

Abstract
Evolutionarily conserved Notch signaling plays a critical role during embryonic and postnatal life. The importance of Notch signaling in the determination of cell fate, and the spatio-temporal regulation of proliferation, differentiation and apoptosis has been demonstrated in various different organ systems. However, how Notch signaling affects the bone development was unknown until now. The in vivo effects of Notch signaling in lineage commitment, bone formation and bone resorption were demonstrated in recent studies. In addition to regulation of osteoblastogenesis, osteoblast directed osteoclastogenesis by Notch signaling revealed a dimorphic effect for this signaling pathway providing another example of such in bone development. Moreover, identification of the cross-talk between the hematopoietic stem cell niche and osteoblasts through Notch signaling also suggested another important role for Notch signaling, i.e., the coupling of cellular components of the bone microenvironment. The association between the gain and loss of function of Notch activity in bone pathology highlights Notch as a potentially novel therapeutic target for the treatment of metabolic bone disease and bone cancer. In this review, we will focus primarily on the regulation of bone cells, i.e., osteoblasts and osteoclasts by Notch signaling. We will also review the importance of Notch in specifying bone-hematopoietic stem cell niche interactions within the bone microenvironment. Finally, we will discuss potential clinical implications and future directions for this field.
Mammalian Notch signaling
Evolutionarily conserved Notch signaling plays an important role in developmental processes and adult tissue homeostasis by regulating cell fate determination, proliferation, differentiation and apoptosis in a spatio-temporal manner. Altered Notch signaling has been associated with many different diseases including cancers of epithelial and hematopoietic origins. The Notch receptor and its ligands are transmembrane proteins whose signaling requires cell to cell contact between neighboring cells. Mammals have four Notch receptors (Notch1-4) each containing an extracellular domain with numerous epidermal growth factor (EGF) like repeats, and three Notch/LIN-12 repeats. A Ram domain, six tandem ankyrin repeats and a PEST sequence are found in the intracellular domain. The mammalian Notch ligands fall into two classes: Delta and Jagged [1]. Notch proteins are synthesized as full-length unprocessed proteins, and following transport through the secretory pathway to the trans-Golgi network, Notch is first cleaved by a furin-like enzyme to generate two non-covalently attached subunits. This heterodimeric transmembrane receptor is then transferred to the cell surface. Here, physical interaction between the EGF repeats of the ligand and Notch receptor results in a cleavage in the extracellular domain of the receptor by the metalloproteinase tumor necrosis factor-α converting enzyme (TACE). After this cleavage, the Notch receptor becomes a substrate for the γ-secretase complex and is cleaved by the γ-secretase complex containing Presenilin1/2, nicastrin, Pen-2 and Aph-1 [2, 3]. While the first proteolytic cleavage does not cause activation of the Notch receptor, the second and third cleavages are necessary for the activation of the receptor [1, 4]. After the third cleavage, the Notch intracellular domain (Notch ICD) is released from the membrane, and translocated to the nucleus.
In the nucleus, Notch ICD binds to the Rbp-Jκ (alias CBF1) transcription factor and converts it from a transcriptional repressor to a transcriptional activator. Mastermind-like (MAML), another transcription factor required for Notch signaling, forms a ternary complex with RBP-Jκ and Notch ICD and recruits transcriptional co-activators to induce the expression of a basic-helix-loop-helix (bHLH) family of genes (Figure 1). In mammals, at least two families of bHLH proteins are downstream of Notch signaling: The hairy/enhancer of split [5] family, and the hairy-related transcription factor (HRT; HEY, HESR) family. Both HES and HRT families can function as transcriptional repressors.
Figure 1. Canonical Notch signaling in mammals.

Upon binding of Notch ligand (Delta-like or Jagged) to Notch receptor (1-4) on an adjacent cell, a series of proteolytic cleavages occurs (TACE, γ-secretase), resulting in the release of the Notch intracellular domain (Notch ICD). Notch ICD subsequently translocates into the nucleus. In the absence of nuclear Notch ICD, the transcription factor Rbp-Jκ is bound to co-repressors (Co-R) and represses transcription. When Notch signaling is activated, nuclear Notch ICD binds to Rbp-Jκ and recruits the nuclear protein Mastermind Like (MAML) to form a ternary complex that functions as a transcriptional activator activating the transcription of Notch target genes including those belonging to the Hes and Hey families.
Bone formation and remodeling
Skeleton forms through two different mechanisms; endochondral and intramembraous ossification [6, 7]. During endochondral ossification, cells located in the center of mesenchymal condensations differentiate into chondrocytes. Later, chondrocytes at the growth plate undergo well-ordered and controlled phases of cell proliferation, maturation, and apoptosis in order to form the future skeletal elements [8]. During intramembranous ossification, mesenchymal cells give rise to pre-osteoblasts to form future bone. Pre-osteoblasts differentiate into functional osteoblasts through the transcription factors Runx2 and Osterix. These mature osteoblasts are responsible for the synthesis of most proteins of the extracellular bone matrix, including expression of the genes that are necessary for mineralization and regulation of osteoclastogenesis.
