

Abstract
Although the outgrowth of micrometastases into macrometastases is the rate-limiting step in metastatic progression and the main determinant of cancer fatality, the molecular mechanisms involved have been little studied. Here, we compared the gene expression profiles of melanoma lymph node micro- and macrometastases and unexpectedly found no common up-regulation of any single growth factor/cytokine, except for the cytokine-like SPP1. Importantly, metastatic outgrowth was found to be consistently associated with activation of the transforming growth factor-β signaling pathway (confirmed by phospho-SMAD2 staining) and concerted up-regulation of POSTN, FN1, COL-I, and VCAN genes—all inducible by transforming growth factor-β. The encoded extracellular matrix proteins were found to together form intricate fibrillar networks around tumor cell nests in melanoma and breast cancer metastases from various organs. Functional analyses suggested that these newly synthesized protein networks regulate adhesion, migration, and growth of tumor cells, fibroblasts, and endothelial cells. POSTN acted as an anti-adhesive molecule counteracting the adhesive functions of FN1 and COL-I. Further, cellular FN and POSTN were specifically overexpressed in the newly forming/formed tumor blood vessels. Transforming growth factor-β receptors and the metastasis-related matrix proteins, POSTN and FN1, in particular, may thus provide attractive targets for development of new therapies against disseminated melanoma, breast cancer, and possibly other tumors, by affecting key processes of metastasis: tumor/stromal cell migration, growth, and angiogenesis.
Metastasis is a complex process comprising multiple steps, including dissemination of cells from a primary tumor into blood or lymph vessels, survival of the cells in these vessels, arrest and extravasation into a new organ, initiation and maintenance of growth, and vascularization of the metastatic tumor (reviewed in 1–3). Even before dissemination, the tumor cells may secrete growth factors and cytokines that induce systemic changes and prime the distant site for metastasis (reviewed in 4, 5). The early steps in metastasis occur efficiently, in contrast to later steps, where only a small subset of cancer cells at a secondary site initiate growth and form pre-angiogenic micrometastases, and, of these, only a tiny proportion continue to become vascularized and progressively growing macrometastases (reviewed in 1, 6). The ability to grow at a distant site is largely dictated by molecular interactions of the cancer cells with the new microenvironment, which may have an even greater impact on cell behavior at a distant site than at the primary tumor location.
Since the growth and spread of metastases is the main (90%) cause of death from solid cancers, better therapies are urgently needed. Strategically, the rate-limiting step of metastasis, the colonization of secondary sites, might be a more potent therapeutic target than the earlier efficient steps of metastasis, requiring inhibition of fewer cells. Moreover, at the time of primary-tumor diagnosis the earlier steps may already have occurred. The problem is, however, that although the mechanisms of the earlier steps of metastasis have been fairly extensively studied (though giving rise to differing mechanistic models, reviewed in 7, 8), the molecules and mechanisms behind the growth of micrometastases to overt macrometastases, especially in actual human cancers, are still little studied and poorly understood.
A good model to study the progression of metastasis is human cutaneous melanoma. It is one of the most aggressive malignancies, and it frequently has already disseminated cells to other organs by the time that the primary tumor diagnosis is made; it has no effective treatment after metastasis. The initial route of metastasis in melanoma is thought to mainly occur through the lymphatics to the first lymph nodes (the sentinel LNs) draining the primary melanoma site. Micro- and macrometastases in melanoma are presently defined by their detection method, according to the American Joint Committee on Cancer staging system.9 The patients are classified as having clinically occult (microscopic, detected by sentinel or elective lymphadenectomy) or clinically apparent (macroscopic, detected by physical or radiological examination) metastases without taking into consideration metastasis size. This procedure does not, however, necessarily reflect any biological properties of the metastases. In fact, several studies have shown that size of a metastasis (2 to 3 mm) or tumor burden analyzed by quantitative reverse transcription (RT)-PCR (qRT-PCR) in sentinel LNs is an efficient predictor of recurrence and survival.10,11,12 These measures may provide a division into pre-angiogenic micrometastases and more persistently growing macrometastases.
In this study, our goal was to identify genes required for the outgrowth of melanoma metastases by comparing gene expression profiles of melanoma micro- and macrometastases from LNs to understand the mechanisms involved and to identify rational targets for therapy. Expression of the genes identified was then analyzed in the metastases by immunohistochemistry (IHC) and confocal microscopy. Our results revealed the metastatic outgrowth to be associated with formation of an intricate network by four specific extracellular matrix (ECM) proteins: collagen-I (COL-I), fibronectin 1 (FN1), periostin (POSTN), and versican (VCAN), which appear to regulate tumor/stromal cell adhesion, migration, growth, and angiogenesis/tubulogenesis. We further studied breast cancer LN metastases, as well as melanoma and breast cancer metastases from other organs, to assess the generality of these findings.
Materials and Methods
Patient Samples
We divided the melanoma LN metastases into micrometastases (n = 42, maximum diameter ≤2.5 mm) and macrometastases (n = 65, maximum diameter >2.5 mm). We based this division on our gene expression clustering analyses, on a systematic study of metastasis diameters (0.3 to 10 mm) identifying 3 mm as the most significant cut-point predictive of survival,11 and on studies using a 2-mm cut-point10—a diameter arbitrarily chosen for clinical breast cancer staging. The results and conclusions were, however, unaffected if we used a 2- or 3-mm cut-point. The breast cancer LN metastases were analyzed both with a 2.0- and 2.5-mm cut-point for macrometastases. The sixth edition of the TNM (tumor-nodes-metastasis) classification defines metastases between 0.2 mm and 2 mm in largest diameter as micrometastases and ≤0.2-mm metastases as isolated tumor cells,13 but these measures are not evidence based and not used in melanoma diagnostics. As micrometastatic deposits are usually not spherical, measurements of the largest diameter alone may not be sufficient for the tumor size estimation. The melanoma metastases from LNs (n = 107), liver (n = 2), and lung (n = 12), and breast cancer metastases from LNs (n = 45), liver (n = 3), and lung (n = 2), as well as noncancerous control LNs (n = 39; see12) were obtained by surgical excision at Helsinki University Central Hospital. Tissue specimens were either fixed in formalin or immediately frozen in liquid nitrogen for the various analysis methods. Protocols for taking the tissue specimens were approved by the Ethics Committees of Helsinki University Central Hospital. Further, all patients gave their informed consent before the operations.
RNA Isolation and Purification
Total RNA was extracted from cells and frozen tissues with the RNeasy kit (Qiagen, Crawley, UK) and pigment, if present, was removed by Bio-Gel P-60 gel filtering, as described.12 Quality of the purified RNA was assessed by gel electrophoresis or by Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA).
Microarray Analysis
Melanoma LN micrometastases (n = 9) and macrometastases (n = 13; 11 detected by palpation and 2 by sentinel LN biopsy; in this group, all patients but one—who had undergone multiple surgical operations—subsequently died within 2 years) were analyzed with the Human Genome U133 Set (Affymetrix, Santa Clara, CA), as described.12 None of the patients received any chemo- or radiotherapy before sample collection. Breast cancer LN metastases (n = 11, 6 ductal and 5 lobular) and normal LNs (n = 7) were analyzed with the Human Genome U133 Plus 2.0 Array (Affymetrix). In addition, the normal LNs (n = 11) analyzed with the HG-U133 Set12 served for control purposes.
Statistical Analysis of Microarray Data
The HG-U133A and -B chips were analyzed separately. The individual probe signals were summarized by the RMA algorithm (RMAExpress version 0.3, http://rmaexpress.bmbolstad.com/). To reveal all of the gene expression changes in melanoma cells and stromal cells, the expression data from the micro- and macrometastatic lymph nodes were first quantile–normalized together.14 Gene probe sets with a mean difference <100 and a fold-change <1.5 between micro- and macrometastases were filtered off (to remove probe sets with low signals and with small expression level changes, which are not measured reliably). The remaining probe sets were ordered by significance analysis of microarrays (SAM) 3.015 (http://www-stat.stanford.edu/∼tibs/SAM/), and their class specificity was evaluated by 5000 random permutations. Delta was chosen so that the false discovery rate was <1%. The list of significant genes was then filtered for a fourfold difference. In addition, the data were normalized to melanocyte marker genes (SOX10, MLANA, TYR) to find genes with potentially specific up-regulation in melanoma cells.
To confirm the reproducibility of the list of differentially expressed genes, SAM analysis was also performed using data preprocessed with MAS 5.0 algorithm (Affymetrix). Further, filtering of RMA-summarized data before SAM analysis was performed in various ways: by removing only probe sets with absent calls in all samples, probe sets with expression signal values <20 in all samples, or probe sets with a mean difference <100 between the groups. In addition to SAM analysis, we produced a list of differentially expressed genes using fold change-ranking combined with t-statistics (Volcano plot).16 Similar lists of top-ranked genes were obtained with all methods.
Additionally, the genes (with a mean difference ≥100 and a fold-change ≥1.5 between groups) were clustered with Pearson correlation measurement and average linkage as implemented in GeneSpring GX 7.3 (Agilent Technologies) to explore the coordinate expression of selected genes in melanoma and breast cancer metastases. We also subjected the SAM-ordered probes (mean difference ≥ 100 and fold-change ≥ 1.5) to gene set enrichment analysis.17 The Molecular Signature Database (MSigDB, v2.5, Broad Institute, MA, http://www.broad.mit.edu/gsea/msigdb/index.jsp) gene set collections C2 to C5 were analyzed (C2 comprised of 1892 curated gene sets). The data were also analyzed by GenMAPP 2.1 and Ingenuity Pathway analysis tools (Ingenuity Systems, Redwood City, CA).
Semi-Quantitative RT-PCR and DNA Sequencing
One μg of total RNA was reverse-transcribed into cDNA, and PCR was performed essentially as described.12 POSTN primers were designed to cover the C-terminal region of POSTN mRNA (NM_006475), which is known to be differentially spliced. The forward primer was 5′-GTTTGTTCGTGGTAGCACCT-3′ and reverse 5′-TGTTGGCTTGCAACTTCCTCAC-3′, amplifying from the full-length POSTN mRNA a fragment of 500 bp. PCR was performed in a DYAD DNA engine (MJ Research, Waltham, MA) under the following conditions: 94°C for 15 s, 57°C for 30 s, and 72°C for 1 minute 10 seconds for 32 cycles. β-actin (ACTB) served as the control. PCR products were separated by electrophoresis on a 2% agarose gel, visualized by SYBR Green I staining, and documented by a video camera system.
For sequencing, PCR fragments (separated on high resolution MetaPhor agarose, Cambrex/Lonza, Basel, Switzerland) were purified with Ultrafree-DA centrifugal filter units (Millipore, Billerica, MA) and with the Illustra GFX PCR DNA and gel band purification kit (GE Health care, Waukesha, WI). DNA sequencing of the PCR products was performed with the BigDye Terminator v3.1 kit and 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA).
