

Abstract
Abnormal TDP-43 aggregation is a prominent feature in the neuropathology of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration. Mutations in TARDBP, the gene encoding TDP-43, cause some cases of ALS. The normal function of TDP-43 remains incompletely understood. To better understand TDP-43 biology, we generated mutant mice carrying a genetrap disruption of Tardbp. Mice homozygous for loss of TDP-43 are not viable. TDP-43 deficient embryos die about day 7.5 of embryonic development thereby demonstrating that TDP-43 protein is essential for normal prenatal development and survival. However, heterozygous Tardbp mutant mice exhibit signs of motor disturbance and muscle weakness. Compared with wild type control littermates, Tardbp+/− animals have significantly decreased forelimb grip strength and display deficits in a standard inverted grid test despite no evidence of pathologic changes in motor neurons. Thus, TDP-43 is essential for viability, and mild reduction in TDP-43 function is sufficient to cause motor deficits without degeneration of motor neurons.
Introduction
Abnormal TDP-43 protein inclusions are part of the neuropathology of amyotrophic lateral sclerosis (ALS) [32], Alzheimer's disease (AD) [2, 30], frontotemporal lobar dementia-ubiquitin type (FTLD-U) [32], and other neuro-degenerative diseases. In these disorders, collectively known as TDP-43 proteinopathies, aggregated ubiquinated TDP-43 is observed in neurons and in glial cells. Recently, a consensus conference recommended that FTLD-U now be referred to as FTLD-TDP [27]. In ALS, TDP-43 deposits occur in the spinal cords of almost all patients, with the notable exception of ALS caused by SOD1 mutations [26, 32, 43]. Some rare cases of familial ALS are caused by mutations in TARDBP, the gene encoding TDP-43 [16, 21, 38, 45, 51]. Thus, abnormal TDP-43 is not merely a marker for ALS, but is causal in some cases. In FTLD-TDP, TDP-43 deposits occur in cases caused by mutations in either progranulin (PGRN) or in vasolin containing protein (VCP). In AD, approximately 20–50% of cases have TDP-43 deposits [2, 27]. Thus, TDP-43 inclusions are clearly implicated in the pathogenesis of multiple neurodegenerative disorders.
TDP-43, a ubiquitously expressed nuclear protein, was originally identified as a 414 amino acid protein that binds the TAR DNA motif of the human immunodeficiency virus type 1 (HIV-1) (for recent review of TDP-43 function see [8]). The DNA binding activity of TDP-43 regulates HIV-1 gene expression in vivo [33]. TDP-43 also acts as a tran-scriptional regulator of the mouse spermatogenesis protein SP-10 encoding gene [1]. Other work showed TDP-43 binds RNA and controls RNA splicing of transcripts from the cystic fibrosis transmembrane conductance regulator (CFTR) [9], survival of motor neuron (SMN) [7], and apolipoprotein A-II (APOA2) [28] genes. TDP-43 also binds a sequence in the 3′ UTR region of neurofilament light chain (NEFL) mRNA and potentially affects the cytoplasmic stability of this message [41]. TDP-43 has both nuclear export and import signals and shuttles between the nucleus and the cytoplasm, although in normal cells its distribution is primarily nuclear [49]. In addition to these trafficking sequences, TDP-43 has two RNA-binding motifs [9] followed by a glycine-rich region near the car-boxyl terminus. Cross-species comparisons of TDP-43 proteins from human, Drosophila, and C. elegans show a high degree of amino acid sequence conservation with the greatest identity found in the RNA recognition motifs (RRMs). Indeed, this sequence conservation translates into functional conservation as proteins from man, worm, and fly all share equivalent RNA and DNA recognition specificities [5]. Therefore, TDP-43 is a multifunctional nuclear protein involved in several different cellular processes, but its functions are not yet fully understood.
In TDP-43 proteinopathies, aggregates of this protein occur in the nucleus, the cytoplasm, or in neurites (for recent reviews of TDP-43 pathology see [15, 23]). In some cells where cytoplasmic inclusions are seen, the nucleus is cleared of TDP-43 [20, 32]. These observations led to the hypothesis that compromised neuronal function may be caused by the depletion of available TDP-43 protein due to sequestration into aggregated protein inclusions. This hypothesis is viable if TDP-43 is indeed required for neuronal function. To test this hypothesis, we generated mice lacking TDP-43 and found that homozygous loss of TDP-43 protein is embryonic lethal while heterozygous loss of TDP-43 causes defects in motor function.
Materials and Methods
Strains and cells
The BayGenomics ES cell line RB030 was obtained from the mutant mouse regional resource center (MMRRC, University of California-Davis). Targeting of the Tardbp gene by the gene trapping vector pGT1lxf was reconfirmed by sequencing of Tardbp-specific RT-PCR products. Also the genetrap insertion site was confirmed by DNA sequencing of the Tardbp genomic region flanking the genetrap. C57BL/6 mice were obtained from Jackson Labs. The TardbpGt(RB030)Byg chimeric mice were generated in the Murine targeted genomics laboratory at the MMRRC from the RB030 ES cells described above. The TardbpGt(RB030)Byg heterozygous mice were backcrossed to C57BL/6 mice five times.
Genotyping
Mice were genotyped using DNA prepared from tail clips for live mice and whole embryo or liver tissue of killed animals. The Tardbp gene was genotyped in a duplexed PCR using primers: TDP intron 2-23129S (AACTGCGCT AGCCCAAGTCTTGAGT), TDP-intron2 23511A (CCCA CCTTCTATTTCCTGCCTCAGC), and pGT_pmsA_501(CCA TCCACTACTCAGTGCAGTGCAGT) yielding a 400-bp product for the wild-type Tardbp allele and a 635-bp product for TardbpGt(RB030)Byg. Single-cell PCR (reviewed in [3]) was conducted using Hotstar PCR reagents (Qiagen) as recommended by the manufacturer to genotype 3.5 day old embryos. Early post-implantation embryos cannot be reliably genotyped as separating maternal tissue from the embryo is very difficult; therefore, genotypes were inferred from Tardbp expression of day 5.5–9.5 embryos. For day 12.5 embryos, embryonic tissue was dissected away from maternal tissue, subjected to DNA extraction using Qiagen DNAeasy reagents, and genotyped using the same scheme as above using Hotstar PCR reagents (Qiagen).
