

Abstract
Introduction
Parenteral OKT3 is used to treat transplant rejection and a humanized anti-CD3 Mab has shown positive clinical effects in new onset diabetes. Oral administration of anti-CD3 has not been tested in humans, but suppresses autoimmunity in animal models. Beta-glucosylceramide enhances NKT cell and regulatory T cell activity and enhances the effects of oral anti-CD3 in animals.
Materials and methods
Fifteen healthy volunteers (three per group) received orally administered OKT3 over a dose range of 0.2 to 5.0 mg daily with or without beta-glucosylceramide 7.5 mg for 5 days. Safety and immune parameters were measured on days 5, 10, and 30.
Results and discussion
Oral OKT3 enhanced T cell proliferation, suppressed Th1 and Th17 responses by 43% and 41%, respectively, increased TGF-β/IL-10 expression and decreased IL-23/IL-6 expression by dendritic cells, and affected the IgG repertoire as measured by antigen arrays. Co-administration of oral beta-glucosylceramide induced similar effects. No side effects were observed and no subjects developed human anti-mouse antibodies.
Conclusion
These findings demonstrate that oral anti-CD3 monoclonal antibody is safe and biologically active in humans and presents a new avenue for the treatment of autoimmune diseases.
Keywords: Anti-CD3, immunotherapy, mucosal tolerance, dendritic cells, IL-17, TGF-beta
Introduction
Parenteral administration of CD3-specific antibody induces an immunomodulatory effect in animal models of autoimmunity [1] as well as in humans with autoimmune diabetes [2, 3]. In addition, murine anti-CD3 monoclonal antibody (OKT3) is approved therapy for acute transplant rejection, although side effects, some of which can be severe, and the development of HAMA, limit its chronic use [4].
We have shown in animal models that oral administration of anti-CD3 monoclonal antibody is biologically active and suppresses animal models of autoimmunity including EAE, and autoimmune diabetes [5, 6]. Nasal and oral anti-CD3 is effective in suppressing animal models of lupus erythematosus [7, 8]. In mice, oral anti-CD3 antibody is rapidly taken up by gut-associated lymphoid tissue (GALT) and induces CD4+CD25-LAP+ regulatory T cells in the mesenteric lymph nodes that function to suppress EAE and diabetes in a TGF-β dependent fashion [5, 6]. A dose-dependent effect of oral anti-CD3 was observed in animals with suppressive effects best seen at intermediate doses.
Beta-glycosphingolipids are naturally occurring glycolipids which are metabolic intermediates in the anabolic and catabolic pathways of glycosphingolipids [9]. Administering beta-glucosylceramide (GC) exerts NKT- and Treg-dependent immune regulation in ConA-mediated hepatitis, colitis, and models of insulin resistance [10-14].We have found that beta-glucosylceramide given orally enhances the effect of oral anti-CD3 in some animal models (unpublished). Based on these findings, we investigated the safety of oral anti-CD3 in healthy human volunteers and whether oral anti-CD3 induced measurable immunologic effects in peripheral blood tested over a dose range of 0.2 mg, 1.0 mg, and 5.0 mg. We also investigated the effect of co-administration of beta-glucosylceramide.
Methods
Patient population
Healthy males (≥18 years) not on therapy for medical or other illnesses were enrolled in accordance with the guidelines of the Hebrew University-Hadassah Institutional Committee for Human Clinical Trials, and the approval of the Israel Ministry of Health Committee for Human Trials. Of 35 potential study subjects screened, 18 met inclusion and exclusion criteria and were randomized to one of the six treatment groups.
OKT3 and β-Glucosylceramide
OKT3 (Orthoclone OKT-3) was purchased from Ortho Biotech Inc. (New Jersey, USA) and GC from Avanti Polar Lipids, Inc., Alabaster, Alabama, USA.
Drug administration
Nine subjects (three per group) received 0.2 mg, 1.0 mg or 5.0 mg of oral OKT3 daily for 5 days. Six subjects (three per group) received 7.5 mg of beta-glucosylceramide in combination with 0.2 mg or 1.0 mg of OKT3 and three subjects received GC alone. Immune parameters were measured on days 5, 10, and 30 (Fig. 1). All subjects were treated with 20 mg of omeprazole (a proton pump inhibitor) during the 5 days of dosing. Dosing occurred in the morning before breakfast at the study site following an 8-h fast. We administered a proton pump inhibitor to protect the antibody from the acidic environment of the gut.
Fig. 1.
