

Abstract
Lower respiratory tract infections caused by the paramyxoviruses human metapneumovirus (hMPV) and respiratory syncytial virus (RSV) are characterized by short-lasting virus-specific immunity and often long term airway morbidity, both of which may be the result of alterations in the antigen presenting function of the lung which follow these infections. In this study, we investigated whether hMPV and RSV experimental infections alter the phenotype and function of dendritic cells (DC) subsets which are recruited to the lung. Characterization of lung DC trafficking demonstrated a differential recruitment of plasmacytoid DC (pDC), conventional DC (cDC) and interferon-producing killer DC (IKDC) to the lung and draining lymph nodes after hMPV and RSV infection. In vitro infection of lung DC indicated that in pDC, production of IFN-α, TNF-α, and CCL5 was induced only by hMPV while CCL3 and CCL4 were induced by both viruses. In cDC, a similar repertoire of cytokines was induced by hMPV and RSV, except for IFN-β, which was not induced by RSV. The function of lung pDC was altered following hMPV or RSV infection in vivo, as we demonstrated a reduced capacity of lung pDC to produce IFN-α as well as other cytokines including IL-6, TNF-α, CCL2, CCL3 and CCL4 in response to TLR9 agonist. Moreover, we observed an impaired capacity of cDC from infected mice to present Ag to CD4+ T cells, an effect that lasted beyond the acute phase of infection. Our findings suggest that acute paramyxovirus infections can alter the long term immune function of pulmonary DC.
Keywords: Dendritic cells, Lung, Viral, Cytokines, Cell trafficking
INTRODUCTION
Human metapneumovirus (hMPV) is a recently discovered pathogen first identified in respiratory specimens from young children suffering from clinical respiratory syndromes, ranging from a mild to severe lower respiratory tract illness (1). Seroprevalence studies have shown that by the age of 5 years, approximately 70% of all children have developed antibodies against hMPV (2). hMPV has a seasonal distribution, being usually isolated during the winter time, and is associated with both upper and lower respiratory tract infections in children and adults (3–5). hMPV is responsible for 10% of all hospitalizations of elderly patients with respiratory tract infections and those immunocompromised are also more susceptible to hMPV respiratory tract infections (5,6). hMPV is an RNA virus of the Paramyxoviridae family and part of the Pneumovirinae subfamily along with human respiratory syncytial virus (RSV) (7). In young children, clinical symptoms associated with hMPV infections are virtually indistinguishable from those caused by RSV (8,9), although some but not all studies have reported a lower severity of disease compared to RSV (5,10). Infections caused by both hMPV and RSV are characterized by short-lasting immunity and as consequence reinfections occur throughout life (11). Moreover, both infections have been associated with long-term airway morbidity, including the development of wheezing and asthma (12,13).
Dendritic cells play a central role as immunological sentinels (14,15). They can efficiently sense invading pathogens by a set of pattern recognition receptors and because of their strategic localization at mucosal sites they are involved in response to viral infections (15,16). After detection, uptake and degradative processing of invading pathogens, DC undergo maturation and migration to lymphoid tissue where they present processed viral antigen to lymphocytes (15,17). Respiratory tract dendritic cells are present at high frequency within airway epithelium, submucosa and associated lung parenchyma tissue under resting conditions (18). At least three subsets of pulmonary DC have been described in mice: 1) the CD11cintB220+Ly6C+ plasmacytoid DC (pDC), which are major producers of type I IFN in response to stimulation with enveloped viruses and hence are key effectors in the innate immune system (19,20); 2) the CD11chiMHCIIhi myeloid DC (cDC), which are the primary antigen-presenting cells; and 3) the CD11cintB220+CD49b+ NK interferon-producing killer DC (IKDC) which express cell surface markers of DC as well as NK cell markers (21,22). IKDC could be considered as NK-like DC or DC-like NK cells that might play a major role as a distinct population of innate effectors against viral pathogens (21,23,24). All of these cell types participate in the innate immune response and also are involved in either the generation or modulation of the adaptive immune response.
Despite the critical role of DC in the antiviral immune response, there are not data available regarding the response of lung DC upon hMPV infection, whether this infection results in a distinct response compared to RSV, and whether DC function may be altered beyond the period of acute infection, thus possibly affecting immune response in the convalescence period or even for longer period of times. Therefore, in this study we investigated the effect of hMPV infection on trafficking and activation of lung DC in a mouse model of infection and compared it with RSV. We show that the recruitment and activation of lung DC were different following infection with RSV or hMPV. Moreover, we show that hMPV and RSV infections resulted in impaired ability of lung pDC to produce IFN-α and other cytokines in response to TLR9 agonist, and cDC to present antigen to CD4+ T cells. These data suggest that such subversion of pulmonary DC function may play an important role in the pathogenesis of acute infections caused by RSV and hMPV and possibly their long term consequences, such as failure to develop anti-viral immunologic memory, increased susceptibility to other infections, and altered response to bystander antigens.
MATERIAL AND METHODS
RSV and hMPV preparations
RSV A2 was grown in HEp-2 cells (American Type Culture Collection, Manassas, VA.) and purified by polyethylene glycol precipitation, followed by centrifugation on 35 to 65% discontinuous sucrose gradients as described elsewhere (25). The hMPV strain CAN97-83 was obtained from the Centers for Disease Control (CDC), Atlanta, GA, with permission from Dr. Guy Boivin at the Research Center in Infectious Diseases, Regional Virology Laboratory, Laval University, Quebec City, Canada. Virus was propagated and titrated in LLC-MK2 cells (ATCC, Manassas, VA) in MEM (without serum) containing 1.0 μg trypsin/ml (Worthington, Lakewood NJ), as described elsewhere (26).
Infection of mice and treatment protocol
Female, 8–10-week-old BALB/c mice were purchased from Harlan (Houston, TX) and were housed under pathogen-free conditions in the animal research facility of the University of Texas Medical Branch (UTMB), Galveston, TX, in accordance with the National Institutes of Health and UTMB institutional guidelines for animal care. Under light anesthesia, mice infected intranasally (i.n.) with 50μl of RSV or hMPV diluted in PBS (final administered dose: 107 PFU) (27). Control mice were inoculated with the same volume of PBS (referred to hereinafter as mock infection). To monitor the progression of disease, daily determination of body weight was assessed.
Preparation of lung and lymph node cells
Lungs and lung draining lymph nodes (LN) were harvested at different time points up to 14 days after hMPV, RSV or mock infection. At the indicated time points (see Results) mice were anesthetized with an intraperitoneal injection of ketamine and xylazine before the thoracic cavity was opened. Mice were exanguinated and the trachea opened by incision of the cricothyroid membrane. Prior to harvest, lungs were gently perfused via the right ventricle with 5 ml of PBS containing 2 mM EDTA to remove blood cells from the pulmonary circulation. They were then cut into small pieces and incubated with collagenase A (0.5 mg/ml PBS; Sigma-Aldrich) and type IV bovine pancreatic DNase (20 μg/ml PBS; Sigma) for 1 h at 37°C. After digestion, lung cells were dispersed by shearing through a 20-gauge needle, followed by filtration through a nylon screen cell strainer (100 μm). Single cell suspensions were washed and contaminating erythrocytes were lysed using ACK lysis buffer (Biosource). Lung draining LN were consistently collected and digested to obtain single cell suspensions as mentioned above for lung tissue.
Flow cytometry
Lung and LN cells were incubated with anti-FcγRII/FcγRIII mAb (24G2; BD Biosciences, San Diego, CA). After washing, cells were stained with the following anti-mouse antibodies: anti-CD11c, anti-I-A/I-E (MHC-II), anti-CD11b, anti-CD49b/pan-NK (DX5), anti-Ly6C (Gr-1), anti-CD45R/B220, anti-CD103 (all from BD-Pharmingen, San Diego, CA) and anti-mPDCA1 (Miltenyi Biotec). In a separate set of experiments, lung cells were stained with anti-CD11c, anti-MHC-II in combination with anti-CD40, anti-CD80, anti-CD86, anti-programmed death-1 ligand (PD-L1) and anti-PD-L2 (BD-Pharmingen, San Diego, CA). Samples were stained at 4°C in PBS with 1% FBS and analyzed with a FACS Canto flow cytometer equipped with BD FACSDiva software (both from Becton Dickinson Immunocytometry Systems, San Jose, CA). Analysis was performed using FlowJo software (Tree Star V7.2.2, Ashland, OR).
Cell sorting
Lung cells were first enriched by MACS for specific CD11c+ and mPDCA-1+ populations (Automacs, Miltenyi Biotec), followed by FACS sorting isolation for cDC and pDC, respectively. CD11c+ cells were stained with Cy7-conjugated CD11c and FITC-conjugated MHC-II antibodies and the mPDCA1+ cells were stained with FITC-conjugated CD11c, PerCP-conjugated B220, and PE-conjugated Gr-1 (RB6-8C5) antibodies (BD-Pharmingen). CD11chiMHC-IIhi (cDC), and CD11cintB220+Gr1+ cells (pDC) were sorted using FACS Aria instrument (Becton Dickinson Immunocytometry Systems, San Jose, CA). Routine post-sorting analysis was performed to ensure that that the purity was > 97%. DC were cultured in RPMI-1640 supplemented with 10% FCS, 2 mmol/L L-glutamine, 1 mM sodium pyruvate, and HEPES (complete medium).
