

Abstract
Neutrophil-induced coronary microvascular barrier dysfunction is an important pathophysiological event in heart disease. Currently, the precise cellular and molecular mechanisms of neutrophil-induced microvascular leakage are not clear. The aim of this study was to test the hypothesis that rho kinase (ROCK) increases coronary venular permeability in association with elevated endothelial tension. We assessed permeability to albumin (Pa) in isolated porcine coronary venules and in coronary venular endothelial cell (CVEC) monolayers. Endothelial barrier function was also evaluated by measuring transendothelial electrical resistance (TER) of CVEC monolayers. In parallel, we measured isometric tension of CVECs grown on collagen gels. Transference of constitutively active (ca)-ROCK protein into isolated coronary venules or CVEC monolayers caused a significant increase in Pa and decreased TER in CVECs. The ROCK inhibitor Y-27632 blocked the ca-ROCK-induced changes. C5a-activated neutrophils (106/ml) also significantly elevated venular Pa, which was dose-dependently inhibited by Y-27632 and a structurally distinct ROCK inhibitor, H-1152. In CVEC monolayers, activated neutrophils increased permeability with a concomitant elevation in isometric tension, both of which were inhibited by Y-27632 or H-1152. Treatment with ca-ROCK also significantly increased CVEC monolayer permeability and isometric tension, coupled with actin polymerization and elevated phosphorylation of myosin regulatory light chain on Thr18/Ser19. The data suggest that during neutrophil activation, ROCK promotes microvascular leakage in association with actin-myosin-mediated tension development in endothelial cells.
Keywords: microvascular permeability, neutrophil-endothelium interaction, signal transduction, cytoskeleton
The microvascular barrier regulates fluid and solute exchange between blood and the tissues. During inflammation, inflammatory mediators can cause excessive microvascular leakage, resulting in edema and tissue dysfunction. Endothelial cells play an active role in the regulation of microvascular permeability (21, 51). Electron microscopy studies by Majno and Palade (26) suggested that increases in permeability are attributed to elevated paracellular flux of solutes through gaps formed between endothelial cells. Subsequently, several reports have suggested that inflammatory mediator-induced gap formation between endothelial cells is often caused by two concomitant actions: 1) increased actomyosin-driven contraction of endothelial cells and 2) decreased adhesiveness between endothelial cells (12, 13, 32, 33, 50). Thus regulation of the endothelial barrier involves a dynamic balance between acto-myosin-dependent centripetal forces and adhesion-dependent centrifugal forces in individual endothelial cells. The specific mechanisms regulating these events, however, are not clear.
Polymorphonuclear leukocytes (PMN), predominantly neutrophils, play an important role in inflammatory responses by releasing various hyperpermeability factors (6, 9, 25, 57). Our laboratory has previously demonstrated that C5a-activated PMN increase permeability in both cell monolayer and isolated venule models (8, 42, 43, 52). The permeability increases were associated with increased dual phosphorylation of myosin regulatory light chains (MLC), actin stress fiber formation, and tyrosine phosphorylation of the adherens junction protein β-catenin (14, 42, 43, 52).
The role of the signaling protein rho kinase (ROCK) in microvascular permeability regulation has recently become an interesting topic, particularly because the precise role of ROCK is controversial. Several studies using cultured endothelial cell models report that ROCK promotes increased permeability in response to various edemagenic stimuli (10, 27, 45, 46, 48, 58). However, other studies utilizing rodent mesenteric microvessels report that ROCK is not involved in permeability regulation (1, 2). Recently, we reported that C5a-activated PMN cause an increase in RhoA activity in endothelial cells and that inhibition of ROCK attenuated PMN-stimulated hyperpermeability (8). However, this study was limited to an endothelial cell monolayer model and did not involve an assessment of permeability in microvessels. The biomechanical basis for ROCK-mediated hyperpermeability remains to be established.
Thus the first objective of this study was to test the hypothesis that ROCK promotes venular hyperpermeability. Keeping in mind that actin-myosin-mediated contraction is thought to regulate permeability (13, 28, 29, 31, 47), at least in part, and that ROCK is involved in the regulation of actin (3, 4), the second objective was to test the hypothesis that ROCK-mediated increases in venular permeability are associated with elevated endothelial cell isometric tension. We applied an integrative approach to study these objectives, using both isolated venule and venular endothelial cell monolayer models. We used the protein transfection technique that we developed before (40, 44), complemented by a pharmacological approach, to specify the role of ROCK in mediating inflammatory hyperpermeability.
MATERIALS AND METHODS
Materials
Human C5a, Y-27632, and H-1152 were obtained from Calbiochem (San Diego, CA). Recombinant ROCK II protein (contains the catalytic domain and a His tag) was from Upstate (Lake Placid, NY). Endothelial basal medium and endothelial growth medium (EGM) were obtained from Clonetics (San Diego, CA). FITC-albumin, FITC-phalloidin, Hoechst 33342, and cytochalasin D were from Sigma (St. Louis, MO). Goat anti-His tag and anti-goat IgG-FITC antibodies were from Santa Cruz (Santa Cruz, CA). Rabbit anti-phospho-MLC, rabbit anti-MLC, and anti-rabbit horseradish peroxidase antibody were from Cell Signaling (Beverly, MA). TransIT-LT1 was from Mirus (Madison, WI). Vectashield mounting medium was from Vector Laboratories (Burlingame, CA).
Isolated coronary venule permeability model
All animal protocols were performed with approval from the University of California at Davis and Texas A&M Institutional Animal Care and Use Committees and in accordance with the National Research Council’s Guide for the Care and Use of Laboratory Animals. This technique has been described previously in detail (53, 55, 56). Briefly, Yorkshire pigs (male or female, randomly selected), ~5 wk old and weighing 9–13 kg, were sedated with Telazol (tiletamine-zolazepam; 4.4 mg/kg im) and xylazine (2.2 mg/kg im), anesthetized with pentobarbital sodium (30 mg/kg iv), and heparinized (250 U/kg iv). A left thoracotomy was performed, and the heart was excised and placed in 4°C physiological saline. For each experiment, a venule 30–60 μm in diameter was dissected and cannulated with a micropipette on each end, with a third smaller pipette inserted into the inflow micropipette. In transfection experiments, a fourth smaller pipette was inserted into the outflow pipette. Each micropipette was connected to a reservoir to allow independent control of intraluminal perfusion pressure and flow. The vessel was interchangeably perfused with either physiological salt solution through the outer pipette or the same perfusate containing FITC-albumin through the inner inflow pipette. Transfection reagent (TransIT-LT1) and ca-ROCK protein were administered via the inner outflow pipette by reversing flow. C5a and isolated PMN were added to the bath solution. The permeability of the vessel was quantified by measuring the ratio of transvascular flux to the transmural concentration difference of the tracer (53). The apparent solute permeability coefficient of albumin (Pa) is calculated as Pa = (1/ΔIf)(dIf/dt)0(r/2), where ΔIf is the initial step increase in fluorescent intensity, (dIf/dt)0 is the initial rate of gradual increase in intensity as solutes diffuse out of the vessel, and r is the venular radius. For every experiment, venules were perfused at a constant perfusion pressure of 10 cmH2O at a flow velocity of 7 mm/s.
Protein transfection
Transference of ca-ROCK into endothelial cells was performed using a previously described protein transfection method (39, 40, 44). Briefly, ca-ROCK (3 μg/ml) is combined with the polyamine transfection reagent TransIT-LT1 (10 μl/ml) and then incubated with coronary venular endothelial cells (CVEC) or perfused through an isolated venule for the times indicated.