Quantitative RT-PCR and Statistical Analyses
Real-time qRT-PCR analysis for POSTN mRNA was performed with the ABI PRISM 7700 Sequence Detection System instrument and software (Applied Biosystems) using a Taqman Gene Expression Assay Hs00170815_m1 for POSTN and a Taqman pre-developed assay for β-actin. POSTN and ACTB cDNA levels were measured in triplicate. Standard curves were generated for both genes, and the relative POSTN expression values were normalized to that of ACTB.
Immunohistochemistry
Paraffin-embedded sections (5 μm) were deparaffinized, rehydrated through a graded ethanol series, and washed in distilled water. Antigen retrieval was performed by trypsin treatment (0.5% in PBS) at 37°C for 30 minutes, except for POSTN and phospho-SMAD2 (pSMAD2) staining, where the samples were heated in a microwave oven at 850 W for 3 minutes and at 170 W for 7 minutes in 10 mmol/L citric acid, pH 6.0. For both paraffin-embedded and frozen sections (5 μm), the endogenous peroxidase activity was blocked with 1% H2O2 in methanol for 30 minutes. After blocking in 1 to 10% goat serum in PBS or in CAS-block reagent (Zymed Laboratories, San Francisco, CA), the sections were incubated with the primary antibodies (see supplemental Table S1 at http://ajp.amjpathol.org/) diluted in the blocking buffer at 4°C overnight. Immunodetection was performed with the StreptABComplex/HRP Duet kit (Dako, Glostrup, Denmark) according to the manufacturer’s protocol. The chromogen was either 3,3′-diaminobenzidine or 3-amino-9-ethylcarbatzole. Slides were counterstained with Mayer’s hematoxylin and mounted with Mountex (Histolab Products, Göteborg, Sweden) or Aquamount (BDH Laboratories, Poole, UK). Images were obtained with a Nikon Eclipse E800 or 80i microscope, Nikon DXM1200 digital camera, or Digital Sight DS-5M camera, and the ACT-1 or NIS-Elements F 2.20 software (Nikon, Tokyo, Japan).
Confocal Immunofluorescence Microscopy
Frozen tissue samples were embedded in Tissue-Tek optimal cutting temperature compound (Miles Inc., Elkhart, IN) and cut into 40- to 80-μm thicknesses. Sections were mounted onto SuperFrost Plus slides (Menzel-Glaser, Braunschweig, Germany) and air-dried at room temperature for 1 hour. The slides were washed with PBS pH 7.4, pre-incubated with a blocking mixture containing 0.25% Triton X-100 and 10% normal horse serum in PBS (1 hour, room temperature), and incubated overnight at room temperature with the primary antibodies (see supplemental Table S1 at http://ajp.amjpathol.org/). The polyclonal and monoclonal antibodies to FN1 showed similar staining patterns. After incubation with primary antibodies, the slides were washed with PBS and incubated with Alexa-conjugated (488, 568, 647) secondary antibodies (Invitrogen, Carlsbad, CA) diluted 1:500 in PBS containing Triton X-100 and 10% normal horse serum for 1 hour at room temperature. The slides were then washed with PBS and mounted with glycerol and PBS (1:1).
Specimens were examined with a Leica DM RXA epifluorescence microscope and Leica TCS MP SP confocal microscopy system. The emission wavelengths were set up for Alexa 488 at 495 to 550 nm, for Alexa 568 at 600 to 650 nm, and for Alexa 647 at 660 to 750 nm. The fluorophores were excited by use of an argon-krypton laser beam at 488 nm (Lasos, Jena, Germany) or by an 85 YCA-series diode-pumped solid-state yellow laser beam at 561 nm and a helium-neon laser at 633 nm (Omnichrome, Melles Griot, Carlsbad, CA). Acquisition of data and the three-dimensional reconstruction imaging were performed with Leica TCS NT/SP Scanware software.
Surface Plasmon Resonance Analysis
Binding of the 90-kDa full-length recombinant human POSTN (R&D Systems, Minneapolis, MN), the 75-kDa fragment of human POSTN (BioVendor, Heidelberg, Germany), human plasma FN (pFN, Chemicon International, Temecula, CA), human cellular FN (cFN, United States Biological, Swampscott, MA), COL-I (Sigma-Aldrich, St. Louis, MO), human tenascin C (TNC, Chemicon International), and human secreted phosphoprotein 1/osteopontin (SPP1, R&D Systems) to each other was analyzed with the Biacore 2000 surface plasmon resonance-based biosensor (Biacore, Uppsala, Sweden). The purity of all proteins was >90%, mostly >95%. CM5 sensor chip surfaces were coupled with pFN/cFN, COL-I, and full-length POSTN to final resonance unit values of 3900, 3700, and 1800, respectively (in the experiment shown), using the standard amine coupling kit (Biacore) according to the manufacturer’s protocol. Flow cell 1 was activated and blocked without any protein for controlling any nonspecific binding.
Binding experiments were performed in PBS at a flow rate of 10 μl/minute. All proteins served as the ligand coupled to the sensor surface and as the soluble analyte injected over the sensor surfaces, at 50 to 200 nmol/L concentrations for 2 minutes (followed by monitoring of dissociation) with five separate chips. The responses obtained from a blank control surface were subtracted from the data from the protein-coated surfaces, and the sensorgrams were analyzed with the BIAevalution 3.1 Software (Biacore). The chip surface was regenerated after each injection by adding 100 mmol/L HCl for 30 s, followed by a re-equilibration with PBS.
Cell Culture
Primary human melanocytes and melanoma cells were isolated and cultured as described.18 Melanoma cell lines WM793 and WM239 were kindly provided by Dr. M. Herlyn (Wistar Institute, Philadelphia, PA), and primary human adult and embryonic skin fibroblasts by Dr. A-M. Ranki and Dr. A. Vaheri (University of Helsinki, Finland), respectively. Breast cancer cell line MB-MDA-231 was from ATCC (Teddington, UK). Cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and antibiotics. Primary human microvascular endothelial cells (HMVECs, adult dermis, Invitrogen) were cultured in growth factor–supplemented Medium 131 (Invitrogen).
Short Hairpin RNA Lentiviral Particles Transduction
WM793, WM239, HMVECs, and primary human embryonic and adult fibroblasts were transduced with short hairpin RNA (shRNA) lentiviral particles (Santa Cruz Biotechnology, Santa Cruz, CA) targeting FN1 (sc-29315-V), POSTN (sc-61324-V), or with negative (scrambled) control shRNA particles (sc-108080) according to the manufacturer’s instructions. Transductions were performed twice in duplicates in 12- or 24-well plates, and puromycin-resistant pools of cells were used in the assays (to avoid clonal variation).
Cell Adhesion Assay
Flat-bottomed 96-well plates were incubated with 10 μg/ml bovine serum albumin (fatty acid-free, Sigma-Aldrich), pFN, cFN, full-length POSTN, POSTN fragment, TNC, COL-I, or combinations of these proteins for 2 hours at 37°C and washed three times with PBS. Cells were suspended in serum-free medium at a density of 2 × 105 cells/ml, and 0.1 ml of the cell suspension was added to each well of the coated plate. After 1 hour of incubation at 37°C, attached and spread cells were photographed and counted. Experiments were repeated three to eight times (depending on the coated protein). Adhesion of cells expressing the control, FN1, or POSTN shRNAs was studied the same way but using uncoated plates.
Cell Migration Assay
Falcon cell culture inserts (8 μm, BD Biosciences, Franklin Lakes, NJ) were coated on the outer surface with 45 μg/ml cFN or COL-I (40 μl; to enable attachment of the migrated cells in serum-free medium) and on the inner surface with 90 μg/ml cFN or COL-I (40 μl), or with a mixture (40 μl) of 45 μg/ml cFN or COL-I and 45 μg/ml full-length POSTN or POSTN fragment. Cells were suspended in the medium at a density of 1.5 × 105 cells/ml, and 0.2 ml of the cell suspension was added to each insert. The lower chamber was filled with 800 μl of serum- and growth factor-free medium specifically to measure spontaneous cell migration (rather than chemoinvasion). After a 16- to 18-hour incubation (depending on the cell type), the insert filters were fixed in 3.5% paraformaldehyde and washed three times with PBS. The fixed cells were stained with 0.5% crystal violet (in 20% methanol) for 2 hours and washed with distilled water. Cells on the upper face of the filter were scraped off, and migrated cells on the lower face of the filter were photographed and counted. Experiments were repeated three to six times, and the results were analyzed by two-tailed t-test.
Matrigel Invasion Assay
The effect of POSTN and cFN on melanoma cell invasion was assayed in thick three-dimensional growth factor–reduced Matrigel (BD Biosciences, Franklin Lakes, NJ) essentially as described.19 The lower and upper Matrigel layers as well as the growth medium were supplemented or not with POSTN, cFN, or both at 0.1, 1, 2.5, or 10 μg/ml. Then, 20,000 WM793 cells in 100 μl of RPMI-1640 were plated on top of the Matrigel and allowed to adhere for 1 hour at 37°C. Excess medium was removed, and 250 μl Matrigel (supplemented or not with POSTN, cFN, or both at 0.1, 1, 2.5, or 10 μg/ml) was layered above the cells. Finally, 500 μl of growth medium without or with POSTN, cFN, or both (0.1, 1, 2.5, or 10 μg/ml) was added on top of the Matrigel matrix. The growth medium was replenished every third day. The growth pattern of the cells in Matrigel was followed daily by microscopy and photography.
Results
Microarray Analysis of Melanoma Micro- and Macrometastases
It has become increasingly evident that metastatic growth is not only dependent on the tumor cells, but also on the activation or suppression of resident stromal cells (fibroblasts, inflammatory and immune cells, and endothelial cells), and/or recruitment of various progenitor cells from the bone marrow. To identify the genes associated with the macroscopic growth of melanoma metastases, we performed gene expression analyses of micro- and of macrometastatic LNs with metastasis diameters of ≤2.5 mm and of >2.5 mm. Specifically, we were interested to discover any common denominator(s) for the metastatic growth. Significance analysis of microarrays resulted in 166 probe sets representing 131 genes significantly overexpressed (Table 1, and supplemental Tables S2 and S3 at http://ajp.amjpathol.org/) and 145 probe sets representing 121 genes underexpressed ≥fourfold (see supplemental Tables S4 and S5 at http://ajp.amjpathol.org/; containing mostly immune cell-specific genes) in the macrometastases compared with micrometastases; for genes with two- to fourfold changes, see supplemental Tables S6 and S7 at http://ajp.amjpathol.org/. Inspection and analysis of the gene lists by several methods revealed no universal up-regulation of any single growth factor or cytokine in melanoma macrometastases. However, SPP1, which has cytokine-like properties, was frequently up-regulated. In individual cases some other factors as well, such as GDF15 (see supplemental Table S3 at http://ajp.amjpathol.org/) and CXC chemokines, were highly increased. These factors may be of importance for macrometastatic growth,20,21,22,23 although, in our preliminary studies, recombinant SPP1 and GDF15 alone or together did not induce proliferation of melanoma cells in serum-free culture (data not shown).
Table 1.