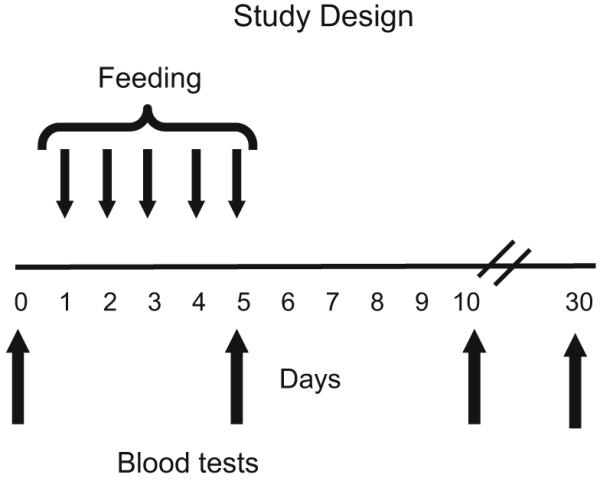
Study design. Nine subjects (three per group) were orally administered 0.2 mg, 1.0 mg, or 5.0 mg of OKT3 daily for 5 days. Six subjects (three per group) received 7.5 mg of beta-glucosylceramide in combination with 0.2 mg or 1.0 mg of OKT3. Subjects were followed for 30 days and immune parameters were measured on days 5, 10, and 30
Clinical and laboratory follow-up
All patients underwent a full medical history and physical examination, including review of adverse effects on days 1, 5, 10, and 30, along with complete blood counts, differential, electrolytes, liver and kidney function tests, lipid profile, C-reactive protein, and sedimentation rates. Antibodies to OKT3 were evaluated on day 30 using the human anti-mouse antibodies (HAMA)-ELISA kit (MEDAC, Hamburg, Germany)
Immune Assays
1. Standardization of assays in untreated subjects
for the purpose of standardization, and in order to establish that immune changes we observed were related to treatment with oral anti-CD3 and not related to fluctuations in the assays, we performed the same assays that were used to measure immune function in treated subjects in untreated healthy controls longitudinally on days 0, 5, and 10. We observed no changes in any of the immune assays over the three time points measured, thus allowing us to ascribe immune changes observed in subjects given oral anti-CD3 as being related to treatment. An example for proliferation, 17 production and cell surface staining is shown in Fig. 2.
Fig. 2.

Immune measures in untreated subjects. For the purpose of standardization, and in order to establish that immune changes we observed were related to treatment with oral anti-CD3 and not related to fluctuations in the assays, we performed the same assays that were used to measure immune function in treated subjects in untreated healthy controls longitudinally days 0, 5, and 10. Proliferation, examples of cytokine production and cell surface staining are shown. No significant changes were observed
2. Antibodies and reagents
CD16/CD32-specific (FcBlock); FITC, PE, or APC-conjugated CD4-specific (L3T4); and, PE-conjugated CD25-specific (PC61) were purchased from BD PharMingen (San Jose, CA, USA). Affinity-purified biotinylated goat anti-LAP specific polyclonal antibody was purchased from R&D Systems (Minneapolis, MN, USA), and strepavidin-APC was used as secondary reagent for detecting the biotinylated primary antibody (R&D). 7-AAD for staining dead cells was purchased from Sigma-Aldrich (St. Louis, MO, USA). Alexa Fluor-647 FOXP3 kit was purchased from BioLegend (San Diego, CA, USA). FITC-conjugated CD4 or CD8, PE-conjugated CD25 and their compensation controls, were from eBioscience (San Diego, CA, USA). Other markers as APC-conjugated CD11c and PE-conjugated CD40, CD80, CD83, CD86, and HLA-DR were also from eBioscience. FITC-conjugated Lin1 and APC-conjugated CTLA4 was purchased from BD PharMingen (San Jose, CA, USA).
3. Proliferation and cytokine assays
PBMCs were isolated from blood samples using Ficoll Hypaque solution and 2×105 cells per well were cultured in triplicate in RPMI 1640 medium with 5% FBS (Biological Industries, Israel), 100 units/ml penicillin, 100 μg/ml streptomycin, 1% glutamine, 1% non-essential fatty acids, 1% sodium pyruvate and β-mercapto ethanol (Biological Industries, Israel). Cells were stimulated with 5 μg/ml soluble anti-CD3 mAb (eBioscience, CA, USA). For TGF-β, 2×105 cells per well were cultured in serum free media (Biotarget, Biological Industries, Israel). Supernatants were collected at 48 h. Enzyme-linked immunosorbent assays for IL-13, IL-17, IFN-γ, and TGF-β were performed according to the manufacturer’s protocol (R&D Systems, Minneapolis, MN). After collecting the supernatants, 1 μCi [3H]thymidine (Amersham Biosciences, UK) was added to each well, and cells were harvested 18 h later. Proliferation was measured by scintillation counting.
4. FACS analysis and sorting of frozen lymphocytes
frozen PBMCs were used for sorting dendritic cells and for surface staining. Cell sorting was performed by thawing 20×106PBMCs at room temperature (RT) and washing them twice in medium at RT in 50-ml tubes in a 10-ml volume at 1,400 rpm. Cells were suspended to 20×106/ml and stained with seven colors using Fc block, Lin-FITC, CD11c-APC, CD123-PE, CD3-Amcyan, CD4-Alexa700, CD25-Pacific Blue, and 7-AAD. Myeloid DCs were sorted on a FACS Aria (BD Biosciences), put in 350 μl of RLT (lysis buffer), and frozen at -70°C in Eppendorff microtubes.
For surface staining, freshly isolated PBMCs were suspended at 8–10×106 cells/ml. Surface two to three color staining of cells were done with the following surface antibodies: CD4-FITC/CD25-PE, CD8-FITC/CD25-PE, CD3-APC/CD69-PE,CD11c-APC/Lin-FITC/CD86-PE, CD11c-APC/Lin-FITC/CD83-PE,CD11c-APC/Lin-FITC/HLAdr-PE. For LAP staining cells were preincubated with rLAP/control antibody for 20 min, and stained with CD4-FITC and CD25-PE or CD8-FITC. For Foxp3 surface staining, surface two-color staining of cells using CD4-FITC and CD25-PE was performed, followed by intracellular staining with IC/Foxp3-Alexa 647-APC or with CTLA4-APC.