Isolation and activation of porcine neutrophils
Porcine neutrophils (PMN) were isolated as previously described (42, 43, 52). Briefly, porcine arterial blood was centrifuged for 20 min at 300 g to separate plasma and blood cells. The plasma was centrifuged for 10 min at 2,500 g to obtain platelet-poor plasma (PPP). The buffy coat was removed, and the pellet containing red blood cells and PMN was incubated in a solution containing 2% gelatin and 20% PPP in Hanks’ balanced salt solution at 37°C for 45 min. The supernatant was centrifuged at 300 g for 10 min, and PMN were further purified from the supernatant by hypotonic hemolysis. Viability and normal chemotactic function were verified as previously reported (54, 57).
Endothelial cell culture and monolayer permeability assay
CVEC, harvested from the bovine heart (37), were routinely maintained in EGM supplemented with 10% fetal bovine serum. For permeability assays, cells were grown 4–5 days on gelatin-coated Costar Transwell membranes (VWR, Houston, TX) as previously described (8, 43). CVEC were treated with PMN (106/ml) immediately followed by 10−8 M C5a in the presence of FITC-albumin for the indicated times. The 106 PMN/ml suspension was chosen based on a standard procedure used by many laboratories and to ensure that concentrations of mediators secreted by PMN would be consistent between experiments. A 106 PMN/ml suspension translated into PMN:CVEC ratios ranging from 1:1 to 5:1, depending on the cultureware required for each experiment. These ratios are within the range used by others (20, 22). For experiments with inhibitors, the indicated agent was added 20 min before the addition of PMN. Samples were collected from both the upper (luminal) and lower (abluminal) chambers for fluorometry analysis. Albumin concentrations were determined using a standard curve, and Pa was calculated as Pa = [A]/t × 1/A × V/[L], where [A] is the abluminal albumin concentration, t is time in s, A is the area of the membrane in cm2, V is the volume of the abluminal chamber, and [L] is the luminal albumin concentration.
Assessment of barrier function by transendothelial electrical resistance
Transendothelial electrical resistance (TER) was measured using an electrical cell motility system (CET, Iowa City, IA) as previously reported (15, 23, 28, 34). Briefly, CVEC were cultured (105 cells/cm2) on a small gold electrode array (Applied Biophysics, Troy, NY). Medium served as the electrolyte, and barrier function was dynamically measured by determining the impedance of a cell-covered electrode. A 1-V, 4,000-Hz alternating current signal was supplied through a 1-MΩ resistor to approximate a constant-current source. The in-phase voltage (proportional to resistance) and the out-of-phase voltage (proportional to capacitive resistance) were measured. Endothelial monolayers with baseline resistance values below 5,000 Ω were excluded from the study. Endothelial barrier function is expressed as the fractional change in TER.
Measurement of cell monolayer isometric tension
Isometric tension of endothelial cells was dynamically measured using methods based on previously described systems (7, 16, 24). Briefly, cells were grown to confluence on collagen gels affixed to polyethylene bars on two sides. The polyethylene bars on either side of the gel were connected to isometric force transducers (Harvard Apparatus, Natick, MA) using spinal needles bent 90° at each end. The force transducers were oriented in opposite vectors so that both measured increasing force as the gel contracts. Voltage output from each transducer was directed through a 4-Hz low-pass filter (National Instruments, Austin, TX) and then to a computer via a data-acquisition board (National Instruments). Measurements were made while the cell-covered collagen gel floated freely in serum-free medium containing 1% fatty acid-free BSA. Baseline was monitored for 60 min to make sure there was no drift in baseline tension. Before addition of drugs, sham experiments with vehicle only were performed to ensure that observed effects of drugs were not due to nonspecific mechanical disturbance of the collagen gels. In addition, sham experiments with no cells on the collagen gel were performed to exclude the possibility that drugs or PMN altered tension by directly acting on the collagen (data not shown). Mean changes in isometric tension are presented as force per units of surface area of gel covered by cells or as a percentage of the baseline tension.
Fluorescence microscopy
CVEC were grown to confluence on gelatin-coated, round No. 1 coverslips (VWR). After treatment with PMN and/or drugs, the cells were fixed in 4% paraformaldehyde in PBS for 5 min and permeabilized with 0.5% Triton X-100 for 5 min. Transfected ca-ROCK protein (which contains a His tag) was labeled with goat anti-His antibody followed by an anti-goat IgG-FITC secondary antibody. F-actin was labeled with FITC-phalloidin, and nuclei were labeled with Hoechst 33342. The coverslips were mounted on glass slides with Vectashield. Images were obtained using a Zeiss Axiovert 300M fluorescence microscope equipped with an AxioCam MRm black-and-white digital camera (Zeiss, Thornwood, NY). Digital images were collected using Zeiss Axiovision 4.0 software.
Data analysis
Pa and tension (means ± SE) are presented either as absolute values or as a percentage of baseline or control. ANOVA followed by Tukey’s honestly significantly difference test was used to evaluate the significance of intergroup differences. Significance was accepted at P < 0.05.
RESULTS
To test the hypothesis that ROCK promotes increased microvascular leakage, we assessed Pa in isolated coronary venules during transfection of ca-ROCK protein. After the addition of the ca-ROCK transfection mixture, Pa steadily increased in magnitude and was significantly higher than baseline 50 min after the initiation of transfection. Pa remained significantly elevated until the 100-min time point, after which Pa remained slightly (but insignificantly) higher than baseline (Fig. 1). Treatment of isolated coronary venules with the transfection reagent (TransIT-LT1) alone does not cause any changes in Pa compared with baseline (42, 52). When ca-ROCK transfection was performed in the presence of the ROCK inhibitor Y-27632, Pa increased slightly but did not differ significantly from baseline (Fig. 1).
Fig. 1.

Transference of constitutively active rho kinase (ca-ROCK) protein increases coronary venular permeability to albumin (Pa). Venules were transfected with 3 μg/ml ca-ROCK protein for 120 min in the absence (solid line, ■, n = 6) or presence (dashed line, ▲, n = 4) of the ROCK inhibitor Y-27632 (5 × 10−6 M). Y-27632 was added to the bath 20 min before the addition of ca-ROCK. Transfection was initiated at 0 min and continued throughout the time course. *P < 0.05 vs. baseline permeability (0-min time point). †P < 0.05 vs. ca-ROCK transfection when Y-27632 is present.
We next evaluated whether pharmacological inhibition of ROCK could attenuate PMN-induced increases in coronary venular Pa. C5a-activated PMN caused a significant increase in Pa within 7 min after the initiation of treatment and remained significantly elevated through the 20-min time point (Fig. 2A). Pretreatment of isolated venules for 20 min with Y-27632 significantly diminished the PMN-stimulated increase in Pa (Fig. 2A) in a concentration-dependent manner (Fig. 2B). The novel and structurally distinct ROCK inhibitor H-1152 also significantly attenuated PMN-induced hyperpermeability (time course data not shown) in a concentration-dependent manner (Fig. 2C).
Fig. 2.

Inhibition of ROCK diminishes neutrophil-induced increases in Pa in isolated coronary venules. A: time course of polymorphonuclear leukocytes (PMN)-induced hyperpermeability in the absence (●, n = 4) and presence (▲, n = 6) of 5 × 10−6 M Y-27632. *P < 0.05 vs. baseline Pa. †P < 0.05 vs. PMN + Y-27632 group. B and C, respectively, show that Y-27632 and H-1152 inhibit PMN-induced increases in venular Pa in a concentration-dependent manner (10-min time point). Hatched and solid bars, respectively, represent the absence and presence of C5a-activated PMN. *P < 0.05 vs. paired baseline. †P < 0.05 vs. C5a-activated PMN applied without inhibitor. No. of replicates for each group is shown in parentheses.
