

Abstract
Placental Alkaline Phosphatase (PLAP) is a tissue-restricted isozyme of the Alkaline Phosphatase (AP) superfamily. PLAP is an oncodevelopmental enzyme expressed during pregnancy and in a variety of human cancers, but its biological function remains unknown. We report here a series of catechol compounds with great affinity for the PLAP isozyme and significant selectivity over other members of the AP superfamily. These selective PLAP inhibitors will provide small molecule probes for the study of the pathophysiological role of PLAP.
Keywords: Enzyme inhibitor, Placental Alkaline Phosphatase, cancer, oncodevelopmental
Introduction
Alkaline phosphatases (APs) are found in many organisms from bacteria to humans.1,2 APs are a subgroup of a superfamily of metalloenzymes with similar metal-binding sites and predicted conserved protein structural folds.3 In humans, APs are encoded by four genes traditionally named after the tissues where they are predominantly expressed. The tissue-nonspecific AP (TNAP) gene is expressed at greatest levels in liver, bone, kidney, in the placenta during the 1st trimester of pregnancy, and at lower levels in numerous other tissues.1,2 The other three isozymes, (i.e., placental AP (PLAP), germ cell AP (GCAP), and intestinal AP (IAP)), show a much more restricted tissue expression, generally referred to as tissue-specific APs. Tissue-specific PLAP, GCAP and IAP are 90–98% homologous while TNAP is only 50% identical with the other three APs. Most of what we know about function comes from studies of the biological roles of TNAP and more recently IAP. TNAP is essential for proper skeletal mineralization. Its role is to hydrolyze inorganic pyrophosphate, a potent calcification inhibitor, via its pyrophosphatase activity.4-7 Lack of TNAP leads to infantile hypophosphatasia, a lethal form of rickets and osteomalacia in mice, rescued by enzyme replacement therapy.8 IAP appears to have a role in fat absorption in the gastrointestinal tract9,10 and as a gut mucosal defense factor11 where its expression coincides with the stage of colonization by commensal bacterial flora.12,13 In contrast, relatively little is known about the functions of PLAP and GCAP largely due to the fact that these genes evolved only recently, having appeared during a late evolutionary duplication event that preceded the divergence of Old World Monkeys and Apes.1
PLAP is expressed in large amounts in the syncytiotrophoblast cells of the placenta from about the eighth week of gestation throughout pregnancy.14 PLAP was one of the first enzymes recognized as an oncofetal protein expressed in a variety of cancers.15,16 GCAP is also found expressed in the placenta although at levels 50-fold lower than PLAP17 and it is re-expressed in testicular germ cell tumors of the testis, particularly seminomas.18,19 PLAP and GCAP are often co-expressed in ovarian cancer20 and it has been suggested that transformation of normal to malignant trophoblast might be associated with a switch from PLAP to GCAP expression.21 Thus, the ability to discriminate between these AP isozymes might facilitate studies aimed at uncovering their pathophysiological function.
Several isozyme-selective inhibitors of APs have been reported (Figure 1), including L-Phe, L-Trp, L-Leu22, 23, L–homoarginine24 and also some unrelated compounds, such as L-Leucinamide, Levamisole, the L-stereoisomer of tetramisole25, and Theophylline (a 1,3-dimethyl derivative of xanthine).26 Inhibition by these agents is of a rare uncompetitive type27 and the IC50 of these inhibitors are reported to be above 5 micromolar for all three APs and poorly selective.28, 29 Because of the great homology between PLAP, GCAP and IAP, no selective inhibitors of the PLAP enzymes have been reported until now. In this report, we describe the development of potent PLAP inhibitors that possess selectivity over TNAP and IAP and moderate selectivity over GCAP.
Figure 1.

Examples of AP inhibitors
A library of 95,857 compounds30 was tested in a luminescent HTS assay31 for discovery of chemical inhibitors of PLAP. Initially, 192 “hits” (i.e., 0.2%) were identified as having at least 50% inhibition of PLAP functional activity in an assay at 20 μM. Eighty two of these hits were confirmed in dose-response experiments for inhibition of PLAP function.31 In parallel studies, the 82 confirmed hits were also tested for inhibition selectivity against TNAP functional activity. A few confirmed hits contained a catechol moiety and were selected as a lead series. The catechol series was chemically stable and showed early trends of a structure activity relationship (SAR). Compound 1 (Fig. 2) was the most potent inhibitor “hit” from the PLAP inhibition screening campaign. 1 possessed an IC50 of 2.5 μM but was weakly selective over TNAP (IC50 = 8.6 μM) and IAP (IC50 = 49.5 μM) (Fig. 2).
Figure 2.

Depiction of the three regions of screening “hit” 1 explored by chemical synthesis: Left Hand Side (LHS), Right Hand Side (RHS) and Linker region (encircled).
A SAR was developed around 1 by varying all three regions (LHS, RHS and linker) of the catechol “hit” 1 (Fig. 2) to obtain relatively selective PLAP inhibitors over the other members of the AP family.
Chemistry
For variation of the LHS and RHS regions of the original hit 1, compounds 3-20 were readily synthesized using a one step synthesis by combining commercially available aryl substituted 2-chloroacetophenones 2 with various amines or thiols as shown in Scheme 1. Typically, the desired substituted 2-chloroacetophenone 2 was treated with one equivalent of a nucleophile in dioxane either at room temperature or by heating the mixture up to 80 °C. Products 1, 3-20 precipitated from the reaction mixture.
Scheme 1.

Synthetic approach to explore the SAR of the LHS and RHS of screening “hit” 1; preparation of analogs 3-20, X is nitrogen or sulfur. (a) 1 equiv. HXR2R3, dioxane.
