

Abstract
A crucial question in the field of gene regulation is whether the location at which a transcription factor binds influences its effectiveness or the mechanism by which it regulates transcription. Comprehensive transcription factor binding maps are needed to address these issues, and genome-wide mapping is now possible thanks to the technological advances of ChIP-chip and ChIP-Seq. This review discusses how recent genomic profiling of transcription factors gives insight into how binding specificity is achieved and what features of chromatin influence the ability of transcription factors to interact with the genome, and also suggests future experiments to further our understanding of the causes and consequences of transcription factor-genome interactions.
Introduction
Understanding how genomic information is translated into gene regulation has been the subject of intense scientific investigation for the last several decades. Until recently, most studies focused on detailed characterization of a particular gene or gene family. These studies resulted in the development of general principles of gene regulation, but genome-scale studies are now prompting re-examination of some of these principles.
The established view of transcriptional regulation is that cis regulatory elements, such as promoters and enhancers, and proteins that bind to these elements control different levels of transcription of different genes1, 2. Promoters are composed of common sequence elements, such as a TATA box and initiator, and binding sites for other transcription factors, which work together to recruit the general transcriptional machinery to the transcriptional start site (TSS). Enhancers also contain binding sites for transcription factors but are located some distance from the site of transcription initiation. Transcriptional activity resulting from the general factors binding to the core promoter is usually quite low but can be increased by site-specific factors binding to proximal promoter regions, which can help to recruit or stabilize the interaction of the general factors at the core promoter. Promoter activity can be further stimulated by factors binding to distal enhancer regions and subsequent recruitment of a histone modifying enzyme that creates a more favorable chromatin environment for transcription, or a kinase that induces a bound initiation complex to begin elongation (Figure 1). Transcription can also be modulated by repressive factors that bind to upstream repressing sequences and/or silencers, which can interfere with activator binding (and thus prevent recruitment of the general transcriptional machinery) or recruit histone modifying complexes that create repressive chromatin structure.
Figure 1. Transcriptional regulation by promoters and enhancers.
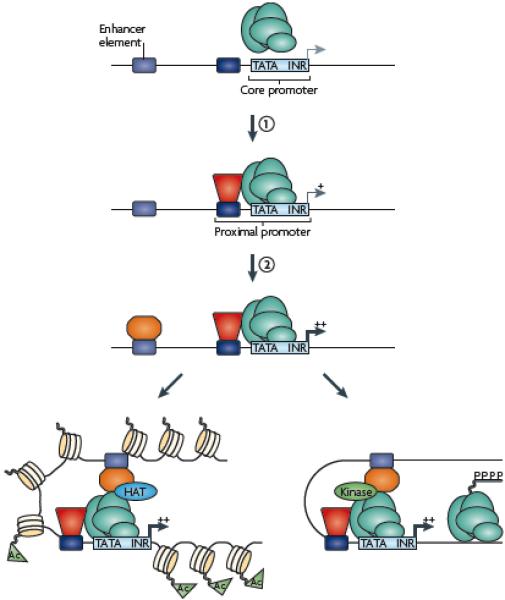
General transcription factors (green) bind to core promoter regions via recognition of common elements such as TATA boxes (TATA) and initiators (INR). However, these elements on their own provide very low levels of transcriptional activity due to unstable interactions of the general factors with the promoter region. Promoter activity can be increased by site-specific DNA binding factors (red) interacting with cis elements in the proximal promoter region and stabilizing the recruitment of the transcriptional machinery through direct interaction of the site-specific factor and the general factors (step 1). Promoter activity can be further stimulated to higher levels by site-specific factors (orange) binding to enhancers (step 2). The enhancer factors can stimulate transcription by (A) recruiting a histone-modifying enzyme (for example a histone acetyltransferease, HAT) to create a more favorable chromatin environment for transcription (acetylated histones, Ac) or (B) recruiting a kinase that can phosphorylate the C terminal domain of RNA polymerase II and stimulate elongation.
Recent genome-scale studies have enabled more precise definition of thousands of promoters for known genes and identified many previously unrecognized transcription units, revealing that some previous assumptions about transcriptional regulation are not correct. For example, based on the detailed characterization of a small subset of promoters, a typical RNA polymerase II (RNAPII) promoter was thought to contain a TATA box located 30 bp upstream of the TSS. However, we now know that TATA-driven promoters are the exception and not the rule 3, 4. Other recent genomic studies suggest that ~50% of human genes have alternative promoters 5, indicating that regulatory sequences for a particular gene can be spread over a considerable distance. Clearly, access to large datasets documenting RNA expression and transcription factor binding on a genome-wide scale now provides an exciting opportunity for investigators to reevaluate previous models of transcriptional regulation. Of particular interest is the role of site-specific DNA binding factors, which is the focus of this review.
It has been estimated that there are 200-300 transcription factors, in humans, that can be considered components of the general transcriptional machinery that bind to core promoter elements (for example, subunits of RNA polymerases and complexes such as TFIID that are required for transcription of most protein-coding genes), and perhaps 1400 transcription factors that have sequence-specific DNA binding properties and thus regulate only a subset of genes by binding to site-specific cis elements 6-8. Interestingly, the site-specific factors tend to be either expressed in all or most tissues or instead are expressed in only one or two tissues, suggesting either a very broad or very specific function 7. Alterations in gene expression caused by the inappropriate level, structure, or function of a transcriptional regulator have been associated with a diverse set of human diseases, including cancers and developmental disorders 9. For example, 164 transcription factors have been shown to be directly responsible for 277 diseases 7. This is undoubtedly a large underestimate of the importance of transcription factors in human disease due to the fact that most human transcription factors are essentially uncharacterized 7. Because of the paucity of our knowledge concerning the function of transcription factors and the likelihood that increased knowledge of transcription factors will lead to increased insight into the causes of human diseases, it is of utmost importance to expand our understanding of how site-specific transcription factors contribute to gene regulation. Crucial questions that need to be addressed are: where do transcription factors bind in the genome; how is specificity of binding achieved; what features of the chromatin can influence the ability of transcription factors to stably interact with the genome; and how is binding of the factor related to its subsequent function in respect to regulation of a nearby gene?
Fortunately, recent advances in the techniques of chromatin immunoprecipitation followed by microarray (ChIP-chip) or by sequencing (ChIP-seq) (Box 1), and similar techniques such as DamID now allow investigators to create a global map of specific protein-DNA interactions in a given cell type in a single experiment10-18 19. Binding sites identified from these ChIP studies 20-28 are categorized relative to genomic features such as the nearest gene, frequency of binding relative to gene structure (for example a promoter, enhancer, exon, or intron), and the type of chromatin domain. The cost of ChIP-Seq depends partly on the depth of sequencing, but an estimate is that 10-12 million uniquely mapped reads should be sufficient for most human transcription factors, which can obtained in 1 or 2 lanes of sequencing, for a cost of one to two thousand dollars. As multiple DNA microarrays are needed to cover the entire human genome, comprehensive studies by ChIP-chip are more expensive. However, for certain applications (such as detailed analyses of a protein complex binding to a small segment of a genome), a focused ChIP-chip experiment currently remains more cost-effective than a genome-wide ChIP-seq analysis.
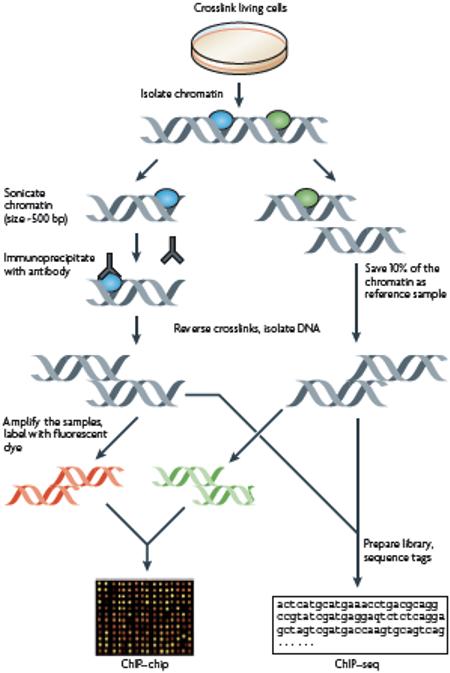
Box 1: Chromatin immunoprecipitation methods.