Bone remodeling is a process of continuous resorption and neo-synthesis of bone that determines bone structure and quality during adult life. Bone formation and bone resorption are coupled processes. Osteoblasts (that form the bone) can also regulate osteoclast formation by expressing the negative regulator Osteoprotegrin (Opg), or positive regulators RANKL and MSCF. Imbalances of bone remodeling can result in severe perturbations in skeletal structure and function such as osteoporosis, osteosclerosis, and osteopetrosis. Osteoporosis is a disease of bone in which the bone mineral density (BMD) is reduced, the bone micro architecture is disrupted, and the amount of non-collagenous proteins in bone is altered due to an imbalance in bone remodeling. Osteoporotic bones are more at risk of fracture. Another bone related disease osteopetrosis is a disease of osteoclastic dysfunction characterized by failure of bone resorption. Despite this excess bone formation, individuals with osteopetrosis tend to have bones that are more brittle than normal. In contrast, osteosclerosis is a bone disorder characterized by an abnormal hardening and progressive increase in bone mass of the skeleton resulting from increased bone formation. Unlike osteopetrosis, the primary defect this disorder causing increased bone mass results from altered osteoblast function. Hence, bone mass is regulated by a balance of bone formation by osteoblasts vs. bone resorption by osteoclasts. Differentiation, proliferative, apoptotic, and/or functional defects of either or both of these cell types can lead to bone diseases.
Notch signaling during skeletogenesis
Until recently, the role of Notch signaling during skeletogenesis has mainly been limited to its role in patterning and somitogenesis. The involvement of the Notch signaling pathway in somitogenesis was first revealed by the finding that Notch1 and its ligand Dll1 were highly expressed in the presomitic mesoderm (PSM) of mouse embryos. Subsequently, additional cycling genes, such as Lunatic fringe (Lfng), Hes7, and Hes1 were identified in different species to be components of this pathway [9, 10]. All of these genes are linked to the Notch signaling pathway suggesting that Notch-signaling activity in the PSM is itself oscillating and is either controlled by the segmentation clock or is a central component of it. In support of this, Notch1 null mouse embryos exhibited significantly delayed and disorganized somitogenesis[11]. The Rbp-Jκ null embryos displayed slightly more severe somitogenesis due to complete loss of the Notch sigaling [12]. Knock-out models and spontaneous mutations of Notch signaling components such as Delta-like 1(Dll1), Dll3, Presenilin1, Lunatic fringe and Hes7 also led to somitic phenotypes (Table 1). Together, these studies clearly implicate Notch signaling in the direct regulation of segmentation. Consistent with this, two human disorders, Spondylocostal dysostosis (SCD) and Alagille syndrome (AGS), caused by mutations in Notch pathway genes, exhibit vertebral column defects indicating that correct, cyclic function of the Notch pathway within the vertebrate segmentation clock is essential for proper somitogenesis in both mice and humans [13-15].
Table 1.
Summary of mouse mutant phenotypes of Notch signaling components and related human diseases.
| Gene | Protein Type | Loss of Function | Mutations in Human |
|---|---|---|---|
| Notch 1 | Receptor | Lethal before E11.5 | T cell neoplasm |
| Notch 2 | Receptor | Lethal before E11.5 | |
| Notch 3 | Receptor | Normal and fertile | CADASIL |
| Notch 4 | Receptor | Normal and fertile | Schizophrenia |
| Delta-like3 | Ligand | Lethal at birth or P10 | Spondylocostal Dysostosis |
| Jagged 2 | Ligand | Lethal right after birth | |
| Jagged 1 | Gamma secretase | Lethal at E10 | Alagille Syndrome |
| PS1 | Gamma secretase | Lethal right after birth | Alzheimer |
| PS2 | Gamma secretase | Normal and fertile | Alzheimer |
| PS1/PS2 | Gamma secretase | Lethal at E9.5 | Alzheimer |
| RBP-Jκ | Transcription factor | Lethal before E10.5 |
Apart from its role in somitogenesis, several in vitro studies with conflicting results implicated the Notch pathway in the regulation of osteoblast differentiation. In these studies, the expression of Notch1 in MC3T3-E1 osteoblastic cells at early differentiation stages was detected. When Notch1 ICD was delivered by an adenovirus vector to osteoblastic MC3T3-E1 cells, a significant increase in calcified nodule formation was observed. Similarly, when the C3H10T1/2 multi-potent mesenchymal cell line was infected with an adenovirus expressing EGFP-Notch1ICD, stimulation of osteoblastic differentiation and inhibition of adipogenesis was observed, suggesting that osteoblastic differentiation was influenced by Notch [16]. In support of this, the Notch target gene Hes-1 was shown to interact with Runx2 and increase its transactivation effect on an osteoblast specific enhancer [17]. On the other hand, while these studies support a role for Notch in the stimulation of osteoblastogenesis, other groups have reported the opposite results also in vitro [18-20]. In these studies, over expression of Notch1 in stromal and mesenchymal cells impaired osteoblastic differentiation.
The possibility of Notch regulation of osteoclastogenesis was also investigated in an in vitro study, in which osteoclastogenesis was shown to be inhibited by an immobilized Notch ligand, Delta-1 [21]. Constitutively active Notch1-transfected stromal cells showed increased expression of RANKL and OPG genes, and strong inhibition of M-CSF expression, resulting in negative regulation of osteoclastogenesis thus providing the first clue of cross talk between osteoblasts and osteoclasts that might be mediated by Notch signaling.