Exploration of the SAR of linker region of 1 (Fig. 2) necessitated using different starting materials. Compound 22 (Scheme 2) was prepared with standard reductive-amination conditions by treating a mixture of 3,4-dihydroxybenzaldehyde 21 and 2-amino-4,6-dimethylpyrimidine in the presence of triacetoxyborohydride in dichloroethane (DCE). Compounds 25 and 26 (Scheme 2) were obtained by treating 3,4-dihydroxybenzylamine 23 and 3,4-dihydroxyphenylethylamine 24, respectively, with 4,5-dimethyl-2-furoic acid in the presence of standard peptide coupling conditions (HBTU, DIEA).
Scheme 2.

Synthetic approach to explore the linker region of screening “hit” 1: preparation of analogs 22, 25 and 26. (a) 2-amino 4,6-dimethyl pyrimidine, NaBH(OAc)3, DCE; (b) 4,5-dimethyl 2-furoic acid, HBTU, DIEA, DMF
Results and Discussion
To gain a quick understanding of the SAR requirements, analogs 27-33 (Fig. 3) were purchased and a sublibrary of 27 compounds (Tables 1-4) was synthesized according to the procedure illustrated in Schemes 1 and 2. Chemical re-synthesis of compound 1 (Table 1) was done according to Scheme 1 to compare an authentic sample with the commercially purchased compound in the bioassays used. Resynthesized 1 was found to have similar PLAP inhibitory potency as the library compound. Compounds 3-33 were compared to 1 in the enzyme inhibition assays (i.e., PLAP, TNAP) to characterize enzyme inhibition potency and selectivity. The most active compounds in the PLAP assay were also tested against IAP and GCAP.
Figure 3.

Examples of commercially available analogs 27-33 purchased and their IC50 values in the PLAP assay.
Table 1.
IC50 values for PLAP inhibition when the left hand side was varied
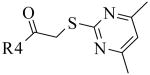 |
 |
 |
 |
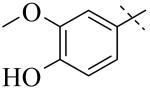 |
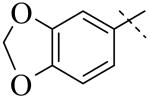 |
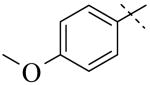 |
|---|---|---|---|---|---|---|
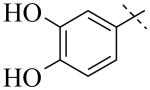
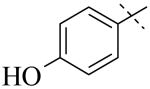
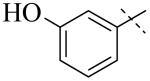
| Cmpd number | 1 | 3 | 4 | 5 | 6 | 7 |
| PLAP potency (IC50) μM a | 2.5 ± 0.1 | >100 | >100 | >100 | >100 | >100 |
Values are the mean ± SD of three experiments.
Table 4.
IC50 values for inhibition of PLAP, TNAP and IAP: SAR of the right hand side of lead structure 1.
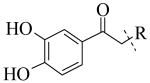 |
 |
 |
 |
 |
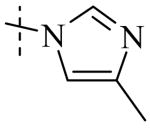 |
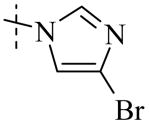 |
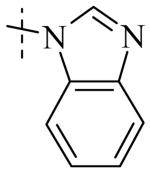 |
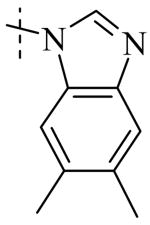 |
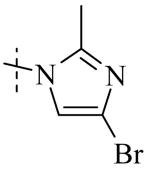 |
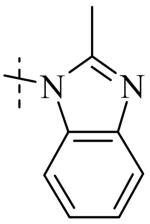 |
|---|---|---|---|---|---|---|---|---|---|---|
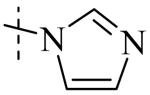
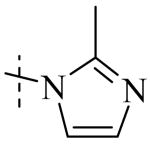
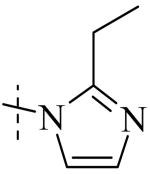
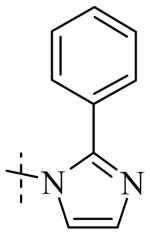
| Cmpd number | 8 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 |
| PLAP potency a IC50 μM | 2.1 ± 0.2 | 3.3 ± 0.2 | 4.2 ± 0.3 | 17.2 ± 1.3 | 5.8 ± 0.2 | 15.8 ± 1 | 0.8 ± 0.05 | 5.8 ± 0.6 | 4.9 ± 0.6 | 1.2 ± 0.1 |
| TNAP (IC50) μM Selectivity a (TNAP/PLAP) |
>100 (>32) |
78.3 ± 12.9 (25) |
>100 (>27) |
>100 (>6) |
41.1 ± 4.1 (7) |
47.5 ± 5.5 (3) |
18.0 ± 1.7 (9) |
>100 (>17) |
8.7 ± 0.7 (1.7) |
26.3 ± 2.8 (8) |
Values are the mean ± SD of three experiments.
Based on several purchased analogs (three representative examples, 27, 31-33, are shown in Figure 3), it was rapidly recognized that the 3,4-dihydroxyl arrangement of the catechol in the LHS seemed optimal for inhibition of alkaline phosphatase. Compound 27 (Fig. 3) had an IC50 of 6.7 μM whereas 28 (Fig. 3) and 29 (Fig. 3) that did not have a 3,4-dihydroxyl group were not potent (IC50 >100 μM) PLAP inhibitors.
A more thorough exploration of the SAR of the LHS aryl substituent of compound 1 (Table 1) confirmed that the 3,4-dihydroxy phenyl substituent was crucial for inhibition of PLAP functional activity. Compared to lead compound 1, removing one of the hydroxyl groups (i.e., 3, 4), capping one (i.e., 5, 7), or capping both hydroxyl groups (i.e., 6) afforded compounds with no PLAP inhibitory potency at 100 μM (Table 1).