The Most Overexpressed Genes (≥fourfold) in Melanoma LN Macrometastases Compared with Micrometastases by Significance Analysis of Microarrays (SAM)
| Gene | Gene description | Probe set ID | Mean ± SD
|
Fold macros versus micros | SAM* score macros versus micros | |
|---|---|---|---|---|---|---|
| Micro-metastases | Macro-metastases | |||||
| SOX10 | SRY (sex determining region Y)-box 10 | 209842_at | 148.0 ± 75.7 | 1070.6 ± 257.3 | 7.2 | 6.902 |
| MLANA | Melan-A | 206427_s_at | 454.6 ± 575.0 | 4274.5 ± 1848.6 | 9.4 | 5.573 |
| TYR | Tyrosinase | 206630_at | 363.6 ± 245.8 | 2954.0 ± 1259.3 | 8.1 | 5.475 |
| PRAME | Preferentially expressed antigen in melanoma | 204086_at | 93.9 ± 84.3 | 888.7 ± 324.8 | 9.5 | 5.084 |
| COL1A2 | Collagen, type I, alpha 2 | 202404_s_at | 235.0 ± 141.3 | 1860.5 ± 849.2 | 7.9 | 4.887 |
| PLP1 | Proteolipid protein 1 | 210198_s_at | 58.0 ± 28.3 | 814.3 ± 328.7 | 14.0 | 4.864 |
| FN1 | Fibronectin 1 | 211719_x_at | 594.0 ± 262.3 | 4093.5 ± 2032.1 | 6.9 | 4.786 |
| FN1 | Fibronectin 1 | 210495_x_at | 626.4 ± 254.0 | 4024.2 ± 2033.9 | 6.4 | 4.645 |
| GPM6B | Glycoprotein M6B | 209170_s_at | 94.1 ± 62.0 | 1084.3 ± 512.5 | 11.5 | 4.547 |
| FN1 | Fibronectin 1 | 216442_x_at | 610.2 ± 243.4 | 3910.9 ± 2021.7 | 6.4 | 4.539 |
| LGALS3BP | Lectin, galactoside-binding, soluble, 3 binding protein | 200923_at | 361.2 ± 158.5 | 1675.6 ± 729.5 | 4.6 | 4.476 |
| SPP1 | Secreted phosphoprotein 1 | 209875_s_at | 80.4 ± 61.3 | 1732.9 ± 1028.0 | 21.5 | 4.232 |
| IGSF3 | Immunoglobulin superfamily, member 3 | 202421_at | 109.8 ± 28.7 | 651.8 ± 247.9 | 5.9 | 4.220 |
| FN1 | Fibronectin 1 | 212464_s_at | 495.6 ± 210.0 | 3654.7 ± 2106.2 | 7.4 | 4.187 |
| MYO10 | Myosin X | 201976_s_at | 108.6 ± 81.2 | 1059.7 ± 543.7 | 9.8 | 4.157 |
| ETV5 | Ets variant 5 | 203349_s_at | 167.6 ± 61.8 | 718.3 ± 257.7 | 4.3 | 4.141 |
| GPM6B | Glycoprotein M6B | 209167_at | 87.5 ± 65.6 | 845.1 ± 409.5 | 9.7 | 4.128 |
| POSTN | Periostin | 210809_s_at | 126.1 ± 108.6 | 2095.0 ± 1288.4 | 16.6 | 4.114 |
| MLPH | Melanophilin | 218211_s_at | 339.1 ± 116.4 | 1389.2 ± 648.2 | 4.1 | 3.965 |
| MLANA | Melan-A | 206426_at | 157.2 ± 136.1 | 1521.2 ± 884.5 | 9.7 | 3.962 |
| COL1A1 | Collagen, type I, alpha 1 | 202310_s_at | 638.1 ± 462.4 | 2930.2 ± 1617.2 | 4.6 | 3.804 |
| ERBB3 | v-erb-b2 erythroblastic leukemia viral oncogene homolog 3 (avian) | 202454_s_at | 72.3 ± 39.7 | 742.4 ± 404.0 | 10.3 | 3.703 |
| SNCA | Synuclein, alpha | 204466_s_at | 134.2 ± 92.8 | 1061.2 ± 610.1 | 7.9 | 3.689 |
| MITF | Microphthalmia-associated transcription factor | 207233_s_at | 106.6 ± 22.6 | 1068.5 ± 647.1 | 10.0 | 3.668 |
| EDNRB | Endothelin receptor type B | 204271_s_at | 329.3 ± 69.9 | 1761.9 ± 1039.6 | 5.4 | 3.631 |
| PON2 | Paraoxonase 2 | 210830_s_at | 116.5 ± 26.6 | 487.0 ± 169.8 | 4.2 | 3.622 |
| CALU | Calumenin | 200757_s_at | 297.3 ± 73.0 | 1291.7 ± 684.5 | 4.3 | 3.608 |
| WBP5 | WW domain binding protein 5 | 217975_at | 89.8 ± 33.2 | 374.2 ± 100.9 | 4.2 | 3.560 |
| TUBB2A | Tubulin, beta 2A | 204141_at | 200.2 ± 59.6 | 986.7 ± 526.2 | 4.9 | 3.538 |
| TFAP2A | Transcription factor AP-2 alpha | 204653_at | 94.0 ± 73.0 | 1018.5 ± 661.6 | 10.8 | 3.451 |
| EDNRB | Endothelin receptor type B | 206701_x_at | 177.4 ± 67.9 | 1022.3 ± 639.3 | 5.8 | 3.245 |
| PFN2 | Profilin 2 | 204992_s_at | 159.7 ± 33.2 | 873.0 ± 530.9 | 5.5 | 3.194 |
| ACOT7 | Acyl-CoA thioesterase 7 | 208002_s_at | 168.0 ± 41.1 | 691.1 ± 354.7 | 4.1 | 3.181 |
| STXBP1 | Syntaxin binding protein 1 | 202260_s_at | 128.4 ± 30.8 | 713.9 ± 415.3 | 5.6 | 3.173 |
| LAPTM4B | Lysosomal protein transmembrane 4 beta | 208029_s_at | 263.6 ± 89.9 | 1065.2 ± 615.1 | 4.0 | 3.170 |
| PLOD3 | Procollagen-lysine, 2-oxoglutarate 5-dioxygenase 3 | 202185_at | 401.5 ± 111.1 | 1606.5 ± 1026.1 | 4.0 | 3.084 |
| DCT | Dopachrome tautomerase | 205337_at | 50.7 ± 38.5 | 881.9 ± 670.1 | 17.4 | 3.077 |
| SLC7A5 | Solute carrier family 7, member 5 | 201195_s_at | 191.0 ± 76.7 | 981.5 ± 628.5 | 5.1 | 3.076 |
| PHLDA1 | Pleckstrin homology-like domain, family A, member 1 | 217996_at | 54.1 ± 23.4 | 417.5 ± 226.9 | 7.7 | 2.996 |
| VCAN | Versican | 221731_x_at | 99.4 ± 75.6 | 722.4 ± 495.6 | 7.3 | 2.931 |
The FDR q-value for each was <0.001.
Most importantly, SAM analysis revealed a common ECM modulatory/fibroblast response in the macrometastatic tissues. Query of the SAM-ordered probes with the gene set enrichment analysis tool indicated that transforming growth factor β (TGFβ)-regulated genes likely play an important role in the growth of melanoma metastases. For the gene sets TGFBETA_ALL_UP and TGFBETA_EARLY_UP (Pubmed 11279127) (comprising genes up-regulated in skin fibroblasts after TGFβ treatment at any time or at 30 minutes, respectively) the normalized enrichment scores were 2.66 and 2.53; the false discovery rate q-value was <0.001 for both sets. Other interesting enriched gene sets were STEMCELL_NEURAL_UP and STEMCELL_EMBRYONIC_UP (comprising genes up-regulated in mouse neural and embryonic stem cells compared with differentiated brain and bone marrow cells) with normalized enrichment scores of 3.34 and 3.18, and an false discovery rate q-value <0.001 for both sets. However, the classic stem cell markers NANOG, POU5F1/OCT4, and SOX2 (reviewed in 24) or the stem cell markers identified in melanoma PROM1/CD133 and ABCB5 (reviewed in 25), were found neither to be overexpressed in our melanoma macrometastasis samples nor to be present in those stem cell gene sets (except for POUF51). All four gene sets were also selected by gene set enrichment analysis performed on our breast cancer LN metastasis samples (data not shown).
Although gene set enrichment analysis indicated increased TGFβ signaling, individual components of the TGFβ signaling pathway were not picked up by SAM, except for TGFβ receptor 1, which showed a significant 1.6-fold increase at the mRNA level in melanoma macrometastases (data not shown). Nor did analysis of the expression of TGFB1, TGFB2, and TGFB3 in melanoma micro- and macrometastases by RT-PCR reveal any marked general increases (data not shown). But when analyzing the TGFβ receptor-mediated phosphorylation of SMAD2 by IHC in melanoma metastases, we found all macrometastases (n = 30) to show positive pSMAD2-staining in the nuclei of melanoma cells, particularly at the periphery (growing margins) of the metastases (see supplemental Figure S1, E–H and supplemental Table S8 at http://ajp.amjpathol.org/). The cells of the surrounding stroma (fibroblasts) also showed positive and, in some cases, even more prominent pSMAD2 staining than did the melanoma cells (see supplemental Figure S1H and supplemental Table S8 at http://ajp.amjpathol.org/). Of the 26 micrometastases, 8 showed no pSMAD2 staining (see supplemental Figure S1, A and B at http://ajp.amjpathol.org/), whereas the rest showed variable, mostly faint to moderate staining in a portion of the melanoma cells (see supplemental Figure S1, C and D at http://ajp.amjpathol.org/). These results are in line with those of a recent study on breast cancer cells showing that TGFβ activity is more closely associated with metastasis than are the expression levels of the TGFβ pathway components.26
Of the top-scoring genes in the SAM analysis, we have identified many, ie, SOX10, MLANA, TYR, PRAME, PLP1, GPM6B, SPP1, and POSTN, as potential melanoma metastasis markers in sentinel LNs.12 The folds of increase in expression of the melanocyte/melanoma marker genes SOX10, MLANA, and TYR of course reflect the relative increase in melanoma cells in the macrometastatic tissues and serve as internal controls for size of the metastases. Normalization to these marker genes revealed that there are only a few genes with higher folds of overexpression, ie, potentially up-regulated in the melanoma cells in all metastases (data not shown). The same conclusion can be derived from the data in Table 1. We further checked by microarray analyses the expression levels of all of the listed genes in Table 1 in primary human melanocytes, nevus cells, and melanoma cells (isolated from vertical growth phase melanomas or LN metastasis;12 unpublished data) to determine whether they were of melanoma cell or stromal origin. Of the genes expressed by melanoma cells, SPP1 appeared to show a slightly higher increase in expression in the macrometastatic lymph nodes than did the melanoma marker genes. This indicates that SPP1 expression is maintained at a high level or is even up-regulated in the melanoma cells during metastatic growth, strengthening earlier findings on its importance in melanoma progression.12,27 In the same analyses, we found the TGFβ-inducible genes COL1A2 (scoring very high in the SAM analysis), POSTN, COL1A1, and VCAN (28–30 and our unpublished data) to show markedly lower expression in the melanoma cells than in the metastatic tissues (see supplemental Figure S2 at http://ajp.amjpathol.org/), indicating that they are expressed by other than melanoma cells. In fact, these genes are known to be expressed by fibroblasts (28–30 and our unpublished data), indicating an important role for stromal fibroblasts in regulation of melanoma metastasis growth. FN1, which is also a TGFβ target gene (30 and our unpublished data), was, in turn, highly expressed in both fibroblasts and melanoma cells (see supplemental Figure S2 at http://ajp.amjpathol.org/, and our unpublished data). We then subjected these four genes (scoring high in our SAM analysis) to more detailed study, paying special attention to the gene showing the highest increase in expression, POSTN.