The distribution of Notch receptors and their ligands during articular cartilage development has also been reported [22]. In vitro studies suggested that Notch negatively regulates the initiation of pre-chondrogenic condensation and nodule formation, and later differentiation and proliferation of early chondrogenic cells are suppressed by Notch [23-25]. Notch signaling has been shown to be required for the chondrogenic specification of mouse mesencephalic neural crest cells. The expression of Sox9, a transcription activator of collagen type II, was up-regulated by Notch activation and this activation of Notch signaling thereby promoted differentiation of proliferative and prehypertrophic chondrocytes [26]. On the other hand, transduction of human mesenchymal stem cells hMSCs with an adenovirus expressing Jagged-1 activated Hey-1 expression, which resulted in up-regulation of type II collagen expression. Jagged-1-transduced hMSCs, which exposed to continuous elevated expression of Jagged-1, showed a complete inhibition of chondrogenesis [27]. Thus, Jagged-1-mediated Notch signaling in hMSC was necessary to initiate chondrogenesis, but it must be switched off for chondrogenesis to proceed. In vivo, in chick, Notch ligand Delta-1 negatively regulates the transition from pre-hypertrophic to hypertrophic chondrocytes during cartilage formation [28]. The exact role of Notch and its temporal effects in chondrogenesis and osteoblastogenesis remains to be further demonstrated. We will discuss recently created animal models which shed light on Notch function in the two primary types of bone cells, namely osteoblasts and osteoclasts.
Notch regulates osteoblast commitment, proliferation and differentiation
The importance of Notch function in osteoblast was demonstrated in vivo by studies in which tissue specific gain and loss of function mutants were generated. We generated an osteoblast specific over-expression of Notch1 ICD by using 2.3 kb collagen type 1 (Col1a1) promoter to drive the expression of activated Notch receptor specifically in committed osteoblasts [29]. These transgenic mice showed a dramatic increase in osteoblast number, proliferation and formation resulting in a severe osteosclerotic phenotype. Histological analyses of these mice indicated highly disorganized woven bone formation suggesting a maturation defect in early osteoblastic precursors. This was confirmed by decreased expression of mature osteoblastic markers in the calvarial osteoblasts of the mutant mice. Mechanistically, we showed that Notch1 ICD activates the Osterix promoter and up-regulates expression of cell cycle proteins Cyclin D and Cyclin E explaining the massive expansion of this early osteoblastic compartment. Our data also demonstrated that Notch1 ICD can physically interact with Runx2 and represses its function including, transactivation of the Osteocalcin gene [14]. While these findings helped to explain the maturation defects in the transgenic mice, it also raised the question of whether the early function of Notch signaling in bone marrow mesencyhmal stem cells was to maintain undifferentiated/ uncommitted progenitor pool via Runx2 repression.
Interestingly, in a subsequent study, expression of Notch1 ICD under the control of an earlier expressing 3.6 kb Col1a1 promoter caused inhibition of differentiation and low bone mass i.e. osteopenia [30]. The differences in the phenotypes were explained by the differential activation of the 2.3- and 3.6-kb fragments of the type I collagen promoter, thus the arrest of osteoblastic cell differentiation at different stages of maturation. It was suggested in this study that Notch ICD over-expression under the control of the 2.3-kb type I collagen promoter repressed the terminal differentiation of osteoblasts allowing for the proliferation of immature cells, thus leading to the increased formation of woven bone, while Notch ICD over-expression under the control of the 3.6-kb type I collagen promoter repressed osteoblastogenesis at an earlier stage of cell differentiation, leading to a decreased number of mature osteoblasts and an osteopenic phenotype.
To evaluate the loss of function of Notch signaling in committed osteoblasts, we deleted Presenilin1 and Presenilin2 using the 2.3 kb Col1a1-Cre mice. Deletion of Presenilins, which are required for the activation of all four Notch receptors, provided an indirect model for the loss of function of Notch in committed osteoblasts. Double knock-out mice did not show any bone phenotype in the immediate postnatal period. However, as they age they started to show a significant low bone mass phenotype due to increased osteoclastogenesis mediated by decreased Opg levels [29]. While our study demonstrated Notch’s effects in committed osteoblasts through gain and loss of function analyses, a complementary study to ours elucidated the function of Notch during early skeletal formation. In this study, Presenilin1 and Presenilin2 were deleted by using Prx-Cre mice to abolish Notch function in osteoblast progenitors/committed osteoblasts [31]. Unlike Col1a1 mediated deletion of Presenilins, Prx-Cre deletion of Presenilin1 and Presenilin2 (PPS mice) led to a high bone mass phenotype at 2 months of age. Deletion of Notch 1 and Notch2 receptors via Prx-Cre (PNN mice) mice mimicked the high bone mass phenotype of PPS mice. However, with aging these mice showed a significant bone loss due to reduction of the density of osteoblasts and increased bone resorption (Table 2). The bone marrow mesenchymal progenitors from PNN mice produced fewer CFU-f and CFU-ob and fewer differentiated adipocytes consistent with a reduction in bone marrow mesenchymal progenitors. This suggested that during early osteoblastogenesis, Notch signaling maintains mesenchymal stem cells in an undifferentiated stage and decreases osteoblast differentiation. Together, these studies show that during the early stages of osteogenesis Notch maintains mesenchymal progenitor cells in an undifferentiated stage. Upon loss of Notch signaling, these cells adopt an osteoblastic fate. Once committed, Notch signaling induces osteoblast proliferation and inhibits osteoblast maturation. This leads to the maintenance of the early osteoblastic pool. In a pathological context, increased expression of activated Notch in committed osteoblasts resulted in abnormal proliferation of immature osteoblasts and a high bone mass phenotype (Figure 2).
Table 2.