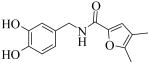
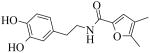
A brief SAR survey of the linker region was done by synthesis and it was observed that synthetic modification of the linker region of 1 led to a significant decrease of potency of PLAP inhibition. Thus, shortening the linker by one atom spacer and removing the ketone moiety afforded a 25-fold decrease in potency of inhibition (e.g., compound 22, Table 2). Compounds 25 and 26 (Table 2) that had a 4,5-dimethylfuran in place of the 4,6-dimethylpyrimidine as well as a different linker resulted in significant loss of inhibition potency.
Table 2.
IC50 values for PLAP inhibition when the linker region of 1 was varied.
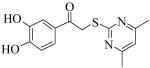 |
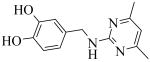 |
 |
 |
|
|---|---|---|---|---|
| Cmpd number | 1 | 22 | 25 | 26 |
| PLAP activity (IC50) μMa | 2.5 ± 0.1 | 51.2 ± 5.7 | > 100 | >100 |
Values are the mean ± SD of three experiments.
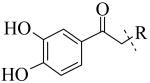
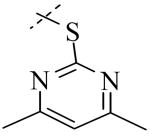
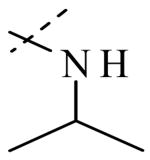
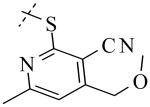
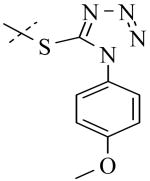
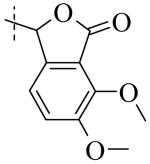
Structural variation of the RHS side chain provided SAR information for further refinement of lead molecule 1. Compound 30 (Table 3) and other RHS alkyl-substituted side-chains (not shown) suggested that an aromatic ring of the RHS was necessary for potent PLAP inhibition. The isopropyl amine analog 30 was the most potent of the alkyl substituted side-chains with an IC50 of 39.3 μM for PLAP inhibition (Table 3). Additional SAR studies with compounds maintaining a catechol group in the LHS showed that compounds tolerated a large number of RHS heteroaromatic rings (i.e., substituted thiopyridine 31, substituted tetrazole 32, benzofuranone 33, imidazole 8, triazole 9, pyrazole 10, 2-amino benzimidazole 11, Table 3) and linkers (i.e., sulfur 1, 31, 32; carbon 33; nitrogen 30, 8-11, Table 3) in place of the pyrimidine moiety. However, such heteroaromatic compounds were also generally poor selective inhibitors of PLAP compared to TNAP (i.e., 2-, 4-, and 10-fold selectivity IC50 PLAP / IC50 TNAP for 31, 32, 33 and 11 respectively, Table 3).
Table 3.
IC50 values for PLAP and TNAP inhibition: Variation of the right hand side of the lead.
 |
 |
 |
 |
 |
 |
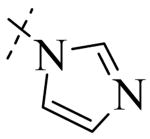 |
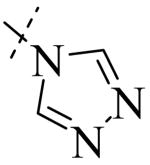 |
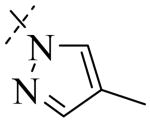 |
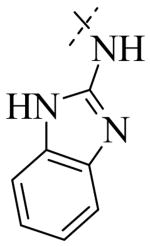 |
|---|---|---|---|---|---|---|---|---|---|
| Cmpd number | 1 | 30 | 31 | 32 | 33 | 8 | 9 | 10 | 11 |
| PLAP inh. potency a (IC50)μM | 2.5 ± 0.1 | 39.3 ± 4.3 | 2.2 ± 0.1 | 2.0 ± 0.1 | 3.0 ± 0.2 | 2.1 ± 0.2 | 13.4 ± 1.1 | 2.7 ± 0.1 | 9.1 ± 0.6 |
| TNAP inh. potency a (IC50) μM | 8.6 ± 0.5 | >100 | 4.8 ± 0.2 | 8.0 ± 0.6 | 29.2 ± 1.4 | >100 | >100 | >100 | 93.8 ± 16.2 |
Values are the mean ± SD of three experiments.
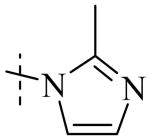
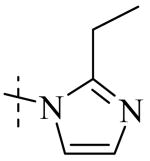
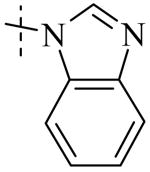
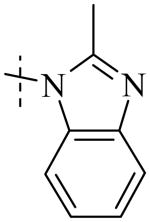
Exploration of the SAR of the RHS region of compound 1 with heterocycles such as imidazole, triazole or pyrazole directly attached to the core structure led to moderately potent PLAP inhibitors (IC50 < 14 μM) that possessed PLAP inhibition selectivity over TNAP (IC50 > 100 μM) (i.e., compounds 8-10, Table 3). Encouraged by the large inhibition selectivity of 8-10 for inhibition of PLAP compared to TNAP, a more thorough examination of the SAR of the RHS was explored using substituted imidazoles. Keeping the catechol moiety (LHS, Fig. 2) of compound 1 unchanged, a library of substituted imidazoles was prepared and tested as inhibitors of all 3 isozymes (i.e., PLAP, TNAP and IAP) for inhibition potency and selectivity. Results are summarized in Table 4. The presence of a hydrogen (8) or a small alkyl group (i.e., methyl, 12 or ethyl, 13) at the 2-position of the imidazole had little influence on PLAP inhibitory potency. All three compounds, 8, 12 and 13, had IC50 values of approximately 3 μM. With the introduction of a bulkier group at the 2-position (i.e., phenyl, 14), potency decreased 5-fold (IC50 of 14 = 17.2 μM). Substitution at the 4 position of the imidazole ring with a small group such as methyl (e.g., 15, Table 4), maintained similar inhibitory potency as the unsubstituted imidazole, 8. However, introduction of a larger group such as a bromine atom into the 4 position of the imidazole (i.e., 16, Table 4) led to a decrease in potency by about 7-fold (i.e., IC50 = 15.8 μM). Addition of a methyl group at the 2 position of compound 16 (i.e., compound 19, Table 4) increased inhibitory potency 3-fold. Compound 19 had an IC50 value of 5.0 μM compared to an IC50 of 15.8 μM for 16. The 2-benzimidazole analog (i.e., 17) was one of the most potent inhibitors with an IC50 of 0.8 μM and this result suggested that aromatic pi-pi interaction with the enzyme may be important. Extending the steric bulk of compound 17 by introduction of an alkyl group (i.e., 5,6-dimethylbenzimidazole) to afford 18 decreased the potency by about 3-fold (i.e., IC50 of 18 = 5.8 μM). Addition of a methyl at the 2 position of the benzimidazole of 17 to afford 20 maintained similar potency of inhibition compared with 17.