Periostin Expression in Fibroblasts, Melanoma Cells, and Metastatic Melanoma Tissues
The increased expression of POSTN in the macrometastases (seen in the microarray analyses) was verified by qRT-PCR designed to measure the total expression of all known POSTN isoforms. These assays revealed a 35-fold higher average expression of POSTN mRNA (two-tailed t-test, P = 0.0357) in the macrometastatic than in micrometastatic LNs (Figure 1A).
Figure 1.

POSTN gene expression in different normal cells, tissues, and melanoma cells. A: POSTN expression in normal, micrometastatic, and macrometastatic LNs. POSTN and β-actin (ACTB) cDNA levels were measured by qRT-PCR in triplicate for each sample. POSTN/ACTB ×103 was calculated as (cDNA POSTN/cDNA ACTB) ×103. Horizontal bars represent the mean expression values. B: Expression of POSTN C-terminal splice-variants in different primary cells, cell lines, and tissues as detected by semiquantitative RT-PCR. ACTB served as a control. C: Schematic drawing of the C-terminal structures of the POSTN splice variants identified. The last nucleotide in exon 16 starts a new codon, resulting in one new triplet of nucleotides (which causes a single amino acid change, marked by asterisks) in the beginning of the next exon compared with the full-length protein. The sizes of exons are indicated above the full-length transcript.
We additionally performed semiquantitative RT-PCR analyses of the metastatic tissues (and different cell types) with primers that encompassed the C-terminal region of POSTN (known to be subject to alternative splicing) to see whether the increase in POSTN expression in macrometastases is associated with synthesis of some specific isoforms (and to discover their source). In our analyses of primary human fibroblasts, melanocytes, and melanoma cells from primary tumors and LN metastases, melanoma cell lines, normal LNs, and melanoma LN metastases, we found a total of five splice-variants in addition to full-length POSTN (Figure 1B). Sequencing of the PCR fragments revealed that those exons subjected to alternative splicing were 17, 18, 19, and 21 (Figure 1C). Notably, alternative splicing in the POSTN C-terminus results, in addition to exon deletions, in a codon change (causing a single amino acid change) in the beginning of the following exon joined to exon 16 (Figure 1C). Primary fibroblasts, especially embryonic ones, expressed high levels of POSTN variants 1, 2, 3, and lower levels of variant 5. Normal human melanocytes and primary melanoma cells from two primary tumors expressed only the variant 3 or 4 at low levels. The continuous cell line WM793, established from an early vertical growth phase melanoma, showed a broadly similar result but in addition expressed small amounts of variants 1 and 2. Melanoma cells isolated from metastases showed either a similar expression pattern to that of the melanoma cells isolated from primary tumors (data not shown) or expressed elevated levels of variants 1, 2, 3, and 5 (Figure 1B). The WM239 melanoma cell line, established from a LN metastasis, was found to express full-length POSTN and the variants 1, 2, 3, and 4. Normal LNs expressed low levels of variants 1, 2, 3, and 5. Metastatic melanoma LNs expressed all of the variants, especially variant 2, at elevated levels in all six samples analyzed. Macrometastases with the highest POSTN expression appeared to express relatively more of the larger isoforms, including the full-length POSTN.
To unravel the processes with which POSTN might be associated in metastatic growth, we searched for genes coordinately expressed with POSTN by hierarchical gene clustering and by Pearson correlation statistics. Several genes encoding ECM proteins showed coordinate expression with POSTN in both melanoma and breast cancer metastases: FN1, VCAN, and the collagen genes COL1A1, COL1A2, and COL5A2 (Figure 2A and B, and supplemental Tables S9 and S10 at http://ajp.amjpathol.org/). These genes, known to be expressed by fibroblasts, encode proteins associated with cell adhesion and migration. An additional interesting gene expressed coordinately with POSTN in melanoma metastases was RAC1, which encodes a Rho-like GTPase associated with reorganization of the actin cytoskeleton and cell migration. To further confirm the coordinate expression of these ECM protein-encoding genes in metastatic growth, we performed Pearson correlation analyses on two independent data sets of 21 melanoma and 81 breast cancer LN metastases, and found FN1, VCAN, and COL1A2 to be coordinately expressed with POSTN in both sets (see supplemental Tables S11 and S12 at http://ajp.amjpathol.org/).
Figure 2.

Genes expressed coordinately with POSTN in metastatic LNs. A: Genes showing coordinate expression with POSTN in melanoma metastases. First, genes with a <100 mean difference and a <1.5 fold-change between micro- and macrometastatic LNs were filtered off. The remaining 2211 probe sets were analyzed by hierarchical gene clustering in nine melanoma LN micro- and 13 macrometastases. B: Genes showing coordinate expression with POSTN in breast cancer metastases. Genes with a <100 mean difference and a <1.5 fold-change between normal LNs and breast cancer LN metastases were filtered off. The remaining 4547 probe sets were analyzed by hierarchical gene clustering in 11 breast cancer metastases.
Periostin, Fibronectin, Collagen-I, and Versican Colocalize in Metastatic Tissues
Next, we assessed by IHC the protein expression levels and tissue distribution of POSTN, FN1, COL-I, and VCAN in consecutive sections from 88 melanoma LN metastases (55 macro- and 33 micrometastases) and 22 normal or metastasis-free sentinel LNs. In macrometastases, we found POSTN, FN1, VCAN, and pro-collagen-I (PCOL-I, stained instead of mature COL-I to visualize the newly synthesized COL-I) to colocalize in extracellular strand and ring structures that surrounded individual melanoma cells or cell nests (Figure 3, B, C, F, G, J, K, N, and O). These fibrillar strands were visible inside the metastases and most prominently at the tumor-stroma interface (Figure 4, D and H; see supplemental Figure S3 at http://ajp.amjpathol.org/). The structures were detected in virtually all macrometastases though with varying intensity, diameter, and abundance (Figure 3 and Figure 4), with the centers of large tumors being frequently fairly negative (see supplemental Figure S3 at http://ajp.amjpathol.org/). It remains to be elucidated whether the fibrillar networks could be transient/dynamic in nature, for instance, being formed during the active phase of metastatic growth and then digested/remodeled by the melanoma or stromal cell-produced proteases (known to degrade many of these ECM proteins) (see supplemental Table S13 at http://ajp.amjpathol.org/). Micrometastases, in turn, showed staining patterns varying from no staining of POSTN and marginal staining of FN1 to faint or moderate staining of both proteins (Figure 4, A–C and E–G). In this series of micro- and macrometastases, the staining intensity of POSTN seemed to correlate with the extent of pSMAD2 staining better than did that of FN1 (Figure 4, A–L), the TGFβ stimulation of which may also be SMAD-independent.31 It will be interesting to see whether these staining patterns can predict the growth potential of micrometastases to macrometastases. Another important, more difficult question to study is whether the expression of these proteins could also be associated with the growth of isolated tumor cells to “true micrometastases,” ie, escape from cellular dormancy.32,33
Figure 3.

Immunohistochemical staining of POSTN, FN1, VCAN, and PCOL-I in melanoma and breast cancer LN metastases. A–P: POSTN, FN1, VCAN, and PCOL-I staining in control LNs (A, E, I, and M), melanoma metastases (B, C, F, G, J, K, N, and O) with (C, G, K, and O) showing the boxed area from (B, F, J, and N, respectively), and breast cancer metastases (D, H, L, and P). POSTN, VCAN, and PCOL-I are shown in red and FN1 in brown. Note the specific organization of the PCOL-I-synthesizing fibroblasts along the fibrillar structures surrounding the melanoma cell nests in (O). Scale bars = 200 μm.
Figure 4.

Immunohistochemical staining of POSTN, FN1, and phosphorylated SMAD2 in melanoma LN micro- and macrometastases. A–L: POSTN, FN1, and pSMAD2 staining in melanoma micrometastases from three patients (A–C, E–G, and I–K) and in a melanoma macrometastasis (D, H, and L). FN1 is shown in brown and other proteins in red. Scale bars = 100 μm (A–C, E–G, and I–K) and 200 μm (D, H, and L).
Among the four proteins analyzed, the most conspicuous staining was that of FN1 and POSTN inside and at the periphery of the tumors. Their relative staining intensities varied slightly between the macrometastases, but the staining patterns were strikingly superimposable in all of the samples (Figure 3). VCAN staining, in turn, showed a slightly different (less demarcated) pattern, but localized frequently with POSTN and FN1 strands around tumor cell nests (Figure 3) and at the invasion fronts (see supplemental Figure S3 at http://ajp.amjpathol.org/). PCOL-I staining was visible in thin fibrillar strands (rapidly assembled into mature collagen fibers) as described.34 Or staining appeared intracellularly in PCOL-I-synthesizing fibroblasts organized along the fibrillar strands (Figure 3O). In most of the normal LNs and tumor-free sentinel LNs, POSTN, FN1, VCAN, and PCOL-I staining was visible only in the LN capsules and trabeculae (Figure 3, A, E, I, and M).
As POSTN, FN1, VCAN, and COL-I showed coordinate expression also in breast cancer metastases, we analyzed the expression pattern of the respective proteins in consecutive tissue sections from breast cancer LN micro- and macrometastases (n = 19, ≤2.0 mm; n = 23, >2.0 mm in diameter; as defined in the sixth edition of the TNM classification), as well. POSTN, FN1, VCAN, and PCOL-I colocalized again in extracellular ring and strand structures in all macrometastases and in some of the micrometastases (Figure 3, D, H, L, and P and data not shown), similarly to the melanoma metastases. Two breast cancer metastases of 2.2 and 2.4 mm in size (the latter comprised of incoherent cell clusters) showed similar staining patterns to the ≤2 mm micrometastases. Staining of pSMAD2 showed 17 of the micrometastases to be negative and 2 faintly positive. Of the macrometastases, in turn, the majority (12 of 16) showed moderate to strong pSMAD2 staining. The four pSMAD2-negative macrometastases imply that, in breast cancer LN metastases, some pathway other than the SMAD2 signaling pathway may also be associated with the production of the four fibrillar proteins. We further extended the analyses of FN1, POSTN, VCAN, and COL-I to distant melanoma and breast cancer metastases in other organs (5 liver and 14 lung metastases). Again, these four proteins colocalized in the fibrillar structures surrounding the tumor cell nests (see supplemental Figure S4 at http://ajp.amjpathol.org/).