Phenotypes of different mouse models of Notch signaling in bone. Gain and loss of function mutants of Notch signaling players were listed. Osteoclast number per bone volume (N.Oc/BV), osteoclast number per bone surface (N.Oc/BS), osteoclast surface per bone surface (Oc.S/BS), osteoclast number per mm (N.Ob/mm)
| Col1a1 2.3Kb Cre Ps1f/f;PS2-/- | Prx-Cre Ps1f/f;PS2-/- | Prx-Cre Notch1-/f;Notch2f/f | Col1a1 2.3 kb Notch1 ICD | Col1a1 3.6 kb Notch1 ICD | |
|---|---|---|---|---|---|
| Osteoblasts | Osteoblast parameters were unchanged | Increased trabecular bone were reported | N.Ob/mm2 were increased | Increased proliferation, woven bone, osteosclerosis | Inhibition of differentiation, osteopenia |
| Osteoclasts | N.Oc/BS, N.Oc/BV, Oc.S/BS were increased | Not reported | N.Oc/mm2 and Oc.S/BS were increased | N.Oc/BS and N.Oc/BV were not different | N.Oc/BS and N.Oc/BV were not different |
| Osteoporosis | Age-related | Age-related and late | Present | ||
| Osteosclerosis | Early | Early | Present |
Figure 2. Function of Notch in bone homeostasis.
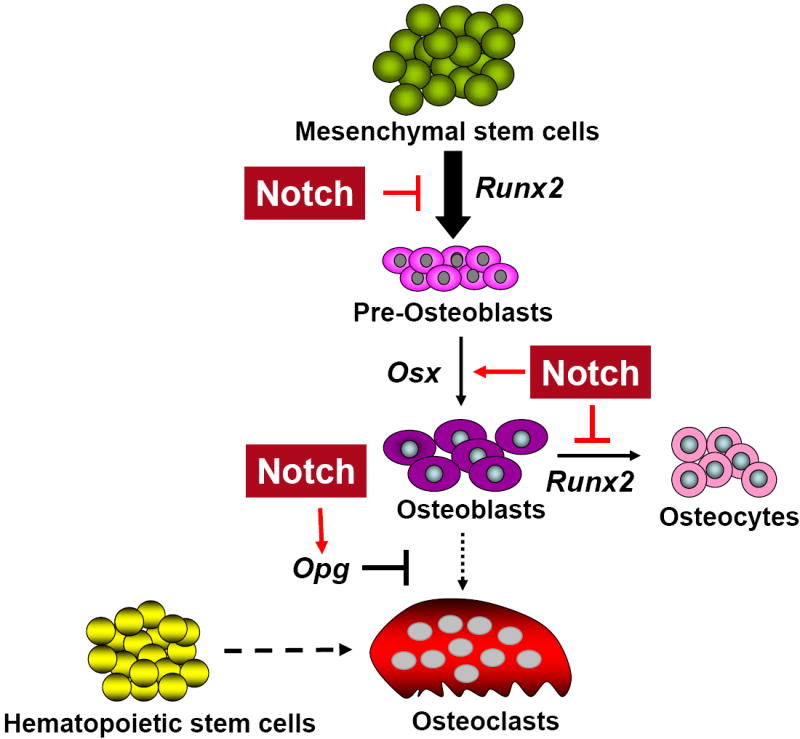
Prior to the commitment to osteoblastic lineage, Notch maintains mesenchymal stem cells in an undifferentiated stage by repressing Runx2. In established osteoblastic lineages, pathological gain of Notch function activates expansion of the immature osteoblastic pool by increasing transcription of Osx, Cyclin D, and Cyclin E and by repressing the function of Runx2 via direct interaction and inhibition of its binding. Physiologically, it inhibits osteoclastogenesis by increasing Opg production over Rankl production.
Notch regulates osteoclastogenesis directly and indirectly
The animal models generated above indicated that Notch could also regulate osteoblast dependent formation of osteoclasts. It has been shown that in PNN mice while RANKL expression was up regulated, Opg levels were decreased. Osteoblast specific deletion of Presenilins via the 2.3kb Col1a1-Cre resulted in decreased levels of Opg mRNA and protein as well. This demonstrates that Notch signaling may regulate osteoclastogenesis in a non- cell autonomous manner through Opg regulation in osteoblasts (Figure 3). However, whether this Opg dysregulation is due to direct regulation by Notch1 ICD or through its target transcription factors is unknown. One possible mechanism for Opg regulation by Notch could be its negative regulatory effect on Runx2. Opg has putative Runx2 binding elements in its promoter region, and Runx2 transgenic mice under the control of type I collagen promoter show decreased Opg expression [32]. In support of this, Runx2 mediated suppression of Opg expression was demonstrated in a study [33]. Thus, in the absence of Notch, increased Runx2 may lead to decreased Opg levels although this needs to be demonstrated experimentally.
Figure 3. Multiple roles of Notch in bone marrow environment.