Within the imidazole series (Table 4), substitution at the 2-position did not seem to have a large effect on TNAP potency as the TNAP IC50 for compounds 8, 12-14 was greater than 78 μM leading to selectivity for PLAP inhibition over TNAP over 25 fold for an unsubstituted imidazole (8) or imidazoles bearing a small alkyl group such as methyl (12) or ethyl (13). Steric bulk at the 4- and or 5-position of the imidazole with compounds such as 15, 16 or 19 and the benzimidazoles 17 and 20 showed a beneficial effect on the compound's ability to inhibit TNAP. The IC50 for 15-17, 19 and 20 were 41.1, 47.5, 18, 8.7 and 26.3 μM, respectively, with bromine being the optimum group size for the compounds tested (IC50 for 19 = 8.7 μM). A larger group such as the 5,6-dimethylbenzimidazole (18) was not tolerated by the TNAP active site probably due to steric factors. The IC50 for 18 was above 100 μM.
In summary, of the compounds examined (Table 4), the unsubstituted imidazole 8 was a considerably more selective inhibitor of PLAP compared with TNAP and had an IC50 TNAP / IC50 PLAP of over 32-fold.
The most potent inhibitors of PLAP (i.e., 1, 8, 11-13, 17 and 20, Table 5) were also tested as inhibitors of IAP and GCAP, the two other isozymes of the Alkaline Phosphatase family. Results are summarized in Table 5. Substitution at the 2 position of the imidazole appeared to be critical for selectivity of IAP inhibition. When the 2-substitutent was changed from a hydrogen to a methyl to an ethyl group, the IC50 for IAP inhibition increased, leading to increasingly less potent compounds (IC50 = 30.2 μM, 64.6 μM, >100 μM respectively, for 8, 12, 13). While the number of compounds examined was limited, a similar trend was also observed for the benzimidazole analogs 17 and 20. Compound 17 had an IC50 of 14.8 μM in the IAP assay while its 2-methylated version (i.e., compound 20), did not show any detectable inhibition in the IAP assay (IC50 > 100 μM) that lead to a selectivity of over 80-fold for IAP over PLAP. Compounds shown in Table 5 were also tested for their inhibitory potential against GCAP. All of them were moderately potent inhibitors of GCAP with IC50 values < 10 μM. The IC50 value for 1, 8, 11 and 17 was 0.6 μM, 1.2 μM, 6.2 μM and 2.7 μM, respectively. Also, compared to the inhibitory potency of compounds 1, 8, 11 and 17 on PLAP and GCAP (Fig. 4), compounds 1, 8 and 11 can partially discriminate between PLAP and GCAP; the discrimination being optimal in the range around 1 μM while compound 12, 13, 17 and 20 had the same inhibitory potency for both isozymes at all inhibitor concentrations tested (graphs not shown for 12, 13 and 20).
Table 5.
Inhibitory activity for IAP and GCAP enzymes for compounds 1, 8, 10-13, 17 and 20.
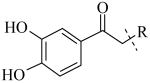 |
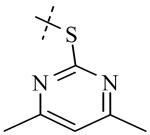 |
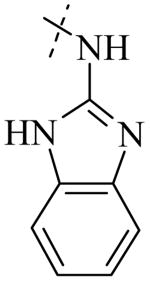 |
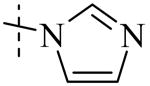 |
 |
 |
 |
 |
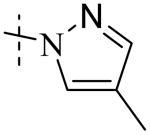 |
|---|---|---|---|---|---|---|---|---|
| Cmpd number | 1 | 11 | 8 | 12 | 13 | 17 | 20 | 10 |
| PLAP potency IC50 μM a | 2.5 ± 0.1 | 9.1 ± 0.6 | 2.1 ± 0.2 | 3.3 ± 0.2 | 4.2 ± 0.3 | 0.8 ± 0.05 | 1.2 ± 0.1 | 2.7 ± 0.1 |
| IAP (IC50) μM a Selectivity (IAP/PLAP) |
49.5 ± 6.1 (20) |
Nd | 30.2 ± 2.5 (15) |
64.6 ± 7.5 (20) |
>100 (>24) |
14.8 ± 6.8 (18) |
>100 (>80) |
53.3 ± 3.1 (25) |
| GCAP (IC50) μM a Selectivity (GCAP/PLAP) |
0.6 (0.2) |
6.2 (0.7) |
1.2 (0.6) |
* (2.1) |
* (2.1) |
2.7 (3.4) |
* (1.4) |
Nd |
Values are the mean ± SD of three experiments.