We next sought to investigate biochemical and biophysical mechanisms that may be involved in ROCK-mediated hyperpermeability, which required the use of cultured endothelial cells. We verified that transfected ca-ROCK enters into CVEC by immunofluorescence labeling of the His tag contained in the ca-ROCK protein (Fig. 3, A and B) and investigated whether cultured CVEC functionally behave in a similar manner as isolated coronary venules. CVEC monolayers were ~10 times more permeable to albumin than isolated venules, with baseline Pa values averaging 1.38 × 10−5 cm/s. Transference of ca-ROCK into CVEC monolayers increased Pa (Fig. 3C) and decreased TER (Fig. 3D). Addition of the fresh, low-serum medium with no additives (control), or medium containing TransIT alone, TransIT plus ca-ROCK protein, or TransIT plus ca-ROCK protein and 5 × 10−6 M Y-27632, all transiently decreased TER. In all cases except the addition of TransIT plus ca-ROCK, TER quickly recovered to a level near baseline. Subsequent washout and addition of serum-free medium also transiently decreased TER, followed by a slow recovery toward baseline for the control, TransIT, and TransIT + ca-ROCK + Y-27632 groups. On the other hand, the mean TER of the TransIT + ca-ROCK group remained diminished and was significantly lower than the other groups even 300–360 min after the initiation of transfection (Fig. 3D and Table 1).
Fig. 3.

Transference of ca-ROCK into venular endothelial cells causes barrier dysfunction. A: immunofluorescence labeling of coronary venular endothelial cells (CVEC) with an anti-His tag antibody shows a background nuclear localization. Bar, 10 μm. B: when CVEC are transfected with 3 μg/ml ca-ROCK for 2 h, there is also localization of the His tag in the cytoplasm. Images are representative of 3 separate experiments. C: transfection of CVEC monolayers with ca-ROCK for 2 h causes a significant increase in Pa vs. control and the transfection reagent (10 μl/ml TransIT-LT1) alone (*P < 0.05; n = 4 for all groups). D: the transendothelial electrical resistance (TER) of CVEC monolayers decreased after transfection with ca-ROCK but remained stable in the control, transfection reagent alone, and ca-ROCK + 5 × 10−6 M Y-27632 groups (n = 4 for all groups). Shaded areas represent SEs as follows: control, medium-dark gray; TransIT alone, medium-light gray; ca-ROCK, lightest gray; ca-ROCK + Y-27632, darkest gray.
Table 1.
Mean normalized resistance of CVEC monolayers at selected time points after transfection with ca-ROCK
| Time, min | Control (n = 4) | TransIT (n = 4) | ROCK (n = 4) | ROCK + Y-27632 (n = 4) |
|---|---|---|---|---|
| 0 | 1.00±0.02 | 1.00±0.03 | 1.00±0.04 | 1.00±0.04 |
| 30 | 0.99±0.02 | 0.95±0.04 | 0.89±0.05 | 0.84±0.04 |
| 60 | 0.99±0.02 | 1.00±0.05 | 0.89±0.06 | 1.02±0.02 |
| 90 | 1.01±0.01 | 1.04±0.06 | 0.82±0.05* | 1.03±0.02 |
| 120 | 1.04±0.01 | 1.06±0.06 | 0.83±0.04* | 1.03±0.03 |
| 180 | 0.93±0.01 | 0.95±0.04 | 0.85±0.03 | 0.96±0.02 |
| 240 | 0.95±0.04 | 0.92±0.05 | 0.83±0.02 | 0.90±0.02 |
| 300 | 0.94±0.03 | 0.95±0.04 | 0.82±0.03* | 0.94±0.02 |
| 360 | 0.97±0.04 | 0.95±0.03 | 0.81±0.01* | 0.97±0.03 |
Values are means ±SE. CVEC, coronary venular endothelial cell; ca-ROCK, constitutively active rho kinase.
P < 0.05 vs. control. See Fig. 3D for further details.
We also evaluated the response of CVEC monolayers to C5a-activated PMN. In our previous study, the PMN were incubated with C5a before their addition to CVEC monolayers (8). However, in the current study we added PMN (10−6 cells/ml) to the monolayer followed by 10−8 M C5a, in a similar fashion as in isolated venule experiments. C5a-activated PMN caused a significant increase in Pa 10 min after their addition (Fig. 4A). Blockade of ROCK activity with either Y-27632 or H-1152 inhibited PMN-induced hyperpermeability in CVEC monolayers (Fig. 4B). We further examined CVEC barrier function using TER. The addition of PMN and C5a to CVEC monolayers caused a transient decrease in TER with peak change in TER ~10 min after their addition. Addition of C5a alone did not change TER, and addition of PMN without C5a actually caused a transient increase in TER (Fig. 5A). Both ROCK inhibitors, Y-27632 and H-1152, attenuated the PMN-induced decreases in TER (Fig. 5, B and C, and Table 2). These inhibitors did not significantly alter baseline TER when applied alone (data not shown).
Fig. 4.
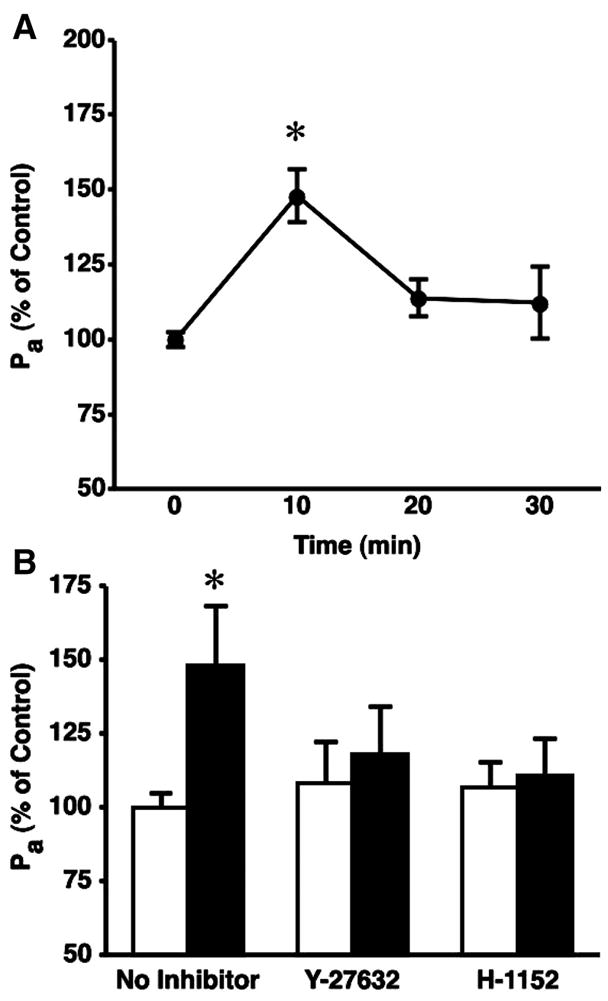
C5a-activated neutrophils cause barrier dysfunction in cultured endothelial cell monolayers. A: time course of PMN-induced increases in Pa. *P < 0.05 vs. baseline (0 min); n = 4 for each time point. B: both 5 × 10−6 M Y-27632 and 10−6 M H-1152 inhibit PMN-induced increases in CVEC monolayer Pa (10-min time point). Open bars, Pa in the absence of PMN; solid bars, Pa when activated PMN are present. *P < 0.05 vs. paired baseline.