Briefly, chromatin immunoprecipitation (ChIP) (illustrated in the figure) involves crosslinking DNA-binding proteins to DNA by treatment of cells with formaldehyde and preparation of chromatin by sonication or enzymatic digestion. An immunoprecipitation of the crosslinked chromatin is performed using an antibody that recognizes a specific transcription factor or histone isoform, resulting in the collection of all the binding sites in the genome for the factor of interest. After purification of the precipitated fragments, the sample can be analyzed by PCR to study particular genes. However, genome-wide analysis can be performed by microarray (ChIP-chip) or sequencing (ChIP-Seq). For ChIP-chip, the immunoprecipitated sample and input DNA, as a control, are labeled with fluorescent dyes and hybridized to microarrays. Binding sites are identified by the intensity of signal of the ChIP sample in relation to the signal of the input sample at each probe on the microarray using various ChIP-chip peak-calling programs 21 22. For a single ChIP-chip experiment, most investigators use between 106 and 107 cells, however recent methodological improvements using amplification methods have enabled successful ChIP-chip experiments with as few as 104 cells 77-80. For ChIP-seq, the immunoprecipitated sample is used to create a library that is analyzed using high throughput next generation sequencers. Binding sites are identified using various ChIP-seq peak calling programs 16 27 81 26 82, all of which identify target sites based on the number of sequenced tags from the ChIP library corresponding to each position in the genome. For a ChIP-seq experiment designed to map binding of a site-specific factor, most investigators use 107 to 108 cells, although 104 to 105 cells is sufficient for the ChIP-seq analysis of certain histone modifications 83. It is important to note that because ChIP assays require such large cell numbers, the observed peaks in either ChIP-chip or ChIP-seq represent an average of binding of a factor at a particular site in the cell population. Thus, a small peak could represent very strong binding in only a subset of the cells (for example, cells at one stage of the cell cycle) or modest binding in the entire cell population. ChIP-seq experiments, which allow binding to be analyzed at all unique overlapping oligomers of a certain length (usually 27-50 nts are sequenced per fragment) in the genome, can provide very high resolution mapping of transcription factor binding sites. For example, three-fourths of all the ChIP-Seq peak positions for the DNA binding proteins CTCF, NRSF and STAT1 are within 18, 27 and 51 bp, respectively, of the nearest motif for that factor 82. In general, genome-scale ChIP-chip experiments are less precise in mapping the exact location of a binding site because the oligomers on the array are not overlapping but are spaced approximately 35-100 nt apart, due to the large number of arrays that would be required if overlapping oligomers were used.
This review summarizes recent discoveries provided by genome-wide profiling of site-specific transcription factors and how they have led to new insights regarding patterns of transcription factor binding, how binding specificity is achieved, and what features of the chromatin can influence the ability of transcription factors to interact stably with the genome. The focus will be on the human genome, although relevant insights from other organisms are also incorporated (in particular when studies using model organisms are more advanced than similar studies of the human genome) as it is likely that the implications of transcription factor recruitment for gene regulation will be similar across all eukaryotes. Importantly, genome-wide studies have not only provided new information, they have also created new challenges in our understanding of gene regulation, such as why certain transcription factors bind to so many places in the genome and why so much of the regulation appears to be via steps that occur after recruitment of the site-specific factor to the DNA. Therefore, this review concludes with suggestions for future experiments that are needed to further our understanding of the causes and consequences of specific transcription factor-genomic interactions.
Localization of binding sites
Two decades ago, investigators were using in vitro assays or reporter constructs, to define cis elements necessary for basal transcriptional activity or regions that control cell type-specific, hormonal, or environmental transcriptional responses. In most cases, relatively small promoter segments (from 500 bp to perhaps 10 kb upstream of a TSS) were used as the starting point for mutational analyses. One common observation was that severe truncation of a fragment could cause large changes in promoter activity but that incremental deletion of the 5′ end of the fragment resulted in only minor changes in activity, suggesting that multiple transcription factor binding sites were scattered throughout the analyzed region (for example ref. 29). In contrast, other studies found that hormonal regulation or cell type-specific transcription from a promoter could not be reproduced using reporter assays (for example ref. 30). Such results raised two important questions that are now being addressed by genome-wide binding analyses: do different transcription factors bind in clusters near each other and are most of the binding sites for a given transcription factor located in proximal promoter regions?
Binding to proximal promoters
Transcription factors have been categorized into those that bind proximal promoters and those that bind enhancers1, 2. However, in most past work, a single binding site, or in some cases a small set of sites, was studied for a particular factor. Such focused analyses do not allow general conclusions to be drawn as to whether a factor usually binds near or distal to a promoter region. Thus, accurate categorization of factors is not possible without genome-wide analysis of binding sites. Knowing the location, relative to the TSS, at which a factor binds is of interest as it can provide insight into the mechanisms by which it regulates transcription (Figure 1). For example, factors that bind close to TSSs have been proposed to regulate transcription by stabilizing general transcription factors at the core promoter elements; factors that bind to distal regions, either upstream or downstream of a gene, might regulate transcription by mediating protein-protein contacts between distal complexes and the general transcriptional machinery bound at start sites (that is, by a looping mechanism). Thus, comprehensive location analysis of a factor can not only allow the development of a genomic map but can also provide insight into the mechanisms by which it regulates transcription.
Initial large-scale analyses of transcription factor binding, by ChIP-chip, focused on the identification of binding sites near CpG islands or within 1-5 kb of the TSS of known genes 15, 31-34. Although these studies identified hundreds, and in some cases thousands, of promoters that were bound by a particular transcription factor they were limited to target sites in proximal promoter regions so it was not known whether the identified sites were representative of the majority of the genomic binding sites for a given factor. Analyses of 1% of the human genome as part of the ENCODE pilot project, which are being continued both by the ENCODE Consortium and others 3, 22-24, 35, 36, have now shown that transcription factors that bind almost exclusively at proximal promoters might be the exception, not the rule. Some factors, for example E2F transcription factor family members, are almost always bound in proximal promoter regions (Figure 2A). In fact, it is often difficult to distinguish E2F binding patterns from the binding patterns of general transcription factors such as RNAPII or the TATA box binding protein-associated factor TAF1 15, 22. However, other factors that have recently been analyzed by genome-wide ChIP-chip or ChIP-seq, such as GATA1 and ZNF263, bind to diverse regions of the genome (Figure 2B), including extragenic regions distant from the TSS and intragenic regions (including both introns and exons). Other examples of transcription factors that have wide-spread binding patterns include p53, p63, the estrogen receptor, FoxA2, and TCF4 13 10, 24, 36, 37.
Figure 2. Location analysis of transcription factors.
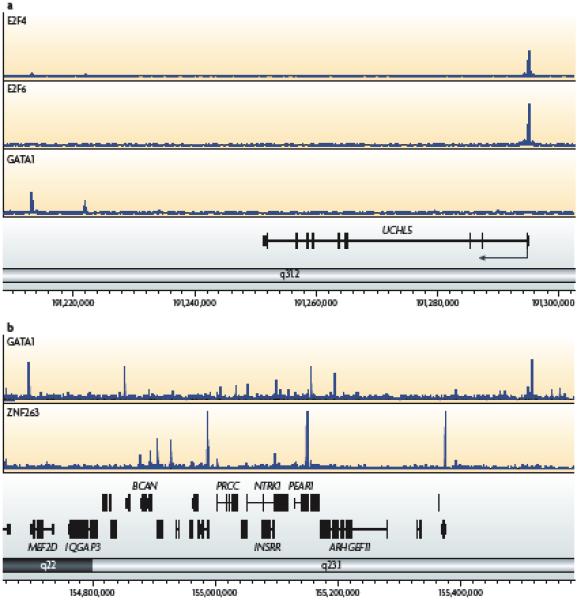
(A) Localization analysis reveals two classes of binding patterns for transcription factors. Shown is the analysis of binding sites identified using ChIP-seq for E2F4, E2F6, and GATA1 for a region of chromosome 1 containing the UCHL5 gene (the direction of transcription is shown by the arrow beginning at the start site). E2F4 and E2F6 bind to the promoter region whereas GATA1 binds downstream of the gene. (B) Shown is the analysis of binding sites identified using ChIP-seq for GATA1 and ZNF263 for a region of chromosome 1. The binding sites for these two factors do not cluster at the same genomic locations.