Cytokine production
Lung cDC or pDC isolated from uninfected mice were infected in vitro with RSV or hMPV at multiplicity of infection (MOI) of 3 for 24h in 96-well plates (105 cells/well) in a total volume of 200 μl. Cell-free supernatant was collected and tested for multiple cytokines using the Bio-Plex Mouse Cytokine 23-Plex panel (Bio-Rad Laboratories, Hercules, CA), according to the manufacture’s instructions. The panel included the following cytokines: IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12 p40, IL-12 p70, IL-13, IL-17, TNF-α, GM-CSF, IFN-γ, G-CSF, KC, CCL3 (MIP-1α), CCL4 (MIP-1β), CCL11 (Eotaxin), CCL2 (MCP-1) and CCL5 (RANTES). IFN-α and IFN-β were measured by ELISA (PBL Biomedical Laboratories, Piscataway, NJ). The IFN-α ELISA recognizes the αA, α1, α4, α5, α6, and α9 isoforms. In a separate set of experiments, pDC were isolated from mock-, hMPV-, and RSV-infected mice after 3 days of infection. 105 cells/well were stimulated with 1 μg/ml of stimulatory oligodeoxinucleotide (ODN) with mouse-specific CpGs. We used ODN 1826 sequence (5′-tccatgacgttcctgacgtt-3′, Invivogen, San Diego, CA).
T cell Proliferation
For T cell proliferation assays, FACS sorted cDC from virus-infected or uninfected mice (104 cells/well) were cultured for 3 days with CD4+ T cells from DO11.10 mice (105 cells/well) in 96-well round bottom microtiter plates in complete medium. cDC were loaded with 1 μM of OVA peptide (323–339) 1h prior to co-culture with T cells. Cells were pulsed with 1 μCi/well of [3H]-thymidine for the last 18 h. Cells were harvested onto filters using a cell harvester (Brandel, Gaithersburg, MD) and incorporation of [3H]-thymidine to DNA was determined using a Wallac 1450 Microbeta/Trilux liquid scintillation counter (Perkin Elmer, Boston, MA).
Statistical analysis
Statistical analysis was performed using the InStat 3.05 biostatistics package (GraphPad, San Diego, CA) using a one way ANOVA to ascertain differences between groups, followed by a Tukey-Kramer test to correct for multiple comparisons. Unless otherwise indicated, results are expressed as mean ± SEM.
RESULTS
Trafficking of pDC, cDC and IKDC after hMPV or RSV infection
Lung DC are the primary immunological sentinels that can efficiently sense invading pathogens by a set of pattern recognition receptors (14,15). For an effective immune response to occur, DC must be able to sample the peripheral microenvironment and migrate through the afferent lymph to the lymph nodes (LN) where they prime naïve T cells (28). Herein, we determined the recruitment of the pDC, cDC and IKDC to the lung and LN after hMPV infection and compared to that induced by RSV. Mice were infected i.n. with hMPV, RSV or mock inoculum and assessed daily for body weight loss to evaluate the progression of the disease. At different time points, mice were sacrificed to analayze the number of DC tissue in lung and LN. As shown in Fig. 1A, hMPV-infected mice showed a biphasic body weigh loss at days 1 (modest) and 7 and total recovery by day 11. On the other hand, maximum body weight loss in RSV-infected mice occurred at day 2 after infection with gradual recovered by day 7. Recruitment of pDC was evaluated by identifying cells expressing CD11cint/B220+/Gr-1+ (Fig. 1B). Following hMPV infection the number of pDC rapidly increased from 1 ± 0.1 × 105 cells/lung (in mock-infected mice) to 4.7 ± 0.8 × 105 cells/lung by day 1 and reaching a maximum of 8.4 ± 1.6 × 105 cells/lung on day 8 (Fig. 1C). By day 14, number of lung pDC returned to levels similar to those of mock-infected mice. On the other hand, in RSV-infected mice, pDC peaked in the lung at day 3 after infection (8.2 ± 1.2 × 105 cells/lung) and returned to basal levels by day 8. In regard to the trafficking of pDC to LN, we generally observed an increase in size and total number of cells in LN from infected mice as early as day 1 after infection. Number of pDC in LN was similar for both hMPV and RSV infections, although pDC were recruited in slightly higher numbers in hMPV-infected mice (Fig. 1D). pDC were rapidly recruited to LN by day 1 and peaked by day 6 (3.9 ± 0.3 × 104) after hMPV infection and started to decline by day 8. Similar results were observed when pDC were identified by their expression of mPDCA1 and B220 (data not shown). Overall, the data show that in this murine model both hMPV and RSV induced a distinct kinetics of pDC recruitment to the lung that appeared overall to parallel the body weight curve. Pattern of pDC recruitment to the draining LN was similar for both viruses.
Figure 1. pDC recruitment to the lung in hMPV- and RSV-infected mice.
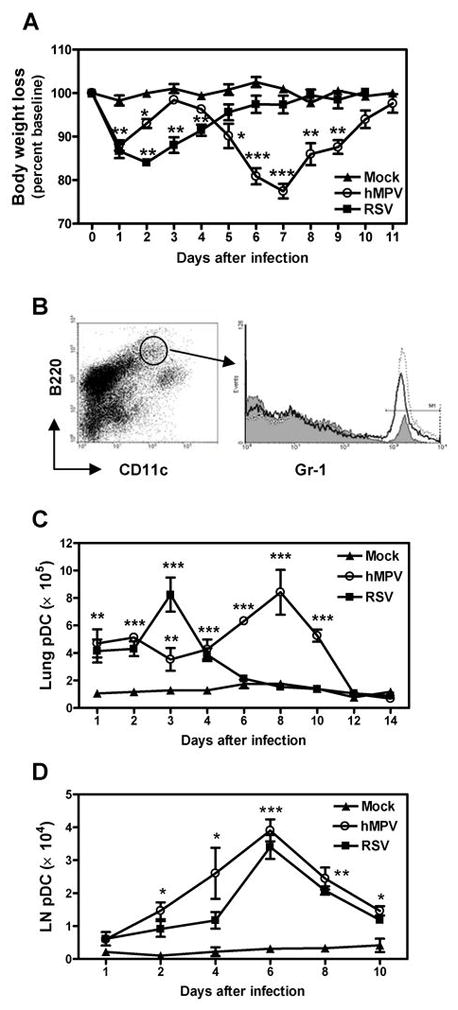
BALB/c mice were infected with hMPV or RSV (107 PFU/mouse). Mock-infected mice were inoculated with 50 μl of PBS. Mice were monitored daily for body weight loss. Lungs and lymph nodes (LN) were removed at different time points and tissue was dispersed by collagenase digestion. Cells were stained with anti-B220-PerCP, anti-CD11c-PE-Cy7 and anti-Gr-1-PE. Cells were acquired in a FACS Canto and data were analyzed by FlowJo software. A) Body weight curve. B) Dot plot indicates gated population of B220+/CD11cint cells. pDC were identified by the additional expression of Gr-1. pDC from mock- (gray shaded histogram), RSV- (open histogram) and hMPV- infected mice (dotted line histogram) are shown. C) Total lung pDC. D) Total LN pDC. Graphs represent mean ± SEM (n = 9 – 16 mice/group/time point). * P < 0.05, ** P < 0.01, *** P < 0.001, compared with mock-infected mice. Data are representative of 3 independent experiments with similar results.
Recruitment of cDC to the lung and LN during hMPV or RSV infection was examined by the expression of CD11chi and MHC-IIhi (Fig. 2A). As shown in Fig. 2B, number of cDC increased from 1.6 ± 0.2 × 105 cells/per lung on day 1 to a maximum of 32.8 ± 7.9 × 105 cells/lung on day 10 after infection with hMPV. Thereafter, the number of lung pDC declined slowly, yet remained significantly elevated until day 18 (12.2 ± 1.3 × 105 cells/lung) compared to mock-infected mice (2.2 ± 0.1 × 105 cells/lung). Similar recruitment pattern was observed when mice were infected with RSV but lung cDC peaked earlier since a maximum of 32.3 ± 1.7 × 105 cells/lung were observed at day 8 after infection. Trafficking of cDC to mediastinal LN was almost identical in hMPV or RSV infections, with an increasing time-dependent number of cells and a peak at day 6 after infection (Fig. 2C).
Figure 2. cDC recruitment to the lung in hMPV- and RSV-infected mice.
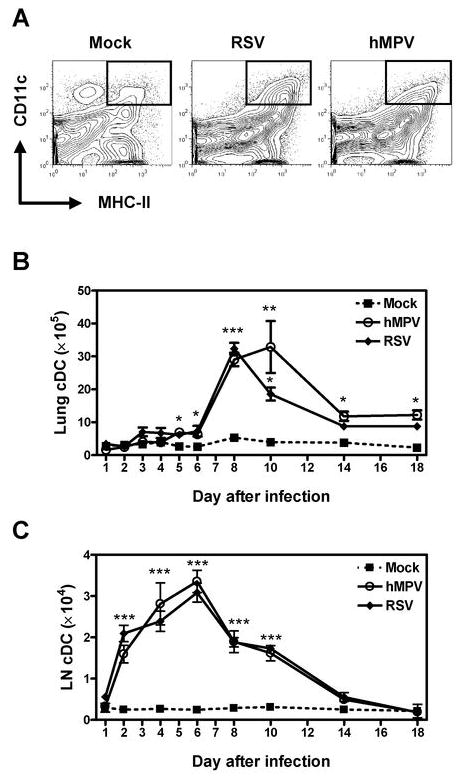
Cells from lung and LN of hMPV-, RSV- or mock-infected mice were stained with anti-MHC-II-FITC and anti-CD11c-PE-Cy7. Cells were acquired in a FACS Canto and data were analyzed by FlowJo software. A) Dot plots indicate gated cDC identified as CD11chi/MHC-IIhi. B) Total lung cDC. C) Total LN cDC. Graphs represent mean ± SEM (n = 8 – 16 mice/group/time point). * P < 0.05, ** P < 0.01, *** P < 0.001, compared with mock-infected mice. Data are representative of 4 independent experiments.