RNA blotting
Adult Tardbp+/− or Tardbp+/+ mice were euthanized and brains dissected. Whole brain was homogenized in Trizol reagent according to manufacturer's recommendations (Invitrogen) for purification of RNA. Poly(A)+ RNA was selected using poly(A) purist MAG reagents as recommended by the manufacturer (Ambion). Four micrograms of poly(A)+ RNA was glyoxylated, resolved, transferred, and analyzed as previously described [34] using a TDP-43 exon 1 specific and beta-geo specific 32P radiolabeled probe.
Immunoblotting
Adult Tardbp+/− or Tardbp+/+ mice were euthanized and internal organs dissected. Tissue samples were homogenized in homogenization buffer (0.1 M Tris, 0.05 M NaCl, 0.001 M EDTA, pH 7.4) by sonication. Protein concentration was measured by Bradford assay as recommended (Biorad). Protein concentrations were normalized and resolved on a precast 4–15% gradient SDS PAGE gel (Biorad), and transferred to PVDF membrane (Biorad). Immunoblots were probed with polyclonal pan-TDP-43 antibody (ProteinTec Group 1:1,500), N-terminal TDP-43 (1:1,000), C-terminal TDP-43 antibody (1:1,000) [20], and lacZ (beta-geo1:1,000.) specific antibody (DSHB).
Grip strength and endurance measurements
Subjects were male and female Tardbp+/− heterozygous mice (n= 18) and control Tardbp+/− littermates (n= 22), ranging in age from 342 to 840 days at the time of testing. The two groups had well-matched age distributions, with a mean age of 536 and 581 days for control and Tardbp+/− groups, respectively. The grip strength of the animals was tested in two ways: the inverted grid test and specific forelimb strength measurements. The inverted grid test was performed by placing each animal onto a metal grid which was held 12 in. above a padded surface and slowly inverted. The amount of time each animal gripped the grid was measured, up to a maximum of 2 min. Each animal was given three trials, with a rest period of 30 min between trials. Forelimb strength was measured using a digital force gage (Extech Instruments) fitted with a wire grid for animals to grip. Each animal was allowed to grip the grid with forepaws only and was gently pulled by the tail to measure maximum grip strength. Data represent the average of three trials for each animal. Data for both experiments were initially analyzed by ANOVA using sex and genotype as grouping factors, with effects of α ≤ 0.05 considered significant. No interactions with sex were identified, and thus the sexes were collapsed into a single group for subsequent analyses. Linear regression analysis was used to examine potential correlations between grip strength measurements and age or body weight.
Gait analysis
A subset of the animals that were tested for grip strength as described above were also tested for gait abnormalities (n= 11 Tardbp+/−; n= 18 controls). Animals were trained to walk down a narrow alley with a white paper floor and a darkened goal box at the opposite end. After they had successfully demonstrated the ability to walk straight down the alley into the goal box, the hindpaws of each animal were dipped in nontoxic paint to produce a record of their footprints on the paper. The distance between sequential footprints on the same side was measured, to obtain the average stride length. The distance between sequential footprints on opposite sides was measured, to obtain the average hindbase width.
Histology and immunohistochemistry
Mouse uteri/embryos on days E5.5, 6.5, 7.5, 8.5, and 12.5 of gestational age were fixed overnight in 10% neutral buffered formalin, paraffin embedded and serially sectioned at 6 μm. Further, brains and spinal cords from eight Tardbp+/− mice and seven normal littermate controls were similarly fixed, embedded, and sectioned. Study mice for histology were as follows: Tardbp+/− mice were 13, 13, 18, 24, 24, 24, 27, and 28 months of age while normal control animals were 13, 16, 18, 24, 24, 25, and 28 months of age. Mice used for histology and IHC had been tested for grip strength, endurance, and gait analysis prior to killing for histology. Sections from the uteri/embryos, brains and spinal cords at all time points were stained with hematoxylin and eosin (H&E) and immunohistochemistry (IHC) was performed on E7.5, 8.5, and 12.5 uteri/embryos as described [26]. Briefly, after antigen retrieval with citrate buffer (Vector Antigen Unmasking Solution H3300) using the Biogenex EZ-Retriever with microwave treatment for 15 min at 99°C, sections were stained with an anti-TDP-43 polyclonal antibody (1:2,000; ProteinTec Rabbit anti-TDP-43, Catalog# PTG10782) followed by a light hematoxylin counterstain. For the brain, spinal cord and muscle of adult Tardbp+/− and wild-type mice, additional antibodies to synaptic, cytoskeletal, and disease proteins were used including: phospho-TDP43 at 1:1,000 with the same microwave antigen retrieval protocol used for the polyclonal TDP43 antibody [31]; synaptophysin (Abcam cat # 8049 mouse monoclonal) at 1:200; MAP 2 1:2,000 (rabbit polyclonal in house antibody); GFAP (Dako cat #Z0334 rabbit polyclonal) at 1:10,000; NFL (in house rabbit polyclonal) at 1:2,000; alpha synuclein (in house SYN 303 mouse monoclonal) at 1:4,000 with 5 min antigen retrieval in 88% formic acid and phosphorylated tau (AT8 Innoge-netics cat #90206 mouse monoclonal) at 1:2,000 as described in previous publications [14, 31, 52].
Results
Deletion of mouse Tardbp
To generate mice lacking Tardbp, we employed a genetrap insertion strategy. We obtained mouse ES cell line RB030 containing a putative insertion in intron 2 of the Tardbp gene. RB030 is derived from a genetrap library and contains an integrated copy of the genetrap vector pGT1lxf [42]. We sequenced Tardbp DNA fragments from RB030, and confirmed the insertion of the pGT1lxf genetrap cassette within intron 2 of Tardbp at base pair 1226 (Fig. 1a). This insertion should disrupt the production of normally spliced TDP-43 and generate an in frame fusion of Tardbp exon 2 with the genetrap-encoded beta-galactosidase/neo-mycin (beta-geo) marker (Fig. 1b). This insertion should result in the termination of the transcript prior to Tardbp exons 3-5. The protein encoded by the TardbpGt(RB30)Byg mutant allele should include only the first 65 amino acids of TDP-43 fused in frame to beta-geo (reviewed in [40]) and lack all TDP-43 functional domains and sequences including the nuclear localization and export signals, RNA binding domains, and glycine-rich domains required for normal function (Fig. 1c).
Fig. 1.