Although it is difficult to make accurate comparisons of binding patterns generated by different research groups using different experimental platforms, genome-wide profiles for a large number of factors were compared in the ENCODE pilot project 3. This study found that less than 10% of the factors tested had greater than 50% of their binding sites within 2.5 kb of a transcription start site (see Figure 6 and Figure S31 of ref 28). Another study, which analyzed 13 site-specific factors in mouse ES cells using ChIP-seq, also found that many binding sites were located outside of proximal promoter regions 38. Clearly, a typical reporter or in vitro assay cannot monitor the contribution to promoter activity of sites distant from the proximal promoter. The new findings of the distribution of factors throughout the genome might explain many of the failed attempts in the past to demonstrate accurate regulation of a target gene using reporter assays or transgenic constructs. Also, the distributive pattern of binding seen for many factors has important implications for subsequent functional analyses. For example, it is not easy to link enhancers to specific promoters if the enhancer is between two genes, but at a great distance from both; this is discussed in more detail below.
Figure 6. Revised model for transcriptional regulation.
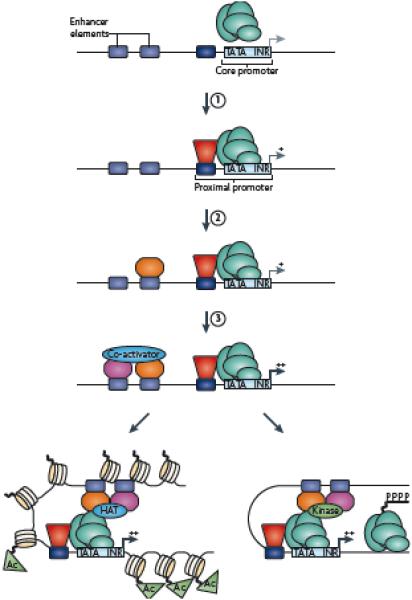
ChIP-chip and ChIP-seq studies have confirmed that RNA polymerase II (RNAPII) and other factors are bound to thousands of promoters that are on at low levels, thus supporting step 1 (as shown in Figure 1). That is, promoter activity can be increased by site-specific DNA binding factors interacting with cis elements in the proximal promoter region and stabilizing the recruitment of the transcriptional machinery through direct interaction of the site-specific factor and the general factors. However, these studies have also revealed that binding of a factor to an enhancer region may be necessary, but not sufficient, for high levels of promoter activity (step 2), thus leading to the inclusion of a new step in the model (step 3): the binding of a cell type-specific partner protein that allows the recruitment of a coactivator to provide for cell type-specific function of a constitutively bound factor. Currently, the projected later steps remain as shown in Figure 1: the enhancer factors can stimulate transcription by (A) recruiting a histone-modifying enzyme to create a more favorable chromatin environment for transcription or (B) recruiting a kinase that can phosphorylate the C terminal domain of RNA polymerase II and stimulate elongation.
Binding to enhanceosomes
Early studies of Drosophila melanogaster development identified regulatory regions that are bound by combinations of different transcription factors, leading to the concept that transcription factors can cluster near each other to regulate transcription cooperatively 39. For example, enhancers that regulate D. melanogaster segmentation contain a module that typically receives input from multiple transcription factors and has multiple binding sites for each of the factors; in many cases the binding sites are clustered within a small interval of 0.5-1 kb. Recently, large-scale profiling of the binding patterns of a set of D. melanogaster transcription factors revealed binding hotspots, each 1-5 kb in length and spaced ~50 kb apart 40. The D. melanogaster genome is one-tenth the size of the human genome and therefore it is not yet clear if the same sort of clustering will be commonly found for human transcription factors.
Owing to the large size of the human genome and the large number of transcription factors (~ 1400), most investigations of the concept of clustered binding sites creating a regulatory element have used computational tools 41. As detailed below, bioinformatic analyses are not sufficient to determine which of all possible binding sites are actually occupied by a transcription factor in vivo. However, there is some experimental evidence that at least a few binding hotspots do exist in the human genome. An extensively studied mammalian enhancer is the interferon beta enhanceosome 42, 43 in which 8 transcription factors bind to overlapping elements within a 55 bp region upstream of the interferon beta gene (IFNB1). This enhancer was characterized over many years using classical mutational analyses of a single regulatory element. Although very few regions of the human genome have been characterized in as much detail as the IFNB1 enhancer, several other enhancer regions have been fairly well-studied, including the mouse and chicken beta-globin locus control regions and the human growth hormone and MHCII enhancer regions 44.
Chen et al analyzed a set of factors that work together to mediate pluripotency and maintain self-renewal properties of mouse ES cells 38. They found that some regions (termed MTL for multiple transcription factor-binding loci) were bound by several factors. Specifically, clusters of Nanog, Oct4, and Sox2 sites were identified outside of promoter regions, suggesting that these regions might be enhancers, and a subset of MTL showed strong enhancer activity in follow-up experiments. Identification of these MTL may have been facilitated by the fact that Nanog, Oct4, and Sox2 were previously known to cooperate in regulating the mouse ES cell transcriptome.
Unfortunately, to date only a handful of human factors (very few of which have been implicated in regulating the same sets of genes) have been analyzed using ChIP-seq and these factors do not seem to show a large degree of overlap in binding at locations outside of promoter regions (Figure 2B). However, it is hard to know if the lack of observed clustering is because there are in fact no hotspots for binding in the human genome or because the correct combinations of factors have not yet been studied. Knowledge of the extent of clustered binding in mammalian genomes must await the collection of more ChIP-seq data. Genome-wide analyses of enhancers based on specific histone modification patterns have also recently been initiated 45, 46. However, identifying a potential enhancer region based on histone patterns does not reveal how many site-specific factors bind to the region. If clusters of binding sites are found in mammalian genomes, they could correspond to enhancesomes similar to the one at IFNB1, with multiple factors all working together to mediate transcriptional activation. Alternatively, they could represent nonfunctional “storage bins” for excess transcription factors, provide functional redundancy that decreases the chances that a gene might be turned off due to mutation, or allow activation of a gene by multiple different signaling cascades.
Do consensus motifs specify binding?
In vitro studies, such as CASTing (cyclic amplification and selection of targets), and sequence comparisons of small sets of promoters known to be bound by a factor have allowed the derivation of consensus binding motifs for some transcription factors 47. Subsequent bioinformatic analyses that search the human genome using consensus motifs or position weight matrices – a collection of motifs similar, but not identical, to the consensus motif - allow the identification of all locations in the genome to which a transcription factor might bind 41, 48. This approach provides the set of all possible locations for a given factor; however, in a mammalian genome there are clearly many more occurrences of a consensus motif for a given factor than there are binding sites 37, 49. Also, the utility of bioinformatic studies relies on the assumption that transcription factors are recruited to the genome in vivo via motifs similar to identified in vitro studies. These caveats have led to uncertainties as to the importance of consensus motifs for in vivo binding. ChIP-chip and ChIP-seq studies have allowed investigators to address two important questions concerning motif usage: what percentage of binding sites contain a consensus motif and what influences whether a specific motif is in fact bound by a particular factor?
Motif enrichment within binding regions
Although some factors appear to be recruited to a majority of their binding sites via a common motif, other factors seem to have a more diverse set of recruitment mechanisms. For example, members of the E2F family appear to lack a requirement for a specific motif for binding in vivo 49. In contrast, the set of binding sites for factors such as p63, STAT1 and NRSF show high enrichment for a specific motif 16, 20, 37. It should be stressed that binding detected at sites that lack a consensus motif is not due to a general, low affinity DNA binding activity. ChIP-chip and ChIP-seq measure DNA-protein interactions as an average of individual binding events in millions of cells and a peak at a site without a motif can be as high and as sharp as a peak located over a consensus motif, which is inconsistent with random protein-DNA interaction.
Several mechanisms have been proposed to explain how specific recruitment can occur in the absence of a consensus motif (Figure 3). These include: binding at a distal site that contains a consensus motif and looping to the site in question via protein-protein interactions (perhaps via a co-activator or co-repressor); ‘piggyback’ binding mediated by protein-protein interactions with a second factor, with no contribution of the DNA binding domain of the first factor; or assisted binding to a site that is somewhat similar to the consensus site, enhanced by protein-protein interaction with another site-specific DNA binding factor or with a specifically modified histone. Clearly, the greater the contribution of protein-protein interactions to the genomic localization of a factor, the greater is the difficulty of using a strictly bioinformatic approach to identify in vivo binding sites.