IKDC were identified by expression of CD11cint/B220+/DX5+. Figure 3A shows the morphology of sorted lung IKDC stained by Wright-Giemsa. As shown in Fig. 3B, in hMPV-infected mice IKDC recruitment resembled that of pDC, i.e. as early as day 1 and with a first peak at day 2 (3.4 ± 0.4 × 105 cells/lung) and a second peak at day 8 after infection (4.0 ± 0.1 × 105 cells/lung). By day 10, the numbers of IKDC have returned to similar levels as those observed in mock-infected mice (1.5 ± 0.4 × 105 cells/lung). On the other hand, in RSV-infected animals, IKDC peaked at day 3 (4.7 ± 0.7 × 105 cells/lung) and returned to control levels by day 8 after infection (Fig. 3B). Recruitment of IKDC to LN was similar in both hMPV and RSV infections. As shown in Fig. 3C, IKDC started to be recruited by day 2 with a peak of 4.2 × 104 cells at day 6 after infection, to rapidly decline to 0.7 × 104 cells by day 8 after infection. Thus, IKDC trafficking after hMPV and RSV infection resembles that of pDC. Overall, these data indicate that IKDC is the smallest DC population recruited to the airways upon paramyxovirus infection, followed by pDC and being the cDC the predominant DC subset recruited to the lung.
Figure 3. IKDC recruitment to the lung in hMPV and RSV-infected mice.
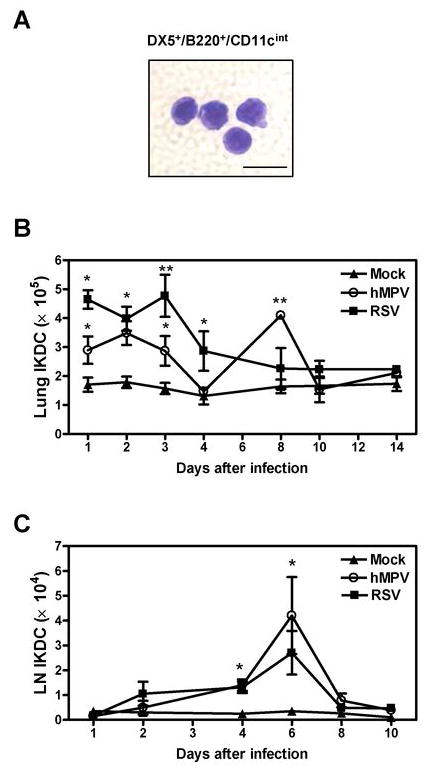
Three-color flow cytometry studies using anti-B220-PerCP, anti-CD11c-PE-Cy7 and anti-DX5-PE were performed on lung and LN cells from hMPV-, RSV- or mock-infected BALB/c mice. IKDC were identified as DX5+/B220+/CD11cint by flow cytometry analysis using FlowJo software. A) Sorted IKDC were stained with Wright-Giemsa staining. Scale bar shows 10 μm. B) Total lung IKDC. C) Total LN IKDC. Graphs represent mean ± SEM (n = 4 – 11 mice/group/time point). * P < 0.05, ** P < 0.01, compared with mock-infected mice. Data are representative of 4 independent experiments.
Production of cytokines by ex-vivo infected lung DC
We have previously shown that hMPV and RSV infections induce a distinct profile of cytokines in human peripheral blood pDC and monocyte-derived DC (29). Since the response of lung DC to these paramyxoviruses has not been investigated, we next examined the ability of lung pDC and cDC to produce cytokines upon hMPV and RSV infection. We isolated lung pDC (CD11cint/B220+/Gr-1+/mPDCA1+) and cDC (CD11chi/MHC-IIhi) from naïve mice (Figs. 4A and 5A) by cell sorting. Isolation of sufficient numbers of lung IKDC from naïve mice to carry out this set of experiments was not feasible. Cells were infected for 24 h and cell-free supernatants were tested for the presence of various cytokines including IFN-α and IFN-β. A highly purified pDC population was isolated from lungs after cell-sorting (Fig. 4A). Exposure of pDC to hMPV resulted in production of significant amount of IFN-α, while very low to undetectable levels were observed upon RSV infection (Fig. 4B). In addition, hMPV but not RSV was able to induce TNF-α and CCL5 production, although their concentrations did not reach statistical significance. Production of IL-12p40 was induced by both viruses, with hMPV being a stronger inducer than RSV. No significant difference in CCL3 and CCL4 production was observed after infection with either hMPV or RSV. Overall, RSV failed to induce cytokine production by lung pDC, with the exception of CCL3 and CCL4. Concentrations of IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12 p70, IL-13, IL-17, GM-CSF, IFN-γ, G-CSF, KC, CCL11, and CCL2 were at the lower limit of detection in control or virus-infected lung pDC (data not shown). These results collectively indicate that pDC produce a limited repertoire of cytokines in response to viral infection and that hMPV is a stronger inducer of cytokines compared to RSV.
Figure 4. Ex-vivo cytokine production by lung pDC upon hMPV and RSV infection.
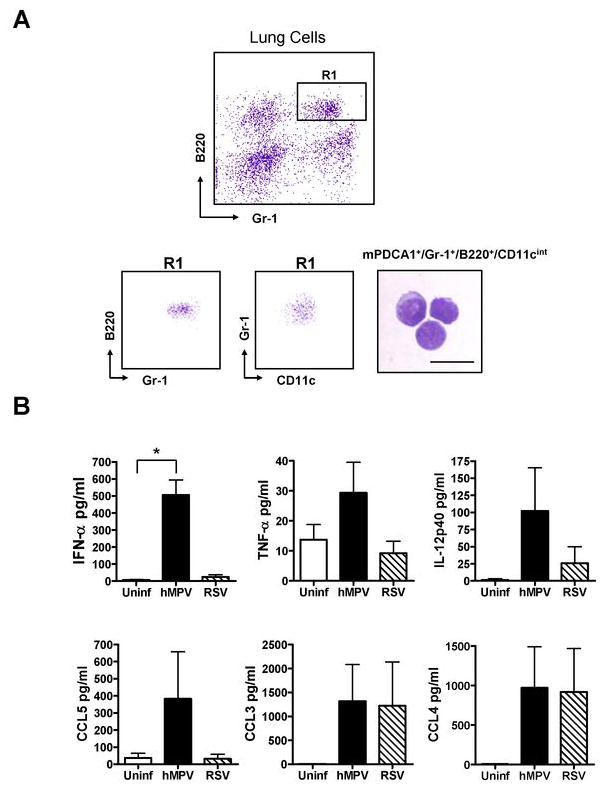
Pulmonary pDC were isolated from naïve BALB/c mice by collagenase digestion. Enriched mPDCA1+ populations by MACS were stained with anti-B220-PerCP, anti-CD11c-PE-Cy7 and anti-Gr-1-PE and sorted by FACS. pDC (105 cells/well) were infected with hMPV or RSV at an MOI of 3 for 24 h. Concentration of IFN-α was determined by ELISA and other cytokines by Bio-Plex assay. A) Upper dot plot represent lung cells before FACS sorting indicating R1 region as mPDCA1+/B220+/Gr-1+ cells. Lower dot plots show R1 region after FACS sorting where pDC were identified as mPDCA1+/Gr-1+/B220+/CD11cint. Sorted pDC were stained with Wright-Giemsa. Scale bar shows 10 μm. B) Cytokine production in lung pDC after hMPV and RSV infection. Bar graphs represent mean ± SEM for n = 3 independent experiments. *P < 0.05.
Figure 5. Ex-vivo cytokine production by lung cDC upon hMPV and RSV infection.
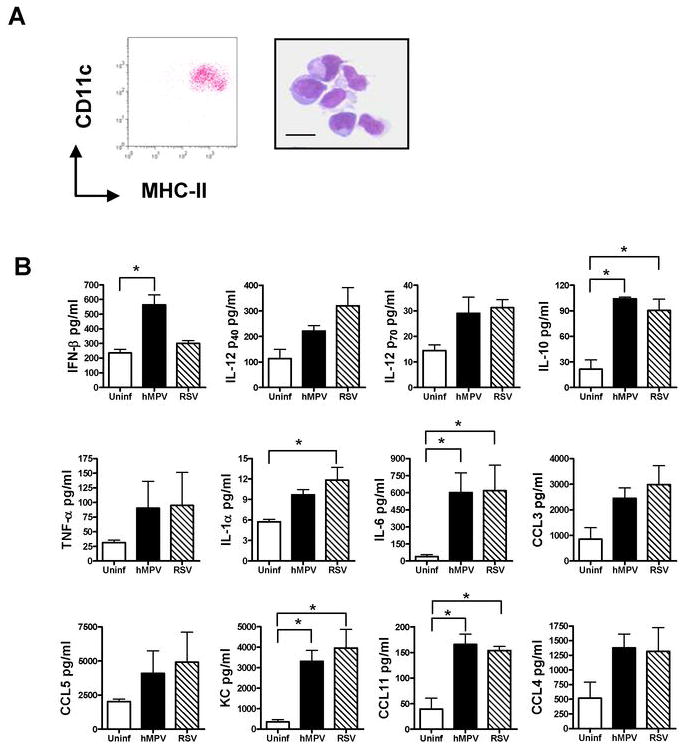
cDC were isolated lungs of naïve mice. Enriched CD11c+ populations by MACS were stained with anti-MHC-II-FITC and anti-CD11c-PE-Cy7 and sorted by FACS. cDC (105 cells/well) were infected with hMPV or RSV at an MOI of 3 for 24 h. Concentration of IFN-β was determined by ELISA and other cytokines by Bio-Plex assay. A) Dot plot represents cDC (CD11chi/MHC-IIhi) after FACS sorting. Sorted cDC were stained with Wright-Giemsa. Scale bar shows 10 μm. B) Cytokine production in lung cDC after hMPV and RSV infection. Bar graphs represent mean ± SEM for n = 3 independent experiments. *P < 0.05.
