. P. (2009) Coarse-grained MD simulations and protein-protein interactions: The cohesin-dockerin system. J. Chem. Theory Comput. 5, 2465–2471. [DOI] [PubMed] [Google Scholar]
- Haider S.; Khalid S.; Tucker S.; Ashcroft F. M.; Sansom M. S. P. (2007) Molecular dynamics simulations of inwardly rectifying (Kir) potassium channels: A comparative study. Biochemistry 46, 3643–3652. [DOI] [PubMed] [Google Scholar]
- Huang C. L.; Feng S. Y.; Hilgemann D. W. (1998) Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature 391, 803–806. [DOI] [PubMed] [Google Scholar]
- Zhang H. L.; He C.; Yan X. X.; Mirshahi T.; Logothetis D. E. (1999) Activation of inwardly rectifying K+ channels by distinct PtdIns(4,5)P-2 interactions. Nat. Cell Biol. 1, 183–188. [DOI] [PubMed] [Google Scholar]
- Lopes C. M. B.; Zhang H. L.; Rohacs T.; Jin T. H.; Yang J.; Logothetis D. E. (2002) Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron 34, 933–944. [DOI] [PubMed] [Google Scholar]
- Enkvetchakul D.; Jeliazkova I.; Bhattacharyya J.; Nichols C. G. (2007) Control of inward rectifier K channel activity by lipid tethering of cytoplasmic domains. J. Gen. Physiol. 130, 329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon M. A. (2007) Pleckstrin homology (PH) domains and phosphoinositides. Biochem. Soc. Symp. 74, 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlugosz M.; Trylska J. (2009) Aminoglycoside Association Pathways with the 30S Ribosomal Subunit. J. Phys. Chem. B 113, 7322–7330. [DOI] [PubMed] [Google Scholar]
- Jiang Y.; Lee A.; Chen J.; Cadene M.; Chait B. T.; MacKinnon R. (2002) The open pore conformation of potassium channels. Nature 417, 523–526. [DOI] [PubMed] [Google Scholar]
- Grottesi A.; Domene C.; Sansom M. S. P. (2005) Conformational dynamics of M2 helices in KirBac channels: Helix flexibility in relation to gating via molecular dynamics simulations. Biochemistry 44, 14586–14594. [DOI] [PubMed] [Google Scholar]
- Rosenhouse-Dantsker A.; Logothetis D. E. (2006) New roles for a key glycine and its neighboring residue in potassium channel gating. Biophys. J. 91, 2860–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam A.; Jiang Y. (2007) High-resolution structure of the open NaK channel. Nat. Struct. Mol. Biol. 16, 30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jogini V.; Roux B. (2005) Electrostatics of the intracellular vestibule of K+ channels. J. Mol. Biol. 354, 272–288. [DOI] [PubMed] [Google Scholar]
- Tai K.; Haider S.; Grottesi A.; Sansom M. S. P. (2009) Ion channel gates: Comparative analysis of energy barriers. Eur. Biophys. J. 38, 347–354. [DOI] [PubMed] [Google Scholar]
- Praprotnik M.; Delle Site L.; Kremer K. (2005) Adaptive resolution molecular-dynamics simulation: Changing the degrees of freedom on the fly. J. Chem. Phys. 123, 224106. [DOI] [PubMed] [Google Scholar]
- Li Z.; Venable R. M.; Rogers L. A.; Murray D.; Pastor R. W. (2009) Molecular dynamics simulations of PIP2 and PIP3 in lipid bilayers: Determination of ring orientation, and the effects of surface roughness on a Poisson-Boltzmann description. Biophys. J. 97, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sands Z. A.; Sansom M. S. P. (2007) How does a voltage-sensor interact with a lipid bilayer? Simulations of a potassium channel domain. Structure 15, 235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjelkmar P.; Niemela P. S.; Vattulainen I.; Lindahl E. (2009) Conformational changes and slow dynamics through microsecond polarized atomistic molecular simulation of an integral Kv1.2 ion channel. PLoS Comput. Biol. 5, . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunte C.; Richers S. (2008) Lipids and membrane protein structures. Curr. Opin. Struct. Biol. 18, 406–411. [DOI] [PubMed] [Google Scholar]
- Schmidt D.; Jiang Q. X.; MacKinnon R. (2006) Phospholipids and the origin of cationic gating charges in voltage sensors. Nature 444, 775–779. [DOI] [PubMed] [Google Scholar]
- Marius P.; Zagnoni M.; Sandison M. E.; East J. M.; Morgan H.; Lee A. G. (2008) Binding of anionic lipids to at least three nonannular sites on the potassium channel KcsA is required for channel opening. Biophys. J. 94, 1689–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.