Figure 3. Models for recruitment of factors to sites that lack consensus motifs.
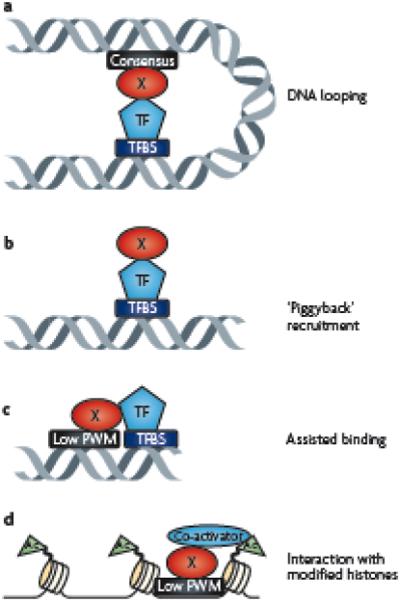
(A) Transcription factor X could bind to its consensus motif (black box) and loop via protein-protein interactions to another transcription factor (TF) bound to a different binding site (TFBS) that is located at a distant region of the chromosome. In this case, because formaldehyde can create both protein-DNA and protein-protein crosslinks, ChIP assays for Factor X would enrich for a region containing its own consensus motif and a region bound by the other factor. (B) Factor X could be recruited to a sequence via protein-protein interactions with another transcription factor (TF) in a manner completely independent of its DNA binding abilities. In this case, ChIP assays would detect binding of Factor X at a region that has no match to its consensus or position weight matrix (PWM). Factor X could bind to a sequence that has a low match to its PWM and be anchored on the genome via protein-protein interactions with a nearby factor (C) or with a specifically modified histone (D). In both of these cases, ChIP assays would detect binding of Factor X at a region that contains a low match to its PWM.
Sorting binding sites for a factor into subsets that contain or lack a specific motif might eventually provide insight into alternative recruitment or regulatory mechanisms mediated by that factor; the ability of a factor to be recruited to the genome in more than one way might allow a factor to participate in multiple different signaling pathways. For example, serum response factor (SRF) is ubiquitously expressed but its activity is modulated at several levels, including protein-protein interaction 50 51. Perhaps recruitment of SRF via a consensus motif allows for regulation of one set of targets in many cell types, whereas stabilized binding via protein-protein interaction to sites lacking the consensus motif allows the constitutively expressed SRF to also have some cell type-specific functions. It should be noted that even factors that prefer to bind to regions containing a specific motif can also have subsets of binding sites that lack that motif 52, 53. A recent study has shown that the ability of a factor to bind to more than one motif is not necessarily due to protein-protein interactions but instead can be observed using purified proteins and in vitro assays. Using protein binding microarrays, Badis et al. 54 found that about half of a set of 104 mouse DNA binding proteins recognized multiple different sequence motifs. Such studies suggest that motif analysis of ChIP-seq data should be performed under the assumption that more than one motif can be present in the set of identified binding regions.
Do epigenetic modifications influence motif usage?
As discussed above, a major difficulty with using a bioinformatics motif-driven approach to identify binding sites is that it is clear that only a small percentage of all occurrences of a motif are actually bound by that factor. Therefore, the majority of regions in the genome that contain a consensus motif for a given factor are not occupied. Lack of binding to the genome in certain regions could be due to chromatin structure (inaccessibility due to close packing of the nucleosomes in heterochromatin) or to DNA methylation (reduced binding affinity due to methylation of a critical residue in the recognition motif). However, a comparison of unoccupied E2F consensus sites in a human breast cancer cell line to sites of repressive chromatin (that is, histone H3 trimethylated on lysine 9 or lysine 27 (H3K9me3 or H3K27me3)) and DNA methylation showed that neither repressive histone marks nor DNA methylation appeared to account for the lack of E2F binding 49. An alternative possibility is that specific histone modifications enhance transcription factor recruitment to certain genomic regions. For example, recent ChIP-chip and ChIP-seq studies have shown that histone H3 monomethylated at lysine 4 (H3K4me1) is localized at enhancer regions 45, 46. Of course, it is not known whether the histone modification or the binding of a factor comes first, but it is possible that certain factors might have an affinity for a specific histone modification. For example, PHD finger domains in several proteins, such as the TAF3 subunit of TFIID, BPTF, and ING2, can mediate a specific high affinity interaction with histone H3 trimethylated on lysine 4 55 56 57, 58, which is highly localized to promoter regions 3 46. PHD domains in site-specific factors or co-activators could help localize DNA binding factors to consensus motifs located in proximal promoters; other domains might mediate interaction of transcription factors or co-activators with H3K4me1, resulting in preferential occupancy of motifs located in enhancer regions (Figure 3D).
Although each of the models presented in Figure 3 are possible, it is generally not clear why some consensus motifs are occupied and others are not. Perhaps once we have binding maps for hundreds of factors, it will become obvious that binding of a factor to one motif commonly prevents another motif from being occupied by a different factor. For example, an ETS and an E2F binding site overlap in the MYC promoter and it is only after mutation of the E2F site that ETS1 can bind in vivo 59. Alternatively, as described above, we might find that stable binding is rarely mediated by a single DNA-protein interaction but requires cooperative binding between adjacent site-specific factors either through direct interaction between the two site-specific factors or indirect interaction through a platform such as a co-activator or co-repressor 60.
Are all occupied binding sites important?
The discovery of thousands of binding sites by genome-wide profiling has raised two important questions: can a factor occupy a certain site in many cell types but regulate transcription via binding to that site in only one (or a few) cell types, and is functional redundancy a built-in safeguard for maintaining accurate regulation of the genome?
Understanding gene expression data
Several recent studies have attempted to assess the functional importance of each of the thousands of binding sites for a given factor by altering the level of that factor in the cell. A frequent finding is that changing the level of a factor alters expression of 1-10% of the potential target genes 12, 37, 61, 62. One interpretation of these results is that most binding is not functional. There are, however, several caveats to this conclusion. First, the assignment of a specific binding site to a target gene is not always accurate. Investigators use the most expedient approach, which is to assign the binding site to the nearest known gene, but this can lead to false binding site - target gene pairing due to long-range regulation, undiscovered genes, or alternative upstream promoters. Changes in expression of a gene that does not have a nearby binding site for the factor that is altered might initially be interpreted as indicative of indirect regulation, but might be owing to direct regulation by a site many thousands of kb away (Figure 4a). Second, altering expression of a human transcription factor is fraught with problems. Down regulation of a transcription factor in human cells is usually accomplished using small interfering RNAs (siRNAs or shRNAs). However, loss of expression is rarely complete; it is possible that a reduction of 90% of the protein might not have functional consequences if there is a 10-fold excess of the factor under normal conditions. Many studies are performed in cancer cell lines that can have, as shown by western blot, a massive increase in the amount of particular transcription factor compared to a normal cell. Thus, what appears to be an efficient knockdown in a cancer cell line might leave sufficient levels of the factor for normal regulation (Figure 4B). Very few studies have actually shown reduced binding of a transcription factor in knockdown cells by ChIP-chip or ChIP-seq. To deal with this problem, mouse knockouts can be used. However, cells from these mice could undergo compensation for loss of a factor during development, resulting in related proteins being selected to regulate the targe genes. Third, closely related family members might bind to the same sites and have the same function. Thus, elimination of one family member could allow more binding of another family member (Figure 4C). Finally, only a small proportion of the binding sites for a factor might be functional in a given cell type. For example, if a cell type-specific partner needs to be recruited for transcriptional activity, then binding of the site-specific factor is necessary but not sufficient for transcription of a target gene (Figure 4D). Thus, knockdown of a factor in 10 cell types may show 10 different subsets of affected target genes. To address this possibility, one would have to collect ChIP-chip or ChIP-seq data and gene expression data before and after knockdown of the factor in a diverse set of cell lines. However, most transcription factors have been studied on a genome-wide scale in only one cell type. The ENCODE consortium (http://www.genome.gov/10005107) has chosen a set of different cell types for thorough characterization of binding of a large number of site-specific factors and initial studies appear to show that factors can be grouped into those that show very little cell type specificity in binding, such as E2F4 and YY1 (H. O’Geen and P. Farnham, unpublished observations) and those that show considerable cell type-specific binding, such as JunD (D. Raha and M. Snyder personal communication) and the estrogen receptor 15, 63, 64. Continuing studies will address whether factors that have small numbers of cell type-specific binding sites show regulation of a large percentage of their target genes in a given cell type compared to factors that show constitutive binding to a large number of sites and might regulate only a subset of target genes in each cell type.