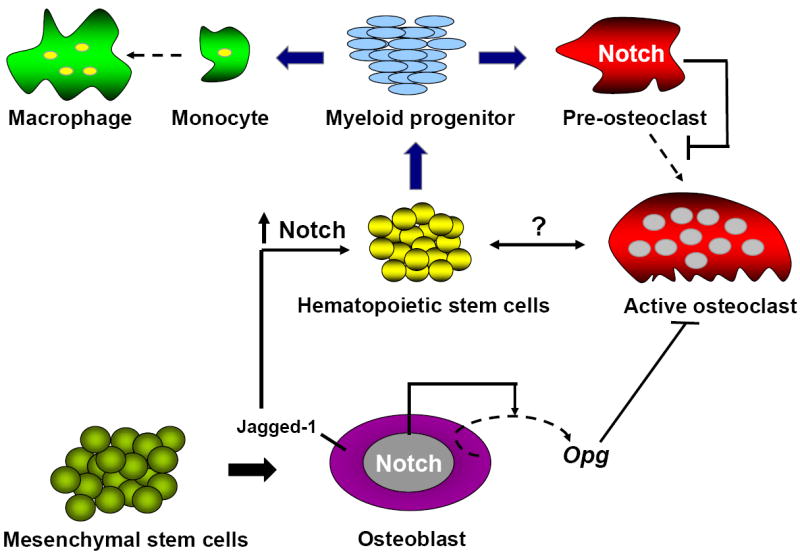
Osteoblasts act as signal sending cells and activate Notch signaling in HSCs leading to increased expansion of these cells. If osteoclasts or other cell types of bone marrow micro-environment also trigger a Notch dependent response remain to be elucidated.
In another study exemplifying an osteoblast dependent regulation of osteoclastogenesis, RANKL was shown to induce expression of Jagged1 and Notch2 in bone marrow macrophages during osteoclast differentiation. While suppression of Notch signaling by a selective γ-secretase inhibitor or Notch2 short hairpin RNA suppressed RANKL-induced osteoclastogenesis, induction of Notch signaling by Jagged1 or by ectopic expression of intracellular Notch2 enhanced NFATc1 promoter activity and expression leading to increased osteoclastogenesis [34].
In addition to osteoblast dependent regulation of osteoclastogenesis by Notch, it has been shown that Notch signaling can also regulate osteoclast precursor differentiation in a cell-autonomous fashion. Deletion of Notch1-3 in bone marrow macrophages directly promoted their commitment to the osteoclast phenotype. These osteoclast precursors proliferated more rapidly than the wild type in response to macrophage colony-stimulating factor [35].
The function of Notch signaling in bone is a rare example of a signaling pathway capable of regulating both osteoblastic and osteoclastic lineages. Another in vivo example for this is Ephrin B2 signaling where reverse signaling through ephrin B2 ligand expressed by osteoclasts suppress osteoclast precursors, whereas forward signaling through EphB4 receptor expressed by osteoblasts enhances osteoblast formation [36, 37].
Osteoblast regulation of hematopoietic stem cells via Notch signaling
In bone marrow, osteoblasts of the trabecular bone are in close physical association with hematopoietic stem cells and blood vessels, suggesting that the bone marrow microenvironment may provide regulatory signals for hematopoietic cells. Emerging data support the notion that osteoblasts lining the endosteal surface of trabecular bone can provide important cues to regulate/support the hematopoietic stem cell (HSC) niche. Transgenic mice expressing Parathyroid Hormone Receptor (PTH1R) under the control of a 2.3 kb Col1a1 promoter had increased osteoblast numbers with increased production of Notch ligand Jagged-1, and increased numbers of HSCs with activated Notch1 [38]. Addition of PTH to stromal cultures increased the osteoblast number and their ability to support hematopoietic cells while treatment with gamma-secretase inhibitors abrogated the expansion of HSCs in vitro. These data demonstrated that osteoblastic cells are a regulatory component of the hematopoietic stem cell niche and that it affects stem cell function through activation of Notch signaling.
Are osteoblasts the only cellular constituent of bone marrow that can regulate HSCs expansion? Recent demonstration of Notch regulation of osteoclastogenesis both in a cell autonomous and non-cell autonomous manner raises the possibility of osteoclastic regulation of HSCs. Indeed, activation of osteoclasts by RANKL or stress response resulted in mobilization of hematopoietic progenitor cells into the circulation [39]. Moreover, treatment of mice with strontium (Sr), a bone anabolic agent that enhances osteoblast function and inhibits osteoclast activity, increased osteoblast number, bone volume, and trabecular thickness. However, administration of Sr had no influence on primitive HSCs, and hematopoietic recovery was delayed in mice after bone marrow transplantation [40]. These data suggest that an increase in osteoblast number may not necessarily be sufficient alone to expand HSCs and that activation of specific signaling pathways within the osteoblasts is required. It also implies that osteoblasts may require other cellular components of bone marrow microenvironment such as stromal cells, osteoclasts, and/or endothelial cells in order to regulate the HSC niche. It is possible that when interacting with these cells osteoblasts may act as both sending and receiving cell with respect to Notch. Consistent with this, while gain and loss of function of Notch1 receptor and Presenilins in osteoblasts results in a HSC phenotype, increased production of Notch ligand Jagged-1 in osteoblasts to due to over expression of PTH1R results in Notch1 activation in HSCs and leads to increased number of these cells [38].
Notch as a therapeutic target for bone diseases
Loss of function studies of Notch signaling indicated an age dependent osteoporotic phenotype in mice. There are few anabolic bone agents for the treatment of osteoporosis, with most therapies targeted at inhibition of bone resorption. Up-regulation of Notch signaling may represent a potential approach for increasing bone formation over bone resorption as well as for inhibiting osteoclastogenesis. However, Notch’s effects on other cellular compartments such as the mesenchymal stem cell pool would have to be considered, i.e., Notch inhibition of Runx2 function could inhibit mesenchymal stem cell commitment to the osteoblastic lineage. Moreover, the described in vivo studies clearly support both a context-dependent and time-dependent consequence of Notch signaling. Activating Notch signaling early vs. late in the same cell lineage may have opposite tissue consequences.