5. Expression of cytokines by dendritic cells
total RNA was isolated from frozen sorted DC pellets using the RNA easy Mini Kit (Qiagen). RNAwas stored at −80°C. First-strand cDNA synthesis was performed for each RNA sample from 0.5–1 μg of total RNA using Taqman reverse transcription reagents (Applied Biosystems). cDNA was amplified using sequence-specific primers (for IL-23, IL-10, IL-6, and TGF-β) and real-time PCR mix (Applied Biosystems) on an ABI7500 cycler. The GAPDH gene was used as an endogenous control to normalize for differences in the amount of total RNA in each sample. All values were expressed as fold-increase or -decrease relative to the expression of GAPDH.
6. Antigen arrays
a panel of self and non-self proteins, peptides and lipids were spotted onto Epoxi slides (TeleChem, CA, USA) as described [15], a complete list of the antigens included in the arrays is provided in supplementary Table 1. Since several of the self-antigens included in our antigen microarrays are targeted by natural antibodies [16, 17], we could detect both the up- and down-regulation of preexisting IgG and IgM reactivities. Sera from OKT3-treated subjects were assayed at a 1/10 dilution and the IgG or IgM reactivities displaying significant changes upon treatment were identified. Briefly, antigens were spotted in replicates of six, the microarrays were blocked for 1 h at 37°C with 1% bovine serum albumin, and incubated for 2 h at 37°C with a 1:200 dilution of the test serum in blocking buffer. The arrays were then washed and incubated for 45 min at 37°C with a 1:500 dilution of detection antibodies: a goat anti-mouse IgG Cy3-conjugated antibody or a goat anti-mouse IgM Cy5-conjugated antibody (Jackson ImmunoResearch, West Grove, PA). The arrays were scanned with a ScanArray 4000X scanner (GSI Luminomics, Billerica, Massachusetts, USA) and the IgM and IgG results were recorded separately.
To compare the results from different arrays the raw data were normalized [15] and analyzed using the GeneSpring software (Silicon Genetics, Redwood City, CA). Antigen reactivity was defined by the mean intensity of binding to the replicates of that antigen on the microarray. To identify antibody reactivities that were consistently up- or down-regulated following treatment with OKT3 the data were analyzed with the Wilcoxon rank-sum test, a non-parametric test robust to outliers. The multidimensional nature of the data produced by the antigen arrays results in an increased probability of picking up “false positives”, this is controlled by using a False Discovery Method [18]. We used the Benjamini and Hochberg false discovery method with a p value of 0.2 to determine significance [18]. To identify antibody reactivities that show a similar behavior in response to OKT3 administration we used a hierarchical clustering method. The hierarchical clustering we performed using a pairwise average linkage algorithm based on Pearson’s correlation as a distance measure [18].
Statistical analysis
Differences were analyzed using Student’s t test. When there were more than two groups used for comparison, differences were analyzed by the one-way analysis of variance test. p values<0.05 were considered to be significant.
Results
Safety of oral OKT3 and GC
The treatment was well tolerated by all subjects and no systemic effects were observed at any doses including changes in vital signs (temperature, pulse, blood pressure), and liver, kidney or hematologic measures (complete blood counts including differential), during treatment or follow-up (30 days post treatment). The oral OKT3 did not cause any GI symptoms such as diarrhea constipation, or irritable bowel disease.
Effect of oral OKT3 on cell surface CD3, lymphocyte count, and proliferation
Unlike what has been reported for treatment with IV anti-CD3 (which is given at a dose of 5 mg per day for 5 days), we observed no decrease in the CD3+ lymphocyte count or modulation of CD3 from the T cell surface Fig. 3. In addition, no subject developed anti-OKT3 (HAMA) antibodies. As shown in Fig. 4, we observed an increase in proliferation to anti-CD3 stimulation on day 5 that reverted to baseline by day 10. Figure 4 shows the proliferative responses in the three subjects that received 1.0 mg OKT3 orally (p<0.001) for day 5 vs. 0, and p<0.0013 for day 10 vs. 5). No significant differences in proliferation were observed in subjects dosed with 0.2 mg or 5.0 mg of OKT3. No additional effects were observed in the GC-treated groups.
Fig. 3.

Oral OKT3 does not affect surface expression of CD3. Surface staining for CD3 was performed on day 5 after oral OKT3 at all doses fed (0.2 mg, 1.0 mg. and 5.0 mg) using anti-CD3 from eBioscience. No changes in surface expression of CD3 were observed
Fig. 4.