When we determined the response of cDC to both hMPV and RSV, we observed that hMPV but not RSV was able to induce significant levels of IFN-β (Fig. 5B). Both viruses were able to induce significant amounts of IL-1α, IL-6, IL-10, KC and CCL11. Other cytokines including IL-12p40, IL-12p70, TNF-α, and chemokines CCL3, CCL4, and CCL5 were also similarly induced by hMPV and RSV but did not reach statistical significance when compared with uninfected cells. Levels of IFN-α were below the lower limit of detection in control or viral-infected cDC (data not shown). These data indicate that, with the exception of IFN-β production, hMPV induce a similar profile of cytokines than RSV in pulmonary cDC.
hMPV and RSV inhibit CpG-induced cytokine production by lung pDC
Evidence is accumulating that viral pathogens can use DC as a conduit for subverting the immune response and establishing infection in the host (30). We and others have previously shown that both RSV and hMPV have the ability to inhibit the production of cytokines such as IFN-α by human pDC in response to TLR9 agonists (29,31). However, it is unknown whether hMPV and RSV are capable of inhibiting cytokine production in lung pDC after infection in vivo. Therefore, BALB/c mice were infected with hMPV, RSV or mock for 3 days (when sizable numbers of pDC are recruited at the site of infection, see Fig. 1B). Lung pDC were isolated by cell sorting as described in Material and Methods. Cells (105 pDC/well) were stimulated with 1 μg/ml of CpG-ODN 1826 and cell-free supernatant was collected after 24 h. As shown in Fig. 6, infection with hMPV or RSV significantly inhibited the capacity of lung pDC to produce IFN-α, IL-6, TNF-α, CCL3, CCL4, and CCL5 (by RSV infection) in response to CpG-ODN. We also observed a modest reduction in the production of CCL11, CCL5 (by hMPV infection) and CCL2 and a marginal change in IL-12p70 by any of the two infections in vivo. No change was observed in the production of IL-12p40 in any of the three cultures. These data demonstrate that both hMPV and RSV are able to interfere with the ability of lung pDC to respond to a secondary stimulus for the production of IFN-α and other cytokines.
Figure 6. Cytokine production by lung pDC in response to TLR9 agonist.
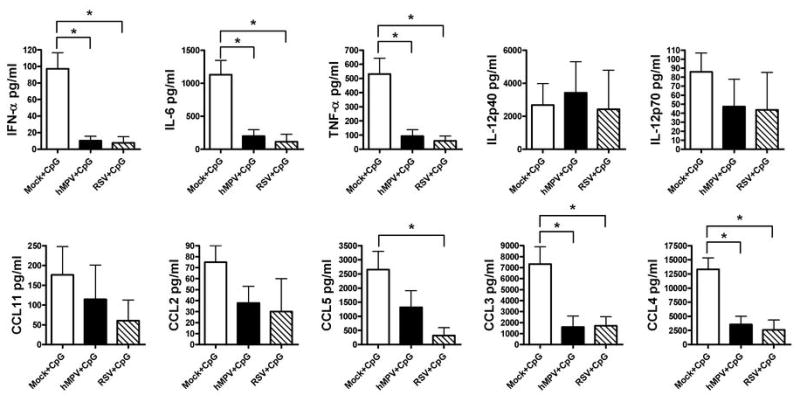
pDC were isolated from lungs of hMPV-, RSV- or mock-infected mice at day 3 after infection. pDC (105 cells/well) were stimulated with 1 μg/ml of CpG-ODN 1826 for 24 h. Concentration of IFN-α was assessed by ELISA and other cytokines by Bio-Plex assay. Bar graphs represent mean ± SEM for n = 3 independent experiments. *P < 0.05.
Impaired antigen presenting capacity by lung cDC after viral infection
We have previously shown that RSV and hMPV are able to inhibit the ability of human monocyte-derived DC (moDC) to stimulate T cell proliferation in vitro (29). To investigate the effect of hMPV and RSV on the antigen presenting capacity of pulmonary cDC, we isolated lung cDC from infected and mock-infected mice at different time points after infection. cDC isolated from lungs at 1, 2, 3, and 6 weeks after hMPV, RSV or mock infection were loaded with OVA peptide and co-cultured with purified CD4+ T cells from DO11.10 mice. Co-cultures of pulmonary cDC from mock-infected mice with DO11.10 T cells resulted in a robust T-cell proliferation (Fig. 7). At the 1 week time point, lung cDC from hMPV- or RSV-infected mice were more efficient in stimulating CD4+ T cell proliferation than cDC isolated from mock mice. However, at week 2, we observed that infection with RSV significantly decreased the ability of cDC to stimulate CD4+ T cell proliferation by ~50%. cDC from hMPV-infected were also deficient in the induction of OVA specific T cell proliferation but in a lower extent (~20 %). At week 3 after infection, both viruses were able to decrease in a similar degree (~70%) the cDC stimulatory capacity when compared to cDC from mock-infected mice. By week 6 after infection, cDC from both RSV- and hMPV-infected mice were able to stimulate T cell proliferation as efficient as those from mock mice. When expression of RSV or hMPV antigens was assessed in lung cDC at 2 or 3 weeks after infection, RT-PCR assays indicated that cDC isolated from infected mice did not express viral antigen (data not shown). These findings indicate RSV and hMPV infection transiently impairs the ability of pulmonary cDC to present antigen to T cells.
Figure 7. CD4+ T cell proliferation induced by cDC from hMPV- and RSV-infected mice.
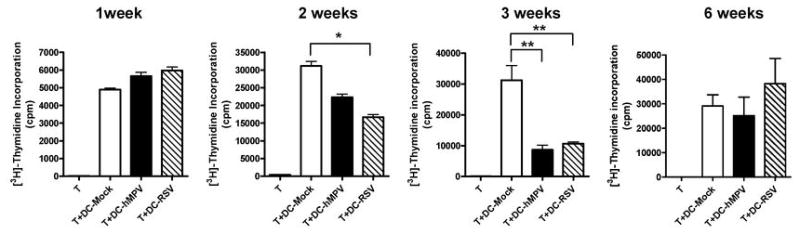
cDC were isolated from lungs of hMPV-, RSV- or mock-infected mice at different time points. cDC were loaded with 1 μM of OVA peptide for 1h before co-culture with CD4+ T cells from D011.10 mice at ratio 1:10. T cell proliferation was analyzed on day 3 of culture by [3H]-Thymidine incorporation. Data are expressed as cpm (mean ± SEM). A representative experiment from three similar experiments is shown. *P<0.05, **P<0.01.
Surface molecule expression by lung cDC after hMPV and RSV infection
The observed inhibition of T cell proliferation by lung cDC isolated from infected mice may be the result of altered expression of APC costimulatory molecules. Positive and negative costimulatory molecules individually or collectively regulate T cell activation thresholds. To examine the possibility that viral infection altered the surface molecule expression, lung cDC (CD11chi/MHC IIhi) were isolated from hMPV-, RSV- and mock-infected mice and were stained with anti-CD40, anti-CD80, anti-CD86, anti-PD-L1, and anti-PD-L2 antibodies. As shown in Fig. 8, expression of CD80 gradually increased over a three week period after RSV or hMPV infection, compared to mock-infrected mice. Similarly, PD-L1 expression increased progressively over the time of infection. Eight weeks after infection, expression of both CD80 and PD-L1 by cDC was comparable to that observed in mock-infected mice. Expression of CD86 was slightly increased by week 2 after RSV infection but not hMPV infection compared to cDC from mock-infecetd mice. CD40 and PD-L2 were not affected by viral infection. These findings indicate that both RSV and hMPV are able to induce activation of pulmonary cDC.
Figure 8. Cell surface molecule expression by lung cDC from hMPV and RSV infected mice.
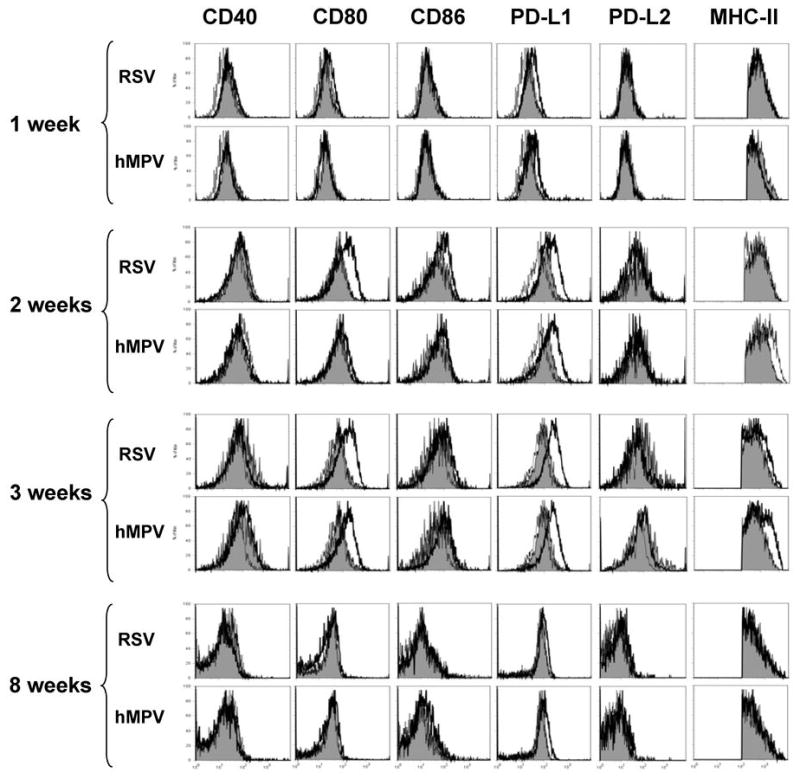
Three-color flow cytometry studies using anti-CD11c and anti-MHC-II in combination with anti-CD40, anti-CD80, anti-CD86, anti-PD-L1 or anti-PD-L2 were performed on lung cells from hMPV-, RSV- or mock-infected BALB/c mice. Gated cDC (CD11chi/MHC-IIhi) were analyzed for the expression of each additional surface molecule expression. Isotype control (doted histograms), cDC from mock-infected mice (shaded histograms) and cDC from virus-infected mice (open histograms) are shown. A representative experiment from three similar experiments is shown.