Compounds 12, 13 and 20 were tested for GCAP and compared to PLAP on a different occasion. The enzyme concentrations were different. IC50 values for PLAP and GCAP were 14.3, 30.5 μM for 12; 0.8, 1.7 μM for 13 and 6.6, 9.4 μM for 20, respectively.
Figure 4.

Effect of increasing concentrations of compound 1, 8, 11 and 17 on the inhibition of PLAP (●) and GCAP (○).
In summary, a series of 3,4-dihydroxy substituted catechols was developed that maintained inhibitory potency of the original screening hit 1 as an inhibitor of PLAP. Molecular modification of 1 provided PLAP inhibitors with greater inhibition selectivity over two other related isozymes, TNAP and IAP (Tables 4 and 5). Compound 13, possessing a 2-ethylimidazole substituent, was over 27-fold more selective as an inhibitor of PLAP compared to TNAP and IAP and maintained reasonable PLAP inhibitory potency (IC50 = 4.2 μM). Compound 10 was more than 50- and 25-fold selective as an inhibitor of PLAP over TNAP and IAP, respectively (Figure 5). Finally, compound 20 was 10- and 40-fold more selective an inhibitor of PLAP over IAP and TNAP, respectively. All three compounds (i.e., 10, 13 and 20) were more potent inhibitors of PLAP than previously reported isozyme-selective inhibitors of APs (Figure 1) and considerably more selective. We were also able to improve upon the selectivity of the screening hit 1, especially with regard to TNAP selectivity. Compounds 1, 8 and 11 are also somewhat selective for PLAP over GCAP in the one micromolar range. Because of their inhibitory selectivity, these catechol compounds may be useful as tools to understand the physiological role of PLAP in biology and pharmacology and may have applications for treating cancer or in cancer diagnostics because elevated levels of TNAP are found in ovarian and testicular cancer patients.
Figure 5.

Summary of the IC50 values for selective PLAP inhibitors 10, 13 and 20.
Experimental Section
General Methods and Materials: Reagents, starting materials, and solvents were purchased from commercial suppliers in the highest quality available and were used as received. To concentrate refers to evaporation under vacuum using a Büchi rotory evaporator. Reaction products were purified, when necessary, by chromatography on silica gel (40-63 μm) with the solvent system indicated. Proton nuclear magnetic resonance data were recorded on a Varian Mercury 300 MHz Spectrometer using TMS as the internal standard. Carbon nuclear magnetic resonance data were recorded on a Brucker 300 MHz Spectrometer using TMS as the internal standard. Compounds were assessed for purity by HPLC-MS and were at least 85% pure at 2 wavelengths (224nM and 254nM). All tested compounds were characterized by 1H NMR and purity assessed by LC/MS-1 (Platform: Agilent: equipped with an autosampler, an UV detector (220 nm and 254 nm), a MS detector (APCI); HPLC column: Waters XTerraMS C18, 3.0 × 250 mm; HPLC gradient: 1.0mL/min., from 10% acetonitrile in water to 90% acetonitrile in water in 46 min. maintaining 90% for 7.0 min. Both acetonitrile and water have 0.025% TFA.) or LC/MS-2 (Platform: Agilent 1100 series: equipped with an auto-sampler, an UV detector (230 nm and 254 nm), a MS detector (APCI); HPLC column: Phenomenex Synergi-Max RP, 2.0 × 50 mm column; HPLC gradient: Solvent C is 6mM Ammonium Formate in water, solvent D is 25% Acetonitrile in Methanol. The gradient runs from 5% D (95% C) to 95% D (5% C) in 6.43min with a 1.02 min hold at 95% D followed by a return and hold at 5% D for 1.52 min.) (tR is retention time (TIC % purity).
TNAP, PLAP and IAP Luminescent assays31
Briefly, the TNAP assay was performed in 384-well white plates (784075, Greiner, Monroe, NC) at 50 μM CDP-star substrate in assay buffer containing 100 mM DEA-HCl (pH 9.8), 1 mM MgCl2, and 20 μM ZnCl2. The luminescence signal was measured after a 30-min incubation at room temperature on an EnVision plate reader (PerkinElmer, Waltham, MA). Analogous luminescent assays were optimized and used for PLAP, IAP and GCAP with the CDP-star concentration adjusted to their respective Km values, 85 μM, 177 μM and 10μM respectively.
Synthesis of compounds 1, 3-20
In a typical procedure, 2-chloro-acetophenone (0.27 mmol) in dioxane (1 mL) was added to the desired substituted amine or thiol (0.27 mmol). The solution was stirred overnight in a sealed vial at room temperature. The reaction was monitored by TLC (dichloromethane/methanol, 90:10, v/v) and the reaction product that precipitated from the solution was filtered, washed with a minimum amount of dioxane and then dried under high vacuum to afford the product as a solid in 30 to 50 % yield.
Cmpd 1: 1H NMR (CD3OD) δ 7.55 (dd, J = 8.3 and 2.3 Hz, 1H), 7.47 (d, J =2.3Hz, 1H), 7.12 (s, 1H), 6.84 (d, J= 8.3Hz, 1H), 4.77 (s, 2H), 2.38 (s, 6H); LC/MS-1: tR= 1.41 (100%). MS: m/z 290.9 [M+H]+, expected 291 [M+H]+.
Cmpd 3: 1H NMR (DMSO) δ 7.95 (d, J=3Hz, 2H), 7.10 (s, 1H), 6.85 (d, J=3Hz, 2H), 4.57 (s, 2H), 2.32 (s, 6H); LC/MS-1: tR= 1.83 (100%). MS: m/z 275.4 [M+H]+, expected 275.4 [M+H]+.