Increased Notch activity causes abnormal proliferation of osteoblasts. Thus, in an opposing fashion, inhibition of Notch signaling could be used as a therapeutic strategy for the proliferative diseases of bone such as osteosclerosis and bone cancer. It is intriguing that Notch may play a role in proliferative diseases of bone such as osteosarcomas, since its tumorigenic and oncogenic role is well established in various cancer types [41-45]. In a recent paper, expression of the Notch target gene HES1 was associated with invasive and metastatic potential of osteosarcoma cell lines. Blockade of Notch signaling with gamma secretase inhibitors eliminated invasion in Matrigel without affecting cell proliferation, survival, or anchorage-independent growth [5]. While in this study proliferative effects of Notch was not observed, we found that Notch signaling components are significantly up regulated in primary human osteosarcomas samples. Inhibition of Notch signaling by γ-secretase inhibitors or by using lentiviral mediated expression of dominant negative Mastermind-like protein (DN-MAML) decreased osteosarcoma cell proliferation in vitro. In vivo, tumor xenografts in nude mice showed decreased tumor growth after chemical or genetic inhibition of Notch signaling. Moreover, transcriptional profiling of osteosarcomas from p53 mutant mice confirmed up-regulation of Notch1 target genes Hes1, Hey1 and its ligand Dll4 suggesting that activation of Notch signaling contributes to the pathogenesis of human osteosarcomas and its inhibition may be a therapeutic approach for the treatment of this mesenchymal tumor [46].
All these data indicate that Notch is an important signaling pathway for embryonic and post-natal bone development and in the pathogenesis of bone diseases. Thus, temporally and spatially controlled application of Notch activators or Notch inhibitors might provide a novel therapeutic option for the treatment of bone diseases.
Future Directions
Notch has a dual function in bone development in regulating both osteoblastogenesis and osteoclastogenesis. Cells in osteoclast and osteoblast lineages communicate with each other through cell to cell contact. Although osteoblast dependent activation of osteoclasts through Notch signaling has been demonstrated, whether there is reverse signaling between these two types of bone cells needs to be further investigated. Such cross talk and coupling might also exist with in the bone marrow microenvironment between the HSC and osteoblasts/osteoclasts. Considering that all of these cells express both ligands and receptors, what molecular cues identifies one cell as a receiver and the other as a sender in the context of bidirectional signaling remains to be elucidated.
In adults, the potential of Notch as both anabolic and anti-resorptive therapies is intriguing. HSC expansion is important for hematopoietic cell transplantation and is often required after cancer and leukemia treatments. Targeting Notch signaling in bone cells may provide an approach to support HSC niche and its expansion. Although effects of osteoblasts on HSCs were demonstrated through increased Jagged-1 expression, functions of other components of this signaling pathway need to be further studied.
In addition to the role of Notch in bone development, emerging new data suggest that it may play an important role during cartilage development as well. Analyses of Notch regulation of chondrocytes will require the generation of tissue specific gain and loss of function animal models. Considering the well established role of Notch in cell fate determination, it is possible that the fate choice of mesenchymal stem cells into either the osteoblast or chondrocyte lineage might be regulated by Notch. Finally, most work has focused on the established pathway that can be characterized as canonical Notch signaling. Not surprisingly, Drosophila studies have suggested the existence of non-canonical Notch signaling that may be independent of Rbp-Jκ. The demonstration of this in mammals and importance in skeletogenesis remains to be studied.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mumm JS, Kopan R. Notch signaling: from the outside in. Dev Biol. 2000;228:151–165. doi: 10.1006/dbio.2000.9960. [DOI] [PubMed] [Google Scholar]
- 2.De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 3.Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, Haass C. Reconstitution of gamma-secretase activity. Nat Cell Biol. 2003;5:486–488. doi: 10.1038/ncb960. [DOI] [PubMed] [Google Scholar]
- 4.Leong KG, Karsan A. Recent insights into the role of Notch signaling in tumorigenesis. Blood. 2006;107:2223–2233. doi: 10.1182/blood-2005-08-3329. [DOI] [PubMed] [Google Scholar]
- 5.Zhang P, Yang Y, Zweidler-McKay PA, Hughes DP. Critical role of notch signaling in osteosarcoma invasion and metastasis. Clin Cancer Res. 2008;14:2962–2969. doi: 10.1158/1078-0432.CCR-07-1992. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Provot S, Schipani E. Molecular mechanisms of endochondral bone development. Biochem Biophys Res Commun. 2005;328:658–665. doi: 10.1016/j.bbrc.2004.11.068. [DOI] [PubMed] [Google Scholar]