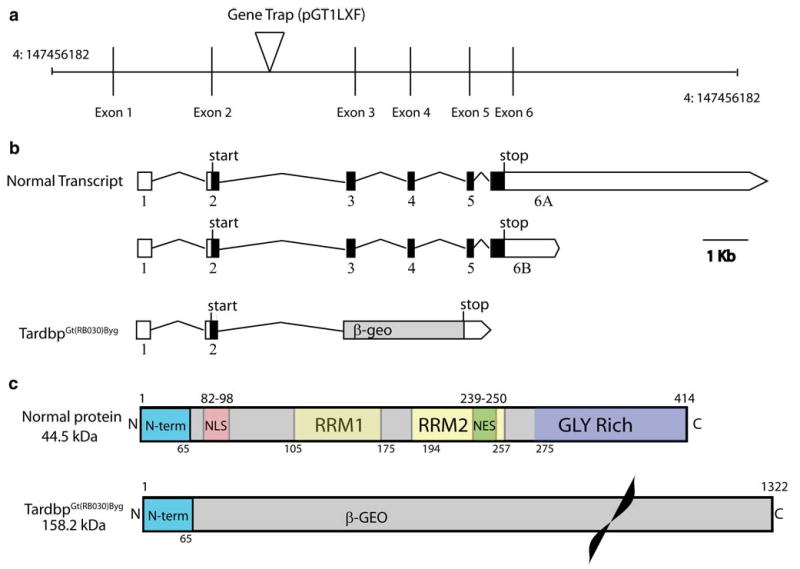
Genetrap targeting of the Tardbp gene. a The genetrap insertion site on mouse chromosome 4 is depicted. Vertical bars are exons, intervening lines represent introns. The position on chromo-some 4 is indicated. b The predicted mRNAs encoding TDP-43 and the truncated genetrap product are indicated. c The protein domain structure of normal TDP-43 and the TDP-43/beta-geo fusion protein are indicated
To generate mice with a targeted disruption of Tardbp, ES cell line RB030 was injected into blastocysts subsequently implanted into pseudopregnant female mice. Twenty chimeric pups resulted, as determined by coat color assessment ranging from 5 to 80% chimerism. Among these were two male mice exhibiting 80% chimeric coat color, which were mated to C57BL/6 females. Germline transmission was achieved from one of the highly chimeric males resulting in heterozygous TardbpGt(RB030)Byg progeny. For purposes of simplicity, we refer to the TardbpGt(RB030)Byg allele as Tardbp–, and the wild-type Tardbp allele as Tardbp+.
Tardbp+/− heterozygous mice are viable
To confirm that Tardbp+/− mice do indeed contain a targeted allele, we examined the Tardbp locus by southern blotting with TDP-43 and genetrap-specific probes (Fig. S1). Findings from southern blotting are consistent with a single genetrap insertion within the Tardbp locus in the genome of Tardbp+/− mice.
We also examined expression of TDP-43 mRNA and protein in these animals. Using northern blot analysis of brain mRNA with an exon 1 specific probe, we observed two prominent bands (2.7 and 7.7 kb) corresponding to the predicted mRNA transcripts both encoding full length TDP-43 with either a long or short 3′UTR in wild-type mice [46, 47]. In heterozygous Tardbp+/− mice we observed an additional band of 5.4 kb corresponding to the predicted Tardbp exon 2/beta-geo fusion resulting from the TardbpGt(RB030)Byg allele, which also labels with a beta-geo specific probe (Fig. 2).
Fig. 2.
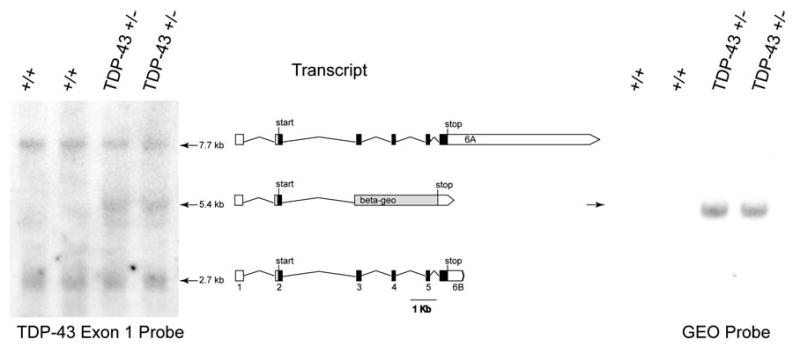
Expression of Tardbp mRNA. Northern blot of brain mRNA purified from Tardbp+/+ and Tardbp+/− animals. The normal short and long 3′ UTR forms of the TDP-43 encoding message are indicated (7.7 and 2.7 kb products). The genetrap truncated mRNA is the intermediate 5.4 Kb band only present in Tardbp+/− animals. The exon 1 targeted radio labeled probe is indicated (left panel). Genetrap specific probe binds beta-geo coding sequence (right panel)
To examine expression of TDP-43 protein in these mice, we analyzed tissue extracts by immunoblotting with TDP-43 specific antibodies (Fig. 3a). Here, we see a prominent TDP-43 band at the predicted size (∼44.5 kDa) in both Tardbp+/+ and Tardbp+/− animals as expected for the major brain isoform of TDP-43. No significant change in TDP-43 protein levels are apparent in Tardbp+/− animals as indicated by quantitative western blotting (n= 5, Tardbp+/− levels of TDP are 99 ± 15% of wiltype, p= 0.98). This lack of reduction in TDP-43 in Tardbp+/− animals was also very recently reported in other models of TDP-43 loss of function [35, 50].
Fig. 3.