As the monoclonal FN1 antibody used for the paraffin sections recognizes both pFN and cFN, and because a substantial fraction of FN1 in tissues may derive from plasma,35 we used a monoclonal antibody specifically detecting cFN (the extra domain A of FN1) to stain frozen sections from three melanoma and three breast cancer metastases. The staining of cFN and POSTN showed superimposed localization, once again: in the fibrillar ECM structures (shown for breast cancer in Figure 5, A–D). Interestingly, strong cFN and POSTN as well as COL-I staining was also visible in the intimal layer of immature tumor blood vessels, “colocalizing” with the endothelial cell markers von Willebrand factor and CD31 (Figure 5, J–N). In addition, strong FN1 staining was also detected in the mural cell layers of mature tumor arteries (Figure 5D), in agreement with earlier studies.36 Blood vessels within normal LNs also showed moderate to strong staining for COL-I, but no detectable staining for POSTN (except for the adventitia) and only faint staining for cFN (Figure 5, E–I and data not shown). Additionally, in normal LNs, faint cFN and COL-I staining appeared in the fibroblastic reticular cell network (having a fine mesh size; reactive for ER-TR7 antibody; data not shown), which regulates lymphocyte entry, migration, and territoriality in LNs (37 and references therein).
Figure 5.

Immunohistochemical staining of POSTN, cFN, COL-I, von Willebrand factor, and CD31 in melanoma and breast cancer LN metastases. A–D: POSTN (A and C) and cFN (B and D) staining in breast cancer metastases (Scale bars = 400 μm). E–N: POSTN, cFN, COL-I, von Willebrand factor, and CD31 staining in control LNs (E–I) and melanoma metastases (J–N; panels L and N are from the same block as J, K, and M but not consecutive sections) (Scale bars = 100 μm). A–N: cFN is shown in brown and other proteins in red.
To unequivocally confirm the colocalizations seen in IHC and to gain a deeper conception of the organization of the FN1, POSTN, and COL-I fibrillar structures in native tissues, we studied these proteins in nonfixed metastases by confocal immunofluorescence microscopy. By triple-staining, we found FN1, POSTN, and COL-I again to colocalize (to be intertwined in the same fibrillar structures) and assemble in intricate three-dimensional fibrillar networks (Figure 6, A–C) surrounding melanoma cell nests (Figure 7A). As our IHC analyses have revealed TNC to colocalize with FN1 in invasive melanomas,34 we additionally studied the localization of TNC in macrometastases by confocal microscopy (although its expression was only increased <2-fold) and found it to colocalize with FN1 and POSTN in the fibrillar networks (see supplemental Figure S5 at http://ajp.amjpathol.org/). On closer examination of the organization of the fibrillar structures, FN1 and COL-I appeared to form straighter and more extended fibrils than did POSTN (Figure 7B, and supplemental Movies S1 and S2 at http://ajp.amjpathol.org/).
Figure 6.

Confocal fluorescence imaging and three-dimensional reconstructions of a melanoma metastasis section stained for FN1, POSTN, and COL-I. A and B: Triple staining of FN1, POSTN, and COL-I showing their colocalization in an abundant fibrillar network (A) and in a crossing point of the network (B). C: Double staining of FN1 and POSTN showing their colocalization in a ring-like fibrillar structure. Scale bars = 100 μm.
Figure 7.

Channel-like organization of the fibrillar protein structures around melanoma cell nests and interactions of FN1, POSTN, and COL-I in vivo. A: Confocal image of a melanoma metastasis triple-stained for FN1, POSTN, and MLANA, showing organization of the fibrillar tubular structures formed by FN1 and POSTN around melanoma cell nests. B: Confocal images of the spatial organizations/interactions of FN1, POSTN, and COL-I in the fibrillar strand structures. Scale bars = 100 μm.
Periostin, Fibronectin, and Collagen-I Bind Directly to Each Other in Vitro
The colocalization of POSTN, FN1, VCAN, and COL-I prompted us to test whether these proteins could directly interact. We thus investigated by surface plasmon resonance the binding of full-length POSTN, cFN, pFN, and COL-I to each other. Unfortunately, purified VCAN was not commercially available for analysis. All of the other proteins were studied both as soluble (injected analyte) and as immobilized onto the chip surface. Specific, high affinity binding was apparent between all of the proteins (see supplemental Figure S6 at http://ajp.amjpathol.org/, and data not shown) in both situations except for cFN, which as an analyte showed no significant binding to POSTN, possibly due to the fact that soluble cFN has a compact conformation.38 The POSTN fragment (containing amino acids 22 to 669 of human POSTN, lacking the signal sequence and the C-terminus), in turn, did not bind specifically to any of these proteins. We also tested the binding of TNC and SPP1 to these proteins, but found no direct interactions. Consistent with this, nor did Ingham et al39 find full-length TNC and FN1 to interact directly in solution.
Full-Length Periostin Has an Anti-Adhesive Effect on Tumor and Stromal Cells
As POSTN is thought to act as a cell adhesion molecule, we wanted to explore its adhesive properties relative to other adhesion molecules on primary human melanoma cells, melanoma cell lines (WM793, WM239, and SKMEL28), breast cancer cells (MDA-MB-231), HMVECs, and also on human fibroblasts, which appeared to be the main producers of POSTN. FN1 (both cFN and pFN) and COL-I supported well the adhesion of all of the cell types, as expected. But, surprisingly, we found full-length POSTN uniformly to have an anti-adhesive effect resulting in even lower adhesion than that of the bovine serum albumin serving as a control for nonspecific binding (Figure 8, A and B and data not shown). In all cases, the anti-adhesive action of POSTN was much stronger than that of TNC, the effect of which, moreover, was cell type–dependent (Figure 8A). In contrast, the truncated form of POSTN supported the adhesion of WM793 cells and HMVECs to about 50% and particularly that of the fibroblasts to about 80% of that obtained with FN1. The truncated form failed, however, to support the adhesion of primary human melanoma cells, of the WM239 melanoma cell line, and of the MBA-MD-231 breast cancer cell line (Figure 8, A and B).
Figure 8.

The ability of various ECM molecules to support adhesion of different types of cells. A and B: Effect of ECM protein coatings on adhesion of primary fibroblasts, HMVECs, and melanoma cells (A), and the cell lines WM793, WM239, and MBA-MD-231 (B). C: Adhesion of WM239 cells expressing control, POSTN, and FN1 shRNAs on uncoated tissue culture plates.
To further test the effect of POSTN on cell adhesion, we generated transfectants of WM793, WM239, HMVECs, and primary fibroblasts expressing lentiviral vectors encoding POSTN, FN1, or control shRNAs. Down-regulation of POSTN and FN1 was confirmed by Western blotting (see supplemental Figure S7 at http://ajp.amjpathol.org/). Down-regulation of POSTN resulted in increased adhesion, whereas down-regulation of FN1 expectedly inhibited adhesion (shown for WM239, Figure 8C).
We further studied the effects of POSTN applied in a coating together with other proteins (at ratios from 1:4 to 4:1) on cell adhesion. When coated at a 1:1 (or higher) ratio with FN1 (or with COL-I, data not shown), POSTN did not block adhesion of WM793 cells or of HMVECs, but induced a more elongated, spindle-like phenotype (see supplemental Figure S8 at http://ajp.amjpathol.org/). During extended culture in serum-free medium, the cells also detached earlier from the protein mixture than from the mere FN1 or COL-I coating, suggesting that POSTN reduces the cell-binding affinities to these adhesion proteins. In addition, soluble POSTN (and TNC) was found to reduce adhesion of melanoma cells to FN1-coated plates (data not shown).
Full-Length Periostin Combined with Fibronectin or Collagen-I Increases Migration of Melanoma Cells, Endothelial Cells, and Fibroblasts
The spindle-like phenotype induced by the mixture of POSTN and FN1 or COL-I on WM793 melanoma cells and HMVECs prompted us to investigate the effect of POSTN combined with FN1 or COL-I on cell migration. Migratory activity was assayed by use of cell culture inserts coated with POSTN and cFN or POSTN and COL-I (1:1) and cFN or COL-I each alone. WM793 cells exhibited a 5.3-fold higher migration through filters coated with cFN and POSTN than with cFN (Figure 9A). When combined with COL-I, POSTN enhanced migration 12.1-fold, as compared with COL-I (which supported migration less efficiently than did FN1) (Figure 9A). Experiments with HMVECs (Figure 9B) and human fibroblasts (data not shown) gave similar results. In comparison, the POSTN fragment coated together with cFN or COL-I showed no enhancement of cell migration compared with that of cFN or COL-I alone (Figure 9A). As a further support of POSTN promoting migration, we found the down-regulation of POSTN by shRNA expression to inhibit the migratory activity of the primary fibroblasts by 30% (two-tailed t-test, P = 0.049). Similar inhibition was obtained with POSTN shRNA expressing WM239 cells.
Figure 9.
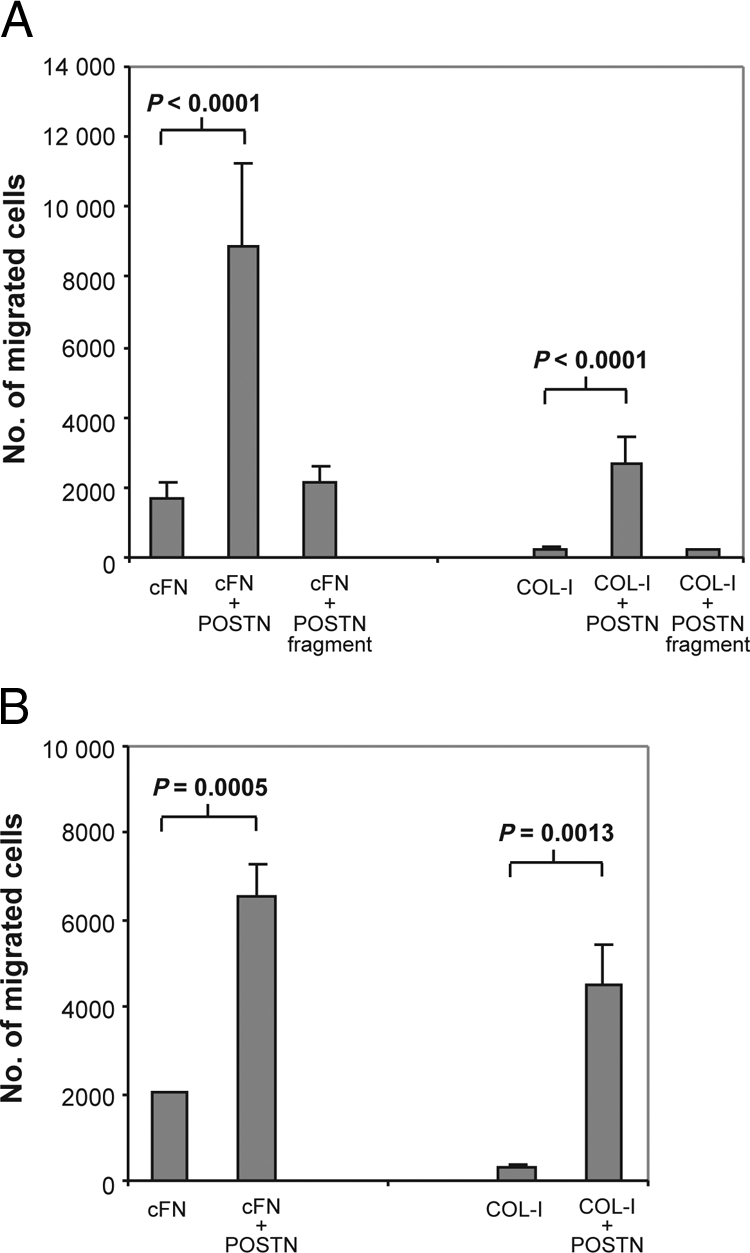
POSTN combined with FN1 or COL-I increases cell migration. A: Effect of coatings of cFN and COL-I alone and combined with full-length POSTN and POSTN fragment on migration of WM793 melanoma cells through transwell inserts. B: Effect of coatings of cFN and COL-I alone and combined with full-length POSTN on migration of HMVECs through transwell inserts.