In addition to the surface molecules described above, we examined whether RSV or hMPV infection altered the phenotype of lung cDC subsets based on CD103 expression. This marker associates with two major cDC populations in mice, CD103+/CD11blow and CD103−/CD11bhi (32), which differ in their ability to prime CD4+ and CD8+ T cells (33), production of proinflammatory cytokines and generation of Foxp3-mediated regulatory function of naïve T cells (34). CD103 (αE) is the α-chain of the αEβ7 integrin, which has been reported to be essential for the adhesion of human and mice intestinal lymphocytes to epithelial cells through the interactions with E-cadherin (35). CD103+ DC in the lung express high amounts of CD11c, MHC-II, and low levels of CD11b (32). It is currently unknown whether respiratory viral infections alter the phenotype of cDC based on expression of CD103. Analysis of cDC CD103+ population was performed by three-color flow cytometry analysis in total lung cells from virus- or mock-infected mice. Gated CD11chi/MHC-IIhi cells were further analyzed for the expression of CD103 (Fig. 9A, upper panels). Consistent with a previous a report, we found that the lung cDC (CD11chi/MHC-IIhi) compartment in uninfected mice is composed by a mixed populations of CD103+ and CD103− cells (32). Moreover, CD11b expression was inversely correlated to CD103 expression since we observed CD103+/CD11b− and CD103−/CD11b+ cells in the cDC subset (data not shown). However, we observed that the percentage of CD103+ cDC decreased from ~50% to ~20% after 7 days of infection, as shown in Fig. 9A (lower panels). Percentage of CD103+ cDC was consistently decreased in infected mice at week 2 and week 3 but returned to normal levels at week 8 after infection (Fig. 9B).
Figure 9. CD103 (αE) expression by lung cDC upon hMPV and RSV infection.
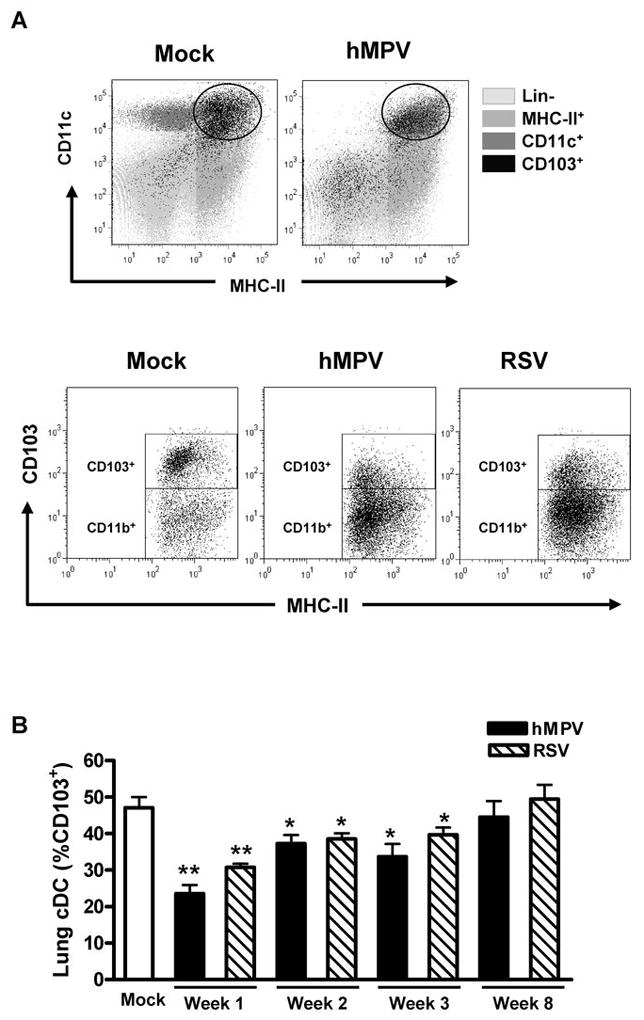
Expression of CD103 in lung cDC from hMPV-, RSV- or mock-infected mice was assessed by three-color flow cytometry studies using anti-CD11c-PE-Cy5 and anti-MHC-II-FITC in combination with anti-CD103-PE. A) Upper dot plots correspond to total lung cells after 7 days after infection, ( ), lineage negative cells, (
), lineage negative cells, ( ), MHC-II+ cells, (
), MHC-II+ cells, ( ), CD11c+ cells and (
), CD11c+ cells and ( ), CD103+ cells. Gated cDC (CD11chi/MHC-IIhi) were analyzed for CD103 expression. Lower dot plots show two major subpopulations of lung cDC as CD103+ and CD103− cells. B) Percentage of lung cDC CD103+ at different time points. Bar graph represents mean ± SEM (n = 4 – 16 mice/group/time point). * P < 0.05, ** P < 0.01, compared with mock-infected mice. Data are representative of 3 independent experiments.
), CD103+ cells. Gated cDC (CD11chi/MHC-IIhi) were analyzed for CD103 expression. Lower dot plots show two major subpopulations of lung cDC as CD103+ and CD103− cells. B) Percentage of lung cDC CD103+ at different time points. Bar graph represents mean ± SEM (n = 4 – 16 mice/group/time point). * P < 0.05, ** P < 0.01, compared with mock-infected mice. Data are representative of 3 independent experiments.
DISCUSSION
This study analyzed the effect of paramyxovirus infections on the immune function of lung dendritic cells. Viruses have developed many ways to evade the host immune response, including interference with the antiviral effects of IFNs, interference with antigen processing and presentation, suppression of the maturation and migration of DC (36). Although previous work has shown that RSV and hMPV are able to impair the DC-mediated immune responses in vitro (29,31,37), this is the first report that demonstrate that RSV and hMPV are able to impair the function of DC compartment in vivo. Following RSV and hMPV infection, we observed an impaired capacity of lung pDC to respond to TLR activation as indicated by the reduced production of IFN-α as well as other cytokines including IL-6, TNF-α, CCL2, CCL3 and CCL4. We also observed an impaired antigen-presenting function, as measured by decreased T-cell proliferation following stimulation of T cells with cDC from infected mice.
The importance of DC during the immune response to RSV and hMPV is supported by our analysis of infected mice. First, we studied the trafficking of pDC, cDC and IKDC after hMPV or RSV infection, as the three types of dendritic cells so far described in the mouse respiratory tract (38). The flow cytometry data show that pDC, cDC and IKDC are rapidly recruited to the lung and draining mediastinal LN after intranasal instillation of RSV and hMPV. In line with theses results there is a study demonstrating that both myeloid DC and pDC numbers were increased in nasal washes of RSV-infected children (39). As demonstrated here, the recruitment of pDC was different between hMPV and RSV infection. RSV induced lung pDC recruitment that peaked at day 3 and returned to basal levels by day 6, while pDC reached the maximum numbers at day 8 in hMPV-infected mice to return to normal numbers by day 12. In contrast with this report, previous work indicate that infection of mice with RSV led to sustained pDC recruitment in the lung of infect mice (40). However, these experiments differ from ours in that pDC were identified as CD11c+/B220+ while we used a more comprehensive set of markers to identify mouse pDC. Specifically, we used a three-color flow cytometry analysis to identify pDC as CD11cint/B220+/Gr-1+ since the expression of CD11c and B220 is shared also by IKDC (21,22,41). The present data were confirmed by identifying pDC by a combined expression of mPDCA1, the specific marker for mouse pDC (42,43), and B220 (data not shown). In fact, a recent report has demonstrated that the heterogeneous population of CD11c+/B220+ can be resolved into three distinct subsets based on the expression of Siglec-H and NK1.1. Siglec-H+/NK1.1− cells are the only true pDC that produce IFN-α in response to TLR stimulation. A second Siglec-H-/NK1.1- subset mostly consists of CD19+/Ig+ B cells. Finally, Siglec-H-/Nk1.1+ cells correspond to IKDC (44). Thus, is possible that the sustained pDC recruitment after RSV infection reported by others may represent recruitment of B cells to the lung. No differences were observed in the recruitment of pDC to the LN between RSV and hMPV infection.
In regard to cDC analysis, our data showed that this DC subset is the most prominent among the DC subsets in the lung after RSV and hMPV infection. The increase in the number of cDC in the lung was sustained up to day 18 after infection, when lungs from infected mice still contained significantly more cDC compared with mock-infected mice. These data are in agreement with those previously reported by Beyer et al. in the lung after RSV infection, where cDC, also identified as CD11chi/MHC-IIhi, were increased and sustained until day 21 (45). In addition, we show that hMPV induce a similar pattern of cDC recruitment than that of RSV infection in both lung and LN.
IKDC was the smallest DC subset recruited to the lung in infected mice. We observed a pattern that resembles that of pDC, including a biphasic and more prolonged recruitment after hMPV infection compared with RSV. Although the classification (21,22,24,44,46), origin (47), as well as the physiological role of IKDC (21,23,41,48,49) remain controversial, our data suggest that this DC subset may play a role in both hMPV and RSV infections. Altogether, these data demonstrate a distinct pattern of DC trafficking to the lung in hMPV or RSV infections. To the best of our knowledge, this is the first study that analyzes the DC response in a mouse model of hMPV infection.