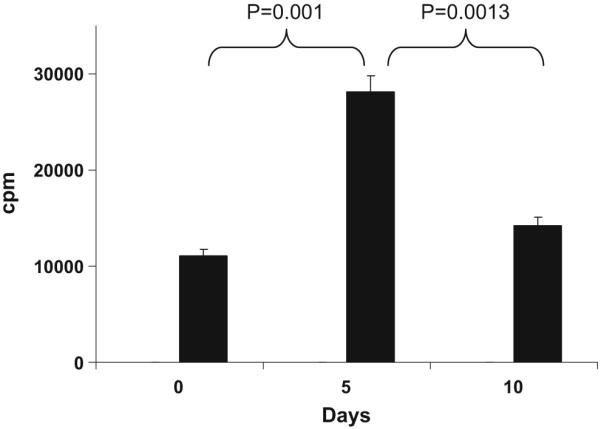
Oral OKT3 increases T cell proliferation. PBMCs were stimulated with anti-CD3 (5μg/ml) for 48 h and proliferation was measured using [3H]thymidine incorporation. Results are presented for the group (n=3) fed with 1.0 mg of OKT3.T cell proliferation was increased on day 5 in subjects orally treated with 1.0 mg of OKT3 (n=3). Graph shows mean±SD of the three subjects
Despite the increase in proliferation on day 5, Th1 and Th17 cytokine secretion was reduced by 43% and 41%, respectively, in the supernatants on day 5 and remained reduced on day 10. As shown in Fig. 5, we observed decreases in IFN-γ (p<0.004 days 5 or 10 vs. 0) and IL-17 (p<0.0044 for days 5 or 10 vs. 0). We observed a trend for an increase of IL-13 and TGF-β on days 5 and 10 but these changes were not statistically significant. We observed no significant changes in proliferation or cytokine production in untreated subjects evaluated at the same schedule and no additional effects in GC-treated subjects. These results demonstrate that OKT3 is biologically active when given orally and decreases the pro-inflammatory profile as measured by IFN-γ and IL-17.
Fig. 5.

Oral OKT3 decreases IFN-γ and IL-17 secretion. PBMCs were stimulated with anti-CD3 (5μg/ml) for 48 h and cytokine production was measured in culture supernatants using ELISA. Results are presented for the group (n=3) fed with 1.0 mg of OKT3. IFN-γ and IL-17 secretion was decreased by 43% and 41% respectively, on days 5 and 10. Graphs show mean±SD of the three subjects
Oral OKT3 increases IL-10/TGF-β and decreases IL-23 and IL-6 expression in dendritic cells
To investigate the effect of oral OKT3 on the innate immune system, we measured the expression of IL-10, TGF-β, IL-23, and IL-6 in dendritic cells by rtPCR. As shown in Fig. 6 for the subjects dosed with 1.0 mg OKT3, we observed an increase in IL-10 and TGF-β (p<0.005 for days 5 or 10 vs. 0). An opposite pattern was observed for IL-23 and IL-6 where we observed a decrease in IL-23 expression (p=0.006) and IL-6 expression (p=0.005) for days 5 or 10 vs. 0. No significant changes were seen in the 0.2- and 5-mg dose groups. In the 1-mg OKT3+GC-treated group, we also found increased expression of IL-10 and TGF-β in DCs which was more apparent in day 5 than in subjects with OKT3 (p<0.005 for day 5 vs. 0; Fig. 7). This is consistent with reports that glycolipids affect the function of DCs [19-22]. We observed no significant changes in DC cytokine profiles in the untreated subjects evaluated at the same schedule. These results demonstrate that oral OKT3 affects the anti-inflammatory profile of DCs.
Fig. 6.

Oral OKT3 increases the expression of IL-10 and TGF-β in dendritic cells and decreases the expression of IL-6 and IL-23. CD11c+DCs were isolated from PBMCs on days 0, 5, and 10 using FACS sorting and IL-10, TGF-β, IL-23, and IL-6 expression was measured by RT PCR. Values are expressed as fold-increase or -decrease relative to the expression of GAPDH. Graphs show mean±SD in subjects treated with 1.0 mg of OKT3 (n=3)
Fig. 7.

Effect of therapy with OKT3 plus ß-glucosylceramide (GC) on cytokine expression by dendritic cells. Three subjects received 7.5 mg of β-glucosylceramide in combination with 1.0 mg of OKT3 orally (n=3). CD11c+DCs were isolated from PBMCs on days 0, 5, and 10 using FACS sorting and IL-10, TGF-β, and IL-23 expression was measured by RT PCR. Values are expressed as fold-increase or -decrease relative to the expression of GAPDH. Graphs show mean±SD
Effect of oral OKT3 on T cell regulatory markers
We measured immunophenotypic markers associated with regulatory T cells, including CD25hi, Foxp3 and CTLA4. We found a significant increase (p<0.005) in Foxp3 expression on CD25hi cells as measured by MFI (Fig. 8a) on day 5. Similarly, CD25hi CTLA4+ cells MFI increased on day 5 (Fig. 8b). We also found an increase in the expression of TGF-β on CD25int/lo T cells (Fig. 8c; p<0.005 for day 5 vs. 10) and an increase in CD8+CD25+cells (Fig. 8d). We were unable to perform functional studies due to limits in cell numbers. No significant changes in regulatory T cell markers were observed in the untreated subjects evaluated at the same schedule. In animals, induction of regulatory T cells is the primary mechanism by which oral anti-CD3 suppresses disease. Although our phenotype data suggests induction of regulatory T cells in humans, demonstrating that oral anti-CD3 induces functional regulatory T cells in humans will require further studies. Of note, we did not observe changes in the surface expression of CD40, CD80, CD83, CD86, HLA-DR, or LAP in subjects orally administered OKT3.
Fig. 8.