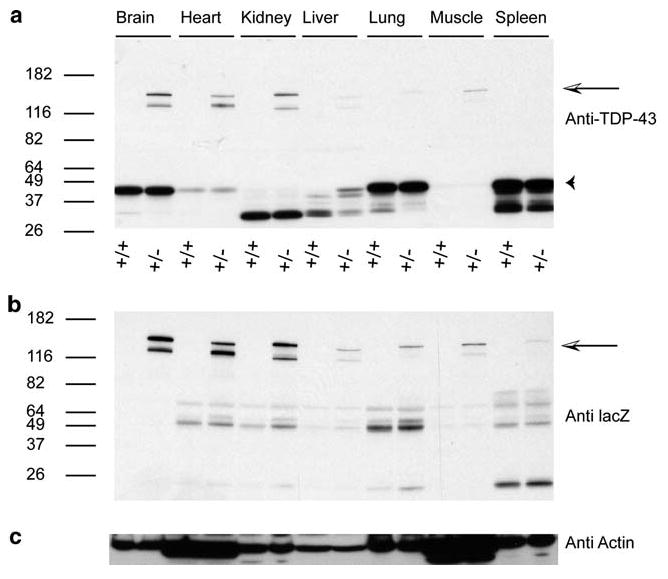
Expression of TDP-43 protein in various tissues. Immunoblotting of brain, heart, kidney, liver, lung, muscle, and spleen tissue protein extracts. Duplicate blots were probed with a pan-TDP-43 specific polyclonal antibodies and b beta-geo specific antibody. Arrow indicates the TDP-43(1–65)/beta-geo fusion protein. Arrowhead indicates the major 43 kDa brain isoform of TDP-43 protein. c beta-Actin specific antibody
We also find other low-molecular-weight non-predicted TDP-43 isoforms in tissues such as kidney and liver. Although the size and origin of these products are not known, we speculate they may arise from alternative splicing and/or proteolytic processing of the TDP-43 mRNA and protein, respectively. The mouse Tardbp gene produces as many as 11 different transcripts encoding a variety of TDP-43 isoforms [46], which may account for the lower molecular weight bands seen here in different tissues. Alternatively, proteolytic processing of full-length normal TDP-43 could occur in these tissues. Furthermore, the mouse genome contains at least one Tardbp-like pseudogene predicted to encode a truncated version of TDP-43. We also observe higher molecular weight bands corresponding to the Tardbp exon 2/beta-geo fusion protein in Tardbp+/− mice, but not wild-type animals. To confirm that the higher molecular weight product does indeed contain the beta-geo genetrap encoded product we probed a duplicate immunoblot with lacZ specific antibody and observed the same high-molecular-weight products only in Tardbp+/− animals (Fig. 3b).
Probing duplicate blots with N-terminal and C-terminal specific TDP-43 antibodies [20] are consistent with these findings (Fig S2). The N-terminal antibody recognizes an epitope between amino acids 6 and 24 of human TDP-43, a region completely conserved between human and mouse. The C-terminal antibody recognizes an epitope between amino acids 394 and 414 of human TDP-43, a region completely conserved between human and mouse. The TDP-43 N-terminal specific antibody recognized the higher molecular weight bands, but the TDP-43 C-terminal specific antibody did not, in keeping with the structure of the predicted Tardbp exon 2/beta-geo fusion product (Fig. 1c).
Homozygous loss of Tardbp causes early embryonic lethality
To obtain mice lacking TDP-43, Tardbp+/− mice were intercrossed. While these intercrosses yielded 79 progeny, none of them were Tardbp+/− suggesting loss of viability in the absence of TDP-43 (Table I). We examined embryos at 3.5 and 12.5 days post-fertilization by PCR genotyping. We observed that 0 out of 31 E12 embryos were Tardbp−/− homozygotes, while 5 out of 20 E3.5 embryos were Tardbp−/−. The failure to obtain any Tardbp−/− E12 embryos or pups suggests that the loss of TDP-43 causes embryonic lethality after day 3.5 but before day 12 of embryogenesis.
Table 1.
Genotype analysis of mouse pups and embryos from Tardbp+/− intercrosses
| Stage | Genotype | Total | ||
|---|---|---|---|---|
| Tardbp+/+ | Tardbp+/− | Tardbp−/− | ||
| E3.5 | 3 | 12 | 5 | 20 |
| E12 | 11 | 20 | 0 | 31 |
| Pup | 22 | 57 | 0 | 79 |
E3.5 embryos were removed pre-implantation and genotyped using single-cell PCR(3). E12 embryos were removed and their DNA extracted. Young mice were genotyped using tail DNA
Degeneration of TDP-43 deficient zygotes
To test the importance of TDP-43 in early embryogenesis, we examined candidate Tardbp−/− homozygous zygotes shortly after implantation. Timed pregnancies were set up by intercrossing heterozygous Tardbp+/− mice. Mated female mice were killed at 5.5, 6.5, 7.5, 8.5, 9.5, and 12.5 days after fertilization. Early post-implantation embryos cannot be reliably genotyped as separating maternal tissue from the embryo is very difficult; therefore, genotypes were inferred from Tardbp expression. Uteri were removed, fixed, and processed for immunohisto-chemistry. Hematoxylin and eosin staining revealed four out of five E12.5 uteri contained viable embryos and the remaining uterus was devoid of embryonic tissue, while five of six E8.5 uteri contained viable embryos. Furthermore, of the nine uteri at E7.5 gestational age, six contained viable embryos and three contained embryos undergoing necrosis. Notably, all of the viable embryos were TDP-43 positive by IHC with pan-TDP-43 antibody, while the embryos undergoing necrosis were TDP-43 negative (Fig. 4). The surrounding uterine decidual tissues served as an internal positive control, and the nuclei in these decidual tissues were strongly positive for TDP-43 in all uteri. In contrast, the E6.5 set of uteri contained six viable embryos and all the embryos in the E5.5 uteri also were viable. There was no evidence of fetal demise or necrotic embryonic tissues on gestational day E6.5 and E5.5. Hence, the findings here are consistent with lethality of homozygous TDP-43 KO embryos at E7.5 with complete elimination of these embryos by E9.5. The appearance of the dying embryos in the uteri shown here is consistent with that reported in other studies examining an in utero lethal phenotype [37].
Fig. 4.