Fig. 5.
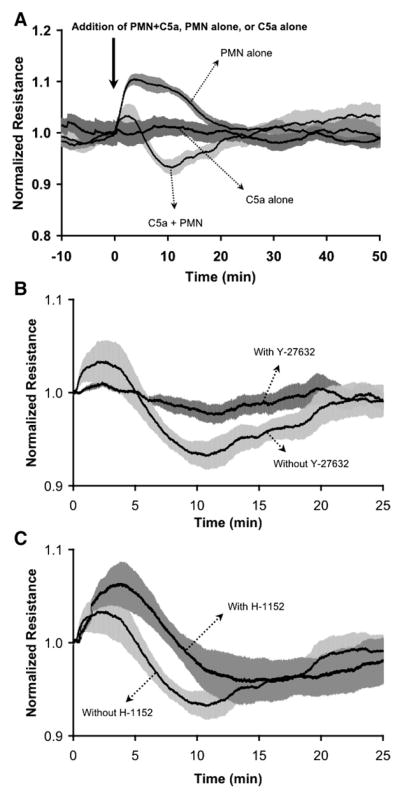
C5a-activated PMN transiently decrease TER of CVEC monolayers. A: addition of 106 PMN/ml immediately followed by 10−8 M C5a causes a transient decrease in TER, while C5a alone and PMN alone do not. B and C show the inhibition of PMN-induced changes in TER by 5 × 10−6 M Y-27632 and 10−6 M H-1152, respectively. Lines depict mean TER, and shaded areas represent SE. Data are normalized to baseline just before addition of PMN. For all groups, n = 4.
Table 2.
Mean normalized resistance of CVEC monolayers at selected time points after treatment with activated PMN.
| Time, min | C5a Alone (n = 4) | PMN Alone (n = 4) | PMN + C5a (n = 4) | PMN + C5a + Y-27632 (n = 4) | PMN + C5a + H-1152 (n = 4) |
|---|---|---|---|---|---|
| 0 | 1.000±0.049 | 1.000±0.034 | 1.000±0.044 | 1.000±0.056 | 1.000±0.017 |
| 2.5 | 0.996±0.002 | 1.096±0.007* | 1.032±0.023 | 1.010±0.003 | 1.054±0.015* |
| 5 | 1.001±0.003 | 1.101±0.013* | 1.001±0.019† | 1.000±0.002 | 1.054±0.023 |
| 7.5 | 1.015±0.013 | 1.094±0.015 | 0.955±0.010*† | 0.990±0.008†‡ | 1.015±0.013†‡ |
| 10 | 1.011±0.005 | 1.087±0.007* | 0.934±0.013*† | 0.982±0.010†‡ | 0.979±0.020† |
| 15 | 1.003±0.011 | 1.043±0.010 | 0.953±0.011*† | 0.988±0.010‡ | 0.960±0.023 |
| 20 | 1.004±0.010 | 1.015±0.009 | 0.983±0.018 | 1.004±0.015 | 0.966±0.021 |
Values are means ± SE.
P < 0.05 vs. C5a alone.
P < 0.05 vs. polymorphonuclear leukocyte (PMN) alone.
P < 0.05 vs. PMN + C5a.
We next investigated the association between endothelial cell tension development and PMN-induced endothelial hyper-permeability. Treatment of CVEC monolayers grown on collagen gels with PMN and C5a caused a transient increase in isometric tension, with an initial peak after approximately 5–6 min, followed by a steady, gradual increase in tension for the duration of the experiment (Fig. 6A, tracing 1). Cytochalasin D abolished the increases in tension, verifying the essential role of cellular actin. Neither C5a alone nor PMN alone caused a change in tension (Fig. 6A, tracings 2 and 3). When the time courses of PMN-stimulated changes in TER and isometric tension are overlayed, there appears to be a temporal relationship between the two events, although they are slightly out of phase. The peak increase in tension occurs ~5 min before the maximal change in TER. A peak in endothelial isometric tension development occurred at 4–6 min into the time course, approximately the same time during the initial drop in TER. Tension decreased but remained elevated as TER continued to drop to its maximal change and on its return to baseline (Fig. 6B). We also tested the involvement of ROCK in PMN-induced increases in endothelial isometric tension. Treatment with either Y-27632 or H-1152 attenuated the maximal PMN-stimulated increase in tension observed in the time frame when Pa was elevated (the first 20 min after addition of PMN and C5a; Fig. 6C).
Fig. 6.
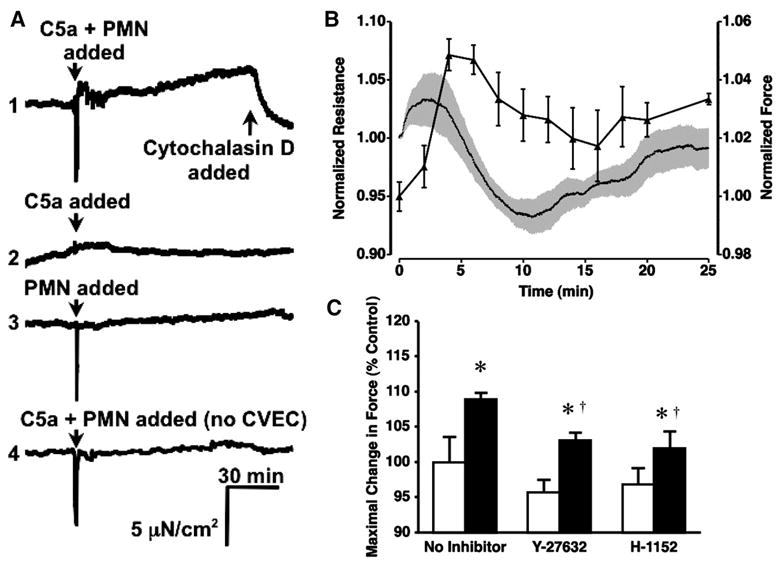
C5a-activated PMN increase endothelial tension through ROCK. A: representative time-course tracings of changes in CVEC monolayer isometric tension after the addition of 1) 10−6 PMN/ml and 10−8 M C5a, 2) 10−8 M C5a alone, 3) 10−6 PMN/ml alone, and 4) 10−6 PMN/ml and 10−8 M C5a applied to a collagen gel with no endothelial cells. Tracings are representative of 3 separate experiments for each group. Cytochalasin D (3 × 10−6 M) was added in tracing 1 to verify that the observed increase in tension was due to actin in endothelial cells. B: overlay of the mean temporal changes in endothelial isometric tension and TER after addition of C5a-activated PMN. C: PMN-induced increases in endothelial isometric tension were inhibited by either 5 × 10−6 M Y-27632 or 10−6 M H-1152. Bars represent the mean maximal change in tension after the addition of sham/inhibitors alone (open bars) or within 20 min after the addition of PMN and C5a (solid bars). *P < 0.05 vs. paired baseline; †P < 0.05 vs. PMN and C5a without inhibitors; n = 3 for all groups.