Oral OKT3 increases the expression of Foxp3 and CTLA4 on CD25high regulatory T cells and TGF-β on CD25int/low T cells. Expression of surface markers on peripheral lymphocytes was measured using flow cytometry on days 0, 5, and 10 in subjects treated with 1.0 mg OKT3 orally (n=3)
Effect of Oral OKT3 on Antigen Arrays
Antigen microarrays constitute a new tool for the study of the immune system in health [15, 23-25] and disease [26, 27]. We used an antigen microarray containing a broad panel of antigens (n=550) that included self and non-self proteins, heat shock proteins, and infectious agents to investigate the effects of oral OKT3 on the immune repertoire. We measured both increases and decreases of immunoglobulin reactivities.
Figure 9a shows that treatment with OKT3 resulted in a dose-dependent change in the T-cell-dependent IgG repertoire. Minimal changes were observed at the 0.2-mg dose (only four reactivities affected) whereas at 1.0 mg, 37 IgG reactivities were affected and at 5.0 mg, 65 IgG reactivates were affected (Fig. 9a). At the 1.0 mg dose there was an equal number of down-regulated (n=19) as up-regulated (n=18) reactivities, whereas at the 5-mg dose, more up-regulated reactivates were observed (47 vs. 18). Figure 9b shows a heatmap of changes in the IgG repertoire following oral administration of 1.0 mg OKT3 in the three individual subjects. No changes occurred in the IgM repertoire. The changes we observed affected a range of reactivities. We did not perform antigen assays in subjects that received OKT3 plus GC.
Fig. 9.

Effect of oral OKT3 on antigen arrays. Sera from subjects treated with OKT3 were assayed at a 1/10 dilution and IgG or IgM reactivities were measured as described. a There was a dose-dependent change in the T cell dependent IgG repertoire following treatment with OKT3 (0.2, 1.0, and 5.0 mg, respectively; n=3 in each group) as measured by numbers of up-or down-regulated IgG reactivities relative to before treatment. b Heatmaps for three subjects showing IgG reactivities at baseline (day 0) and during follow-up (days 5, 10, and 30) after oral treatment with 1.0 mg OKT3 for 5 days
Discussion
Intravenous administration of monoclonal antibodies is used as therapy in human disease conditions [2-4]. We have shown in animal models that oral anti-CD3 is biologically active and suppresses autoimmunity by affecting immune responses [5-8]. To our knowledge, a monoclonal antibody has not been previously administered orally to human subjects. We thus addressed the question of whether oral anti-CD3 given to humans would affect the immune response and whether any toxicity would occur. To test this, we chose to administer OKT3, a murine monoclonal antibody. Based on our animal studies, we hypothesized that even though IV OKT3 given to humans is associated with systemic side effects and development of human anti-mouse responses (HAMA) that this would not occur when OKT3 was given orally. This indeed was the case even when we gave an oral dose of OKT3 (5 mg/dose for 5 days) that was identical to the intravenous dose. We did not observe OKT3 antibody in the blood which is consistent with animal studies in which we demonstrate that oral anti-CD3 acts locally in the gut-associated lymphoid tissue and does not enter the systemic circulation [5, 6]. Consistent with this, we did not observe a decrease of CD3+ T cells in the blood or modulation of CD3 from the cell surface. Furthermore, we observed no HAMA responses 30 days post oral OKT3 at any of the doses tested.
The induction of immune regulation via the oral route is dose-dependent. This has been shown for orally administered antigens [28] and we found similar results for oral anti-CD3 in animals [5-8]. Thus, in animals, the induction of regulatory T cells following oral anti-CD3 is seen at lower, rather than higher doses. A major question in humans is whether a similar dose-dependent effect exists and if so, could one identify a dose that induced regulatory T cells in humans. To address this, we dosed normal human subjects with 0.2, 1.0, and 5.0 mg oral OKT3. Although there were only three subjects per group, we found that the 1.0-mg dose was the most consistent in inducing an anti-inflammatory profile and reducing the pro-inflammatory profile of immune responses, even though some immune effects were observed at all doses fed. Thus, there were effects observed at the 0.2 and 5 mg doses, but they occurred in individual patients, not in the group as a whole. This resulted in few statistically significant effects observed at the 0.2- and 5-mg doses when all subjects treated at those doses were evaluated. The presence of a dose effect was further demonstrated in our antigen array studies.
The immune effects we observed were seen in most instances after 5 days of dosing, though in some instances effects were only observed at 10 days. In some cases (e.g., proliferation), a clear effect was observed at day 5, which was gone by day 10, demonstrating that the immune effect was linked to the time the OKT3 was administered. Further studies are now required to determine whether prolonged administration of oral OKT3 will have longer lasting effects on the immune system.
One of the major avenues being pursued for the treatment of autoimmune diseases such as multiple sclerosis is the induction of regulatory T cells and the development of therapy to decrease production of inflammatory cyto-kines such as IL-17 [29]. We observed such effects in oral anti-CD3 treated subjects. Specifically, we observed decreased production of IL-17 and IFN-γ, and an increase in cells expressing markers for Tregs (Foxp3, CTLA4, TGFβ) following oral OKT3, although we were unable to perform functional assays of suppression.