Cmpd 4: 1H NMR (CD3OD) δ 7.57 (d, J =7.8 Hz, 1H), 7.44 (s, 1H), 7.39 (d, J = 7.8 Hz, 1H). 7.34 (bs, 1H), 7.10 (dd, J = 6.9 and 0.9 Hz, 1H), 4.95 (s, 2H), 2.52 (s, 6H); LC/MS-1: tR= 2.55 (100%). MS: m/z 274.9 [M+H]+, expected 275 [M+H]+.
Cmpd 5: 1H NMR (CDCl3) δ 7.72 (dd, J = 8.1 and 1.8 Hz, 1H), 7.58 (d, J = 1.8 Hz, 1H), 6.96 (d, J= 8.1Hz, 1H), 6.68 (s, 1H), 4.59 (s, 2H), 3.94 (s, 3H), 2.34 (s, 6H); LC/MS-1: tR= 1.57 (100%). MS: m/z 304.9 [M+H]+, expected 305 [M+H]+.
Cmpd 6: 1H NMR (DMSO) δ 7.77 (dd, J = 9.0 and 2.0 Hz, 1H), 7.51 (d, J = 2.0 Hz, 1H), 6.95 (d, J= 9.0Hz, 1H), 6.07 (s, 1H), 4.61 (s, 2H), 2.30 (s, 6H); LC/MS-1: tR= 3.72 (100%). MS: m/z 303.4 [M+H]+, expected 303.5 [M+H]+.
Cmpd 7: 1H NMR (CDCl3) δ 8.05 (d, J = 3 Hz, 2H), 6.95 (d, J = 3 Hz, 2H), 6.71 (s, 1H), 4.57 (s, 2H), 3.87 (s, 3H), 2.32 (s, 6H); LC/MS-1: tR= 3.45 (100%). MS: m/z 288.9 [M+H]+, expected 289 [M+H]+.
Cmpd 8: LC/MS-1: tR= 0.36 (100%). MS: m/z 219.3 [M+H]+, expected 219 [M+H]+.
Cmpd 9: 1H NMR (CD3OD) δ 8.31 (s, 2H), 7.45-7.39 (m, 2H), 6.83 (d, J = 9Hz, 1H), 4.80 (s, 2H). LC/MS-2: tR= 0.65 (100%). MS: m/z 220.1 [M+H]+, expected 220 [M+H]+.
Cmpd 10: 1H NMR (CD3OD) δ 7.48 (dd, J = 8.1 and 1.8 Hz, 1H), 7.42 (d, J = 2.1 Hz, 1H), 7.40 (s, 1H), 7.32 (s, 1H), 6.86 (d, J = 8.1 Hz, 1H), 5.56 (s, 2H), 2.10 (s, 3H); 13C NMR (DMSO) δ 189.6, 151.4, 145.6, 138.9, 132.7, 126.0, 121.8, 116.5, 115.2, 114.7, 58.9, 8.4;); LC/MS-1: tR=2.45 (100%). MS: m/z 234.5 [M+H]+, expected 234 [M+H]+.
Cmpd 11: 1H NMR (CD3OD) δ 7.46-7.41 (m, 2H), 7.29-7.22 (m, 2H), 7.11-7.06 (m, 2H), 6.83 (d, J = 9Hz, 1H). MS (ESI+) m/z 284.2 (M+H)+,
Cmpd 12: 1H NMR (CD3OD) δ 7.55-7.40 (m, 2H), 7.14 (s, 1H), 7.09 (dd, J = 15 and 1.5 Hz, 1H), 6.83 (d, J = 9 Hz, 1H), 4.80 (s, 2H), 2.34 (s, 3H); LC/MS-1: tR= 0.55 (100%). MS: m/z 233.5 [M+H]+, expected 233 [M+H]+.
Cmpd 13: 1H NMR (CD3OD) δ 7.45-7.38 (m, 2H), 7.00 (s, 2H), 6.83 (d, J = 9 Hz, 1H), 4.80 (s, 2H), 2.76 (q, J = 7.8 Hz, 2H), 1.29 (t, J = 7.8 Hz, 3H); 13C NMR (DMSO) δ 190.5, 151.8, 148.6, 145.4, 125.9, 122.8, 121.8, 119.1, 115.3, 114.8, 66.3, 19.4, 11.8; LC/MS-2: tR= 0.74 (100%). MS: m/z 247.1 [M+H]+, expected 247 [M+H]+.
Cmpd 14: 1H NMR (CD3OD) δ 7.84 (d, J = 7.2 Hz, 2H), 7.46-7.34 (m, 5H), 7.14 (s, 2H), 6.83 (d, J = 9 Hz, 1H), 4.78 (s, 2H); LC/MS-1: tR= 1.71 (100%). MS: m/z 295.4 [M+H]+, expected 295.5 [M+H]+.
Cmpd 15: 1H NMR (CD3OD) δ 7.58-7.37 (m, 3H), 7.09 (brs, 1H), 6.84 (d, J = 9 Hz, 1H), 4.80 (s, 2H), 2.32 (s, 3H); MS: m/z 233.3 [M+H]+, expected 233.5 [M+H]+.
Cmpd 16: 1H NMR (CD3OD) δ 7.62 (s, 1H), 7.48-7.39 (m, 2H), 7.11 (s, 1H), 6.83 (d, J = 9 Hz, 1H), 4.80 (s, 2H); LC/MS-1: tR= 1.53 (100%). MS: m/z 299.6 [M+H]+, expected 299.5 [M+H]+.