To further examine the role of ROCK in endothelial tension development, we measured isometric tension in CVEC before and after transference of ca-ROCK protein into CVEC. Endothelial isometric tension began to gradually rise shortly after the addition of the ca-ROCK transfection mixture. This increase was abolished when the actin cytoskeleton was disrupted by cytochalasin D (Fig. 7A). An overlay of the mean changes in isometric tension and TER during ca-ROCK transference shows a temporal correlation between these two events (Fig. 7B). To further examine the mechanism of ca-ROCK transfection in CVEC, we investigated MLC phosphorylation on Thr18/Ser19, a dual-phosphorylation site that promotes actin-myosin association and contraction. Previously, our laboratory has shown that C5a-activated PMN increase MLC phosphorylation and F-actin polymerization in CVEC (8, 52). Phosphorylation of MLC was increased 30 min after the start of ca-ROCK transfection and markedly increased at the 60-min time point (Fig. 7C). In addition, there appeared to be an increase in F-actin stress fibers after transference of ca-ROCK into CVEC (Fig. 7, D and E). In some areas, gaps were visible between endothelial cells as shown in Fig. 7E.
Fig. 7.

Transference of ca-ROCK into CVEC increases actin-myosin mediated isometric tension. A: increase in CVEC monolayer isometric tension occurs shortly after the addition of 3 μg/ml ca-ROCK and 10 μl/ml TransIT-LT1. Addition of 3 × 10−6 M cytochalasin D abolishes the increase in tension. B: an overlay of ca-ROCK-stimulated changes in TER and tension shows that these 2 events occur simultaneously. For TER, n = 4; for tension, n = 3. C: transference of ca-ROCK into CVEC increases phosphorylation of myosin light chain (MLC) on Thr18/Ser19 (pMLC), as determined by Western blot. The lower blot is total MLC. D: F-actin labeling (green) in control cells. Blue areas are nuclear labeling with Hoecsht 33342. Bar, 10 μm. E: F-actin labeling in cells transfected with ca-ROCK for 60 min. Tracings, blots, and images are representative of 3 separate experiments.
DISCUSSION
The data from the present study indicate the involvement of ROCK-mediated endothelial isometric tension development in neutrophil-induced microvascular barrier dysfunction. We demonstrate that neutrophil-induced hyperpermeability is attenuated by pharmacological blockade of ROCK using two distinct inhibitors. In addition, we also show that transfection of a recombinant, constitutively active ROCK protein increases permeability of both isolated coronary venules and CVEC monolayers. The increase in permeability occurs concurrently with an increase in endothelial isometric tension. Moreover, ROCK-mediated increases in permeability and tension are associated with MLC phosphorylation on Thr18/Ser19 and actin stress fiber formation. Together, these data suggest a mechanical coupling between ROCK activation and the disruption of endothelial barrier function.
This study is the first to our knowledge to demonstrate the direct effect of active ROCK on venular permeability and the involvement of ROCK in neutrophil-stimulated coronary microvascular leakage. We chose an integrative approach, including both pharmacological inhibitors and transfection of a constitutively active ROCK protein, because of the inherent strengths and weaknesses associated with each technique. The data provide direct evidence indicating that ROCK promotes increased permeability in coronary venules. In support of our data, previous reports demonstrate that ROCK is involved in endothelial permeability increases stimulated by thrombin, VEGF, transforming growth factor-β, and thermal injury (8, 10, 11, 27, 45, 46, 48, 58). Moreover, we have previously shown that C5a-activated neutrophils cause RhoA activation in endothelial cells (8), and others have shown that inhibition of ROCK attenuates neutrophil migration across endothelial monolayers (35) and neutrophil infiltration after subarachnoid hemorrhage or hepatic ischemia-reperfusion injury (36, 38). In contrast, RhoA and ROCK do not appear to mediate platelet-activating factor and bradykinin-induced hyperpermeability in mouse and rat mesenteric venules (1, 2), and there are conflicting reports regarding histamine-induced hyperpermeability (18, 48). Thus the rhoA/ROCK pathway may not serve as a common mediator for all types of edematous reactions and possibly has varying roles in permeability regulation in different vascular beds.
Another interesting aspect of the ca-ROCK transfection studies was that the elevation in venular permeability lasted only ~100 min, which was shorter than we initially predicted. The reason for this is unknown, although we can speculate that the ca-ROCK protein is inactivated by enzyme cleavage or another unidentified mechanism over time and that the loss of activity allows restoration of the barrier. Alternatively, it is possible that ca-ROCK-mediated disruption of the venular endothelial barrier may indirectly activate other signaling pathways that promote a delayed restoration of the endothelial barrier.
With respect to the changes in TER caused by C5a-activated PMN, it is important to note that neither PMN nor C5a alone caused a decrease in barrier function. In fact, addition of PMN alone caused a transient increase in TER. This is probably due to the fact that the medium serves as the electrolyte and the addition of PMN suddenly creates a suspension of cells, increasing the resistance of this electrolyte. Subsequently, as the cells settle onto the endothelial cell monolayer, the original properties of the electrolyte are gradually restored. A similar initial increase in TER has also been observed when metastatic cells are introduced in the same manner to this type of system (23). This raises the possibility that the observed decrease in TER caused by stimulation with C5a-activated PMN may actually underestimate the PMN-induced decrease in barrier function. However, this issue requires further investigation that is beyond the scope of the current study.
We also report, for the first time to our knowledge, that transference of ca-ROCK protein and C5a-activated PMN both cause an actin-dependent increase in venular endothelial cell monolayer isometric tension. The temporal profile of tension development followed the same pattern as that of MLC phosphorylation. Importantly, the maximal changes in tension occurred concomitantly with the MLC phosphorylation response. We have previously shown that C5a-activated PMN promote the dual phosphorylation of MLC in endothelial cells, as well as actin polymerization (8, 52), events known to cause cell contraction. In support of our data, one previous study showed that PMN activated with N-formyl-L-methionyl-L-lucyl-L-phenylalanine (fMLP) increase MLC phosphorylation and isometric tension of endothelial monolayers (19), in a somewhat similar fashion as we observed in the present study with C5a-activated neutrophils. We also report that inhibition of ROCK attenuates the PMN-stimulated increases in endothelial isometric tension. These data are concordant with our previous reports that ROCK inhibition attenuates PMN-induced actin polymerization in endothelial cells (8) and dual phosphorylation of MLC caused by inflammatory stimuli (41). However, it is also worth noting that we observed a lag between ca-ROCK-induced MLC phosphorylation (noticeably increased at 30 min) and force development (starting at 10 min), indicating that the initial stage of ROCK-mediated contraction may involve other mechanisms that rapidly and transiently affect cell tension. Potential mechanisms include MLC phosphorylation-independent actin polymerization (29), microtubule reorganization, or focal adhesion-mediated force development (50).
We employed an integrative approach to study ROCK-mediated permeability that included both isolated venule and cell monolayer models because 1) obtaining biochemical data from venules is relatively difficult and 2) there is currently no model for measuring endothelial cell isometric tension in intact microvessels. In most cases, we observed similar functional data between these two models of permeability. One minor exception was the time course of ca-ROCK-induced barrier dysfunction, which can be attributed to the fact that endothelial cells grown in vitro are expected to respond to stimuli somewhat slowly compared with intact vessels. Despite this difference, the strength of using this two-model approach is the ability to readily correlate biophysical and biochemical data obtained in cultured endothelial cells with functional data obtained from isolated venules.