The kinetics of DC recruitment demonstrates that one more feature of the immune response is differentially altered by RSV and hMPV, as indicated in previous studies, in which regardless of the amount of infectious dose, a virus-specific response was observed in regard to clinical disease (body weight loss), lung neutrophilia, profile of BAL cytokines/chemokines, and viral clearance. Also, we have recently reported that the contribution of T cell subsets to the pathogenesis of RSV and hMPV infection is clearly distinct, as CD4+ and CD8+ cells appear to be more important in hMPV and RSV disease, respectively (50).
When we determined the effect of hMPV and RSV infection on lung DC, analysis of the cytokine responses of individual DC subsets (Figs. 4 and 5), revealed the potential contribution of pDC and cDC to the production of these mediators during hMPV and RSV infection. pDC isolated from lung were the major producers of IFN-α following ex-vivo infection with hMPV but not RSV. This observation in murine cells differs from previous work in humans, as we and others have shown that RSV is able to stimulate human peripheral blood pDC to produce IFN-α as well as other cytokines (29,51). Apart from potential specie-specific differences (mouse vs human), we speculate that pDC in lung tissue may respond differently to hMPV or RSV than those from peripheral blood. Indeed, the observed lack of IFN-α production by pDC upon RSV stimulation, is in line with recent findings indicating that depletion of pDC in mice did not alter the production of IFN-α in the lung upon RSV infection (52) and that alveolar macrophages rather than pDC are the major producers of IFN-α following RSV infection [D. Kolli and R. P. Garofalo, unpublished observations and (53)]. Our results also show that pDC responded to hMPV but not RSV to produce TNF-α and CCL5. Other chemokines were also induced in pDC by both viruses including CCL3 and CCL4. Analysis of cDC stimulation indicated a very similar profile of cytokines induced by RSV and hMPV. Both viruses were prominent inducers of IL-12p40, IL-10, IL-6, CCL3, CCL5, KC, CCL11 and CCL4, but they induced a modest production of IL-12p70, TNF-α, and IL-1α. Interestingly, only hMPV was able to induce IFN-β, which resembles our observation in human moDC (29), suggesting that hMPV is a stronger inducer of type I IFN than RSV in both human and mouse DC. These data are consistent with our previous reports where we have shown that the profile of cytokines/chemokines in BAL from mice infected with RSV or hMPV differs widely (26). Moreover, we have demonstrated that in human DC, RSV and hMPV induce a differential profile and abundance of cytokines (29). Overall, our results demonstrated that different subsets of isolated lung DC differ both qualitatively and quantitatively in cytokine and chemokine production in response to hMPV and RSV infection and suggest that pDC and cDC play a role as innate immune effectors critical in the host cytokine response to these paramyxovirus infections (26).
The ability of pDC to recognize and respond to viruses is critical to provide a first line of defense at mucosal surfaces. Functional analysis of the effect of RSV and hMPV on lung pDC demonstrated that these viral infections were able to impair the capacity of these cells to respond to TLR agonists. Indeed, infection of mice with hMPV and RSV reduced the ability of their lung pDC to produce IFN-α, IL-6, TNF-α, CCL2, CCL3, CCL4, CCL5 and CCL11 in response to ODN-CpG. The production of IL-12p40 and IL-12p70 were marginally altered. We and others have previously demonstrated that these two viruses inhibit the cytokine response of human DC in vitro (29,31) and that infection of mice with hMPV or RSV prevents the production of IFN-α in response to ODN-CpG (54). Thus, this report provides novel evidence that the innate immune function of lung DC can be impaired by respiratory viral infections in vivo. These findings suggest that lung pDC may only weakly respond in situ to inflammatory stimuli i.e. bacterial infection once they have been exposed to viral infections in the respiratory tract. In line with this report, a recent study has indicated that alveolar macrophages isolated after influenza infection, have impaired NF-κB nuclear translocation to TLR ligation with reduced production of KC, MIP-2α and TNF-α (55). The mechanistic basis for the impairment of lung pDC remains to be defined. It might be induced directly by the viral interaction with pDC or via the interaction with infected epithelial cells or soluble mediators that are released by lung-resident cells. However, no virus was detected in pDC isolated from infected mice (data not shown). Current studies are in progress to determine whether viral infections in vivo may alter the level of the expression of TLR9 in lung pDC.
It is well known that the immune memory against RSV and hMPV is incomplete even in healthy adult individuals (11,56). T cell responses appear to be crucial to clear RSV or hMPV from the infected lung, because patients with immune deficiencies in the T cell arm of the adaptive immune response are unable to clear the virus affectively (57). In fact, recent clinical observations have suggested that T lymphocytes, are virtually absent in lung tissue of fatal cases of RSV-induced lower respiratory tract infections (LRTI) (58), suggesting that certain viral pathogens have developed mechanisms to inhibit cell-mediated immune responses in the airway mucosa. Moreover, we have recently demonstrated in the mouse model that T cells are crucial for antiviral immunity against hMPV (50). The suppressive effects on T cell proliferation in vitro by RSV are well documented (59–62). We and others have previously shown that RSV is able to inhibit the ability of human monocyte-derived DC (moDC) to stimulate T cell proliferation in vitro (29,37,63), and similar trend was observed in moDC infected with hMPV (29). In this work we report for the first time in an in vivo experimental model that infection with either RSV or hMPV impairs the capacity of lung cDC to present antigen to CD4+ T cells. cDC isolated from infected mice at week 2 and 3 induced a significant reduced CD4+ T cell proliferation compared to cDC from mock-infected mice. Moreover, that inhibitory effect seems to be selective for lung cDC since we did not observe any inhibition of the CD4+ T cell proliferation when we used cDC isolated from spleen of infected mice (data not shown). On the other hand, an increased stimulatory capacity of cDC was observed at week 1 after viral infection. These data are in agreement with previous work on RSV that has indicated that lung DC isolated as early as 10 days after infection, led to a significant stimulation of T cells in an alloreaction (45). The mechanisms by which RSV and hMPV impair cDC function have not yet been identified. However, we observed that upon viral infection, several surface molecules were overexpressed in lung cDC from infected mice, including PD-L1. PD-L1 (B7-H1) and PD-L2 (B7-DC), members of the B7 family, are the ligands of PD-1, a member of CD28 receptor family expressed on activated T and B cells (64). PD-L1 is expressed on activated T cells, B cells, monocytes and dendritic cells, as well as non-hematopoetic cells such as keratinocytes and endothelial cells. The expression of PD-L2 has only been described on monocytes and dendritic cells activated with cytokines, particularly IL-4, and on activated human endothelial cells (65–67). Both PD-1/PD-L1 and PD-1/PD-L2 interactions inhibit T cell proliferation, cytokine production, and CTL activity (65,67,68). Studies in vitro have shown that RSV infection is able to up-regulate PD-L1 and PD-L2 expression on epithelial cells (69) and human moDC (data not shown). In this work, however, the overexpression of PD-L1 upon infection suggests that inhibitory molecules may lead to a decreased capacity of lung DC to induce proliferation in antigen-specific CD4+T cells. Current work is under progress to determine whether the expression of PD-L1 is involved in the inhibitory effect of cDC during RSV and hMPV infection.
Alteration in the composition of lung CD103+ cDC was also observed after viral infection. The percentage of cDC CD103+ was reduced as early as day 2 (data not shown) and remained lower than the mock-infected mice until week 3 after infection and returned to control by week 8 (Fig. 9B). Expression of αE(CD103)β7 is associated with important cellular activities of mucosal dendritic antigen presenting cells, such as antigen presentation (32,33,70), production of proinflammatory cytokines, or stimulation of T regulatory cells (Treg) (34) and some mucosal CD8+ T cells (71,72). Overall, several evidences suggest an association of αE(CD103)β7 expression with important immune functions. It is, nevertheless, currently unclear whether and to what extent αE(CD103)β7 itself contributes directly to such cellular functions, so precise role of αE(CD103)β7 in immune regulation still remain largely elusive. Whether in our model of viral infection, the expression of CD103 in cDC plays an important role in the overall anti-viral response against these two viruses remains to be determined.
In summary, our results demonstrate that respiratory infections by hMPV or RSV alter the pulmonary environment and the function of lung DC. These changes last beyond the period of acute infection, are not apparently associated with persistent viral replication, and may significantly alter the host ability to mount an effective immune response against pathogens or bystander antigens in this phase of relatively immunologic anergy.
Acknowledgments
We thank Mark Griffin at the Flow Cytometry and Cell Sorting Core Laboratory, UTMB, for his help with cell sorting analysis and Giovanni Suarez for his assistance with the Bio-Plex assays.
Abbreviations used in this paper
- hMPV
human metapneumovirus
- RSV
respiratory syncytial virus
- DC
dendritic cells
- IKDC
IFN-producing killer DC
- pDC
plasmacytoid DC
- cDC
conventional DC
- ODN
oligodeoxynucleotide
- i.n
intranasal(y)
- LN
lymph nodes
- MOI
multiplicity of infection
- PD-L
programmed death ligand
Footnotes
Grant support
This work was supported by National Institutes of Health Grants P01 AI062885 and N01 AI30039 (to R. P. G.), an Unrestricted Research Grant from the American Thoracic Society and a YCSA Grant from the Fight Attendant Medical Research Institute (to A. G-P.).