Cmpd 17: 1H NMR (CD3OD) δ 7.67-7.25 (m, 6H), 6.98-6.90 (m, 1H), 6.93 (d, J = 9 Hz, 1H), 6.83 (d, J = 9 Hz, 1H), 4.79 (s, 2H); MS: m/z 269.3 [M+H]+, expected 269 [M+H]+.
Cmpd 18: 1H NMR (CD3OD) δ 8.20 (s, 1H), 7.45-7.36 (m, 4H), 6.83 (d, J = 9 Hz, 1H), 4.77 (s, 2H), 2.35 (s, 6H); MS: m/z 297.3 [M+H]+, expected 297.5 [M+H]+.
Cmpd 19: 1H NMR (CD3OD) δ 7.45-7.40 (m, 2H), 6.93 (s, 1H), 6.83 (d, J = 9 Hz, 1H), 4.79 (s, 2H), 2.32 (s, 3H); LC/MS-1: tR= 1.48 (100%). MS: m/z 313.0 [M+H]+, expected 313 [M+H]+.
Cmpd 20: 1H NMR (CD3OD) δ 7.90-7.39 (m, 5H), 6.98-6.90 (m, 1H), 6.93 (d, J = 6 Hz, 1H), 6.83 (d, J = 9 Hz, 1H), 4.79 (s, 2H), 2.68 (s, 3H); 13C NMR (DMSO) δ 189.6, 152.7, 151.4, 151.2, 145.4, 135.4, 126.1, 122.9, 122.3, 121.9, 117.3, 115.2, 113.9, 110.2, 66.3, 13.5; MS: m/z 283.3 [M+H]+, expected 283 [M+H]+.
Cmpd 22: 50 mg (0.36 mmol) of 3,4-dihydroxybenzaldehyde and 44 mg (0.36 mmol) of 2-amino-4,6-dimethylpyridine was stirred in 1 ml DCE in the presence of 153 mg (0.72 mmol) of sodium triacetoxyborohydride and 1 drop of acetic acid at r.t. overnight. The reaction was quenched with 1 ml water. The product was extracted with ethyl acetate and purified by PTLC (ethyl acetate/methanol, 90:10, v/v) to give 25 mg of the product (28% yield). 1H NMR (CD3OD) δ 7.29 (s, 1H), 6.88 (d, J = 9 Hz, 1H), 6.80-6.62 (m, 2H), 4.41 (s, 2H), 2.25 (s, 3H), 1.96 (s, 3H); LC/MS-1: tR= 2.33 (85.13%). MS: m/z 246.4 [M+H]+, expected 246.5 [M+H]+.
Cmpd 25: 50 mg (0.23 mmol) of 3,4-dihydroxybenzylamine 23 was dissolved in 1 ml THF/1 ml DMF. 100 mg (0.27 mmol) of HBTU, 128 μl (0.69 mmol) of DIEA and 32 mg (0.23 mmol) of 1,5-dimethyl-3-furoic acid were added and the mixture was stirred at r.t. overnight. The crude material was purified by PTLC (DCM/MeOH, 90:10, v/v) to give a yellow solid (60 mg, 98% yield)). 1H NMR (CD3OD) δ 6.88 (s, 1H), 6.76 (s, 1H), 6.69 (d, J = 7.8 Hz, 1H), 6.63 (d, J = 7.8 Hz, 1H), 6.27 (s, 1H), 4.35 (s, 2H), 2.25 (s, 3H), 1.96 (s, 3H); LC/MS-1: tR= 2.33 (100%). MS: m/z 262.5 [M+H]+, expected 262.5 [M+H]+.
Cmpd 26: 44 mg (0.23 mmol) of dopamine.HCl 24 was dissolved in 1 ml THF/1 ml DMF. 100 mg (0.27 mmol) of HBTU, 128 μl (0.69 mmol) of DIEA and 32 mg (0.23 mmol) of 4,5-dimethyl-2-furoic acid were added and the mixture was stirred at r.t. overnight. The crude material was purified by PTLC (DCM/MeOH, 90:10, v/v) to give an off-white solid (30 mg, 47% yield)). 1H NMR (CD3OD) δ 6.68 (d, J = 8 Hz, 1H), 6.66 (s, 1H), 6.54 (d, J = 9.4 Hz, 1H), 6.18 (s, 1H), 3.39-3.49 (m, 2H), 2.69 (t, J = 8 Hz, 2H), 2.44 (s, 3H), 2.21 (s, 3H); LC/MS-1: tR= 2.23 (100%). MS: m/z 275.9 [M+H]+, expected 276 [M+H]+.
Cmpds 27-30 and 32-33 were purchased from ChemBridge and 31 from ChemDiv.