One important methodological detail we must note is that while endothelial cells were our target for pharmacological inhibition of ROCK, we cannot exclude the possibility that the pharmacological agents may have also directly influenced neutrophils. However, we think the data principally reflect inhibition of endothelial cell ROCK for a few reasons. First, the isolated venules or endothelial cells were exposed to the inhibitors for 20 min to allow adequate time for the inhibitors to cross cell membranes and bind to their intracellular target, ROCK. In contrast, PMN were only exposed to the pharmacological agents for a matter of seconds before activation with C5a. Second, the endothelial response to activated PMN occurred quickly, and it is questionable as to whether the pharmacological inhibitors of ROCK could have directly acted on the C5a-activated PMN within the same time frame. The third and most convincing reason is our previous work showing that ROCK inhibitors attenuate increases in endothelial permeability caused by supernatant derived from activated PMN after centrifugation (8).
Another issue to consider is whether neutrophil adhesion and transendothelial migration (TEM) play a role in the observed changes in permeability and tension. In the isolated coronary venule model used in this study, the neutrophils were added to the bath and not to the lumen of the vessel, indicating that adhesion and TEM are not requisite for the increases in permeability. With the cell models, PMN and CVEC were in contact with one another, and thus the factors of PMN-endothelial cell adhesion and TEM cannot be excluded. However, in our previous study, the magnitude of permeability changes caused by supernatant taken from activated PMN after centrifugation was not significantly different from those caused by direct application of C5a-activated PMN to CVEC monolayers (8). In addition, it is clear that the CVEC are activated, evidenced by increased RhoA activity, MLC phosphorylation, and changes in the structure of the actin cytoskeleton and adherens junctions (8, 42, 52). Combined, these findings suggest that the changes in permeability and tension observed in this study are mainly due to the active role of endothelial cells on stimulation with factors released from neutrophils, rather than a secondary effect, such as physical disruption of the endothelial barrier during TEM.
After transfection with ca-ROCK, the time course of increased tension appears to be tightly associated with changes in barrier function, suggesting a correlation between ROCK-dependent endothelial tension development and increased permeability. This concept is supported by a previously reported numerical model that describes how tension is coupled with the loss of cell-cell adhesion (28). There are also previous studies that indicate endothelial tension changes play a role in thrombin and phorbol ester-induced endothelial barrier dysfunction (7, 16, 17, 24, 28–30, 33). Several reports have also suggested that increased endothelial cell monolayer permeability is associated with formation of gaps between endothelial cells (13, 49), and we did observe gaps between CVEC treated with ca-ROCK. We concede that these results from cell culture may reflect an exaggeration of changes in intact microvessels, as large gaps (>1 μm) have not been observed in intact microvessels on stimulation with inflammatory mediators, but rather more subtle changes in endothelial cell-cell junctions have been reported (5, 26).
We also show that inhibition of ROCK attenuates neutrophil-induced changes in both endothelial permeability and tension development, suggesting that these changes in barrier function are associated with endothelial tension development. In contrast to ca-ROCK-stimulated changes, PMN-induced changes in tension and barrier function were temporally out of phase, with a peak increase in isometric tension preceding the peak change in barrier function. These data suggest that tension development may not solely account for PMN-induced increases in endothelial permeability. A more likely scenario is that PMN-induced endothelial barrier dysfunction involves multiple signaling pathways and concomitant changes in cell-cell adhesion and cell-matrix adhesion in addition to changes in endothelial cell isometric tension (50, 51). This notion is reinforced by studies showing that the time course of thrombin-induced endothelial barrier dysfunction occurs out-of-phase with the time course of thrombin-induced changes in tension (28, 29).
In summary, we present data suggesting that ROCK-mediated actomyosin contraction promotes increased endothelial cell isometric tension that is temporally associated with changes in permeability. We suggest that ROCK-mediated increases in endothelial cell isometric tension play an important role microvascular leakage during neutrophil stimulation.
Acknowledgments
GRANTS
This work was supported by National Institutes of Health (NIH) Grants HL-61507 (S. Y. Yuan), HL-70752 (S. Y. Yuan), GM-61732 (A. B. Moy), and HL-73324 (M. H. Wu) and a grant from the Scott and White Research and Education Foundation (J. W. Breslin). J. W. Breslin is currently supported by NIH Fellowship F32-HL-76079.
References
- 1.Adamson RH, Curry FE, Adamson G, Liu B, Jiang Y, Aktories K, Barth H, Daigeler A, Golenhofen N, Ness W, Drenckhahn D. Rho and rho kinase modulation of barrier properties: cultured endothelial cells and intact microvessels of rats and mice. J Physiol. 2002;539:295–308. doi: 10.1113/jphysiol.2001.013117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adamson RH, Zeng M, Adamson GN, Lenz JF, Curry FE. PAF-and bradykinin-induced hyperpermeability of rat venules is independent of actin-myosin contraction. Am J Physiol Heart Circ Physiol. 2003;285:H406–H417. doi: 10.1152/ajpheart.00021.2003. [DOI] [PubMed] [Google Scholar]
- 3.Amano M, Chihara K, Kimura K, Fukata Y, Nakamura N, Matsuura Y, Kaibuchi K. Formation of actin stress fibers and focal adhesions enhanced by Rho-kinase. Science. 1997;275:1308–1311. doi: 10.1126/science.275.5304.1308. [DOI] [PubMed] [Google Scholar]
- 4.Amano M, Fukata Y, Kaibuchi K. Regulation and functions of Rho-associated kinase. Exp Cell Res. 2000;261:44–51. doi: 10.1006/excr.2000.5046. [DOI] [PubMed] [Google Scholar]
- 5.Baldwin AL, Thurston G. Changes in endothelial actin cytoskeleton in venules with time after histamine treatment. Am J Physiol Heart Circ Physiol. 1995;269:H1528–H1537. doi: 10.1152/ajpheart.1995.269.5.H1528. [DOI] [PubMed] [Google Scholar]
- 6.Beynon HL, Davies KA, Haskard DO, Walport MJ. Erythrocyte complement receptor type 1 and interactions between immune complexes, neutrophils, and endothelium. J Immunol. 1994;153:3160–3167. [PubMed] [Google Scholar]