Figure 4. Incorrect interpretation of functional assays.
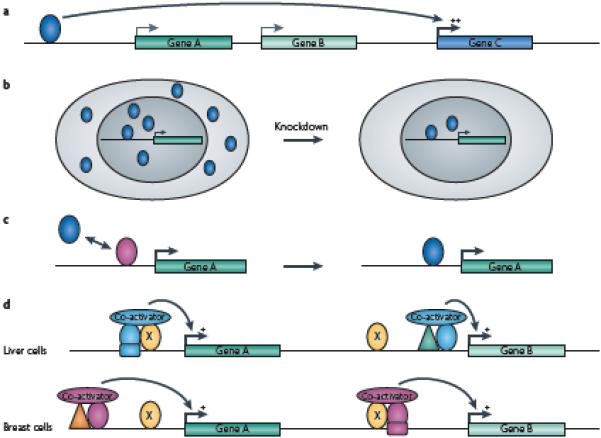
There are a number of reasons, other than a lack of function, why reduction in the level of a transcription factor might not result in a change in expression of the predicted target gene. (A) The transcription factor regulates a gene that is distal to the binding site; therefore, the nearest gene will not show a change in expression upon knockdown of the factor. (B) Knockdown of a factor with an siRNA does not lower the level below that needed for full binding site occupancy; therefore, expression of target genes is not affected. (C) Knockdown of a factor (pink triangle) results in full occupancy by another family member (green triangle) at a site that, under normal conditions, is bound interchangeably by both family members; target gene expression is not affected because the family members are redundant in function. (D) Regulation is dependent on the ubiquitous site-specific factor (yellow oval) in combination with cell type-specific factors. In this example, factor X (yellow oval) is bound to the promoter regions of gene A and gene B in both liver and breast cells and gene A and B are expressed in both tissues. However, in liver, factor X is not involved in regulation of gene B because there is no binding site for the liver-specific factor (blue oval) near the factor X binding site in the gene B promoter. Conversely, in breast cells, factor X regulates gene B through interaction with the breast-specific factor (pink oval) but does not regulate gene A because there is no binding site for the breast-specific factor near the factor X site in the gene A promoter. Thus different subsets of target genes may show changes in expression in different cell types when levels of the ubiquitous site-specific factor are reduced.
Functional redundancy within clusters
Many previous analyses of transcriptional regulation used the assumption that transcription factors act as “individuals”, with a specific assigned role in regulating a particular gene and a specific mechanism of action. However, it is possible that a factor acts as an individual at subset of its sites (perhaps those that show altered regulation of a nearby gene upon loss or enhanced expression of that factor), but has a very different “community” function at other sites. For example, binding of a set of factors in a cluster might regulate transcription throughout a chromatin domain by helping to keep an open chromatin structure, through recruitment of histone acetyltransferases or histone methyltransferases. Loss of single factor would not affect transcription of the nearby genes; it would take the removal of a large proportion of factors bound in the cluster to alter gene regulation (Figure 5a). Alternatively, a cluster of bound factors could serve to define a local genomic search space for a second binding factor. Recent studies have shown that many transcription factors have a very fast dissociation rate in vivo 65. A factor might rebind to the same region of DNA, but in a nonspecific manner, and begin scanning for its high affinity binding site. If the factor moves unimpeded in the wrong direction, there could be a detrimental time lag before it finds another binding site. However, a cluster of bound factors that blocks scanning in the wrong direction might favor release, rebinding, and perhaps scanning in the correct direction. That is, binding of a cluster of factors might affect the expression of a nearby gene whose activation is controlled by an entirely different factor. Again, reduced expression of one of the “bumper proteins” may be fairly inconsequential; loss of several factors from the cluster would be required to cause a significant effect (Figure 5b). Data to support either of these possibilities is not yet available due to the lack of genome-wide binding information for most transcription factors.
Figure 5. Communal action of a set of transcription factors.
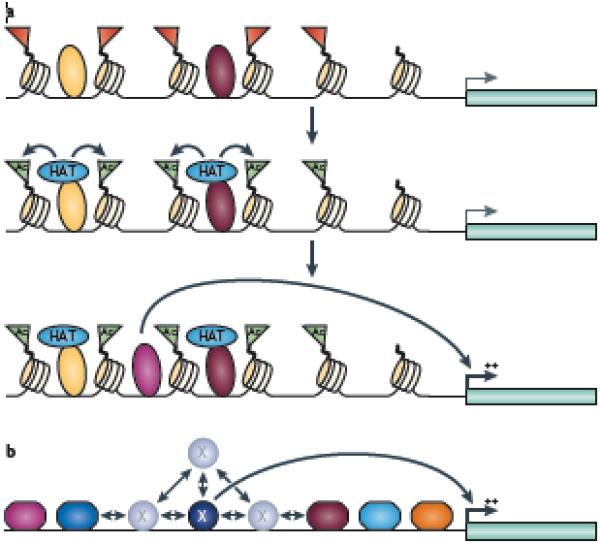
(A) The schematic shows a possible scenario in which two different factors (large yellow and dark red triangles) can bind near to each other on inactive chromatin (represented by the red flags) and each recruit a histone acetyltransferase (HAT), which acetylates histones and creates an open chromatin region (green flags), allowing the binding of another factor (diamond) that stimulates transcription of a gene. In this case, loss of a single factor that recruits a HAT would not result in a major change in regulation of the gene. (B) The schematic shows a possible scenario in which multiple factors (hexagons) bound on either side of factor X (circle) can create a limited search domain for factor X (which is required for activation of a downstream gene). Factor X binds transiently to its binding site; dissociation from the site is followed by localized rebinding and scanning for the high affinity binding site. Transcriptional activation can be enhanced if the scanning is spatially limited by adjacent clusters of other bound factors; loss of a single factor in the cluster would not result in a major change in regulation of the gene.
NEXT STEPS FOR GENOMIC LANDSCAPES
Although enormous progress has been made in mapping transcription factor binding sites throughout the genome and expanding the number of transcription factors for which we have information about global binding patterns is very important, simply collecting genome-wide datasets will not be sufficient to answer all crucial questions. A number of methodological problems now need to be tackled.
Accurate target gene assignment
It is not yet possible to conclusively link a specific binding site with a specific target gene. It remains possible that many binding sites, scattered perhaps tens or hundreds of kb away each other (or perhaps even on different chromosomes), all cooperate to regulate a single target gene. If so, then linking a binding site to the nearest gene is not appropriate and leads both to an incorrect assignment of target genes and to an underestimate of the number of binding sites that contribute to transcriptional regulation. Methods that define features of chromosomal architecture such as transcription factories 66, 67 could aid in defining co-regulated groups of genes, perhaps by collapsing thousands of seemingly unlinked binding sites into a smaller number of interactomes. For example, 3C, a technique which can identify chromosomal loops mediated by multiple, long range protein-protein interactions 68, might reveal a connection between an enhancer binding protein and the promoter of a distant gene, and thereby allow a more accurate interpretation of the regulatory role of that factor in the cell.
Comprehensiveness
Although ChIP-seq can identify all the binding sites for a given factor in a given cell type, the possibility that we must perform ChIP-seq experiments in many different cell types to determine all possible binding sites for a given factor is quite daunting. The ENCODE Consortium is currently performing studies to estimate how many cell types are needed to identify most binding sites for a set of factors. If a limited, but diverse, set of cell types could be identified that are representative of many different human tissues, then perhaps genome-wide analyses will not have to be performed in every possible cell type.