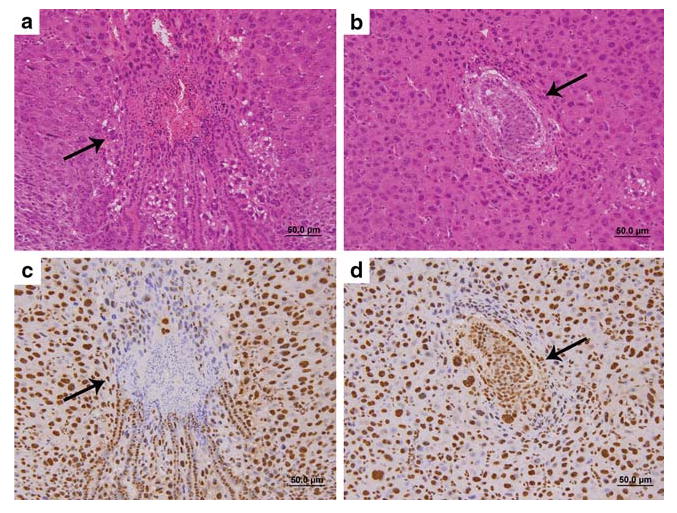
Degeneration of in utero embryos lacking TDP-43 protein. a H&E stained section of E7.5 embryo (arrow) undergoing necrosis; the surrounding tissue is uterine deciduas. b H&E stained section of a normal E7.5 embryo surrounded by uterine decidual tissue. c TDP immunohistochemistry demonstrating lack of TDP staining in the degenerating E7.5 embryo (arrow) surrounded by TDP positive uterine decidual tissue. d TDP immunohistochemistry normal E7.5 embryo (arrow) showing positive staining in the embryo as well as in the surrounding uterine decidual tissue
Tardbp+/− heterozygous mice exhibit motor defects resulting from muscle weakness
Since mutations in the TARDBP gene cause ALS, motor phenotypes may result in mice deficient for TDP-43 function; because Tardbp homozygotes are not viable, we examined motor function in heterozygous mice which should exhibit a partial loss of TDP-43 function. We measured motor function in Tardbp+/− animals using two tests of grip strength and found them to exhibit clear signs of weakness (Fig. 5a, b). Control animals were able to hang onto an inverted wire grid for a significantly longer time than Tardbp+/− animals (p = 0.0002). This measure is partially dependent on the weight of the animals, and we did observe a significant correlation between weight and hang time (r2 =0.3; p= 0.0003). However, there was no significant difference in weight between the groups (46.8 g ± 12.8 Tardbp+/− vs. 46.4 g ± 11.5 controls), indicating that weight differences are not responsible for the genotype effect. We found no significant correlation between hang time and age, in either the control or Tardbp+/− groups. Similarly, we found that Tardbp+/− animals had reduced forelimb grip strength compared with control animals (p= 0.01), as measured by a digital force gage, but did not observe any correlation between grip strength and age in either group. However, gait analysis revealed no difference between Tardbp+/− and controls (Fig. 5c), and we did not observe any evidence of abnormal limb clasping behavior. At 30 days of age, Tardbp+/− animals (n= 16 per genotype) showed no defects in muscle strength, as measured by two different tests, the inverted grid test and forelimb grip strength measured on a dynamometer (Fig. S3). Thus this muscle weakness is an aging-related phenotype.
Fig. 5.
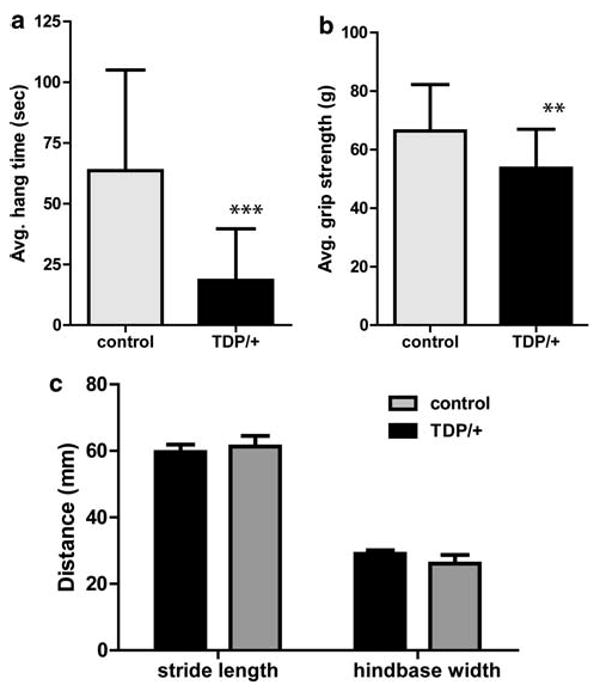
Tardbp+/− animals display deficits in grip strength relative to littermate controls. a Tardbp+/− animals (n = 18) hung on an inverted wire grid for significantly less time than control animals (n = 22) and b also exhibit lower maximum forelimb grip strength. Individual scores are the average of three trials, and the error bars represent standard deviation (**p= 0.01; ***p= 0.0002) c A subset of these animals was also examined for differences in gait, as measured by average stride length and the width between hindpaws while walking. No significant difference between the genotypes was detected
Pathology of Tardbp+/− heterozygous mice
After extensive sampling of brain, spinal cord and muscle of Tardbp+/− heterozygous and wild-type mice, no unequivocal differences were seen between these mice by H&E staining or IHC (see Figs. S4, S5). Thus, while the behavioral testing revealed motor weakness, this is not reflected by evidence of neurodegeneration in the CNS or muscle atrophy in the tissues examined here suggesting subtle non-structural motor neuron deficiencies, metabolic changes, or other systemic abnormalities account for these motor impairments.
Discussion
We generated mice with a targeted disruption of the Tardbp gene, which replaces the normal TDP-43 protein coding sequence with a beta-galactosidase/neomycin resistance fusion protein coding sequence. Mice homozy-gous for the Tardbp disruption do not survive to term and die in utero by day E9.5 of embryogenesis. Immunostain-ing of degenerating embryos shows a pronounced lack of detectable TDP-43 protein in nuclei of necrotic embryos. Because the lethality occurs early in embryogenesis at about E7.5, it is not possible to determine whether the defect is global or if failure to generate specific organ(s) is responsible for embryonic demise. The findings presented here demonstrate an absolute requirement for TDP-43 protein during early to mid embryogenesis. This is not surprising since TDP-43 participates in multiple cellular functions. For example, TDP-43 influences splicing of CFTR, SMN, and APOA2, and probably participates in splicing regulation of numerous other genes. Correct regulation of splicing during embryogenesis is critical, and in adults altered splicing of the tau mRNA causes FTLD-tau [11, 19]. Lack of TDP-43 may also cause early defects in transcriptional regulation of multiple genes including several cell proliferation factors [4]. TDP-43 appears to be localized to a number of nuclear bodies that potentially are complexes important in transcription and splicing [47]. The embryonic lethal phenotype observed demonstrates that TDP-43 is an essential protein and suggests that, at least in embryos, there is no functionally redundant protein equivalent to TDP-43.