- 7.Bodmer JE, Van Engelenhoven J, Reyes G, Blackwell K, Kamath A, Shasby DM, Moy AB. Isometric tension of cultured endothelial cells: new technical aspects. Microvasc Res. 1997;53:261–271. doi: 10.1006/mvre.1997.2011. [DOI] [PubMed] [Google Scholar]
- 8.Breslin JW, Yuan SY. Involvement of RhoA and Rho kinase in neutrophil-stimulated endothelial hyperpermeability. Am J Physiol Heart Circ Physiol. 2004;286:H1057–H1062. doi: 10.1152/ajpheart.00841.2003. [DOI] [PubMed] [Google Scholar]
- 9.Burns AR, Walker DC, Brown ES, Thurmon LT, Bowden RA, Keese CR, Simon SI, Entman ML, Smith CW. Neutrophil transendothelial migration is independent of tight junctions and occurs preferentially at tricellular corners. J Immunol. 1997;159:2893–2903. [PubMed] [Google Scholar]
- 10.Carbajal JM, Gratrix ML, Yu CH, Schaeffer RC., Jr ROCK mediates thrombin’s endothelial barrier dysfunction. Am J Physiol Cell Physiol. 2000;279:C195–C204. doi: 10.1152/ajpcell.2000.279.1.C195. [DOI] [PubMed] [Google Scholar]
- 11.Clements RT, Minnear FL, Singer HA, Keller RS, Vincent PA. RhoA and Rho-kinase dependent and independent signals mediate TGF-β-induced pulmonary endothelial cytoskeletal reorganization and permeability. Am J Physiol Lung Cell Mol Physiol. 2005;288:L294–L306. doi: 10.1152/ajplung.00213.2004. [DOI] [PubMed] [Google Scholar]
- 12.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91:1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 13.Garcia JG, Davis HW, Patterson CE. Regulation of endothelial cell gap formation and barrier dysfunction: role of myosin light chain phosphorylation. J Cell Physiol. 1995;163:510–522. doi: 10.1002/jcp.1041630311. [DOI] [PubMed] [Google Scholar]
- 14.Garcia JG, Verin AD, Herenyiova M, English D. Adherent neutrophils activate endothelial myosin light chain kinase: role in transendothelial migration. J Appl Physiol. 1998;84:1817–1821. doi: 10.1152/jappl.1998.84.5.1817. [DOI] [PubMed] [Google Scholar]
- 15.Giaever I, Keese CR. Micromotion of mammalian cells measured electrically. Proc Natl Acad Sci USA. 1991;88:7896–7900. doi: 10.1073/pnas.88.17.7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goeckeler ZM, Wysolmerski RB. Myosin light chain kinase-regulated endothelial cell contraction: the relationship between isometric tension, actin polymerization, and myosin phosphorylation. J Cell Biol. 1995;130:613–627. doi: 10.1083/jcb.130.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gunduz D, Hirche F, Hartel FV, Rodewald CW, Schafer M, Pfitzer G, Piper HM, Noll T. ATP antagonism of thrombin-induced endothelial barrier permeability. Cardiovasc Res. 2003;59:470–478. doi: 10.1016/s0008-6363(03)00427-9. [DOI] [PubMed] [Google Scholar]
- 18.Hirase T, Kawashima S, Wong EY, Ueyama T, Rikitake Y, Tsukita S, Yokoyama M, Staddon JM. Regulation of tight junction permeability and occludin phosphorylation by Rhoa-p160ROCK-dependent and -independent mechanisms. J Biol Chem. 2001;276:10423–10431. doi: 10.1074/jbc.M007136200. [DOI] [PubMed] [Google Scholar]
- 19.Hixenbaugh EA, Goeckeler ZM, Papaiya NN, Wysolmerski RB, Silverstein SC, Huang AJ. Stimulated neutrophils induce myosin light chain phosphorylation and isometric tension in endothelial cells. Am J Physiol Heart Circ Physiol. 1997;273:H981–H988. doi: 10.1152/ajpheart.1997.273.2.H981. [DOI] [PubMed] [Google Scholar]
- 20.Huang AJ, Manning JE, Bandak TM, Ratau MC, Hanser KR, Silverstein SC. Endothelial cell cytosolic free calcium regulates neutrophil migration across monolayers of endothelial cells. J Cell Biol. 1993;120:1371–1380. doi: 10.1083/jcb.120.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huxley VH, Rumbaut RE. The microvasculature as a dynamic regulator of volume and solute exchange. Clin Exp Pharmacol Physiol. 2000;27:847–854. doi: 10.1046/j.1440-1681.2000.03344.x. [DOI] [PubMed] [Google Scholar]
- 22.Kaslovsky RA, Lai L, Parker K, Malik AB. Mediation of endothelial injury following neutrophil adherence to extracellular matrix. Am J Physiol Lung Cell Mol Physiol. 1993;264:L401–L405. doi: 10.1152/ajplung.1993.264.4.L401. [DOI] [PubMed] [Google Scholar]
- 23.Keese CR, Bhawe K, Wegener J, Giaever I. Real-time impedance assay to follow the invasive activities of metastatic cells in culture. Biotechniques. 2002;33:842–850. doi: 10.2144/02334rr01. [DOI] [PubMed] [Google Scholar]
- 24.Kolodney MS, Wysolmerski RB. Isometric contraction by fibroblasts and endothelial cells in tissue culture: a quantitative study. J Cell Biol. 1992;117:73–82. doi: 10.1083/jcb.117.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kubes P, Grisham MB, Barrowman JA, Gaginella T, Granger DN. Leukocyte-induced vascular protein leakage in cat mesentery. Am J Physiol Heart Circ Physiol. 1991;261:H1872–H1879. doi: 10.1152/ajpheart.1991.261.6.H1872. [DOI] [PubMed] [Google Scholar]
- 26.Majno G, Palade G. Studies on inflammation I. The effect of histamine and serotonin on vascular permeability: an electron microscopic study. J Biophys Cytol. 1961;11:597–605. doi: 10.1083/jcb.11.3.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mehta D, Ahmmed GU, Paria B, Holinstat M, Voyno-Yasenetskaya T, Tiruppathi C, Minshall RD, Malik AB. RhoA interaction with inositol 1,4,5-triphosphate receptor and transient receptor potential channel-1 regulates Ca2+ entry. Role in signaling increased endothelial permeability. J Biol Chem. 2003;278:33492–33500. doi: 10.1074/jbc.M302401200. [DOI] [PubMed] [Google Scholar]
- 28.Moy AB, Blackwell K, Kamath A. Differential effects of histamine and thrombin on endothelial barrier function through actin-myosin tension. Am J Physiol Heart Circ Physiol. 2002;282:H21–H29. doi: 10.1152/ajpheart.2002.282.1.H21. [DOI] [PubMed] [Google Scholar]
- 29.Moy AB, Blackwell K, Wang N, Haxhinasto K, Kasiske MK, Bodmer J, Reyes G, English A. Phorbol ester-mediated pulmonary artery endothelial barrier dysfunction through regulation of actin cytoskeletal mechanics. Am J Physiol Lung Cell Mol Physiol. 2004;287:L153–L167. doi: 10.1152/ajplung.00292.2003. [DOI] [PubMed] [Google Scholar]
- 30.Moy AB, Bodmer JE, Blackwell K, Shasby S, Shasby DM. cAMP protects endothelial barrier function independent of inhibiting MLC20-dependent tension development. Am J Physiol Lung Cell Mol Physiol. 1998;274:L1024–L1029. doi: 10.1152/ajplung.1998.274.6.L1024. [DOI] [PubMed] [Google Scholar]
- 31.Moy AB, Shasby SS, Scott BD, Shasby DM. The effect of histamine and cyclic adenosine monophosphate on myosin light chain phosphorylation in human umbilical vein endothelial cells. J Clin Invest. 1993;92:1198–1206. doi: 10.1172/JCI116690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moy AB, Sheldon R, Lindsley K, Shasby S, Shasby DM. Centripetal tension and endothelial retraction. Chest. 1994;105:107S–108S. doi: 10.1378/chest.105.3_supplement.107s. [DOI] [PubMed] [Google Scholar]