Fibronectin, but Not Periostin, Is Required for Cell Growth
We then tested the effects of POSTN and FN1 on WM793 melanoma cell proliferation in two-dimensional culture and invasion in three-dimensional Matrigel. Neither POSTN nor cFN, alone or together, promoted proliferation or protease-dependent invasive activity (data not shown). However, experiments with WM793, WM239, HMVECs, and primary fibroblasts transfected with POSTN and FN1 shRNA expression vectors revealed that FN1, but not POSTN, is needed for cell growth in the presence of serum growth factors (shown for the primary embryonic fibroblasts, Figure 10A).
Figure 10.

FN1 is required for cell growth. A: Primary human embryonic fibroblasts (4 × 104) transduced with lentiviral particles encoding control, POSTN, or FN1 shRNAs were grown for 20 days. Mean values of the cell number in two parallel cultures are shown. Variation between the two cultures was <12% in all cases. B: Inhibition of growth of WM793 cell and primary fibroblast (1:1) co-cultures by FN1 L8 antibody.
To enable studies on the effects of COL-I, FN1, and POSTN on cell growth in a more physiological three-dimensional cell culture setting, we exploited Puramatrix (BD Biosciences; 0.25% to 0.5% final concentration) supplemented or not with these proteins. None of the proteins alone was again sufficient to promote growth (data not shown). However, in a WM793 melanoma cell and primary human embryonic fibroblast (1:1) co-culture (supporting growth in serum-free medium; Melanocyte Growth Medium M2, Promocell, Heidelberg, Germany), we found FN1 to be essential for the growth of both cell types (Figure 10B), by incubating the cells with a monoclonal FN1-antibody L8 (60 μg/ml, Santa Cruz) preventing FN1 polymerization.
Discussion
The factors and mechanisms involved in the metastatic outgrowth of micrometastases, the rate-limiting step in cancer progression, have remained largely unexplored. Here, we sought to identify genes associated with the growth of metastases by screening first the gene expression profiles of melanoma LN micro- and macrometastases, and then studying melanoma and breast cancer metastases from various organs and preliminarily colon cancer liver metastases with similar results (unpublished data). We especially wanted to see whether there exist changes common to the growth of all macrometastases, which naturally would be of immense importance for therapeutic approaches to cancer. In melanoma metastases, we found no uniform overexpression of any growth factor or cytokine, most likely due to the heterogeneity of melanomas, since individual increases in differing growth factors and cytokines were indeed observable. However, SPP1, a multifunctional protein in tumor growth,20,21 was commonly overexpressed, providing an attractive target for therapeutic interventions in metastatic growth. Another factor of interest may be GDF15, which is already up-regulated in melanoma cells at an earlier phase of melanoma progression (22, 23, and our unpublished data).
Most interestingly, our gene expression analyses revealed a common up-regulation of TGFβ signaling and coordinate overexpression of the TGFβ-inducible ECM protein-encoding genes COL-I, FN1, POSTN, and VCAN (28–30 and our unpublished data). We further found these four proteins to form together elaborate fibrillar networks ensheathing the melanoma and breast cancer cell nests/chains in all LN, liver, and lung macrometastases. TNC also colocalized in these fibrillar structures, in concordance with our previous findings.34 The confocal microscopic movies revealed that COL-I and FN1 fibers were always the most intimately associated and aligned, forming together the core structure around which the POSTN fibrils appeared to deposit. As to the possible mechanisms of the assembly of these structures, it is known that COL-I can self-assemble into fibrils in vitro, but in vivo, FN1 is required for the polymerization.40 In addition, POSTN may regulate COL-I fibrillogenesis in vivo.41 FN1 polymerization, in turn, is dependent on direct interactions with integrin receptors on the cell surface (reviewed in 40). Considering the complicated spatial organization of the different protein fibrils in the meshwork structures, it is difficult to envisage any mechanism for the assembly other than the copolymerization of at least the core proteins (COL-I and FN1). The ability of COL-I, FN1, and POSTN proteins to directly interact with each other (and to bind themselves), as shown by our surface plasmon resonance experiments, is likely to be of key importance for the assembly and polymerization processes.
What is then the functional significance of the fibrillar protein structures formed? FN142,43 and COL-I (40 and references therein) are both well-known cell adhesion proteins regulating many processes such as cell survival, proliferation, and migration. POSTN has similarly been thought to function as an adhesion molecule,44 but surprisingly we found full-length POSTN to be highly anti-adhesive for all of the cell types tested. One explanation for this discrepancy may be that most researchers appear to have used shorter forms of POSTN or its C-terminus-lacking fragment, which we, as well, found to support the adhesion of some cells. It is possible that the shorter forms/fragments of POSTN have a conformation different from that of the full-length protein, which may have its FAS domains inaccessible. However, we found that even the POSTN fragment (comprising the four FAS-adhesion domains) was much inferior to FN1 in promoting adhesion, fitting in with the FAS domains’ being only weak homophilic adhesion domains.45 Further, it is notable that our full-length recombinant POSTN was produced in mammalian cells, whereas others have used recombinant proteins produced in insect or bacterial cells; that may result in different posttranslational modifications and protein conformations. In one study,46 however, full-length POSTN produced in insect cells had an anti-adhesive effect on fibroblasts, in line with our results.
This anti-adhesive property of full-length POSTN changes the current view on the functional roles of POSTN. One interesting possibility is that POSTN, if acting alone, has a dual function in the regulation of cell adhesion—acting either as an anti-adhesive or as a weak adhesive molecule, depending on the alternative splicing of its mRNA. Our data strongly suggest, however, that POSTN function should be examined together with that of other ECM molecules, particularly with COL-I and FN1, which formed, together with POSTN, intricate networks in the tumors. When POSTN was combined with FN1 or COL-I, it modulated their functions, inducing a more spindle-like phenotype on melanoma and endothelial cells and causing a significant increase in migration of cells compared with FN1 or COL-I alone. These data indicate that these ECM proteins can induce an epithelial-to-mesenchymal transition–like morphological change. Hence, the deposition of POSTN and FN1/COL-I also in vivo, especially at tumor invasion fronts, may result in an epithelial-to-mesenchymal transition–like change, making it difficult to differentiate tumor cells from fibroblasts. The fact that the POSTN fragment failed to induce any morphological change or to increase cell migration when coated together with FN1 or COL-I suggests that the anti-adhesive function of full-length POSTN is important for the migratory phenotype. It is plausible that POSTN prevents too tight a cell attachment to FN1 and COL-I, thus enabling the efficient migration that requires sequential attachment and detachment. That POSTN exerts a pro-migratory effect was also supported by our shRNA expression experiments. Similarly to POSTN, VCAN is known to have an anti-adhesive effect and may thus contribute to regulation of cell migration, as well.47
It should also be noted that while most studies have suggested that POSTN promotes tumor cell motility, invasion, and growth, there exist some opposing results.48,49 The reason for this discrepancy remains to be seen. Conceivably, the overexpression of POSTN in cancer cells may show variable effects in different microenvironments, depending on the relative abundance of the other ECM components. Further, it remains interesting to see the importance of the expression of the six POSTN isoforms identified (also reported in a panel of cDNA libraries from normal, fetal, and cancerous kidneys by Castronovo et al50) in the regulation of tumor progression.
Debate has arisen as to which cell types in tumors produce POSTN. A complicating thing is that we still understand very little about the regulation of POSTN expression in vivo and about the factors needed for the maintenance of POSTN expression in tissue culture (as previously discussed in 51). In normal tissues, POSTN is predominantly expressed by mesenchymal stromal cells in connective tissue subjected to constant mechanical stress, including heart valves, tendons, and periodontal ligaments, where its expression may be regulated by mechanical tension and growth factors such as TGFβ (52 and references therein). Analogously in cancer tissues, pressure of the growing tumor mass against the surrounding stroma and increased TGFβ signaling (triggered, for instance, by tumor cells) may be the main inducers of POSTN expression in the cancer-associated fibroblasts. Consistent with this, TGFβ is known to be produced by melanoma cells (reviewed in 53), and TGFβ-transfected melanoma cell lines have been shown to stimulate stromal fibroblasts to produce ECM proteins (collagens and FN1).54 Further, we found the TGFβ/SMAD2 signaling pathway to be activated in all human melanoma macrometastases, both in the melanoma cells (at the tumor edge) and in the surrounding fibroblasts. However, our preliminary RT-PCR analyses show that not all melanoma cells isolated from LN metastases express TGFβ (see also 54), and that primary fibroblasts can even express much higher levels of TGFβ (data not shown), implying that also other factors may initially activate fibroblasts, which are then further stimulated through an autocrine TGFβ loop.
Although fibroblasts appear to be the main producers of POSTN, our RT-PCR analyses of primary tumor cells and cell lines, as well as analyses by others,55 show that POSTN can be produced by some melanoma cells, as well. While we found normal melanocytes and primary melanoma cells to express only low levels of one splice-variant of POSTN, some melanoma cells metastasized to LNs appeared to have acquired the ability to express elevated levels of multiple splice-variants. These cells apparently thereby become less dependent on fibroblasts. However, it appears that in most cases, fibroblasts and the interplay between melanoma cells and fibroblasts play important roles in metastatic growth.
