

Abstract
All four of the C-terminal Eps15 homology domain (EHD) proteins have been implicated in the regulation of endocytic trafficking. However, the high level of amino acid sequence identity among these proteins has made it challenging to elucidate the precise function of individual EHD proteins. We demonstrate here with specific peptide antibodies that endogenous EHD4 localizes to Rab5-, early embryonic antigen 1 (EEA1)- and Arf6-containing endosomes and colocalizes with internalized transferrin in the cell periphery. Knock-down of EHD4 expression by both small interfering RNA and short hairpin RNA leads to the generation of enlarged early endosomal structures that contain Rab5 and EEA1 as well as internalized transferrin or major histocompatibility complex class I molecules. In addition, cargo destined for degradation, such as internalized low-density lipoprotein, also accumulates in the enlarged early endosomes in EHD4-depleted cells. Moreover, we have demonstrated that these enlarged early endosomes are enriched in levels of the activated GTP-bound Rab5. Finally, we show that endogenous EHD4 and EHD1 interact in cells, suggesting coordinated involvement in the regulation of receptor transport along the early endosome to endocytic recycling compartment axis. The results presented herein provide evidence that EHD4 is involved in the control of trafficking at the early endosome and regulates exit of cargo toward both the recycling compartment and the late endocytic pathway.
Keywords: early endosome, EHD1, EHD4, Rab5
The transport of proteins and lipids through the endocytic pathway is a highly complex process in mammalian cells. Key among the regulators of endocytic trafficking events are the Rab family of GTP-binding proteins (1,2). Rab proteins cycle on and off of organellar membranes depending on their nucleotide status (3). GDP-bound ‘inactive’ Rabs localize to the cytosol, and their ‘activation’ is facilitated by specific GTP exchange factors (GEFs) that catalyze GTP loading. After GTP binding, Rab proteins associate with endocytic membranes and recruit a series of functionally diverse Rab effectors that control key trafficking events including vesicle formation, transport, docking and fusion with target organelles. The hydrolysis of Rab–GTP proteins by specific guanosine triphosphatase-activating proteins (GAPs) leads to their return to the cytosol and to the release of their effectors for additional rounds of trafficking.
Over 60 different mammalian Rabs have been identified (1), and a number of them have been implicated in the regulation of various critical steps within the endocytic pathways. Rab5, for example, has been found to play an important role in the fusion of early endosomes as over-expression of a GTP-locked mutant causes aberrantly large early endosomal structures (4,5). Rab5 also acts earlier in controlling internalization events at the plasma membrane (5). A role for Rab4 has been elucidated in the regulation of transport of internalized receptors back to the plasma membrane either directly through early endosomes or indirectly through the endocytic recycling compartment (ERC) (6,7). Another key Rab protein involved in this pathway is Rab11, which controls recycling from the ERC back to the plasma membrane (8,9). In addition to these well-studied Rabs, additional Rab proteins have also been found to affect these pathways, including Rab21 (10,11), Rab22 (12–14) and Rab10 (15–17) in polarized epithelial cells.
While the role of Rab proteins and their effectors in endocytic transport has been studied extensively, additional non-Rab proteins including clathrin, various adaptor proteins and SNARE-family proteins involved in fusion events also play critical roles in trafficking through this pathway (reviewed in 18). One family of proteins recently implicated in endocytic trafficking is the C-terminal Eps15 homology domain (EHD) proteins. This family comprises four highly homologous paralogs in mammalian cells, known as EHD1, EHD2, EHD3 and EHD4 (reviewed in 19). All four proteins contain a C-terminal EHD (20,21), an N-terminal nucleotide-binding loop (22–25) and a central coiled coil involved in oligomerization (24–26). Thus far, EHD1 is the best characterized of these EHD proteins, and the solution structure of its EHD has been recently solved (27). It has been implicated in regulating the recycling of various receptors including transferrin (23,28,29), major histocompatibility complex class I (MHCI) (30), cystic fibrosis transmembrane conductance regulator (31), the insulin-responsive glucose transporter (GLUT4) (32), α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid receptors for long-term potentiation (33) and β1 integrin receptors (34) in addition to controlling cholesterol recycling (35) and homeostasis (36) and facilitating endosome-to-Golgi retrieval (37).