Functional analysis of specific regulatory elements
Most approaches designed to study the relationship between a specific cis element and a potential target gene involve creating a reporter construct that includes the regulatory element of interest 4, 69. Unfortunately, as reporter analyses remove the cis element from its normal genomic context they cannot reveal effects on long-range regulation. Precise mutation or deletion of a single cis element within the genome can be performed in model organisms such as yeast, for which efficient methods to substitute genomic sections have been developed. Theoretically, mutations could be engineered to alter a specific binding site in animal models or human cell lines. However, mutagenesis of specific small regions of the mouse or human genome is not routinely used to study the significance of individual binding sites, due to low frequencies of homologous recombination that limit the efficiency of this technique. New approaches in site-specific targeting of DNAses using artificial zinc fingers 70 might improve the efficiency of genomic replacement, so mutagenesis could become a practical method for dissecting the role of individual cis elements. Also, artificial zinc fingers fused to transcriptional activation or repression domains have been used to specifically regulate cellular promoters 71. It is therefore possible that artificial zinc fingers (without either an activation or repression domain) could be used to simply block access of a factor to single binding site in the genome, but this has not yet been demonstrated successfully. Other possible methods include the use of pyrrole-imidazole polyamides or peptide nucleic acids to bind to (and perhaps also mutate) specific cis elements in the genome72 71 73. Although very few studies have used these methods to target a specific site and even fewer have examined the consequences of such agents on the entire transcriptome, they do hold the promise of providing a method to test the function of a specific binding site in its natural genomic context.
CONCLUSIONS
ChIP-chip and ChIP-seq have greatly advanced our understanding of gene regulation. First, genomic studies have confirmed that RNAPII together with general and site-specific factors are bound to thousands of proximal promoters that are active at very low levels 74-76, thus supporting the first step in the model set out in the introduction. These studies have also revealed that binding of a factor to an enhancer region can be necessary, but not sufficient, for high levels of promoter activity, which leads to the inclusion of a new step in the model (Figure 6, step 3): the binding of a cell type-specific partner protein that allows the recruitment of a coactivator, resulting in cell type-specific function of a constitutively expressed factor. Although the principle that binding of a transcription factor can be necessary, but not sufficient, for regulation of a specific gene was previously established using one-gene-at-a-time approaches, it was not clear whether a cooperative mode of regulation was the exception or the rule for most genes. Recent genome-wide analyses suggest that this type of regulation is very common. For example, of the ~3700 Oct4, ~4500 Sox2 and ~10,000 Nanog binding sites identified in mouse ES cells, only a small number of regions were bound by all three factors and by the co-activator p30038. These studies support the hypothesis that occupancy of an upstream site by a single factor (Oct4) was not functional (as in Figure 6 step 2), but binding of Sox2 and/or Nanog near to the occupied Oct4 site resulted in recruitment of the p300 coactivator (step 3) and transcriptional activation.
Other discoveries, such as the findings that most transcription factors bind to thousands of places in the genome, that binding sites are not localized only in proximal promoter regions, and that some binding sites lack sequences similar to the consensus motif, have also stimulated new ideas concerning long range and combinatorial regulation. However, current genomic studies have not yet determined whether most transcription factors cluster at hotspots in the human genome or with what frequency binding events have a functional outcome. The answers to these two questions will require the genomic profiling of many more factors. It is likely that a true understanding of the role of a given factor at a particular site in the genome will require the identification of all other factors binding nearby and knowledge of histone modifications in that region. These studies will be best performed by a cooperation between large groups such as the ENCODE Consortium (http://www.genome.gov/10005107) and the NIH Roadmap Epigenomics Program (http://nihroadmap.nih.gov/epigenomics/) that can identify binding sites for a large number of transcription factors and develop reference epigenomes in many different cell types and individual investigators who can perform the follow-up functional analyses of the role of a specific factor in a particular cell type. The next several years of large-scale data collecting should provide investigators with a plethora of information that will form the basis for hundreds of follow-up experiments that address important biological questions.
ONLINE SUMMARY.
Alterations in gene expression caused by the inappropriate level, structure, or function of a transcription factor have been associated with a diverse set of human diseases. However, because most human transcription factors are essentially uncharacterized, the role of transcription factors in human health is currently greatly underappreciated.
Technological advances (such as ChIP-chip and ChIP-seq) now allow transcription factor binding to be studied on a genome-wide scale.
Recent discoveries, such as the finding that most transcription factors bind to thousands of places in the genome, that binding sites are not only localized to proximal promoter regions, and that some binding sites lack sequences similar to the consensus motif, have stimulated new ideas concerning long range and combinatorial regulation.
Current genomic studies have not yet determined whether most human transcription factors bind alone or if they cluster at hot spots in the genome. Answers to these questions require the genomic profiling of many more factors.
A critical unanswered question is whether all binding events have a functional outcome (perhaps under some specific condition or in a specific cell type) or whether some transcription factor/genome interactions are simply irrelevant.
Issues that remain to be addressed include the design of comprehensive studies (for example, should all factors be studied in all cell types?) and functional validation (for example, how can we determine the role of one specific binding site in its normal genomic context?).
Acknowledgments
The author thanks Xiaoqin Xu, Henriette O’Geen, and Seth Frietze for providing data used in Figure 2 and the members of the Farnham lab for their insights and discussions.
GLOSSARY
- TATA Box
A consensus sequence within promoters that is enriched in thymine and adenine residues, and is important for the recruitment of the general transcriptional machinery at some promoters.
- Initiator
An element, with a consensus of YYANWYY where A is the transcription start site, which helps to recruit the general transcriptional machinery to promoters.
- Initiation complex
The assembly of RNA Polymerase and associated general factors that binds to the core promoter region.
- CpG island
A sequence of at least 200 bp with a greater number of CpG sites than expected for its GC content. These regions are often GC rich, typically undermethylated, and correspond to promoter regions of many mammalian genes.
- Enhanceosome
A protein complex that binds to an enhancer region (which can be located upstream, downstream, or within a gene); the transcription factors that compose the enhanceosome are thought to work cooperatively to stimulate transcription.
- PHD finger
(Plant Homeo Domain) A 50-80 amino acid domain that contains a Cys4-His-Cys3 motif. It is found in more than 100 human proteins, several of which are involved in chromatin-mediated gene regulation.
- Small interfering RNAs (siRNAs)
Small antisense RNAs (20–25 nucleotides long) that can be directly introduced into cells or be generated within cells from longer dsRNAs. They serve as guides for the cleavage of homologous mRNA in the RNA-induced silencing complex (RISC).
- Transcription factory
A nuclear subcompartment that is rich in RNA polymerases and transcription factors where there is clustering of active genes.
- Interactome
A complete set of macromolecular interactions (physical and genetic). Current use of the word tends to refer to a comprehensive set of protein–protein interactions. However, the protein-DNA interactome (a network formed by transcription factors and their target genes) is also commonly studied.
- Silencer
A DNA sequence capable of binding transcription factors termed repressors, which can negatively influence transcription by preventing recruitment of the general transcriptional machinery or by recruiting histone-modifying complexes that create repressive chromatin structures.
- TFIID
(Transcription Factor II D) A protein complex composed of several subunits called TBP-associated factors (TAFs) and the TATA Binding Protein (TBP). It is one of several complexes that make up the RNA polymerase II initiation machinery.
- Heterochromatin
Chromatin that is characterized by very dense packing of DNA, which makes it less accessible to transcription factors. Certain regions of the genome, such as centromeres and telomeres, are always heterochromatinized (constitutive heterochromatin) whereas other regions are densely packed and repressed only in certain cells (facultative heterochromatin).
- Reporter construct
A plasmid containing a promoter (and sometimes an enhancer) cloned upstream of a reporter gene (often simply called the reporter) which is introduced into cultured cells, animals or plants. Certain genes are chosen as reporters because their products can be easily or quantitatively assayed or used as selectable markers.
- DNA methylation
An epigenetic DNA modification that can be added and removed without changing the original DNA sequence and which is characterized by the addition of a methyl group to the number 5 carbon of the cytosine pyrimidine ring.
- Artificial Zinc finger protein
Chimeras of zinc finger domains – small protein domains that coordinate one or more zinc ions, commonly found in mammalian transcription factors - and an effector domain (for example, activator, repressor, methylase, or nuclease). Linking together six zinc fingers produces a target-site of 18 bp, which is long enough to be unique in all known genomes.
- DamID
An alternate method to ChIP employing a DNA-binding protein fused to a DNA methyltransferase. Adenine methylation of a region identifies it as being located near a binding site.