References
- 1.van den Hoogen BG, de Jong JC, Groen J, Kuiken T, de Groot R, Fouchier RA, Osterhaus AD. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat Med. 2001;7:719–724. doi: 10.1038/89098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamelin ME, Boivin G. Human metapneumovirus: a ubiquitous and longstanding respiratory pathogen. Pediatr Infect Dis J. 2005;24:S203–S207. doi: 10.1097/01.inf.0000188158.27840.7c. [DOI] [PubMed] [Google Scholar]
- 3.Esper F, Boucher D, Weibel C, Martinello RA, Kahn JS. Human metapneumovirus infection in the United States: clinical manifestations associated with a newly emerging respiratory infection in children. Pediatrics. 2003;111:1407–1410. doi: 10.1542/peds.111.6.1407. [DOI] [PubMed] [Google Scholar]
- 4.Falsey AR, Erdman D, Anderson LJ, Walsh EE. Human metapneumovirus infections in young and elderly adults. J Infect Dis. 2003;187:785–790. doi: 10.1086/367901. [DOI] [PubMed] [Google Scholar]
- 5.Williams JV, Harris PA, Tollefson SJ, Halburnt-Rush LL, Pingsterhaus JM, Edwards KM, Wright PF, Crowe JE., Jr Human metapneumovirus and lower respiratory tract disease in otherwise healthy infants and children. N Engl J Med. 2004;350:443–450. doi: 10.1056/NEJMoa025472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pelletier G, Dery P, Abed Y, Boivin G. Respiratory tract reinfections by the new human Metapneumovirus in an immunocompromised child. Emerg Infect Dis. 2002;8:976–978. doi: 10.3201/eid0809.020238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Easton AJ, Domachowske JB, Rosenberg HF. Animal pneumoviruses: molecular genetics and pathogenesis. Clin Microbiol Rev. 2004;17:390–412. doi: 10.1128/CMR.17.2.390-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van den Hoogen BG, van Doornum GJ, Fockens JC, Cornelissen JJ, Beyer WE, de Groot R, Osterhaus AD, Fouchier RA. Prevalence and clinical symptoms of human metapneumovirus infection in hospitalized patients. J Infect Dis. 2003;188:1571–1577. doi: 10.1086/379200. [DOI] [PubMed] [Google Scholar]
- 9.Kahn JS. Human metapneumovirus: a newly emerging respiratory pathogen. Curr Opin Infect Dis. 2003;16:255–258. doi: 10.1097/00001432-200306000-00012. [DOI] [PubMed] [Google Scholar]
- 10.von Linstow ML, Henrik LH, Eugen-Olsen J, Koch A, Nordmann WT, Meyer AM, Westh H, Lundgren B, Melbye M, Hogh B. Human metapneumovirus and respiratory syncytial virus in hospitalized danish children with acute respiratory tract infection. Scand J Infect Dis. 2004;36:578–584. doi: 10.1080/00365540410018166. [DOI] [PubMed] [Google Scholar]
- 11.Hall CB, Walsh EE, Long CE, Schnabel KC. Immunity to and frequency of reinfection with respiratory syncytial virus. J Infect Dis. 1991;163:693–698. doi: 10.1093/infdis/163.4.693. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Garcia ML, Calvo C, Casas I, Bracamonte T, Rellan A, Gozalo F, Tenorio T, Perez-Brena P. Human metapneumovirus bronchiolitis in infancy is an important risk factor for asthma at age 5. Pediatr Pulmonol. 2007;42:458–464. doi: 10.1002/ppul.20597. [DOI] [PubMed] [Google Scholar]
- 13.Webb MSC, Henry RL, Milner AD. Continuing respiratory problems three and a half years after acute viral bronchiolitis. Arch Dis Child. 1985;60:1064–1067. doi: 10.1136/adc.60.11.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 15.Pulendran B, Palucka K, Banchereau J. Sensing pathogens and tuning immune responses. Science. 2001;293:253–256. doi: 10.1126/science.1062060. [DOI] [PubMed] [Google Scholar]
- 16.Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255–258. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 17.Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol. 2006;311:17–58. doi: 10.1007/3-540-32636-7_2. [DOI] [PubMed] [Google Scholar]
- 18.Stumbles PA, Upham JW, Holt PG. Airway dendritic cells: coordinators of immunological homeostasis and immunity in the respiratory tract. APMIS. 2003;111:741–755. doi: 10.1034/j.1600-0463.2003.11107806.x. [DOI] [PubMed] [Google Scholar]
- 19.Barchet W, Cella M, Colonna M. Plasmacytoid dendritic cells--virus experts of innate immunity. Semin Immunol. 2005;17:253–261. doi: 10.1016/j.smim.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 20.Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan CW, Crafton E, Fan HN, Flook J, Yoshimura K, Skarica M, Brockstedt D, Dubensky TW, Stins MF, Lanier LL, Pardoll DM, Housseau F. Interferon-producing killer dendritic cells provide a link between innate and adaptive immunity. Nat Med. 2006;12:207–213. doi: 10.1038/nm1352. [DOI] [PubMed] [Google Scholar]
- 22.Taieb J, Chaput N, Menard C, Apetoh L, Ullrich E, Bonmort M, Pequignot M, Casares N, Terme M, Flament C, Opolon P, Lecluse Y, Metivier D, Tomasello E, Vivier E, Ghiringhelli F, Martin F, Klatzmann D, Poynard T, Tursz T, Raposo G, Yagita H, Ryffel B, Kroemer G, Zitvogel L. A novel dendritic cell subset involved in tumor immunosurveillance. Nat Med. 2006;12:214–219. doi: 10.1038/nm1356. [DOI] [PubMed] [Google Scholar]
- 23.Bonmort M, Dalod M, Mignot G, Ullrich E, Chaput N, Zitvogel L. Killer dendritic cells: IKDC and the others. Curr Opin Immunol. 2008;20:558–565. doi: 10.1016/j.coi.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 24.Chauvin C, Josien R. Dendritic cells as killers: mechanistic aspects and potential roles. J Immunol. 2008;181:11–16. doi: 10.4049/jimmunol.181.1.11. [DOI] [PubMed] [Google Scholar]
- 25.Ueba O. Respiratory syncytial virus: I. concentration and purification of the infectious virus. Acta Med Okayama. 1978;32:265–272. [PubMed] [Google Scholar]
- 26.Guerrero-Plata A, Casola A, Garofalo RP. Human metapneumovirus induces a profile of lung cytokines distinct from that of respiratory syncytial virus. J Virol. 2005;79:14992–14997. doi: 10.1128/JVI.79.23.14992-14997.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haeberle HA, Kuziel WA, Dieterich HJ, Casola A, Gatalica Z, Garofalo RP. Inducible expression of inflammatory chemokines in respiratory syncytial virus-infected mice: role of MIP-1α in lung pathology. J Virol. 2000;75:878–890. doi: 10.1128/JVI.75.2.878-890.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol. 2003;3:867–878. doi: 10.1038/nri1222. [DOI] [PubMed] [Google Scholar]
- 29.Guerrero-Plata A, Casola A, Suarez G, Yu X, Spetch L, Peeples ME, Garofalo RP. Differential response of dendritic cells to human metapneumovirus and respiratory syncytial virus. Am J Respir Cell Mol Biol. 2006;34:320–329. doi: 10.1165/rcmb.2005-0287OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pohl C, Shishkova J, Schneider-Schaulies S. Viruses and dendritic cells: enemy mine. Cell Microbiol. 2007;9:279–289. doi: 10.1111/j.1462-5822.2006.00863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schlender J, Hornung V, Finke S, Gunthner-Biller M, Marozin S, Brzozka K, Moghim S, Endres S, Hartmann G, Conzelmann KK. Inhibition of toll-like receptor 7- and 9-mediated alpha/beta interferon production in human plasmacytoid dendritic cells by respiratory syncytial virus and measles virus. J Virol. 2005;79:5507–5515. doi: 10.1128/JVI.79.9.5507-5515.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sung SS, Fu SM, Rose CE, Jr, Gaskin F, Ju ST, Beaty SR. A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J Immunol. 2006;176:2161–2172. doi: 10.4049/jimmunol.176.4.2161. [DOI] [PubMed] [Google Scholar]
- 33.del Rio ML, Rodriguez-Barbosa JI, Kremmer E, Forster R. CD103− and CD103+ bronchial lymph node dendritic cells are specialized in presenting and cross-presenting innocuous antigen to CD4+ and CD8+ T cells. J Immunol. 2007;178:6861–6866. doi: 10.4049/jimmunol.178.11.6861. [DOI] [PubMed] [Google Scholar]
- 34.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cepek KL, Shaw SK, Parker CM, Russell GJ, Morrow JS, Rimm DL, Brenner MB. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E beta 7 integrin. Nature. 1994;372:190–193. doi: 10.1038/372190a0. [DOI] [PubMed] [Google Scholar]
- 36.Palucka K, Banchereau J. How dendritic cells and microbes interact to elicit or subvert protective immune responses. Curr Opin Immunol. 2002;14:420–431. doi: 10.1016/s0952-7915(02)00365-5. [DOI] [PubMed] [Google Scholar]
- 37.de Graaff PM, de Jong EC, van Capel TM, van Dijk ME, Roholl PJ, Boes J, Luytjes W, Kimpen JL, van Bleek GM. Respiratory syncytial virus infection of monocyte-derived dendritic cells decreases their capacity to activate CD4 T cells. J Immunol. 2005;175:5904–5911. doi: 10.4049/jimmunol.175.9.5904. [DOI] [PubMed] [Google Scholar]
- 38.Grayson MH, Holtzman MJ. Emerging role of dendritic cells in respiratory viral infection. J Mol Med. 2007;85:1057–1068. doi: 10.1007/s00109-007-0212-3. [DOI] [PubMed] [Google Scholar]
- 39.Gill MA, Palucka AK, Barton T, Ghaffar F, Jafri H, Banchereau J, Ramilo O. Mobilization of plasmacytoid and myeloid dendritic cells to mucosal sites in children with respiratory syncytial virus and other viral respiratory infections. J Infect Dis. 2005;191:1105–1115. doi: 10.1086/428589. [DOI] [PubMed] [Google Scholar]