EHD1 (and its orthologs) associates with a number of proteins through its C-terminal EHD (25,28,32,37–41) and with phosphoinositides (58) and plays a key role in the regulation of receptor exit from the ERC, although it has also been reported to be involved in the internalization process of certain receptors (38). EHD3, the closest EHD1 paralog (86% amino acid identity), plays a partially overlapping role in this pathway and appears to act primarily upstream of EHD1 in regulating transport from the early endosomes to the ERC (25,42).
As opposed to EHD3, EHD2 and EHD4 show somewhat less homology to EHD1, with about 70 and 74% amino acid identity, respectively (reviewed in 19). In adipocytes, EHD2 has been localized to GLUT4-containing vesicles (43) and links internalization with the actin microfilaments through an actin-binding protein called EH-binding protein 1 (44). It has also been shown to regulate internalization (45) and recycling (46) of transferrin receptors. On the other hand, the function of EHD4 is not as well understood. It was first described as a component of the extracellular matrix (47), and subsequent studies identified the rat homolog, Pincher, as a regulator of the nerve growth factor receptor internalization (TrkA) in PC12 rat adrenal pheochromocytoma cells (48). EHD4 has also been implicated in interactions with the cell fate determinant, NUMB, and colocalizes with the small GTP-binding protein, Arf6 (49).
In the study herein, we focus on the functional role of endogenous EHD4 and provide evidence that EHD4 is involved in the regulation of transport from early endosomes and that its depletion causes an increase in the levels of active GTP-bound Rab5. Moreover, to the best of our knowledge, we provide the first evidence that endogenous EHD proteins interact in vivo.
Results
Endogenous EHD4 localizes to early endosomes
Progress in elucidating the function of specific C-terminal EHD proteins has been hindered by their high level of homology and the difficulty in obtaining antibodies that exclusively recognize only a single EHD paralog. Accordingly, we identified a unique C-terminal EHD4 sequence and generated rabbit polyclonal peptide antibodies against it. We first tested the affinity-purified anti-EHD4 antibodies by immunoblotting analysis and found that it recognized a band of ~64 kDa from HeLa cell lysates, corresponding to the predicted molecular mass of endogenous EHD4 (Figure 1A, left lane). Incubation of the anti-EHD4 antibodies with the immunizing peptide abolished detection of this band, demonstrating specificity (Figure 1A, right lane). To further define the specificity of the EHD4 antibody, HeLa cells were transfected with green fluorescent protein (GFP)–EHD1, GFP–EHD2, GFP–EHD3 or GFP–EHD4. Lysates from these transfected cells were separated by SDS–PAGE and immunoblotted with the purified anti-EHD4 antibodies (Figure 1B). Expression of each of the four GFP–EHDs was confirmed using an anti-GFP antibody (Figure 1B, right panel). Note that the molecular masses of EHD2 and EHD4 are slightly higher than those of EHD1 and EHD3. When immunoblotted with anti-EHD4 antibodies (Figure 1B, left panel), an ~91-kDa GFP–EHD4 band was detected exclusively in cells transfected with GFP–EHD4. However, despite the high level of homology between EHD proteins, no bands were detected in lysates from cells transfected with the other EHD paralogs (Figure 1B, left panel). On the other hand, ~64 kDa endogenous EHD4 was detected in all the lysates. These experiments demonstrate the high specificity of this antibody for EHD4.
Figure 1. Anti-EHD4 antibody is highly specific and does not cross-react with other EHD family members.