AUTHOR BIOGRAPHY
Dr. Farnham earned a B.A. from Rice University, Houston, USA, a PhD from Yale University, New Haven, USA, and performed postdoctoral work at Stanford University, Palo Alto, USA. She was a faculty member at University of Wisconsin, Madison, USA, from 1987-2004 and moved to University of California Davis, USA, in 2004 where she is Associate Director of Genomics. Dr. Farnham has been a leader in using chromatin immunoprecipitation (ChIP) to study mammalian transcription factors. Currently, she is using ChIP with high throughput sequencing (ChIP-seq) to analyze chromatin structure, as a member of a Reference Epigenome Mapping Center, and for identification of target genes of human transcription factors, as a member of the ENCODE Consortium.
Footnotes
References
- 1.Lee TI, Young RA. Transcription of eukaryotic protein-coding genes. Annu. Rev. Genet. 2000;34:77–137. doi: 10.1146/annurev.genet.34.1.77. A detailed review of transcriptional regulation, general factors, and accessory proteins that control transcription initation and elongation.
- 2.Sandelin A, et al. Mammalian RNA polymerase II core promoters: insights from genome-wide studies. Nat Reviews Genetics. 2007;8:424–436. doi: 10.1038/nrg2026. [DOI] [PubMed] [Google Scholar]
- 3.Consortium TEP. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. Demonstrates how genome-wide studies of transcription, factor binding, chromatin structure, DNA replication, and sequence conservation can synergize.
- 4.Cooper SJ, Trinklein ND, Anton ED, Nguyen L, Myers RM. Comprehensive analysis of transcriptional promoter structure and function in 1% of the human genome. Genome Research. 2006;16:1–10. doi: 10.1101/gr.4222606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kimura K, et al. Diversification of transcriptional modulation: Large-scale identification and characterization of putative alternative promoters of human genes. Genome Res. 2006;16:55–65. doi: 10.1101/gr.4039406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 7.Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. A census of human transcription factors: function, expression and evolution. Nat Reviews Genetics. 2009;10:252–263. doi: 10.1038/nrg2538. A summary of the expression, conservation, and activity of the set of human sequence-specific transcription factors.
- 8.Venter JC, et al. The sequence of the human genome. Science. 2001;291:1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 9.Jimenez-Sanchez G, Childs B, Valle D. Human disease genes. Nature. 2001;409:853–855. doi: 10.1038/35057050. [DOI] [PubMed] [Google Scholar]
- 10.Wederell ED, et al. Global analysis of in vivo Foxa2-binding sites in mouse liver using massively parallel sequencing. Nucleic Acids Res. 2008;36:4549–4564. doi: 10.1093/nar/gkn382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reed BD, Charos AE, Szekely AM, Weissman SM, Snyder M. Genome-wide occupancy of SREBP1 and its partners NFY and SP1 reveals novel functional roles and combinatorial regulation of distinct classes of genes. PLOS Genetics. 2008;4:e1000133. doi: 10.1371/journal.pgen.1000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scacheri PC, et al. Genome-wide analysis of menin binding provides insights to MEN1 tumorigenesis. PLoS Genet. 2006;2:e51. doi: 10.1371/journal.pgen.0020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hatzis P, et al. Genome-wide pattern of TCF7L2/TCF4 chromatin occupancy in colorectal cancer cells. Mol. Cell. Biol. 2008;28:2732–2744. doi: 10.1128/MCB.02175-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Geen H, et al. Genome-Wide Analysis of KAP1 Binding Suggests Autoregulation of KRAB-ZNFs. PLoS Genet. 2007;3:e89. doi: 10.1371/journal.pgen.0030089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu X, et al. A comprehensive ChIP-chip analysis of E2F1, E2F4, and E2F6 in normal and tumor cells reveals iterchangeable roles of E2F family members. Genome Res. 2007;17:1550–1561. doi: 10.1101/gr.6783507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robertson G, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:1–7. doi: 10.1038/nmeth1068. An early demonstration that high throughput sequencing of ChIP samples can be used to identify genome-wide binding sites of site-specific transcription factors.
- 17.Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 18.Rada-Iglesias A, et al. Whole-genome maps of USF1 and USF2 binding and histone H3 acetylation reveal new aspects of promoter structure and candidate genes for common human disorders. Genome Research. 2008;18:380–392. doi: 10.1101/gr.6880908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vogel MJ, Peric-Hupkes D, van Steensel B. Detection of in vivo protein-DNA interactions using DamID in mammalian cells. Nat Protoc. 2007;2:1467–78. doi: 10.1038/nprot.2007.148. [DOI] [PubMed] [Google Scholar]
- 20.Valouev A, et al. Genome-wide analysis of transcription factor binding sites based on ChIP-seq data. Nature Methods. 2008;5:829–834. doi: 10.1038/nmeth.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson DS, et al. Systematic evaluation of variability in ChIP-chip experiments using predefined DNA targets. Genome Research. 2008;18:393–403. doi: 10.1101/gr.7080508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bieda M, Xu X, Singer M, Green R, Farnham PJ. Unbiased location analysis of E2F1 binding sites suggests a widespread role for E2F1 in the human genome. Genome Research. 2006;16:595–605. doi: 10.1101/gr.4887606. Demonstration that some factors bind exclusively to proximal promoters and do not have strict motif requirements for their binding sites.
- 23.Kim TH, et al. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–80. doi: 10.1038/nature03877. An early demonstration that high density oligonucleotide arrays can be used to identify genome-wide binding sites for human transcription factors.
- 24.Cawley S, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116:499–509. doi: 10.1016/s0092-8674(04)00127-8. [DOI] [PubMed] [Google Scholar]
- 25.Nix DA, Courdy SJ, Boucher KM. Empirical methods for controlling false positives and estimating confidence in ChIP-seq peaks. BMC Bioinformatics. 2008;9:523. doi: 10.1186/1471-2105-9-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biology. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fejes AP, et al. FindPeaks 3.1: a tool for identifying areas of enrichment from massively parallel short-read sequencing technology. Bioinformatics. 2008;24:1729–1730. doi: 10.1093/bioinformatics/btn305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rozowsky J, et al. PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat Biotechnol. 2009;27:66–75. doi: 10.1038/nbt.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Michalopoulos GK, Zarnegar R. Structural and functional characterization of the mouse hepatocyte growth factor gene promoter. J Biol Chem. 1994;269:4152–60. [PubMed] [Google Scholar]
- 30.Fujishiro K, et al. Analysis of tissue-specific and PPARalpha-dependent induction of FABP gene expression in the mouse liver by an in vivo DNA electroporation method. Mol Cell Biochem. 2002;239:165–72. [PubMed] [Google Scholar]
- 31.Weinmann AS, Yan PS, Oberley MJ, Huang TH-M, Farnham PJ. Isolating human transcription factor targets by coupling chromatin immunoprecipitation and CpG island microarray analysis. Genes & Dev. 2002;16:235–244. doi: 10.1101/gad.943102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Z, et al. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc Natl Acad Sci U S A. 2003;100:8164–9. doi: 10.1073/pnas.1332764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ren B, et al. E2F integrates cell cycle progression with DNA repair, replication, and G2/M checkpoints. Genes & Dev. 2002;16:245–256. doi: 10.1101/gad.949802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Odom DT, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303:1378–1381. doi: 10.1126/science.1089769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Consortium TEP. The ENCODE (ENCyclopedia of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 36.Carroll JS, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 37.Yang A, et al. Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol. Cell. 2006;24:593–602. doi: 10.1016/j.molcel.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 38.Chen X, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–17. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 39.Mann RS, Carroll SB. Molecular mechanisms of selector gene function and evolution. Curr Opin Genet Dev. 2002;12:592–600. doi: 10.1016/s0959-437x(02)00344-1. [DOI] [PubMed] [Google Scholar]
- 40.Moorman C, et al. Hotspots of transcription factor colocalization in the genome of Drosophila melanogaster. Proc Natl Acad Sci U S A. 2006;103 doi: 10.1073/pnas.0605003103. Demonstrates clustering of transcription factors throughout the Drosophila genome.