Immune effects following oral OKT3 were not only observed in T cells, but occurred in dendritic cells as well. It is well recognized that dendritic cells play a central role in immune responses and that there is cross talk between T cells and dendritic cells [30, 31]. For example, we have shown that TGF-β secreting T cells can condition dendritic cells to induce Tr1 type regulatory T cells [32]. Gut dendritic cells preferentially induce TGF-β-secreting T cells and dendritic cells from the bronchial mucosa preferentially induce IL-10 secreting T cells [33]. Therefore, we measured expression of TGF-β, IL-10 and IL-23 in dendritic cells after oral OKT3. IL-23 is a proinflammatory cytokine that is increased in dendritic cells from patients with multiple sclerosis [34, 35]. We found an increase in the dendritic cell expression of IL-10 and TGF-β, and a decrease in dendritic cell expression of IL-23 and IL-6 following oral administration of 1 mg OKT3. We do not believe that oral OKT3 is working via Fc receptors on dendritic cells, as in animal models we obtain similar results when we mucosally administer Fab’2 fragments [8].
Adjuvants play an important role in mucosal tolerance by altering the type of Tregs induced via skewing of immune function of DCs, or by changing the cytokine milieu and signaling in the mucosa [20]. β-glycosphingolipids can alter the function of both NKT and DCs [19, 21, 22]. However, we did not find that administration of GC in combination with OKT3 significantly enhanced the effect we observed when OKT3 was given alone. Like OKT3, we observed no side effects or HAMA responses in subjects given OKT3 plus GC.
In addition to conventional immune measures, we tested the effect of oral OKT3 using antigen arrays and we found a clear dose-dependent effect when we tested IgG reactivity using antigen arrays. It is known that healthy individuals have endogenous reactivity to a wide range of proteins and we found that these patterns were affected by oral OKT3 and that some of the changes persisted for as long as 30 days. The effects were restricted to the IgG repertoire, but not IgM repertoire. The IgG repertoire is T-cell-dependent whereas the IgM reportoire is not. Thus, our results are consistent with the fact that oral OKT3 targets T cell function. Although the results we obtained with antigen arrays were not related to specific reactivities, they demonstrate a clear biologic effect on the immune system and provide the basis to test antigen arrays directed against specific disease associated proteins if oral OKT3 is tested in subjects with diseases such as lupus erythematosus and multiple sclerosis.
In summary, even though the number of patients tested per dose was small (n=3) we have shown that oral administration of OKT3 in humans is safe over the short term and affects the immune system as measured by proliferation, cytokine secretion by T cells, T cell surface markers, and dendritic cell phenotype. We believe the effect we observe with oral anti-CD3 is initiated by the activation of Tregs in the gut-associated lymphoid tissue. Studies in humans using IV anti-CD3 have for the most part not measured the same parameters we report here (viz., IFN-γ and IL-17 production, changes in dendritic cells). The immune effects reported for IV anti-CD3 in humans include modulation of CD3 from the cell surface, deletion of T cells, and in some instances, re-emergence of regulatory T cells, which are CD8+. We did not observe modulation of CD3 from the cell surface or deletion of T cells. We observed a change in CD8+ T cells, though not as pronounced as observed with IV anti-CD3 and we did not measure functional characteristics of these cells. To our knowledge, this is the first demonstration of the effects of orally administering a monoclonal antibody to human subjects. Given that oral anti-CD3 clearly affects animal models of autoimmunity, the current human study provides the basis to initiate oral dosing of OKT3 in human subjects with autoimmune disease to determine if similar immunologic effects are observed.
Supplementary Material
Contributor Information
Yaron Ilan, Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, 77 Avenue Louis Pasteur, HIM 720, Boston, MA 02115, USA; Department of Medicine, Hebrew University-Hadassah Medical Center, Jerusalem, Israel.
Ehud Zigmond, Department of Medicine, Hebrew University-Hadassah Medical Center, Jerusalem, Israel.
Gadi Lalazar, Department of Medicine, Hebrew University-Hadassah Medical Center, Jerusalem, Israel.
Adi Dembinsky, Department of Medicine, Hebrew University-Hadassah Medical Center, Jerusalem, Israel.
Ami Ben Ya’acov, Department of Medicine, Hebrew University-Hadassah Medical Center, Jerusalem, Israel.
Nila Hemed, Department of Medicine, Hebrew University-Hadassah Medical Center, Jerusalem, Israel.
Ibrahim Kasis, Department of Medicine, Hebrew University-Hadassah Medical Center, Jerusalem, Israel.
Elizabeth Axelrod, Department of Medicine, Hebrew University-Hadassah Medical Center, Jerusalem, Israel.
Lidya Zolotarov, Department of Medicine, Hebrew University-Hadassah Medical Center, Jerusalem, Israel.
Athalia Klein, Department of Medicine, Hebrew University-Hadassah Medical Center, Jerusalem, Israel.
Madi El Haj, Department of Medicine, Hebrew University-Hadassah Medical Center, Jerusalem, Israel.
Roopali Gandhi, Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, 77 Avenue Louis Pasteur, HIM 720, Boston, MA 02115, USA.
Claire Baecher-Allan, Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, 77 Avenue Louis Pasteur, HIM 720, Boston, MA 02115, USA.
Henry Wu, Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, 77 Avenue Louis Pasteur, HIM 720, Boston, MA 02115, USA.
Gopal Murugaiyan, Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, 77 Avenue Louis Pasteur, HIM 720, Boston, MA 02115, USA.
Pia Kivisakk, Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, 77 Avenue Louis Pasteur, HIM 720, Boston, MA 02115, USA.