Acknowledgments
This work was supported by NIH Roadmap Initiatives grant U54HG003916. Ana Maria Simão and José Luis Millán were supported by NIH grants DE12889 and 1X01MH077602-01.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Millán JL. Mammalian alkaline phosphatases. From Biology to Applications in Medicine and Biotechnology. Wiley-VCH Verlag GmbH & Co; Weinheim, Germany: 2006. pp. 1–322. [Google Scholar]
- 2.McComb RB, Bowers GN, Jr, Posen S. Alkaline Phosphatase. New York: Plenum; 1979. [Google Scholar]
- 3.Galperin MY, Bairoch A, Koonin EV. Protein Sci. 1998;7:1829. doi: 10.1002/pro.5560070819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hessle L, Johnson KA, Anderson HC, Narisawa S, Sali A, Goding JW, Terkeltaub R, Millán JL. Proc Natl Acad Sci U S A. 2002;99:9445. doi: 10.1073/pnas.142063399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harmey D, Hessle L, Narisawa S, Johnson KA, Terkeltaub R, Millán JL. Am J Pathol. 2004;164:1199. doi: 10.1016/S0002-9440(10)63208-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harmey D, Johnson KA, Zelken J, Camacho NP, Hoylaerts MF, Noda M, Terkeltaub R, Millán JL. J Bone Mineral Res. 2006;21:1377. doi: 10.1359/jbmr.060619. [DOI] [PubMed] [Google Scholar]
- 7.Murshed M, Harmey D, Millán JL, McKee MD, Karsenty G. Genes Dev. 2005;19:1093. doi: 10.1101/gad.1276205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Millán JL, Narisawa S, Lemire I, Loisel TP, Boileau G, Leonard P, Gramatikova S, Terkeltaub R, Pleshko Camacho N, McKee MD, Crine P, Whyte MP. J Bone Miner Res. 2008;23:777. doi: 10.1359/JBMR.071213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narisawa S, Huang L, Iwasaki A, Hasegawa H, Alpers DH, Millán JL. Mol Cell Biol. 2003;23:7525. doi: 10.1128/MCB.23.21.7525-7530.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakano T, Inoue I, Koyama I, Kanazawa K, Nakamura K, Narisawa S, Tanaka K, Akita M, Masuyama T, Seo M, Hokari S, Katayama S, Alpers DH, Millán JL, Komoda T. Am J Physiol Gastrointest Liver Physiol. 2007;292:1439. doi: 10.1152/ajpgi.00331.2006. [DOI] [PubMed] [Google Scholar]
- 11.Goldberg RF, Austen WG, Jr, Zhang X, Munene G, Mostafa G, Biswas S, McCormack M, Eberlin KR, Nguyen JT, Tatlidede HS, Warren HS, Narisawa S, Millán JL, Hodin RA. Proc Natl Acad Sci USA. 2008;105:3551. doi: 10.1073/pnas.0712140105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bates JM, Akerlund J, Mittge E, Guillemin K. Cell Host and Microbe. 2007;2:371. doi: 10.1016/j.chom.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Narisawa S, Hoylaerts MF, Doctor KS, Fukuda MN, Alpers DH, Millán JL. Am J Physiol Gastrointest Liver Physiol. 2007;293:1068. doi: 10.1152/ajpgi.00073.2007. [DOI] [PubMed] [Google Scholar]
- 14.Fishman L, Miyayama H, Driscoll SG, Fishman WH. Cancer Res. 1976;36:2268. [PubMed] [Google Scholar]
- 15.Fishman WH, Ghosh NK, Inglis NR, Green S. Enzymologia. 1968;34:317. [PubMed] [Google Scholar]
- 16.Fishman WH, Inglis NR, Green S, Anstiss CL, Gosh NK, Reif AE, Rustigian R, Krant MJ, Stolbach LL. Nature. 1968;219:697. doi: 10.1038/219697a0. [DOI] [PubMed] [Google Scholar]
- 17.Povinelli CM, Knoll BJ. Placenta. 1991;12:663. doi: 10.1016/0143-4004(91)90500-f. [DOI] [PubMed] [Google Scholar]
- 18.Wahren B, Holmgren PA, Stigbrand T. Int J Cancer. 1979;24:749. doi: 10.1002/ijc.2910240608. [DOI] [PubMed] [Google Scholar]
- 19.Wahren B, Hinkula J, Stigbrand T, Jeppsson A, Andersson L, Esposti PL, Edsmyr F, Millán JL. Int J Cancer. 1986;37:595. doi: 10.1002/ijc.2910370419. [DOI] [PubMed] [Google Scholar]
- 20.Smans KA, Ingvarsson MB, Lindgren P, Canevari S, Walt H, Stigbrand T, Bäckström T, Millán JL. Int J Cancer. 1999;83:270. doi: 10.1002/(sici)1097-0215(19991008)83:2<270::aid-ijc20>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 21.Ovitt CE, Strauss AW, Alpers DH, Chou JY, Boime I. Proc Natl Acad Sci U S A. 1986;83:3781. doi: 10.1073/pnas.83.11.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fishman WH, Sie HG. Enzymologia. 1971;41:141. [PubMed] [Google Scholar]
- 23.Doellgast GJ, Fishman WH. Clin Chim Acta. 1977;75:449. doi: 10.1016/0009-8981(77)90365-5. [DOI] [PubMed] [Google Scholar]
- 24.Lin CW, Fishman WH. J Biol Chem. 1972;247:3082. [PubMed] [Google Scholar]
- 25.Van Belle H. Clin Chem. 1976;22:972. [PubMed] [Google Scholar]
- 26.Farley JR, Ivey JL, Baylink DJ. J Biol Chem. 1980;255:4680. [PubMed] [Google Scholar]
- 27.Cornish-Bowden A. Fundamentals of Enzyme Kinetics. Third. Chapter 5. London: Portland Press Ltd; 2004. pp. 113–144. [Google Scholar]
- 28.Fishman WH. Clin Biochem. 1987;20:387. doi: 10.1016/0009-9120(87)90003-8. [DOI] [PubMed] [Google Scholar]
- 29.Kozlenkov A, Le Du MH, Cuniasse P, Ny T, Hoylaerts MF, Millán JL. J Bone Miner Res. 2004;11:1862. doi: 10.1359/JBMR.040608. [DOI] [PubMed] [Google Scholar]
- 30.The compound library was supplied by the National Institutes of Health (NIH) Molecular Libraries Small Molecule Repository. MLSMR, http://www.mli.nih.gov/mlsmr.
- 31.Sergienko E, Su Y, Chan X, Brown B, Hurder A, Narisawa S, Millán JL. J Biomol Screen. 2009;14:824. doi: 10.1177/1087057109338517. [DOI] [PMC free article] [PubMed] [Google Scholar]