- 41.Elnitski L, Jin VX, Farnham PJ, Jones SJM. Locating Mammalian Transcription Factor Binding Sites: A Survey of Computational and Experimental Techniques. Genome Research. 2006 Oct 19; doi: 10.1101/gr.4140006. In Advance. [DOI] [PubMed] [Google Scholar]
- 42.Panne D. The enhanceosome. Curr Opin Structural Biol. 2008;18:236–242. doi: 10.1016/j.sbi.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 43.Maniatis T, et al. Structure and function of the interferon-B enhanceosome. Cold Spring Harb Symp Quant Biol. 1998;63:609–620. doi: 10.1101/sqb.1998.63.609. [DOI] [PubMed] [Google Scholar]
- 44.Dean A. On a chromosome far, far away: LCRs and gene expression. Trends in Genetics. 2006;22:38–45. doi: 10.1016/j.tig.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 45.Heintzman ND, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. Identifies specific histone modifications that are associated with cell type specific transcriptional regulation.
- 46.Heintzman ND, et al. Distinct predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature Genetics. 2007 Feb 4; doi: 10.1038/ng1966. published online. 2007. [DOI] [PubMed] [Google Scholar]
- 47.Wright WE, Funk WD. CASTing for multicomponent DNA-binding components. Trends Biochem. Sci. 1993;18:77–80. doi: 10.1016/0968-0004(93)90156-h. [DOI] [PubMed] [Google Scholar]
- 48.Morgan XC, Ni S, Miranker DP, Iyer VR. Predicting combinatorial binding of transcription factors to regulatory elements in the human genome by association rule mining. BMC Bioinformatics. 2007;8:445. doi: 10.1186/1471-2105-8-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rabinovich A, Jin VX, Rabinovich R, Xu X, Farnham PJ. E2F in vivo binding specificity: comparison of consensus vs. non-consensus binding sites. Genome Research. 2008;18:1763–77. doi: 10.1101/gr.080622.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gineitis D, Treisman R. Differential usage of signal transduction pathways defines two types of serum response factor target gene. J. Biol. Chem. 2001;276:24531–24539. doi: 10.1074/jbc.M102678200. [DOI] [PubMed] [Google Scholar]
- 51.Cooper SJ, Trinklein ND, Nguyen L, Myers RM. Serum response factor binding sites differ in three human cell types. Genome Research. 2009;17:136–144. doi: 10.1101/gr.5875007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jin VX, O’Geen H, Iyengar S, Green R, Farnham PJ. Identification of an OCT4 and SRY regulatory module using integrated computational and experimental genomics approaches. Genome Research. 2007;17:807–817. doi: 10.1101/gr.6006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li X, MacArthur S, et al. Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm. PLOS Biology. 2008;6:e27. doi: 10.1371/journal.pbio.0060027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Badis G, et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009;324:1720–3. doi: 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, et al. Molcular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pena PV, et al. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–103. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shi X, et al. Proteome-wide analysis in Saccharomyces cerevisiae identifies several PHD fingers as novel direct and selective binding modules of histone H3 methylated at either lysine 4 or lysine 36. J. Biol. Chem. 2007;282:2450–2455. doi: 10.1074/jbc.C600286200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vermeulen M, et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 59.Albert T, et al. The chromatin structure of the dual c-Myc promoter P1/P2 is regulated by separate elements. J. Biol. Chem. 2001;276:20482–20490. doi: 10.1074/jbc.M100265200. [DOI] [PubMed] [Google Scholar]
- 60.Jin V, Rabinvich A, Squazzo SL, Green R, Farnham PJ. A computational genomics approach to identify cis-regulatory modules from chromatin immunoprecipitation microarray data-a case study using E2F1. Genome Research. 2006;16:1585–1595. doi: 10.1101/gr.5520206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krig SR, et al. Identification of genes directly regulated by the oncogene ZNF217 using chromatin immunoprecipitation (ChIP)-chip assays. J. Biol. Chem. 2007;282:9703–9712. doi: 10.1074/jbc.M611752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martone R, et al. Distribution of NF-kappaB-binding sites across human chromosome 22. Proc Natl Acad Sci U S A. 2003;100:12247–12252. doi: 10.1073/pnas.2135255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krum SA, et al. Unique ERalpha cistromes control cell type-specific gene regulation. Mol. Endocrinol. 2008;22:2393–2406. doi: 10.1210/me.2008-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Raha D, Snyder M. unpublished data. [Google Scholar]
- 65.Voss TC, Hager GL. Visualizing chromatin dynamics in intact cells. Biochem Biophys Acta. 2008;1783:2044–2051. doi: 10.1016/j.bbamcr.2008.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Osborne CS, et al. Active genes dynamicaly colocalize to shared sites of ongoing transcription. Nat Genet. 2004;36:1065–1071. doi: 10.1038/ng1423. [DOI] [PubMed] [Google Scholar]
- 67.Bartlett J, et al. Specialized transcription factories. Biochem Soc Symp. 2006;73:67–75. doi: 10.1042/bss0730067. [DOI] [PubMed] [Google Scholar]
- 68.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–11. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 69.Visel A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. Demonstrates that binding sites identified for p300 by ChIP-seq is a highly accurate means for identifying enhancers that can be shown, using follow-up assays in transgenic mice, to function in a tissue-specific manner.
- 70.Camenisch TD, Brilliant MH, Segal DJ. Critical parameters for genome editing using zinc finger nucleases. Mini Rev Med Chem. 2008;8:669–676. doi: 10.2174/138955708784567458. [DOI] [PubMed] [Google Scholar]
- 71.Bletran A, Liu Y, Parikh S, Temple B, Blancafort P. Interrogating genomes with combinatorial artificial transcription factor libraries: asking zinc finger questions. Assay Drug Dev Technol. 2006;4:317–331. doi: 10.1089/adt.2006.4.317. [DOI] [PubMed] [Google Scholar]
- 72.Faruqi AF, Egholm M, Glazer PM. Peptide nucleic acid-targeted mutagenesis of a chromosomal gene in mouse cells. Proc Natl Acad Sci U S A. 1998;95:1398–1403. doi: 10.1073/pnas.95.4.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Burnett R, et al. DNA sequence-specific polyamides alleviate transcription inhibition associated with long GAA-TCC repeats in Friedreich’s ataxia. Proc Natl Acad Sci U S A. 2006;103:11497–11502. doi: 10.1073/pnas.0604939103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Muse GW, et al. RNA polymerase is poised for activation across the genome. Nat. Genet. 2007;39:1507–1511. doi: 10.1038/ng.2007.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Komashko VM, et al. Using ChIP-chip technology to reveal common principles of transcriptional repression in normal and cancer cells. Genome Research. 2008;18:521–532. doi: 10.1101/gr.074609.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Acevedo LG, et al. Genome-scale ChIP-chip analysis using 10,000 human cells. Biotechniques. 2007;43:791–797. doi: 10.2144/000112625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.O’Neill LP, VerMilyea MD, Turner BM. Epigenetic characterization of the early embryo with a chromatin immunoprecipitation protocol applicable to small cell populations. Nature Genetics. 2006;38:835–841. doi: 10.1038/ng1820. [DOI] [PubMed] [Google Scholar]
- 79.Dahl JA, Collas P. Q2ChIP, a quick and quantitative chromatin immunoprecipitation assay, unravels epigenetic dynamics of developmentally regulated genes in human carcinoma cells. Stem Cells. 2007;25:1037–1046. doi: 10.1634/stemcells.2006-0430. [DOI] [PubMed] [Google Scholar]
- 80.Attema JL, et al. Epigenetic characterization of hematopoietic stem cell differentiation using miniChIP and bisulfite sequencing analysis. Proc. Natl. Acad. Sci. USA. 2007;104:12371–12376. doi: 10.1073/pnas.0704468104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xu H, Wei C-L, Lin F, Sung W-K. An HMM approach to genome-wide identification of differential histone modification sites from ChIP-seq data. Bioinformatics. 2008;24:2344–2349. doi: 10.1093/bioinformatics/btn402. [DOI] [PubMed] [Google Scholar]
- 82.Jothi R, Cuddapah S, Barski A, Cui K, Zhao K. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-seq data. Nucleic Acids Res. 2008;36:5221–5231. doi: 10.1093/nar/gkn488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hoffman BG, Jones SJ. Genome-wide identification of DNA-protein interactions using chromatin immunoprecipitation coupled with flow cell sequencing. J Endocrinol. 2009;201:1–13. doi: 10.1677/JOE-08-0526. [DOI] [PubMed] [Google Scholar]