In Tardbp+/− animals, we observed impaired motor function, expressed as decreased forelimb strength and performance deficits in the inverted grid test. Despite behavioral evidence of motor weakness, comparative examination of the CNS and muscle tissues from Tardbp+/− and wild type revealed no differences that could be ascribed to the Tardbp+/− genotype. The Tardbp+/− mice showed no evidence of neurodegeneration or muscle atrophy, and did not exhibit any gait abnormalities or any sign of limb clasping. This weakness occurs in aged mice, but not in young mice, indicating that the phenotype is associated with the aging process. This leaves us with two possible explanations. First, weakness could be a result of aging-related defects in motor neuron function without detectable pathologic or structural changes. Second, weakness could occur in the context of aging-related metabolic abnormalities. Future studies will distinguish between these possibilities to more fully account for the motor impairments in the Tardbp+/− mice. However, loss of a single copy of Tardbp does not change TDP-43 protein levels in any tissue examined, suggesting that an auto-regulatory mechanism controls TDP-43 protein levels. These findings are consistent with the suggestion that TDP-43 function is critical for cellular homeostasis [4]. Furthermore, at least one TARDBP mutation (Y374X) in ALS is a sporadic mutation causing a premature stop codon in TARDBP [12]. This is consistent with loss of TARDBP driving motor neuron disease in humans, although this is also consistent with TDP-43 truncation being pathological.
Despite the finding that Tardbp+/− animals exhibit approximately wild-type TDP-43 protein levels, we found clear evidence of muscle weakness in these animals. The inverted grid test is a standard test of motor function which has been previously used to characterize a variety of mouse models of neurodegeneration [10, 17, 36]. In particular, the inverted grid test was a sensitive measure of disease progression in the SOD1 mouse model of ALS [48]. This test is a relatively complex measure of the strength of multiple muscle groups, as well as motor coordination and motivation. Thus, we also performed grip strength measurements using a force meter as a more specific test of forelimb strength. Similar tests have been used to characterize motor defects in other models [17, 39]. Our results indicate that loss of function of a single Tardbp allele in mice is sufficient to cause significant defects in adult motor function.
Including this study, three independent knockout mouse models examining loss of function in Tardbp, the gene encoding TDP-43, have been reported [35, 50]. All three models consistently show an early embryonic phenotype in Tardbp−/− mice arising from an early failure in cell division around the time of blastocyst implantation. In all three models the protein levels of heterozygous Tardbp+/− mice are nearly normal suggesting an auto-regulatory mechanism controls TDP-43 protein levels. Preliminary characterization of Tardbp+/− mice from two groups revealed no overt motor phenotypes [35, 50]; however, our analysis of aged animals revealed significant motor dysfunction present in Tardbp+/− mice without obvious neurodegenerative changes. The discrepancy in motor phenotypes among these three models is most likely due to differences in the age of animals examined. It will be interesting to observe what phenotypes materialize in the other two lines of Tardbp+/− animals as they age. Findings from Drosophila suggest TDP-43 is dispensable for viability in the fly [13, 25], which differs from the results presented for mouse Tardbp here and elsewhere [35, 50]. Fly TDP-43 appears to participate in motor neuron function, suggesting that loss of dTDP-43 can drive motor dysfunction, consistent with our observations in Tardbp+/− mice.
TARDBP mutations causing ALS establish that alterations in TDP-43 can cause neurodegeneration [6, 12, 16, 21, 22, 24, 38, 44, 45, 51]. These mutations result in the same type of TDP-43 aggregation seen in ALS cases without TARDBP mutations. The mechanism by which these mutations cause disease could be partial loss of function. However, as shown here (Fig. 3) in mice, loss of one Tardbp allele alters protein levels little in most tissues, presumably due to increased TDP-43 expression from the intact copy of Tardbp. However, even this subtle perturbation in TDP-43 activity is sufficient to cause detectable motor dysfunction in the absence of neurodegeneration. Therefore, a loss-of-function model for ALS mutations is consistent with the experimental data. In TDP-43 protein-opathies, TDP-43 aggregation may result in a more profound reduction in functional TDP-43 levels that cannot be compensated for by increased Tardbp expression. In some neurons where cytoplasmic aggregates are observed, the nuclei are devoid of TDP-43 detectable by immuno-histochemical methods. Nuclear aggregates could act as a sink for TDP-43, leaving no free protein for essential functions. Such a complete loss of TDP-43 may result in death of the cell, a hypothesis supported by the embryonic lethality we observe in our Tardbp−/− embryos. Under this hypothesis, in the case of ALS mutations, both the mutant and the normal TDP-43 would aggregate and be removed from the soluble pool available for normal function.
The cause of TDP-43 aggregation is unknown. In other proteinopathies, mutations can cause increased aggregation rates for the mutated protein (e.g. tau [18, 29]). However, there is no direct evidence that ALS TARDBP mutations change the solubility of TDP-43. Once TDP-43 is depleted, any number of mechanisms could result in motor neuron dysfunction and/or cell death given the number of cellular processes that involve TDP-43. Clearly, further studies are needed, especially the generation of conditional Tardbp knockout mice to address these and other important aspects of the normal biology of TDP-43 and the pathobiology of TDP-43 in neurodegenerative proteinopathies.
Supplementary Material
Acknowledgments
This work was supported by NIH grant AG17586 to GDS, VMYL, and JQT. This work was also supported by grants to BCK from the Office of Research and Development Medical Research Service, Department of Veterans Affairs and NIH (NS064131). This work utilized facilities at the VA Puget Sound Health Care System, Seattle, Washington. We thank Eddie Lee for outstanding assistance examining neuropathology of Tardbp+/− animals and Elaine Loomis, Leojean Anderson, and Harmony Danner for excellent technical assistance. The lacZ monoclonal antibody was obtained from the Developmental Studies Hybridoma Bank maintained by the University of Iowa.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00401-010-0659-0) contains supplementary material, which is available to authorized users.
Contributor Information
Brian C. Kraemer, Email: kraemerb@u.washington.edu, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA, Division of Gerontology and Geriatric Medicine, Department of Medicine, University of Washington, Seattle, WA 98104, USA.
Theresa Schuck, Department of Pathology and Laboratory Medicine, Center for Neurodegenerative Disease Research and Institute on Aging, University of Pennsylvania School of Medicine, Philadelphia, PA 19104-6100, USA.