- 7.Wagner EF, Karsenty G. Genetic control of skeletal development. Curr Opin Genet Dev. 2001;11:527–532. doi: 10.1016/s0959-437x(00)00228-8. [DOI] [PubMed] [Google Scholar]
- 8.Colnot C. Cellular and molecular interactions regulating skeletogenesis. J Cell Biochem. 2005;95:688–697. doi: 10.1002/jcb.20449. [DOI] [PubMed] [Google Scholar]
- 9.Rida PC, Le Minh N, Jiang YJ. A Notch feeling of somite segmentation and beyond. Dev Biol. 2004;265:2–22. doi: 10.1016/j.ydbio.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Cinquin O. Understanding the somitogenesis clock: what’s missing? Mech Dev. 2007;124:501–517. doi: 10.1016/j.mod.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 11.Conlon RA, Reaume AG, Rossant J. Notch1 is required for the coordinate segmentation of somites. Development. 1995;121:1533–1545. doi: 10.1242/dev.121.5.1533. [DOI] [PubMed] [Google Scholar]
- 12.Oka C, Nakano T, Wakeham A, de la Pompa JL, Mori C, Sakai T, Okazaki S, Kawaichi M, Shiota K, Mak TW, Honjo T. Disruption of the mouse RBP-J kappa gene results in early embryonic death. Development. 1995;121:3291–3301. doi: 10.1242/dev.121.10.3291. [DOI] [PubMed] [Google Scholar]
- 13.Turnpenny PD, Alman B, Cornier AS, Giampietro PF, Offiah A, Tassy O, Pourquie O, Kusumi K, Dunwoodie S. Abnormal vertebral segmentation and the notch signaling pathway in man. Dev Dyn. 2007;236:1456–1474. doi: 10.1002/dvdy.21182. [DOI] [PubMed] [Google Scholar]
- 14.Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, Costa T, Pierpont ME, Rand EB, Piccoli DA, Hood L, Spinner NB. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16:243–251. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- 15.Bulman MP, Kusumi K, Frayling TM, McKeown C, Garrett C, Lander ES, Krumlauf R, Hattersley AT, Ellard S, Turnpenny PD. Mutations in the human delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat Genet. 2000;24:438–441. doi: 10.1038/74307. [DOI] [PubMed] [Google Scholar]
- 16.Tezuka K, Yasuda M, Watanabe N, Morimura N, Kuroda K, Miyatani S, Hozumi N. Stimulation of osteoblastic cell differentiation by Notch. J Bone Miner Res. 2002;17:231–239. doi: 10.1359/jbmr.2002.17.2.231. [DOI] [PubMed] [Google Scholar]
- 17.McLarren KW, Lo R, Grbavec D, Thirunavukkarasu K, Karsenty G, Stifani S. The mammalian basic helix loop helix protein HES-1 binds to and modulates the transactivating function of the runt-related factor Cbfa1. J Biol Chem. 2000;275:530–538. doi: 10.1074/jbc.275.1.530. [DOI] [PubMed] [Google Scholar]
- 18.Deregowski V, Gazzerro E, Priest L, Rydziel S, Canalis E. Notch 1 overexpression inhibits osteoblastogenesis by suppressing Wnt/beta-catenin but not bone morphogenetic protein signaling. J Biol Chem. 2006;281:6203–6210. doi: 10.1074/jbc.M508370200. [DOI] [PubMed] [Google Scholar]
- 19.Sciaudone M, Gazzerro E, Priest L, Delany AM, Canalis E. Notch 1 impairs osteoblastic cell differentiation. Endocrinology. 2003;144:5631–5639. doi: 10.1210/en.2003-0463. [DOI] [PubMed] [Google Scholar]
- 20.Zamurovic N, Cappellen D, Rohner D, Susa M. Coordinated activation of notch, Wnt, and transforming growth factor-beta signaling pathways in bone morphogenic protein 2-induced osteogenesis. Notch target gene Hey1 inhibits mineralization and Runx2 transcriptional activity. J Biol Chem. 2004;279:37704–37715. doi: 10.1074/jbc.M403813200. [DOI] [PubMed] [Google Scholar]
- 21.Yamada T, Yamazaki H, Yamane T, Yoshino M, Okuyama H, Tsuneto M, Kurino T, Hayashi S, Sakano S. Regulation of osteoclast development by Notch signaling directed to osteoclast precursors and through stromal cells. Blood. 2003;101:2227–2234. doi: 10.1182/blood-2002-06-1740. [DOI] [PubMed] [Google Scholar]
- 22.Hayes AJ, Dowthwaite GP, Webster SV, Archer CW. The distribution of Notch receptors and their ligands during articular cartilage development. J Anat. 2003;202:495–502. doi: 10.1046/j.1469-7580.2003.00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fujimaki R, Tezuka K. [Notch signaling in chondrogenesis] Clin Calcium. 2006;16:1374–1379. [PubMed] [Google Scholar]
- 24.Fujimaki R, Toyama Y, Hozumi N, Tezuka K. Involvement of Notch signaling in initiation of prechondrogenic condensation and nodule formation in limb bud micromass cultures. J Bone Miner Metab. 2006;24:191–198. doi: 10.1007/s00774-005-0671-y. [DOI] [PubMed] [Google Scholar]
- 25.Watanabe N, Tezuka Y, Matsuno K, Miyatani S, Morimura N, Yasuda M, Fujimaki R, Kuroda K, Hiraki Y, Hozumi N, Tezuka K. Suppression of differentiation and proliferation of early chondrogenic cells by Notch. J Bone Miner Metab. 2003;21:344–352. doi: 10.1007/s00774-003-0428-4. [DOI] [PubMed] [Google Scholar]