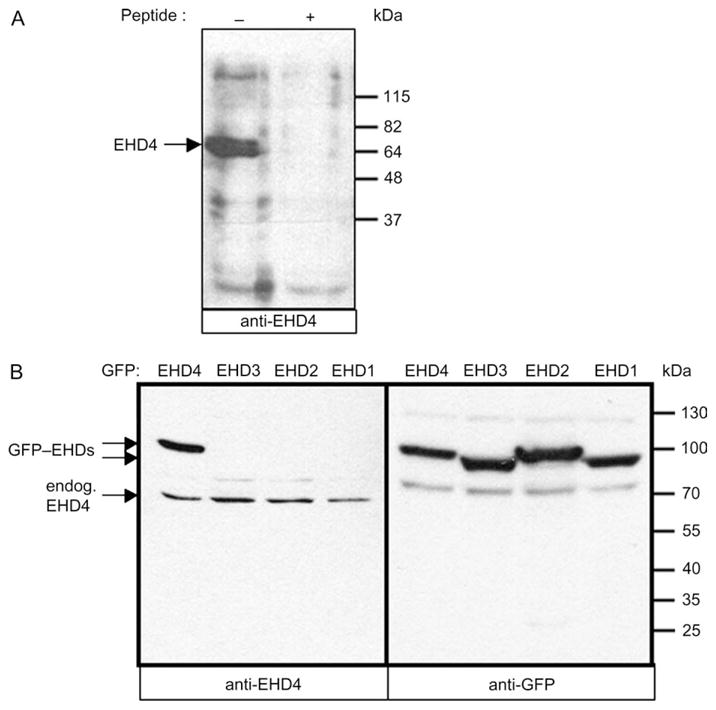
(A and B) Detergent lysates were prepared from untransfected HeLa cells (A) or from cells transiently transfected with GFP–EHD1, GFP–EHD2, GFP–EHD3 or GFP–EHD4 (B). Immunoblotting was performed with affinity-purified polyclonal rabbit anti-EHD4 antibodies (A and B, left panel) or with the same antibody in the presence of 1 μM immunizing peptide (A, right lane) followed by anti-rabbit HRP. GFP–EHD proteins were identified with biotinylated anti-GFP antibody followed by streptavidin–HRP (B, right panel). Enhanced chemiluminescence was used for detection. Endogenous EHD4 (endog. EHD4) and GFP–EHD proteins are denoted by arrows.
We next examined the subcellular localization of endogenous EHD4. As demonstrated, the anti-EHD4 antibody decorated a series of punctate structures, with occasional extended tubular membranes (Figure 2A). This pattern corresponded well with that observed upon overexpression of hemagglutinin-epitope tagged (HA)–EHD4 (Figure 2B) and GFP–EHD4 (data not shown), suggesting that when overexpressed, EHD4 displays a similar subcellular distribution to that of the endogenous protein.
Figure 2. Endogenous EHD4 localizes to an array of tubular–vesicular membranes.
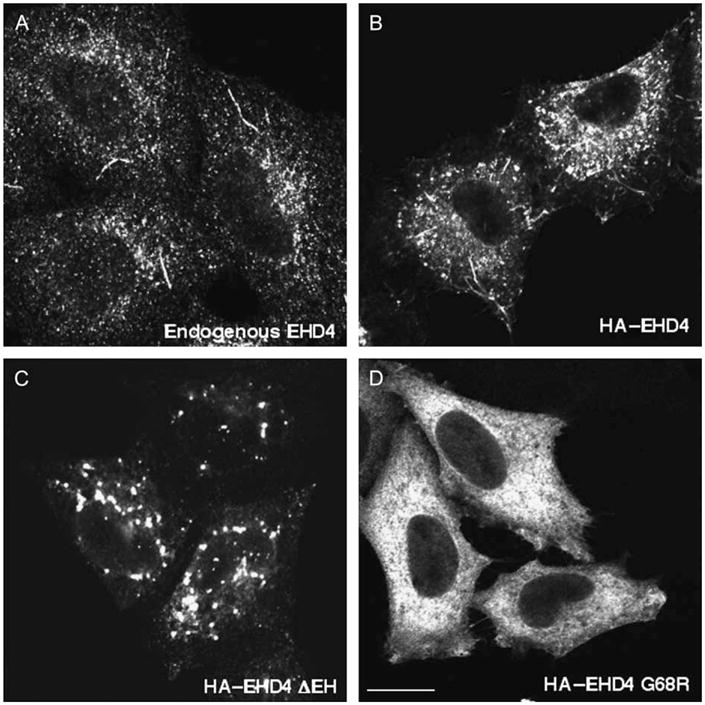
(A) Untransfected HeLa cells on coverslips were fixed and incubated with affinity-purified polyclonal rabbit anti-EHD4 antibodies and then detected using Alexa Fluor 488-conjugated anti-rabbit antibodies. (B–D) HeLa cells on coverslips were transiently transfected with wild-type HA–EHD4 (B), HA–EHD4 ΔEH (C) and HA–EHD4 G68R (D). After 24 h, cells were fixed and incubated with anti-HA antibody followed by Alexa Fluor 488-conjugated goat anti-mouse antibody. Bar, 10 μm.