- 33.Moy AB, Van Engelenhoven J, Bodmer J, Kamath J, Keese C, Giaever I, Shasby S, Shasby DM. Histamine and thrombin modulate endothelial focal adhesion through centripetal and centrifugal forces. J Clin Invest. 1996;97:1020–1027. doi: 10.1172/JCI118493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moy AB, Winter M, Kamath A, Blackwell K, Reyes G, Giaever I, Keese C, Shasby DM. Histamine alters endothelial barrier function at cell-cell and cell-matrix sites. Am J Physiol Lung Cell Mol Physiol. 2000;278:L888–L898. doi: 10.1152/ajplung.2000.278.5.L888. [DOI] [PubMed] [Google Scholar]
- 35.Saito H, Minamiya Y, Saito S, Ogawa J. Endothelial Rho and Rho kinase regulate neutrophil migration via endothelial myosin light chain phosphorylation. J Leukoc Biol. 2002;72:829–836. [PubMed] [Google Scholar]
- 36.Satoh S, Yamamoto Y, Toshima Y, Ikegaki II, Asano T, Suzuki Y, Shibuya M. Fasudil, a protein kinase inhibitor, prevents the development of endothelial injury and neutrophil infiltration in a two-haemorrhage canine subarachnoid model. J Clin Neurosci. 1999;6:394–399. doi: 10.1054/jocn.1999.0089. [DOI] [PubMed] [Google Scholar]
- 37.Schelling ME, Meininger CJ, Hawker JR, Jr, Granger HJ. Venular endothelial cells from bovine heart. Am J Physiol Heart Circ Physiol. 1988;254:H1211–H1217. doi: 10.1152/ajpheart.1988.254.6.H1211. [DOI] [PubMed] [Google Scholar]
- 38.Stasia MJ, Jouan A, Bourmeyster N, Boquet P, Vignais PV. ADP-ribosylation of a small size GTP-binding protein in bovine neutrophils by the C3 exoenzyme of Clostridium botulinum and effect on the cell motility. Biochem Biophys Res Commun. 1991;180:615–622. doi: 10.1016/s0006-291x(05)81110-6. [DOI] [PubMed] [Google Scholar]
- 39.Tinsley JH, De Lanerolle P, Wilson E, Ma W, Yuan SY. Myosin light chain kinase transference induces myosin light chain activation and endothelial hyperpermeability. Am J Physiol Cell Physiol. 2000;279:C1285–C1289. doi: 10.1152/ajpcell.2000.279.4.C1285. [DOI] [PubMed] [Google Scholar]
- 40.Tinsley JH, Hawker J, Yuan Y. Efficient protein transfection of cultured coronary venular endothelial cells. Am J Physiol Heart Circ Physiol. 1998;275:H1873–H1878. doi: 10.1152/ajpheart.1998.275.5.H1873. [DOI] [PubMed] [Google Scholar]
- 41.Tinsley JH, Teasdale NR, Yuan SY. Myosin light chain phosphorylation and pulmonary endothelial cell hyperpermeability in burns. Am J Physiol Lung Cell Mol Physiol. 2004;286:L841–L847. doi: 10.1152/ajplung.00341.2003. [DOI] [PubMed] [Google Scholar]
- 42.Tinsley JH, Ustinova EE, Xu W, Yuan SY. Src-dependent, neutrophil-mediated vascular hyperpermeability and β-catenin modification. Am J Physiol Cell Physiol. 2002;283:C1745–C1751. doi: 10.1152/ajpcell.00230.2002. [DOI] [PubMed] [Google Scholar]
- 43.Tinsley JH, Wu MH, Ma W, Taulman AC, Yuan SY. Activated neutrophils induce hyperpermeability and phosphorylation of adherens junction proteins in coronary venular endothelial cells. J Biol Chem. 1999;274:24930–24934. doi: 10.1074/jbc.274.35.24930. [DOI] [PubMed] [Google Scholar]
- 44.Tinsley JH, Zawieja DC, Wu MH, Ustinova EE, Xu W, Yuan SY. Protein transfection of intact microvessels specifically modulates vasoreactivity and permeability. J Vasc Res. 2001;38:444–452. doi: 10.1159/000051077. [DOI] [PubMed] [Google Scholar]
- 45.Van Nieuw Amerongen GP, Koolwijk P, Versteilen A, van Hinsbergh VW. Involvement of RhoA/Rho kinase signaling in VEGF-induced endothelial cell migration and angiogenesis in vitro. Arterioscler Thromb Vasc Biol. 2003;23:211–217. doi: 10.1161/01.atv.0000054198.68894.88. [DOI] [PubMed] [Google Scholar]
- 46.Van Nieuw Amerongen GP, van Delft S, Vermeer MA, Collard JG, van Hinsbergh VW. Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ Res. 2000;87:335–340. doi: 10.1161/01.res.87.4.335. [DOI] [PubMed] [Google Scholar]
- 47.Waschke J, Curry FE, Adamson RH, Drenckhahn D. Regulation of actin dynamics is critical for endothelial barrier functions. Am J Physiol Heart Circ Physiol. 2005;288:H1296–H1305. doi: 10.1152/ajpheart.00687.2004. [DOI] [PubMed] [Google Scholar]
- 48.Wojciak-Stothard B, Potempa S, Eichholtz T, Ridley AJ. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J Cell Sci. 2001;114:1343–1355. doi: 10.1242/jcs.114.7.1343. [DOI] [PubMed] [Google Scholar]
- 49.Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol. 2002;39:187–199. doi: 10.1016/s1537-1891(03)00008-9. [DOI] [PubMed] [Google Scholar]
- 50.Yuan SY. Protein kinase signaling in the modulation of microvascular permeability. Vascul Pharmacol. 2003;39:213–223. doi: 10.1016/s1537-1891(03)00010-7. [DOI] [PubMed] [Google Scholar]
- 51.Yuan SY. Signal transduction pathways in enhanced microvascular permeability. Microcirculation. 2000;7:395–403. [PubMed] [Google Scholar]
- 52.Yuan SY, Wu MH, Ustinova EE, Guo M, Tinsley JH, De Lanerolle P, Xu W. Myosin light chain phosphorylation in neutrophil-stimulated coronary microvascular leakage. Circ Res. 2002;90:1214–1221. doi: 10.1161/01.res.0000020402.73609.f1. [DOI] [PubMed] [Google Scholar]
- 53.Yuan Y, Chilian WM, Granger HJ, Zawieja DC. Permeability to albumin in isolated coronary venules. Am J Physiol Heart Circ Physiol. 1993;265:H543–H552. doi: 10.1152/ajpheart.1993.265.2.H543. [DOI] [PubMed] [Google Scholar]
- 54.Yuan Y, Fleming BP. A method for isolation and fluorescent labeling of rat neutrophils for intravital microvascular studies. Microvasc Res. 1990;40:218–229. doi: 10.1016/0026-2862(90)90021-i. [DOI] [PubMed] [Google Scholar]
- 55.Yuan Y, Granger HJ, Zawieja DC, Chilian WM. Flow modulates coronary venular permeability by a nitric oxide-related mechanism. Am J Physiol Heart Circ Physiol. 1992;263:H641–H646. doi: 10.1152/ajpheart.1992.263.2.H641. [DOI] [PubMed] [Google Scholar]
- 56.Yuan Y, Granger HJ, Zawieja DC, DeFily DV, Chilian WM. Histamine increases venular permeability via a phospholipase C-NO synthase-guanylate cyclase cascade. Am J Physiol Heart Circ Physiol. 1993;264:H1734–H1739. doi: 10.1152/ajpheart.1993.264.5.H1734. [DOI] [PubMed] [Google Scholar]
- 57.Yuan Y, Mier RA, Chilian WM, Zawieja DC, Granger HJ. Interaction of neutrophils and endothelium in isolated coronary venules and arterioles. Am J Physiol Heart Circ Physiol. 1995;268:H490–H498. doi: 10.1152/ajpheart.1995.268.1.H490. [DOI] [PubMed] [Google Scholar]
- 58.Zheng HZ, Zhao KS, Zhou BY, Huang QB. Role of Rho kinase and actin filament in the increased vascular permeability of skin venules in rats after scalding. Burns. 2003;29:820–827. doi: 10.1016/j.burns.2003.08.004. [DOI] [PubMed] [Google Scholar]