The role of cancer-associated fibroblasts in metastasis is still little studied at the molecular level. In primary tumors, fibroblasts are known to affect several aspects of tumor progression by producing growth factors, chemokines, and proteases and by remodeling ECM (reviewed in 56–58). In vitro, fibroblasts have even been shown to lead the invasion of cancer cells (59 and our unpublished data). The present data show that similar mechanisms may operate to variable extents in the expansive growth of metastases, as well. However, the most important factor, common to the metastatic growth of both melanoma and breast cancer, appears to be the de novo synthesis of specific ECM molecules and the generation of a new microenvironment: a fibrillar protein network likely to regulate many cellular responses and provide a scaffold for the tumor cells to settle on and to efficiently migrate along and spread. Our finding of the requirement of FN1 for tumor and stromal cell growth, as revealed by FN1 shRNA expression and FN1 L8 antibody experiments, is in line with studies with FN1-deficient cells showing that FN1 fibril formation promotes adhesion dependent growth.60
An additional important means by which COL-I, FN1, POSTN, and VCAN can enhance metastasis progression is through promoting the formation of new blood vessels (angiogenesis). COL-I, FN1, and VCAN have been associated with the formation of vascular tumor stroma in primary and metastatic carcinomas of the breast.61 Further, FN1 can enhance vascular endothelial growth factor expression, endothelial cell proliferation and migration, and tubulogenesis.62,63 POSTN has likewise been reported to stimulate vascular cell migration and tubulogenesis64,65 and endothelial cell proliferation by increasing the expression of the vascular endothelial growth factor receptor KDR.66
Interestingly, by IHC we found COL-I, cFN (containing the extra domain A), and POSTN to “colocalize” in the metastases along with endothelial cell markers (von Willebrand factor and CD31) in the forming or newly formed blood vessels without or with a lumen (with COL-I, cFN, and POSTN being likely deposited underneath the endothelial cells). Significantly, little cFN and no POSTN deposition was detectable in the blood vessels of normal LNs. The extra domain A of FN1 has recently been reported to be a vascular marker of liver and lung metastases, as well.67 It is also of note here that FN1 is essential for blood vessel morphogenesis during embryonic development.36 We further found POSTN combined with FN1 or COL-I efficiently to support the migration of endothelial cells and FN1 polymerization to regulate tubulogenesis in fibroblast-endothelial cell co-culture assays (unpublished data). Combined all these data (by our group and others) give us a good reason to hypothesize that fibroblasts, by producing COL-I, cFN, and POSTN, enhance and guide the migration of endothelial cells (like migration of tumor cells) and additionally provide structural components for blood vessel-wall formation in tumors, before endothelization (this study and our unpublished confocal microscopy data). Recently, fibroblasts have also been shown to be essential for carcinoma cell-induced angiogenesis/tubulogenesis in a three-dimensional collagen gel assay, largely through TGFβ-mediated stimulation of vascular endothelial growth factor release.68 Thus, fibroblasts appear to play an important role by providing both soluble growth factors and structural cues and components for efficient tumor angiogenesis, which is essential for macrometastatic growth.
In conclusion, in a search for factors important for the outgrowth of melanoma and breast cancer LN metastases, we found the TGFβ/SMAD2 signaling pathway to be generally activated. Further, we found COL-I, FN1, POSTN, and VCAN—TGFβ target genes—to be consistently up-regulated during metastatic growth (as a result of interplay between cancer cells and fibroblasts), and the encoded proteins to together form elaborate fibrillar networks around tumor cell nests. The same was true for the growth of melanoma and breast cancer liver and lung metastases. Thus, in successful metastasis, the critical question may not be the tumor cells’ adaptability to the new environment but their ability to induce the formation of a new ECM and a milieu conducive for growth. Modeling of these multimolecular structures (in which the relative amounts of the individual components may vary) for functional significance studies in experimental animals is challenging (requiring four regulatable knockouts/knockdowns in specific cell types). Another major problem is that we do not yet know how well the current experimental models of metastasis reflect the behavior of actual human cancers. Our in vitro analyses suggest that these fibrillar protein structures regulate adhesion and migration of melanoma cells, fibroblasts, and endothelial cells, through the anti-adhesive effect of POSTN, which modulated the functions of FN1 and COL-I. In addition, FN1 was found to be required for tumor and stromal cell growth. Further, cFN and POSTN were specifically over-expressed in the newly forming/formed tumor blood vessels. These findings may open up various kinds of new possibilities effectively to treat metastatic disease. Targeting TGFβ type I receptor could be one possibility. Indeed, we have found inhibition of the TGFß type I receptor to efficiently block the invasive growth of melanoma cells co-cultured with fibroblasts in vitro (our unpublished results) which prompts testing such treatments in the therapy of patients with established metastases. However, because TGFβ is implicated in many processes, acting both as a tumor suppressor and tumor promoter, a better strategy may be targeting the metastasis-related ECM proteins—cFN and POSTN in particular—and thereby interfering with key processes of metastasis, tumor/stromal cell migration/spread and growth and tumor angiogenesis.
Acknowledgments
We thank Leena Saikko, Merja Haukka, and Ulla Kiiski for technical assistance. We also thank Dr. Pertti Panula for confocal microscopy facilities, Tomas Strandin for the help with Biacore analyses, and Dr. Adam Riker and Dr. Steven Enkemann for sharing their microarray data.
Footnotes
Address reprint requests to Erkki Hölttä, M.D., Ph.D., Department of Pathology, Haartman Institute, P.O. Box 21 (Haartmaninkatu 3), FI-00014 Helsinki, Finland. E-mail: erkki.holtta@helsinki.fi.
Supported by the Finnish Cancer Organizations, the Academy of Finland, and Helsinki University Central Hospital Research Funds. J.S. is a predoctoral fellow of the Helsinki Graduate School in Biotechnology and Molecular Biology, and P.N. of the Helsinki Biomedical Graduate School.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12:895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Psaila B, Kaplan RN, Port ER, Lyden D. Priming the ‘soil’ for breast cancer metastasis: the pre-metastatic niche. Breast Dis. 2006;26:65–74. doi: 10.3233/bd-2007-26106. [DOI] [PubMed] [Google Scholar]
- Hirakawa S. From tumor lymphangiogenesis to lymphvascular niche. Cancer Sci. 2009;100:983–989. doi: 10.1111/j.1349-7006.2009.01142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- Ma L, Weinberg RA. Micromanagers of malignancy: role of microRNAs in regulating metastasis. Trends Genet. 2008;24:448–456. doi: 10.1016/j.tig.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2004;4:448–456. doi: 10.1038/nrc1370. [DOI] [PubMed] [Google Scholar]
- Balch CM, Buzaid AC, Soong SJ, Atkins MB, Cascinelli N, Coit DG, Fleming ID, Gershenwald JE, Houghton A, Jr, Kirkwood JM, McMasters KM, Mihm MF, Morton DL, Reintgen DS, Ross MI, Sober A, Thompson JA, Thompson JF. Final version of the American Joint Committee on Cancer staging system for cutaneous melanoma. J Clin Oncol. 2001;19:3635–3648. doi: 10.1200/JCO.2001.19.16.3635. [DOI] [PubMed] [Google Scholar]
- Carlson GW, Murray DR, Lyles RH, Staley CA, Hestley A, Cohen C. The amount of metastatic melanoma in a sentinel lymph node: does it have prognostic significance? Ann Surg Oncol. 2003;10:575–581. doi: 10.1245/aso.2003.03.054. [DOI] [PubMed] [Google Scholar]
- Ranieri JM, Wagner JD, Azuaje R, Davidson D, Wenck S, Fyffe J, Coleman JJ., 3rd Prognostic importance of lymph node tumor burden in melanoma patients staged by sentinel node biopsy. Ann Surg Oncol. 2002;9:975–981. doi: 10.1007/BF02574515. [DOI] [PubMed] [Google Scholar]
- Soikkeli J, Lukk M, Nummela P, Virolainen S, Jahkola T, Katainen R, Harju L, Ukkonen E, Saksela O, Holtta E. Systematic search for the best gene expression markers for melanoma micrometastasis detection. J Pathol. 2007;213:180–189. doi: 10.1002/path.2229. [DOI] [PubMed] [Google Scholar]
- Sobin LH, Wittekind C. TNM Classification of Malignant Tumours, 6th Edition. New Jersey: John Wiley & Sons,; 2002:pp. 1–239. [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA: 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Jones WD, Jensen RV, Harris SC, Perkins RG, Goodsaid FM, Guo L, Croner LJ, Boysen C, Fang H, Qian F, Amur S, Bao W, Barbacioru CC, Bertholet V, Cao XM, Chu TM, Collins PJ, Fan XH, Frueh FW, Fuscoe JC, Guo X, Han J, Herman D, Hong H, Kawasaki ES, Li QZ, Luo Y, Ma Y, Mei N, Peterson RL, Puri RK, Shippy R, Su Z, Sun YA, Sun H, Thorn B, Turpaz Y, Wang C, Wang SJ, Warrington JA, Willey JC, Wu J, Xie Q, Zhang L, Zhang L, Zhong S, Wolfinger RD, Tong W. The balance of reproducibility, sensitivity, and specificity of lists of differentially expressed genes in microarray studies. BMC Bioinformatics. 2008;9 Suppl 9:S10. doi: 10.1186/1471-2105-9-S9-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alanko T, Rosenberg M, Saksela O. FGF expression allows nevus cells to survive in three-dimensional collagen gel under conditions that induce apoptosis in normal human melanocytes. J Invest Dermatol. 1999;113:111–116. doi: 10.1046/j.1523-1747.1999.00636.x. [DOI] [PubMed] [Google Scholar]
- Ravanko K, Jarvinen K, Helin J, Kalkkinen N, Holtta E. Cysteine cathepsins are central contributors of invasion by cultured adenosylmethionine decarboxylase-transformed rodent fibroblasts. Cancer Res. 2004;64:8831–8838. doi: 10.1158/0008-5472.CAN-03-2993. [DOI] [PubMed] [Google Scholar]