It has been previously demonstrated that truncation of the EHD of EHD proteins (EHD ΔEH) results in the loss of tubular EHD membranes and the relocalization of EHD proteins exclusively to punctate structures (30,45,46). We analyzed the localization of EHD4 ΔEH and observed a similar phenotype, with EHD4 ΔEH no longer associated with tubular membranes and primarily localized to large punctate structures (Figure 2C). It has also been established that the conserved N-terminal EHD nucleotide-binding motif is required for the association of EHD1 and EHD3 with membranes (24,25). Mutations in the nucleotide-binding motif of EHD4 (G68R) render this protein cytosolic in a similar manner (Figure 2D).
Studies to date have implicated EHD proteins in the regulation of various steps within the endocytic pathways. Armed with this knowledge, and based on the subcellular distribution of the endogenous protein, we first investigated the subcellular compartment to which EHD4 localizes. Our immunofluorescence analysis revealed a significant colocalization of the endogenous early endosomal protein Rab5 with endogenous EHD4 or with HA–EHD4 in transfected cells (Figure 3A–F), suggesting that a considerable pool of EHD4 associates with early endosomes. Endogenous EHD4 also clearly displayed partial overlap with an additional marker for early endosomes, endogenous early embryonic antigen 1 (EEA1) (Figure 3G–I, see insets and arrows for colocalizations), as well as Rab4 (Figure S1G–I) and Sorting Nexins 1 and 2 (Figure S1M–R). Moreover, at the early time-points following internalization, transferrin could also be detected colocalizing with endogenous EHD4 (Figure 3J–L). Endogenous EHD4 also displayed partial colocalization with vesicles and tubules containing Arf6 (Figure 3M–O), suggesting that it may be involved in clathrin-independent endocytic events. On the other hand, little colocalization was observed with Rab11, a protein localized to the recycling compartment (Figure 3P–R), the lysosomal membrane marker, Lamp1 (Figure S1A–C), the cis Golgi membrane protein GM130 (Figure S1D–F) or the late endosomal protein, Rab7 (Figure S1J–L). Taken together, these data indicate that EHD4 localizes to early endosomes.
Figure 3. Partial colocalization of endogenous and transfected EHD4 with early endosomes and Arf6 but not recycling endosomes.