Mauricio F. Farez, Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, 77 Avenue Louis Pasteur, HIM 720, Boston, MA 02115, USA
Francisco J. Quintana, Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, 77 Avenue Louis Pasteur, HIM 720, Boston, MA 02115, USA
Samia J. Khoury, Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, 77 Avenue Louis Pasteur, HIM 720, Boston, MA 02115, USA
Howard L. Weiner, Center for Neurologic Diseases, Brigham and Women’s Hospital, Harvard Medical School, 77 Avenue Louis Pasteur, HIM 720, Boston, MA 02115, USA
References
- 1.Chatenoud L, Bluestone JA. CD3-specific antibodies: a portal to the treatment of autoimmunity. Nat Rev Immunol. 2007;7:622–32. doi: 10.1038/nri2134. [DOI] [PubMed] [Google Scholar]
- 2.Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346:1692–8. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 3.Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352:2598–608. doi: 10.1056/NEJMoa043980. [DOI] [PubMed] [Google Scholar]
- 4.Friend PJ, Hale G, Chatenoud L, Rebello P, Bradley J, Thiru S, et al. Phase I study of an engineered aglycosylated humanized CD3 antibody in renal transplant rejection. Transplantation. 1999;68:1632–7. doi: 10.1097/00007890-199912150-00005. [DOI] [PubMed] [Google Scholar]
- 5.Ishikawa H, Ochi H, Chen ML, Frenkel D, Maron R, Weiner HL. Inhibition of autoimmune diabetes by oral administration of anti-CD3 monoclonal antibody. Diabetes. 2007;56:2103–9. doi: 10.2337/db06-1632. [DOI] [PubMed] [Google Scholar]
- 6.Ochi H, Abraham M, Ishikawa H, Frenkel D, Yang K, Basso AS, et al. Oral CD3-specific antibody suppresses autoimmune encephalomyelitis by inducing CD4+ CD25- LAP+ T cells. Nat Med. 2006;12:627–35. doi: 10.1038/nm1408. [DOI] [PubMed] [Google Scholar]
- 7.Wu H, Center E, Tsokos G, Weiner H. Oral anti-CD3 induces CD4+ CD25-LAP+ regulatory T cells and suppresses murine SLE by downregulatiing pathogenic IL-17+CD4+ICOS+CXCR5+ follicular helper T cells. Lupus. 2009;18(7):586–96. doi: 10.1177/0961203308100511. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu HY, Quintana FJ, Weiner HL. Nasal anti-CD3 antibody ameliorates lupus by inducing an IL-10-secreting CD4+ CD25-LAP+ regulatory T cell and is associated with down-regulation of IL-17+ CD4+ ICOS+CXCR5+ follicular helper T cells. J Immunol. 2008;181:6038–50. doi: 10.4049/jimmunol.181.9.6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stanic AK, De Silva AD, Park JJ, Sriram V, Ichikawa S, Hirabyashi Y, et al. Defective presentation of the CD1d1-restricted natural Va14Ja18 NKT lymphocyte antigen caused by beta-D-glucosylceramide synthase deficiency. Proc Natl Acad Sci U S A. 2003;100:1849–54. doi: 10.1073/pnas.0430327100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lalazar G, Preston S, Zigmond E, Ben Yaacov A, Ilan Y. Glycolipids as immune modulatory tools. Mini Rev Med Chem. 2006;6:1249–53. doi: 10.2174/138955706778742722. [DOI] [PubMed] [Google Scholar]
- 11.Margalit M, Ghazala SA, Alper R, Elinav E, Klein A, Doviner V, et al. Glucocerebroside treatment ameliorates ConA hepatitis by inhibition of NKT lymphocytes. Am J Physiol Gastrointest Liver Physiol. 2005;289:G917–25. doi: 10.1152/ajpgi.00105.2005. [DOI] [PubMed] [Google Scholar]
- 12.Margalit M, Shalev Z, Pappo O, Sklair-Levy M, Alper R, Gomori M, et al. Glucocerebroside ameliorates the metabolic syndrome in OB/OB mice. J Pharmacol Exp Ther. 2006;319:105–10. doi: 10.1124/jpet.106.104950. [DOI] [PubMed] [Google Scholar]
- 13.Safadi R, Zigmond E, Pappo O, Shalev Z, Ilan Y. Amelioration of hepatic fibrosis via beta-glucosylceramide-mediated immune modulation is associated with altered CD8 and NKT lymphocyte distribution. Int Immunol. 2007;19:1021–9. doi: 10.1093/intimm/dxm069. [DOI] [PubMed] [Google Scholar]
- 14.Zigmond E, Preston S, Pappo O, Lalazar G, Margalit M, Shalev Z, et al. Beta-glucosylceramide: a novel method for enhancement of natural killer T lymphoycte plasticity in murine models of immune-mediated disorders. Gut. 2007;56:82–9. doi: 10.1136/gut.2006.095497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quintana FJ, Hagedorn PH, Elizur G, Merbl Y, Domany E, Cohen IR. Functional immunomics: microarray analysis of IgG autoantibody repertoires predicts the future response of mice to induced diabetes. Proc Natl Acad Sci U S A. 2004;101(Suppl 2):14615–21. doi: 10.1073/pnas.0404848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coutinho A, Kazatchkine MD, Avrameas S. Natural autoantibodies. Curr Opin Immunol. 1995;7:812–8. doi: 10.1016/0952-7915(95)80053-0. [DOI] [PubMed] [Google Scholar]