- 26.Nakanishi K, Chan YS, Ito K. Notch signaling is required for the chondrogenic specification of mouse mesencephalic neural crest cells. Mech Dev. 2007;124:190–203. doi: 10.1016/j.mod.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 27.Oldershaw RA, Tew SR, Russell AM, Meade K, Hawkins R, McKay TR, Brennan KR, Hardingham TE. Notch signaling through Jagged-1 is necessary to initiate chondrogenesis in human bone marrow stromal cells but must be switched off to complete chondrogenesis. Stem Cells. 2008;26:666–674. doi: 10.1634/stemcells.2007-0806. [DOI] [PubMed] [Google Scholar]
- 28.Crowe R, Zikherman J, Niswander L. Delta-1 negatively regulates the transition from prehypertrophic to hypertrophic chondrocytes during cartilage formation. Development. 1999;126:987–998. doi: 10.1242/dev.126.5.987. [DOI] [PubMed] [Google Scholar]
- 29.Engin F, Yao Z, Yang T, Zhou G, Bertin T, Jiang MM, Chen Y, Wang L, Zheng H, Sutton RE, Boyce BF, Lee B. Dimorphic effects of Notch signaling in bone homeostasis. Nat Med. 2008;14:299–305. doi: 10.1038/nm1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zanotti S, Smerdel-Ramoya A, Stadmeyer L, Durant D, Radtke F, Canalis E. Notch inhibits osteoblast differentiation and causes osteopenia. Endocrinology. 2008;149:3890–3899. doi: 10.1210/en.2008-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hilton MJ, Tu X, Wu X, Bai S, Zhao H, Kobayashi T, Kronenberg HM, Teitelbaum SL, Ross FP, Kopan R, Long F. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat Med. 2008;14:306–314. doi: 10.1038/nm1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu W, Toyosawa S, Furuichi T, Kanatani N, Yoshida C, Liu Y, Himeno M, Narai S, Yamaguchi A, Komori T. Overexpression of Cbfa1 in osteoblasts inhibits osteoblast maturation and causes osteopenia with multiple fractures. J Cell Biol. 2001;155:157–166. doi: 10.1083/jcb.200105052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Enomoto H, Shiojiri S, Hoshi K, Furuichi T, Fukuyama R, Yoshida CA, Kanatani N, Nakamura R, Mizuno A, Zanma A, Yano K, Yasuda H, Higashio K, Takada K, Komori T. Induction of osteoclast differentiation by Runx2 through receptor activator of nuclear factor-kappa B ligand (RANKL) and osteoprotegerin regulation and partial rescue of osteoclastogenesis in Runx2-/- mice by RANKL transgene. J Biol Chem. 2003;278:23971–23977. doi: 10.1074/jbc.M302457200. [DOI] [PubMed] [Google Scholar]
- 34.Fukushima H, Nakao A, Okamoto F, Shin M, Kajiya H, Sakano S, Bigas A, Jimi E, Okabe K. The association of Notch2 and NF-kappaB accelerates RANKL-induced osteoclastogenesis. Mol Cell Biol. 2008;28:6402–6412. doi: 10.1128/MCB.00299-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bai S, Kopan R, Zou W, Hilton MJ, Ong CT, Long F, Ross FP, Teitelbaum SL. NOTCH1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. J Biol Chem. 2008;283:6509–6518. doi: 10.1074/jbc.M707000200. [DOI] [PubMed] [Google Scholar]
- 36.Zhao C, Irie N, Takada Y, Shimoda K, Miyamoto T, Nishiwaki T, Suda T, Matsuo K. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 2006;4:111–121. doi: 10.1016/j.cmet.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 37.Boyce BF, Schwarz EM, Xing L. Osteoclast precursors: cytokine-stimulated immunomodulators of inflammatory bone disease. Curr Opin Rheumatol. 2006;18:427–432. doi: 10.1097/01.bor.0000231913.32364.32. [DOI] [PubMed] [Google Scholar]
- 38.Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, Milner LA, Kronenberg HM, Scadden DT. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 39.Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y, Tesio M, Samstein RM, Goichberg P, Spiegel A, Elson A, Lapidot T. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med. 2006;12:657–664. doi: 10.1038/nm1417. [DOI] [PubMed] [Google Scholar]
- 40.Lymperi S, Horwood N, Marley S, Gordon MY, Cope AP, Dazzi F. Strontium can increase some osteoblasts without increasing hematopoietic stem cells. Blood. 2008;111:1173–1181. doi: 10.1182/blood-2007-03-082800. [DOI] [PubMed] [Google Scholar]
- 41.Miele L, Golde T, Osborne B. Notch signaling in cancer. Curr Mol Med. 2006;6:905–918. doi: 10.2174/156652406779010830. [DOI] [PubMed] [Google Scholar]
- 42.Grabher C, von Boehmer H, Look AT. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2006;6:347–359. doi: 10.1038/nrc1880. [DOI] [PubMed] [Google Scholar]
- 43.Radtke F, Raj K. The role of Notch in tumorigenesis: oncogene or tumour suppressor? Nat Rev Cancer. 2003;3:756–767. doi: 10.1038/nrc1186. [DOI] [PubMed] [Google Scholar]
- 44.Katoh M, Katoh M. Notch signaling in gastrointestinal tract (review) Int J Oncol. 2007;30:247–251. [PubMed] [Google Scholar]
- 45.Leow CC, Polakis P, Gao WQ. A role for Hath1, a bHLH transcription factor, in colon adenocarcinoma. Ann N Y Acad Sci. 2005;1059:174–183. doi: 10.1196/annals.1339.048. [DOI] [PubMed] [Google Scholar]
- 46.Engin F, Bertin T, Ma O, Jiang MM, Wang L, Sutton RE, Donehower LA, Lee B. Notch Signaling Contributes to the Pathogenesis of Human Osteosarcomas. Hum Mol Genet. 2009;18:1464–1470. doi: 10.1093/hmg/ddp057. [DOI] [PMC free article] [PubMed] [Google Scholar]