Jeanna M. Wheeler, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA, Division of Gerontology and Geriatric Medicine, Department of Medicine, University of Washington, Seattle, WA 98104, USA
Linda C. Robinson, Geriatrics Research Education and Clinical Center, Veterans Affairs Puget Sound Health Care System, Seattle, WA 98108, USA
John Q. Trojanowski, Department of Pathology and Laboratory Medicine, Center for Neurodegenerative Disease Research and Institute on Aging, University of Pennsylvania School of Medicine, Philadelphia, PA 19104-6100, USA
Virginia M. Y. Lee, Department of Pathology and Laboratory Medicine, Center for Neurodegenerative Disease Research and Institute on Aging, University of Pennsylvania School of Medicine, Philadelphia, PA 19104-6100, USA
Gerard D. Schellenberg, Department of Pathology and Laboratory Medicine, Center for Neurodegenerative Disease Research and Institute on Aging, University of Pennsylvania School of Medicine, Philadelphia, PA 19104-6100, USA
References
- 1.Abhyankar MM, Urekar C, Reddi PP. A novel CpG-free vertebrate insulator silences the testis-specific SP-10 gene in somatic tissues: role for TDP-43 in insulator function. J Biol Chem. 2007;282:36143–36154. doi: 10.1074/jbc.M705811200. [DOI] [PubMed] [Google Scholar]
- 2.Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, Graff-Radford NR, Hutton ML, Dickson DW. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007;61:435–445. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aubin JE, Liu F, Candeliere GA. Single-cell PCR methods for studying stem cells and progenitors. Methods Mol Biol. 2002;185:403–415. doi: 10.1385/1-59259-241-4:403. [DOI] [PubMed] [Google Scholar]
- 4.Ayala YM, Misteli T, Baralle FE. TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc Natl Acad Sci USA. 2008;105:3785–3789. doi: 10.1073/pnas.0800546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ayala YM, Pantano S, D'Ambrogio A, Buratti E, Brindisi A, Marchetti C, Romano M, Baralle FE. Human, Drosophila, and C. elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol. 2005;348:575–588. doi: 10.1016/j.jmb.2005.02.038. [DOI] [PubMed] [Google Scholar]
- 6.Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P, Legallic S, Salachas F, Hannequin D, Decousus M, Lacomblez L, Guedj E, Golfier V, Camu W, Dubois B, Campion D, Meininger V, Brice A. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. 2009;65:470–473. doi: 10.1002/ana.21612. [DOI] [PubMed] [Google Scholar]
- 7.Bose JK, Wang IF, Hung L, Tarn WY, Shen CK. TDP-43 overexpression enhances exon 7 inclusion during the survival of motor neuron pre-mRNA splicing. J Biol Chem. 2008;283:28852–28859. doi: 10.1074/jbc.M805376200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buratti E, Baralle FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. 2008;13:867–878. doi: 10.2741/2727. [DOI] [PubMed] [Google Scholar]
- 9.Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001;20:1774–1784. doi: 10.1093/emboj/20.7.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Butchbach ME, Edwards JD, Burghes AH. Abnormal motor phenotype in the SMNDelta7 mouse model of spinal muscular atrophy. Neurobiol Dis. 2007;27:207–219. doi: 10.1016/j.nbd.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D'Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM-Y, Bird TD, Schellenberg GD. Missense and silent tau gene mutations cause front temporal dementia with parkinsonism—chromosome 17 type by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci USA. 1999;96:5598–5603. doi: 10.1073/pnas.96.10.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daoud H, Valdmanis PN, Kabashi E, Dion P, Dupre N, Camu W, Meininger V, Rouleau GA. Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J Med Genet. 2009;46:112–114. doi: 10.1136/jmg.2008.062463. [DOI] [PubMed] [Google Scholar]
- 13.Feiguin F, Godena VK, Romano G, D'Ambrogio A, Klima R, Baralle FE. Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 2009;583:1586–1592. doi: 10.1016/j.febslet.2009.04.019. [DOI] [PubMed] [Google Scholar]
- 14.Forman MS, Lal D, Zhang B, Dabir DV, Swanson E, Lee VMY, Trojanowski JQ. Transgenic mouse model of tau pathology in astrocytes leading to nervous system degeneration. J Neurosci. 2005;25:3539–3550. doi: 10.1523/JNEUROSCI.0081-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geser F, Martinez-Lage M, Kwong LK, Lee VM, Trojanowski JQ. Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP-43 diseases. J Neurol. 2009;256(8):1205–1214. doi: 10.1007/s00415-009-5069-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL, III, Bigio EH, Caselli R, Baker M, Al-Lozi MT, Morris JC, Pestronk A, Rademakers R, Goate AM, Cairns NJ. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63:535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glynn D, Drew CJ, Reim K, Brose N, Morton AJ. Profound ataxia in complexin I knockout mice masks a complex phenotype that includes exploratory and habituation deficits. Hum Mol Genet. 2005;14:2369–2385. doi: 10.1093/hmg/ddi239. [DOI] [PubMed] [Google Scholar]
- 18.Goedert M, Jakes R, Crowther RA. Effects of frontotemporal dementia FTDP-17 mutations on heparin-induced assembly of tau filaments. FEBS Lett. 1999;450:306–311. doi: 10.1016/s0014-5793(99)00508-6. [DOI] [PubMed] [Google Scholar]
- 19.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JBJ, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 20.Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, Clark CM, Elman LB, Miller BL, Grossman M, McCluskey LF, Trojanowski JQ, Lee VM. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol. 2008;173:182–194. doi: 10.2353/ajpath.2008.080003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 22.Kamada M, Maruyama H, Tanaka E, Morino H, Wate R, Ito H, Kusaka H, Kawano Y, Miki T, Nodera H, Izumi Y, Kaji R, Kawakami H. Screening for TARDBP mutations in Japanese familial amyotrophic lateral sclerosis. J Neurol Sci. 2009;284(1– 2):69–71. doi: 10.1016/j.jns.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 23.Kwong LK, Uryu K, Trojanowski JQ, Lee VM. TDP-43 proteinopathies: neurodegenerative protein misfolding diseases without amyloidosis. Neurosignals. 2008;16:41–51. doi: 10.1159/000109758. [DOI] [PubMed] [Google Scholar]