- 17.Quintana FJ, Cohen IR. The natural autoantibody repertoire and autoimmune disease. Biomed Pharmacother. 2004;58:276–81. doi: 10.1016/j.biopha.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 18.Stekel D. Microarray bioinformatics. Cambridge University Press; Cambridge: 2003. [Google Scholar]
- 19.Fujii S, Shimizu K, Hemmi H, Steinman RM. Innate Valpha14(+) natural killer T cells mature dendritic cells, leading to strong adaptive immunity. Immunol Rev. 2007;220:183–98. doi: 10.1111/j.1600-065X.2007.00561.x. [DOI] [PubMed] [Google Scholar]
- 20.Milling SW, Yrlid U, Jenkins C, Richards CM, Williams NA, MacPherson G. Regulation of intestinal immunity: effects of the oral adjuvant Escherichia coli heat-labile enterotoxin on migrating dendritic cells. Eur J Immunol. 2007;37:87–99. doi: 10.1002/eji.200636199. [DOI] [PubMed] [Google Scholar]
- 21.Shimizu K, Kurosawa Y, Taniguchi M, Steinman RM, Fujii S. Cross-presentation of glycolipid from tumor cells loaded with alpha-galactosylceramide leads to potent and long-lived T cell mediated immunity via dendritic cells. J Exp Med. 2007;204:2641–53. doi: 10.1084/jem.20070458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stronge VS, Salio M, Jones EY, Cerundolo V. A closer look at CD1d molecules: new horizons in studying NKT cells. Trends Immunol. 2007;28:455–62. doi: 10.1016/j.it.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 23.Merbl Y, Zucker-Toledano M, Quintana FJ, Cohen IR. Newborn humans manifest autoantibodies to defined self molecules detected by antigen microarray informatics. J Clin Invest. 2007;117:712–8. doi: 10.1172/JCI29943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quintana FJ, Cohen IR. Autoantibody patterns in diabetes-prone NOD mice and in standard C57BL/6 mice. J Autoimmun. 2001;17:191–7. doi: 10.1006/jaut.2001.0544. [DOI] [PubMed] [Google Scholar]
- 25.Quintana FJ, Getz G, Hed G, Domany E, Cohen IR. Cluster analysis of human autoantibody reactivities in health and in type 1 diabetes mellitus: a bio-informatic approach to immune complexity. J Autoimmun. 2003;21:65–75. doi: 10.1016/s0896-8411(03)00064-7. [DOI] [PubMed] [Google Scholar]
- 26.Goldschmidt Y, Sharon E, Quintana FJ, Cohen IR, Brandt A. Adaptive methods for classification of biological microarray data from multiple experiments. 2003. [Google Scholar]
- 27.Hueber W, Kidd BA, Tomooka BH, Lee BJ, Bruce B, Fries JF, et al. Antigen microarray profiling of autoantibodies in rheumatoid arthritis. Arthritis Rheum. 2005;52:2645–55. doi: 10.1002/art.21269. [DOI] [PubMed] [Google Scholar]
- 28.Faria AM, Weiner HL. Oral tolerance. Immunol Rev. 2005;206:232–59. doi: 10.1111/j.0105-2896.2005.00280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roncarolo MG, Battaglia M. Regulatory T-cell immunotherapy for tolerance to self antigens and alloantigens in humans. Nat Rev Immunol. 2007;7:585–98. doi: 10.1038/nri2138. [DOI] [PubMed] [Google Scholar]
- 30.Kabelitz D, Wesch D, Oberg HH. Regulation of regulatory T cells: role of dendritic cells and toll-like receptors. Crit Rev Immunol. 2006;26:291–306. doi: 10.1615/critrevimmunol.v26.i4.10. [DOI] [PubMed] [Google Scholar]
- 31.Kelsall BL, Leon F. Involvement of intestinal dendritic cells in oral tolerance, immunity to pathogens, and inflammatory bowel disease. Immunol Rev. 2005;206:132–48. doi: 10.1111/j.0105-2896.2005.00292.x. [DOI] [PubMed] [Google Scholar]
- 32.Awasthi A, Carrier Y, Peron JP, Bettelli E, Kamanaka M, Flavell RA, et al. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol. 2007;8:1380–9. doi: 10.1038/ni1541. [DOI] [PubMed] [Google Scholar]
- 33.Weiner HL. The mucosal milieu creates tolerogenic dendritic cells and T(R)1 and T(H)3 regulatory cells. Nat Immunol. 2001;2:671–2. doi: 10.1038/90604. [DOI] [PubMed] [Google Scholar]
- 34.Vaknin-Dembinsky A, Balashov K, Weiner HL. IL-23 is increased in dendritic cells in multiple sclerosis and down-regulation of IL-23 by antisense oligos increases dendritic cell IL-10 production. J Immunol. 2006;176:7768–74. doi: 10.4049/jimmunol.176.12.7768. [DOI] [PubMed] [Google Scholar]
- 35.Vaknin-Dembinsky A, Murugaiyan G, Hafler DA, Astier AL, Weiner HL. Increased IL-23 secretion and altered chemokine production by dendritic cells upon CD46 activation in patients with multiple sclerosis. J Neuroimmunol. 2008;195:140–5. doi: 10.1016/j.jneuroim.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.