- 40.Smit JJ, Rudd BD, Lukacs NW. Plasmacytoid dendritic cells inhibit pulmonary immunopathology and promote clearance of respiratory syncytial virus. J Exp Med. 2006;203:1153–1159. doi: 10.1084/jem.20052359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vremec D, O’Keeffe M, Hochrein H, Fuchsberger M, Caminschi I, Lahoud M, Shortman K. Production of interferons by dendritic cells, plasmacytoid cells, natural killer cells, and interferon-producing killer dendritic cells. Blood. 2007;109:1165–1173. doi: 10.1182/blood-2006-05-015354. [DOI] [PubMed] [Google Scholar]
- 42.Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004;21:107–119. doi: 10.1016/j.immuni.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 43.Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol. 2004;5:1219–1226. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 44.Blasius AL, Barchet W, Cella M, Colonna M. Development and function of murine B220+CD11c+NK1.1+ cells identify them as a subset of NK cells. J Exp Med. 2007;204:2561–2568. doi: 10.1084/jem.20070991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beyer M, Bartz H, Horner K, Doths S, Koerner-Rettberg C, Schwarze J. Sustained increases in numbers of pulmonary dendritic cells after respiratory syncytial virus infection. J Allergy Clin Immunol. 2004;113:127–133. doi: 10.1016/j.jaci.2003.10.057. [DOI] [PubMed] [Google Scholar]
- 46.Vosshenrich CA, Lesjean-Pottier S, Hasan M, Richard-Le Goff O, Corcuff E, Mandelboim O, Di Santo JP. CD11cloB220+ interferon-producing killer dendritic cells are activated natural killer cells. J Exp Med. 2007;204:2569–2578. doi: 10.1084/jem.20071451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Welner RS, Pelayo R, Garrett KP, Chen X, Perry SS, Sun XH, Kee BL, Kincade PW. Interferon-producing killer dendritic cells (IKDCs) arise via a unique differentiation pathway from primitive c-kitHiCD62L+ lymphoid progenitors. Blood. 2007;109:4825–4931. doi: 10.1182/blood-2006-08-043810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Caminschi I, Ahmet F, Heger K, Brady J, Nutt SL, Vremec D, Pietersz S, Lahoud MH, Schofield L, Hansen DS, O’Keeffe M, Smyth MJ, Bedoui S, Davey GM, Villadangos JA, Heath WR, Shortman K. Putative IKDCs are functionally and developmentally similar to natural killer cells, but not to dendritic cells. J Exp Med. 2007;204:2579–2590. doi: 10.1084/jem.20071351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spits H, Lanier LL. Natural killer or dendritic: what’s in a name? Immunity. 2007;26:11–16. doi: 10.1016/j.immuni.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 50.Kolli D, Bataki EL, Spetch L, Guerrero-Plata A, Jewell AM, Piedra PA, Milligan GN, Garofalo RP, Casola A. T lymphocytes contribute to antiviral immunity and pathogenesis in experimental human metapneumovirus infection. J Virol. 2008;82:8560–8569. doi: 10.1128/JVI.00699-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hornung V, Schlender J, Guenthner-Biller M, Rothenfusser S, Endres S, Conzelmann KK, Hartmann G. Replication-dependent potent IFN-alpha induction in human plasmacytoid dendritic cells by a single-stranded RNA virus. J Immunol. 2004;173:5935–5943. doi: 10.4049/jimmunol.173.10.5935. [DOI] [PubMed] [Google Scholar]
- 52.Jewell NA, Vaghefi N, Mertz SE, Akter P, Peebles RS, Jr, Bakaletz LO, Durbin RK, Flano E, Durbin JE. Differential type I interferon induction by respiratory syncytial virus and influenza a virus in vivo. J Virol. 2007;81:9790–9800. doi: 10.1128/JVI.00530-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pribul PK, Harker J, Wang B, Wang H, Tregoning JS, Schwarze J, Openshaw PJ. Alveolar macrophages are a major determinant of early responses to viral lung infection but do not influence subsequent disease development. J Virol. 2008 doi: 10.1128/JVI.02541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guerrero-Plata A, Baron S, Poast JS, Adegboyega PA, Casola A, Garofalo RP. Activity and regulation of alpha interferon in respiratory syncytial virus and human metapneumovirus experimental infections. J Virol. 2005;79:10190–10199. doi: 10.1128/JVI.79.16.10190-10199.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Didierlaurent A, Goulding J, Patel S, Snelgrove R, Low L, Bebien M, Lawrence T, van Rijt LS, Lambrecht BN, Sirard JC, Hussell T. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J Exp Med. 2008;205:323–329. doi: 10.1084/jem.20070891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boivin G, Abed Y, Pelletier G, Ruel L, Moisan D, Cote S, Peret TC, Erdman DD, Anderson LJ. Virological features and clinical manifestations associated with human metapneumovirus: a new paramyxovirus responsible for acute respiratory-tract infections in all age groups. J Infect Dis. 2002;186:1330–1334. doi: 10.1086/344319. [DOI] [PubMed] [Google Scholar]
- 57.Hall CB, Powell KR, MacDonald NE, Gala CL, Menegus ME, Suffin SC, Cohen HJ. Respiratory syncytial viral infection in children with compromised immune function. N Engl J Med. 1986;315:77–81. doi: 10.1056/NEJM198607103150201. [DOI] [PubMed] [Google Scholar]
- 58.Welliver TP, Garofalo RP, Hosakote Y, Hintz KH, Avendano L, Sanchez K, Velozo L, Jafri H, Chavez-Bueno S, Ogra PL, McKinney L, Reed JL, Welliver RC., Sr Severe human lower respiratory tract illness caused by respiratory syncytial virus and influenza virus is characterized by the absence of pulmonary cytotoxic lymphocyte responses. J Infect Dis. 2007;195:1126–1136. doi: 10.1086/512615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schlender J, Walliser G, Fricke J, Conzelmann KK. Respiratory syncytial virus fusion protein mediates inhibition of mitogen-induced T-cell proliferation by contact. J Virol. 2002;76:1163–1170. doi: 10.1128/JVI.76.3.1163-1170.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Preston FM, Beier PL, Pope JH. Identification of the respiratory syncytial virus-induced immunosuppressive factor produced by human peripheral blood mononuclear cells in vitro as interferon-alpha. J Infect Dis. 1995;172:919–926. doi: 10.1093/infdis/172.4.919. [DOI] [PubMed] [Google Scholar]
- 61.Salkind AR, McCarthy DO, Nichols JE, Domurat FM, Walsh EE, Roberts NJ., Jr Interleukin-1-inhibitor activity induced by respiratory syncytial virus: abrogation of virus-specific and alternate human lymphocyte proliferative responses. J Infect Dis. 1991;163:71–77. doi: 10.1093/infdis/163.1.71. [DOI] [PubMed] [Google Scholar]
- 62.Preston FM, Beier PL, Pope JH. Infectious respiratory syncytial virus (RSV) effectively inhibits the proliferative T cell response to inactivated RSV in vitro. J Infect Dis. 1992;165:819–825. doi: 10.1093/infdis/165.5.819. [DOI] [PubMed] [Google Scholar]
- 63.Chi B, Dickensheets HL, Spann KM, Alston MA, Luongo C, Dumoutier L, Huang J, Renauld JC, Kotenko SV, Roederer M, Beeler JA, Donnelly RP, Collins PL, Rabin RL. Alpha and lambda interferon together mediate suppression of CD4 T cells induced by respiratory syncytial virus. J Virol. 2006;80:5032–5040. doi: 10.1128/JVI.80.10.5032-5040.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 65.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, Carter LL, Carreno BM, Malenkovich N, Nishimura H, Okazaki T, Honjo T, Sharpe AH, Freeman GJ. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 66.Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, Shalabi A, Shin T, Pardoll DM, Tsuchiya H. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001;193:839–846. doi: 10.1084/jem.193.7.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, Greenfield EA, Liang SC, Sharpe AH, Lichtman AH, Freeman GJ. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol. 2003;33:3117–3126. doi: 10.1002/eji.200324270. [DOI] [PubMed] [Google Scholar]
- 68.Freeman GJ, V, Boussiotis A, Anumanthan A, Bernstein GM, Ke XY, Rennert PD, Gray GS, Gribben JG, Nadler LM. B7-1 and B7-2 do not deliver identical costimulatory signals, since B7-2 but not B7-1 preferentially costimulates the initial production of IL-4. Immunity. 1995;2:523–532. doi: 10.1016/1074-7613(95)90032-2. [DOI] [PubMed] [Google Scholar]
- 69.Stanciu LA, Bellettato CM, Laza-Stanca V, Coyle AJ, Papi A, Johnston SL. Expression of programmed death-1 ligand (PD-L) 1, PD-L2, B7-H3, and inducible costimulator ligand on human respiratory tract epithelial cells and regulation by respiratory syncytial virus and type 1 and 2 cytokines. J Infect Dis. 2006;193:404–412. doi: 10.1086/499275. [DOI] [PubMed] [Google Scholar]
- 70.Gavin MA, Clarke SR, Negrou E, Gallegos A, Rudensky A. Homeostasis and anergy of CD4(+)CD25(+) suppressor T cells in vivo. Nat Immunol. 2002;3:33–41. doi: 10.1038/ni743. [DOI] [PubMed] [Google Scholar]
- 71.Annacker O, Coombes JL, Malmstrom V, Uhlig HH, Bourne T, Johansson-Lindbom B, Agace WW, Parker CM, Powrie F. Essential role for CD103 in the T cell-mediated regulation of experimental colitis. J Exp Med. 2005;202:1051–1061. doi: 10.1084/jem.20040662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johansson-Lindbom B, Svensson M, Pabst O, Palmqvist C, Marquez G, Forster R, Agace WW. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J Exp Med. 2005;202:1063–1073. doi: 10.1084/jem.20051100. [DOI] [PMC free article] [PubMed] [Google Scholar]