- 24.Lemmens R, Race V, Hersmus N, Matthijs G, Van Den Bosch L, Van Damme P, Dubois B, Boonen S, Goris A, Robberecht W. TDP-43 M311V mutation in familial amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2009;80:354–355. doi: 10.1136/jnnp.2008.157677. [DOI] [PubMed] [Google Scholar]
- 25.Lu Y, Ferris J, Gao FB. Frontotemporal dementia and amyotrophic lateral sclerosis-associated disease protein TDP-43 promotes dendritic branching. Mol Brain. 2009;2:30. doi: 10.1186/1756-6606-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, Eisen A, McClusky L, Kretzschmar HA, Monoranu CM, Highley JR, Kirby J, Siddique T, Shaw PJ, Lee VM, Trojanowski JQ. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 27.Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJ, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DM. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 2009;117:15–18. doi: 10.1007/s00401-008-0460-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mercado PA, Ayala YM, Romano M, Buratti E, Baralle FE. Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res. 2005;33:6000–6010. doi: 10.1093/nar/gki897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nacharaju P, Lewis J, Easson C, Yen S, Hackett J, Hutton M, Yen SH. Accelerated filament formation from tau protein with specific FTDP-17 missense mutations. FEBS Lett. 1999;447:195–199. doi: 10.1016/s0014-5793(99)00294-x. [DOI] [PubMed] [Google Scholar]
- 30.Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, Duda JE, Arnold SE, Siderowf A, Grossman M, Leverenz JB, Woltjer R, Lopez OL, Hamilton R, Tsuang DW, Galasko D, Masliah E, Kaye J, Clark CM, Montine TJ, Lee VM, Trojanowski JQ. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 2007;114:221–229. doi: 10.1007/s00401-007-0261-2. [DOI] [PubMed] [Google Scholar]
- 31.Neumann M, Kwong LK, Lee EB, Kremmer E, Flatley A, Xu Y, Forman MS, Troost D, Kretzschmar HA, Trojanowski JQ, Lee VM. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009;117:137–149. doi: 10.1007/s00401-008-0477-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 33.Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69:3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning : a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, New York: 1989. [Google Scholar]
- 35.Sephton CF, Good SK, Atkin S, Dewey CM, Mayer P, III, Herz J, Yu G. TDP-43 is a developmentally regulated protein essential for early embryonic development. J Biol Chem. 2010;285(9):6826–6834. doi: 10.1074/jbc.M109.061846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shinzawa K, Sumi H, Ikawa M, Matsuoka Y, Okabe M, Sakoda S, Tsujimoto Y. Neuroaxonal dystrophy caused by group VIA phospholipase A2 deficiency in mice: a model of human neurodegenerative disease. J Neurosci. 2008;28:2212–2220. doi: 10.1523/JNEUROSCI.4354-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shu Z, Smith S, Wang L, Rice MC, Kmiec EB. Disruption of muREC2/RAD51L1 in mice results in early embryonic lethality which can Be partially rescued in a p53(−/−) background. Mol Cell Biol. 1999;19:8686–8693. doi: 10.1128/mcb.19.12.8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, de Belleroche J, Mitchell JD, Leigh PN, Al-Chalabi A, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stack EC, Kubilus JK, Smith K, Cormier K, Del Signore SJ, Guelin E, Ryu H, Hersch SM, Ferrante RJ. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington's disease transgenic mice. J Comp Neurol. 2005;490:354–370. doi: 10.1002/cne.20680. [DOI] [PubMed] [Google Scholar]
- 40.Stanford WL, Cohn JB, Cordes SP. Gene-trap mutagenesis: past, present and beyond. Nat Rev Genet. 2001;2:756–768. doi: 10.1038/35093548. [DOI] [PubMed] [Google Scholar]
- 41.Strong MJ, Volkening K, Hammond R, Yang W, Strong W, Leystra-Lantz C, Shoesmith C. TDP43 is a human low molecular weight neurofilament (hNFL) mRNA-binding protein. Mol Cell Neurosci. 2007;35:320–327. doi: 10.1016/j.mcn.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 42.Stryke D, Kawamoto M, Huang CC, Johns SJ, King LA, Harper CA, Meng EC, Lee RE, Yee A, L'Italien L, Chuang PT, Young SG, Skarnes WC, Babbitt PC, Ferrin TE. BayGenomics: a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res. 2003;31:278–281. doi: 10.1093/nar/gkg064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan CF, Eguchi H, Tagawa A, Onodera O, Iwasaki T, Tsujino A, Nishizawa M, Kakita A, Takahashi H. TDP-43 immuno-reactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol. 2007;113:535–542. doi: 10.1007/s00401-007-0206-9. [DOI] [PubMed] [Google Scholar]
- 44.Tomiyama H, Kokubo Y, Sasaki R, Li Y, Imamichi Y, Funayama M, Mizuno Y, Hattori N, Kuzuhara S. Mutation analyses in amyotrophic lateral sclerosis/parkinsonism-dementia complex of the Kii peninsula, Japan. Mov Disord. 2008;23:2344–2348. doi: 10.1002/mds.22262. [DOI] [PubMed] [Google Scholar]
- 45.Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, Steinbart E, McCluskey L, Grossman M, Neumann M, Wu IL, Yang WS, Kalb R, Galasko DR, Montine TJ, Trojanowski JQ, Lee VM, Schellenberg GD, Yu CE. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–416. doi: 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang HY, Wang IF, Bose J, Shen CK. Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics. 2004;83:130–139. doi: 10.1016/s0888-7543(03)00214-3. [DOI] [PubMed] [Google Scholar]
- 47.Wang IF, Reddy NM, Shen CK. Higher order arrangement of the eukaryotic nuclear bodies. Proc Natl Acad Sci USA. 2002;99:13583–13588. doi: 10.1073/pnas.212483099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weydt P, Hong SY, Kliot M, Moller T. Assessing disease onset and progression in the SOD1 mouse model of ALS. Neuroreport. 2003;14:1051–1054. doi: 10.1097/01.wnr.0000073685.00308.89. [DOI] [PubMed] [Google Scholar]
- 49.Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem. 2008;283(19):13302–13309. doi: 10.1074/jbc.M800342200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu LS, Cheng WC, Hou SC, Yan YT, Jiang ST, Shen CK. TDP-43, a neuro-pathosignature factor, is essential for early mouse embryogenesis. Genesis. 2010;48:56–62. doi: 10.1002/dvg.20584. [DOI] [PubMed] [Google Scholar]
- 51.Yokoseki A, Shiga A, Tan CF, Tagawa A, Kaneko H, Koyama A, Eguchi H, Tsujino A, Ikeuchi T, Kakita A, Okamoto K, Nishizawa M, Takahashi H, Onodera O. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol. 2008;63:538–542. doi: 10.1002/ana.21392. [DOI] [PubMed] [Google Scholar]
- 52.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.