- Rittling SR, Chambers AF. Role of osteopontin in tumour progression. Br J Cancer. 2004;90:1877–1881. doi: 10.1038/sj.bjc.6601839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister SS, Gifford AM, Greiner AL, Kelleher SP, Saelzler MP, Ince TA, Reinhardt F, Harris LN, Hylander BL, Repasky EA, Weinberg RA. Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell. 2008;133:994–1005. doi: 10.1016/j.cell.2008.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talantov D, Mazumder A, Yu JX, Briggs T, Jiang Y, Backus J, Atkins D, Wang Y. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin Cancer Res. 2005;11:7234–7242. doi: 10.1158/1078-0432.CCR-05-0683. [DOI] [PubMed] [Google Scholar]
- Riker AI, Enkemann SA, Fodstad O, Liu S, Ren S, Morris C, Xi Y, Howell P, Metge B, Samant RS, Shevde LA, Li W, Eschrich S, Daud A, Ju J, Matta J. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med Genomics. 2008;1:13. doi: 10.1186/1755-8794-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J, Smith A. Capturing pluripotency. Cell. 2008;132:532–536. doi: 10.1016/j.cell.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabierowski SE, Herlyn M. Melanoma stem cells: the dark seed of melanoma. J Clin Oncol. 2008;26:2890–2894. doi: 10.1200/JCO.2007.15.5465. [DOI] [PubMed] [Google Scholar]
- Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, Massague J. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133:66–77. doi: 10.1016/j.cell.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Dai DL, Martinka M, Su M, Zhang Y, Campos EI, Dorocicz I, Tang L, Huntsman D, Nelson C, Ho V, Li G. Osteopontin expression correlates with melanoma invasion. J Invest Dermatol. 2005;124:1044–1052. doi: 10.1111/j.0022-202X.2005.23680.x. [DOI] [PubMed] [Google Scholar]
- Verrecchia F, Chu ML, Mauviel A. Identification of novel TGF-beta /Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J Biol Chem. 2001;276:17058–17062. doi: 10.1074/jbc.M100754200. [DOI] [PubMed] [Google Scholar]
- Horiuchi K, Amizuka N, Takeshita S, Takamatsu H, Katsuura M, Ozawa H, Toyama Y, Bonewald LF, Kudo A. Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor beta. J Bone Miner Res. 1999;14:1239–1249. doi: 10.1359/jbmr.1999.14.7.1239. [DOI] [PubMed] [Google Scholar]
- Ignotz RA, Endo T, Massague J. Regulation of fibronectin and type I collagen mRNA levels by transforming growth factor-beta. J Biol Chem. 1987;262:6443–6446. [PubMed] [Google Scholar]
- Hocevar BA, Brown TL, Howe PH. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent Smad4-independent pathway. EMBO J. 1999;18:1345–1356. doi: 10.1093/emboj/18.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA. Models, mechanisms, and clinical evidence for cancer dormancy. Nat Rev Cancer. 2007;7:834–846. doi: 10.1038/nrc2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- Kaariainen E, Nummela P, Soikkeli J, Yin M, Lukk M, Jahkola T, Virolainen S, Ora A, Ukkonen E, Saksela O, Holtta E. Switch to an invasive growth phase in melanoma is associated with tenascin-C, fibronectin, and procollagen-I forming specific channel structures for invasion. J Pathol. 2006;210:181–191. doi: 10.1002/path.2045. [DOI] [PubMed] [Google Scholar]
- Moretti FA, Chauhan AK, Iaconcig A, Porro F, Baralle FE, Muro AF. A major fraction of fibronectin present in the extracellular matrix of tissues is plasma-derived. J Biol Chem. 2007;282:28057–28062. doi: 10.1074/jbc.M611315200. [DOI] [PubMed] [Google Scholar]
- Astrof S, Hynes RO. Fibronectins in vascular morphogenesis. Angiogenesis. 2009;12:165–175. doi: 10.1007/s10456-009-9136-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajenoff M, Egen JG, Koo LY, Laugier JP, Brau F, Glaichenhaus N, Germain RN. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity. 2006;25:989–1001. doi: 10.1016/j.immuni.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzbicka-Patynowski I, Schwarzbauer JE. The ins and outs of fibronectin matrix assembly. J Cell Sci. 2003;116:3269–3276. doi: 10.1242/jcs.00670. [DOI] [PubMed] [Google Scholar]
- Ingham KC, Brew SA, Erickson HP. Localization of a cryptic binding site for tenascin on fibronectin. J Biol Chem. 2004;279:28132–28135. doi: 10.1074/jbc.M312785200. [DOI] [PubMed] [Google Scholar]
- Kadler KE, Hill A, Canty-Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr Opin Cell Biol. 2008;20:495–501. doi: 10.1016/j.ceb.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris RA, Damon B, Mironov V, Kasyanov V, Ramamurthi A, Moreno-Rodriguez R, Trusk T, Potts JD, Goodwin RL, Davis J, Hoffman S, Wen X, Sugi Y, Kern CB, Mjaatvedt CH, Turner DK, Oka T, Conway SJ, Molkentin JD, Forgacs G, Markwald RR. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J Cell Biochem. 2007;101:695–711. doi: 10.1002/jcb.21224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci. 2002;115:3861–3863. doi: 10.1242/jcs.00059. [DOI] [PubMed] [Google Scholar]
- Hynes R. Molecular biology of fibronectin. Annu Rev Cell Biol. 1985;1:67–90. doi: 10.1146/annurev.cb.01.110185.000435. [DOI] [PubMed] [Google Scholar]
- Kudo Y, Siriwardena BS, Hatano H, Ogawa I, Takata T. Periostin: novel diagnostic and therapeutic target for cancer. Histol Histopathol. 2007;22:1167–1174. doi: 10.14670/HH-22.1167. [DOI] [PubMed] [Google Scholar]
- Clout NJ, Tisi D, Hohenester E. Novel fold revealed by the structure of a FAS1 domain pair from the insect cell adhesion molecule fasciclin I. Structure. 2003;11:197–203. doi: 10.1016/s0969-2126(03)00002-9. [DOI] [PubMed] [Google Scholar]
- Katsuragi N, Morishita R, Nakamura N, Ochiai T, Taniyama Y, Hasegawa Y, Kawashima K, Kaneda Y, Ogihara T, Sugimura K. Periostin as a novel factor responsible for ventricular dilation. Circulation. 2004;110:1806–1813. doi: 10.1161/01.CIR.0000142607.33398.54. [DOI] [PubMed] [Google Scholar]
- Wight TN. Versican: a versatile extracellular matrix proteoglycan in cell biology. Curr Opin Cell Biol. 2002;14:617–623. doi: 10.1016/s0955-0674(02)00375-7. [DOI] [PubMed] [Google Scholar]
- Kim CJ, Yoshioka N, Tambe Y, Kushima R, Okada Y, Inoue H. Periostin is down-regulated in high grade human bladder cancers and suppresses in vitro cell invasiveness and in vivo metastasis of cancer cells. Int J Cancer. 2005;117:51–58. doi: 10.1002/ijc.21120. [DOI] [PubMed] [Google Scholar]
- Yoshioka N, Fuji S, Shimakage M, Kodama K, Hakura A, Yutsudo M, Inoue H, Nojima H. Suppression of anchorage-independent growth of human cancer cell lines by the TRIF52/periostin/OSF-2 gene. Exp Cell Res. 2002;279:91–99. doi: 10.1006/excr.2002.5590. [DOI] [PubMed] [Google Scholar]
- Castronovo V, Waltregny D, Kischel P, Roesli C, Elia G, Rybak JN, Neri D. A chemical proteomics approach for the identification of accessible antigens expressed in human kidney cancer. Mol Cell Proteomics. 2006;5:2083–2091. doi: 10.1074/mcp.M600164-MCP200. [DOI] [PubMed] [Google Scholar]
- Tai IT, Dai M, Chen LB. Periostin induction in tumor cell line explants and inhibition of in vitro cell growth by anti-periostin antibodies. Carcinogenesis. 2005;26:908–915. doi: 10.1093/carcin/bgi034. [DOI] [PubMed] [Google Scholar]
- Wilde J, Yokozeki M, Terai K, Kudo A, Moriyama K. The divergent expression of periostin mRNA in the periodontal ligament during experimental tooth movement. Cell Tissue Res. 2003;312:345–351. doi: 10.1007/s00441-002-0664-2. [DOI] [PubMed] [Google Scholar]
- Javelaud D, Alexaki VI, Mauviel A. Transforming growth factor-beta in cutaneous melanoma. Pigment Cell Melanoma Res. 2008;21:123–132. doi: 10.1111/j.1755-148X.2008.00450.x. [DOI] [PubMed] [Google Scholar]
- Berking C, Takemoto R, Schaider H, Showe L, Satyamoorthy K, Robbins P, Herlyn M. Transforming growth factor-beta1 increases survival of human melanoma through stroma remodeling. Cancer Res. 2001;61:8306–8316. [PubMed] [Google Scholar]
- Tilman G, Mattiussi M, Brasseur F, van Baren N, Decottignies A. Human periostin gene expression in normal tissues, tumors and melanoma: evidences for periostin production by both stromal and melanoma cells. Mol Cancer. 2007;6:80. doi: 10.1186/1476-4598-6-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375–379. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- Egeblad M, Littlepage LE, Werb Z. The fibroblastic coconspirator in cancer progression. Cold Spring Harb Symp Quant Biol. 2005;70:383–388. doi: 10.1101/sqb.2005.70.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- Sottile J, Hocking DC, Langenbach KJ. Fibronectin polymerization stimulates cell growth by RGD-dependent and -independent mechanisms. J Cell Sci. 2000;113 Pt 23:4287–4299. doi: 10.1242/jcs.113.23.4287. [DOI] [PubMed] [Google Scholar]
- Brown LF, Guidi AJ, Schnitt SJ, Van De Water L, Iruela-Arispe ML, Yeo TK, Tognazzi K, Dvorak HF. Vascular stroma formation in carcinoma in situ, invasive carcinoma, and metastatic carcinoma of the breast. Clin Cancer Res. 1999;5:1041–1056. [PubMed] [Google Scholar]
- Khan ZA, Chan BM, Uniyal S, Barbin YP, Farhangkhoee H, Chen S, Chakrabarti S. EDB fibronectin and angiogenesis—a novel mechanistic pathway. Angiogenesis. 2005;8:183–196. doi: 10.1007/s10456-005-9017-6. [DOI] [PubMed] [Google Scholar]
- Zhou X, Rowe RG, Hiraoka N, George JP, Wirtz D, Mosher DF, Virtanen I, Chernousov MA, Weiss SJ. Fibronectin fibrillogenesis regulates three-dimensional neovessel formation. Genes Dev. 2008;22:1231–1243. doi: 10.1101/gad.1643308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner V, Wang Q, Conley BA, Friesel RE, Vary CP. Vascular injury induces expression of periostin: implications for vascular cell differentiation and migration. Arterioscler Thromb Vasc Biol. 2005;25:77–83. doi: 10.1161/01.ATV.0000149141.81230.c6. [DOI] [PubMed] [Google Scholar]
- Siriwardena BS, Kudo Y, Ogawa I, Kitagawa M, Kitajima S, Hatano H, Tilakaratne WM, Miyauchi M, Takata T. Periostin is frequently overexpressed and enhances invasion and angiogenesis in oral cancer. Br J Cancer. 2006;95:1396–1403. doi: 10.1038/sj.bjc.6603431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao R, Bao S, Bai X, Blanchette C, Anderson RM, Dang T, Gishizky ML, Marks JR, Wang XF. Acquired expression of periostin by human breast cancers promotes tumor angiogenesis through up-regulation of vascular endothelial growth factor receptor 2 expression. Mol Cell Biol. 2004;24:3992–4003. doi: 10.1128/MCB.24.9.3992-4003.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak JN, Roesli C, Kaspar M, Villa A, Neri D. The extra-domain A of fibronectin is a vascular marker of solid tumors and metastases. Cancer Res. 2007;67:10948–10957. doi: 10.1158/0008-5472.CAN-07-1436. [DOI] [PubMed] [Google Scholar]
- Noma K, Smalley KS, Lioni M, Naomoto Y, Tanaka N, El-Deiry W, King AJ, Nakagawa H, Herlyn M. The essential role of fibroblasts in esophageal squamous cell carcinoma-induced angiogenesis. Gastroenterology. 2008;134:1981–1993. doi: 10.1053/j.gastro.2008.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]