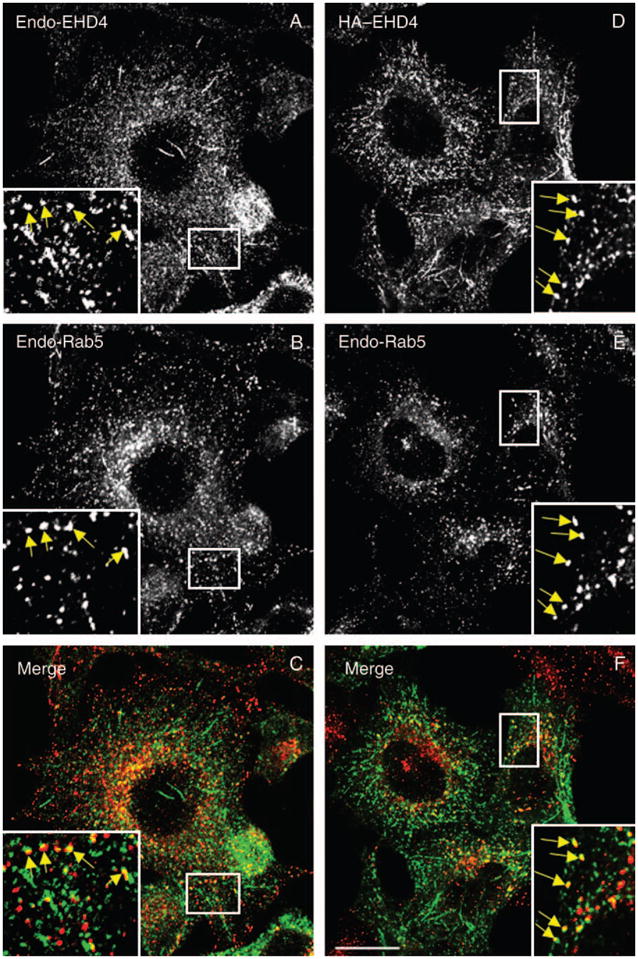
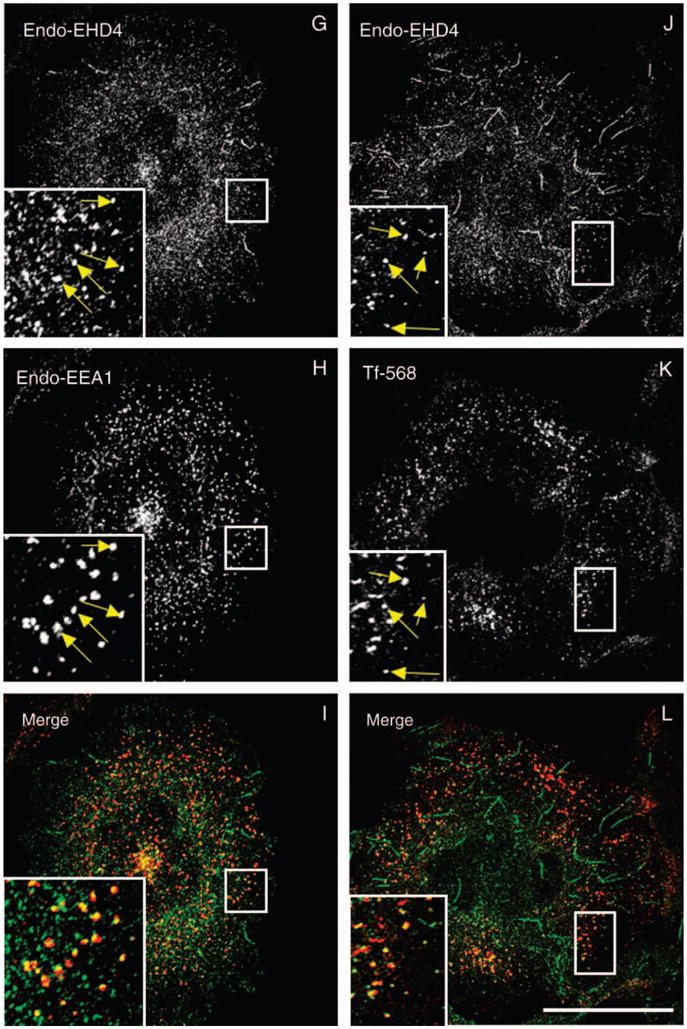
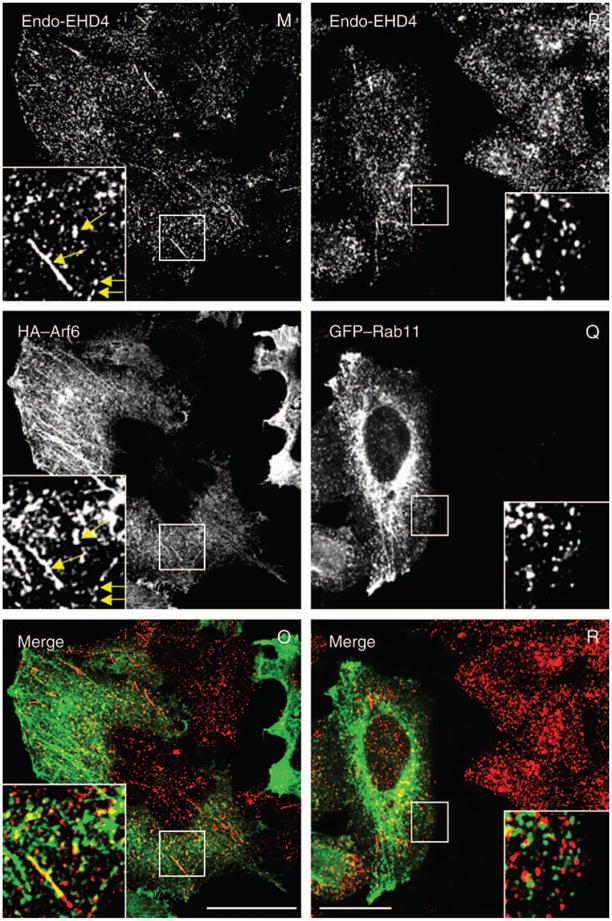
(A–C and G–L) Untransfected HeLa cells grown on coverslips were fixed and coincubated with affinity-purified polyclonal rabbit anti-EHD4 antibodies (A and G) and antibodies directed at either Rab5 (B) or EEA1 (H) followed by the corresponding secondary antibodies. (D–F and M–R) HeLa cells were transiently transfected with HA–EHD4 (D–F), HA–Arf6 (M–O) or GFP–Rab11 (P–R). After 24 h, cells were fixed and coincubated with rabbit anti-EHD4 antibodies (D, M and P) and mouse anti-Rab5 antibodies (E) or mouse anti-HA antibodies (N) followed by the corresponding secondary antibodies. (J–L) Untransfected HeLa cells on coverslips were pulsed with Tf-568 for 5 min at 37°C and then chased in complete media for another 5 min at 37°C (K). After fixation, cells were incubated with affinity-purified polyclonal rabbit anti-EHD4 antibodies (J) followed by Alexa Fluor 488-conjugated anti-rabbit antibodies. Insets (A–R) depict higher magnifications of the boxed areas, and arrows (A–O) show vesicles positive for organelle markers with either endogenous EHD4 or transfected EHD4. Bars, 10 μm.
EHD4 controls traffic at the early endosome
Given the localization of EHD4 to early endosomes, we next sought to gain functional information on its potential role at this stage of the endocytic pathway by determining the effect of its depletion. Accordingly, by small interfering RNA (siRNA) knock-down, we were consistently able to reduce EHD4 expression by 60–75% (Figure 4A). We were unable to increase the efficacy beyond this level, possibly because of the long half-life of the EHD4 protein (data not shown). EHD4–siRNA knock-down was specific for EHD4 as it had no significant effect on the expression levels of EHD1, EHD2 and EHD3 (Figure 4A). EHD4–siRNA- and mock-treated cells were fixed after 72 h and subjected to immunostaining with Rab5 and EEA1 (Figure 4B,C,E,F, both markers of the early endosome) and Rab11 (Figure 4D,G, a marker for recycling endosomes). Depletion of EHD4 had a striking effect on the morphology of early endosomes, which appeared to be larger, clustered and in some cases even fused into much larger structures that were less prevalent in the periphery of the cell (Figure 4E,F). On the other hand, the subcellular distribution of Rab11 did not change upon loss of EHD4 expression (Figure 4D,G).
Figure 4. Loss of EHD4 expression leads to generation of enlarged early endosomes.
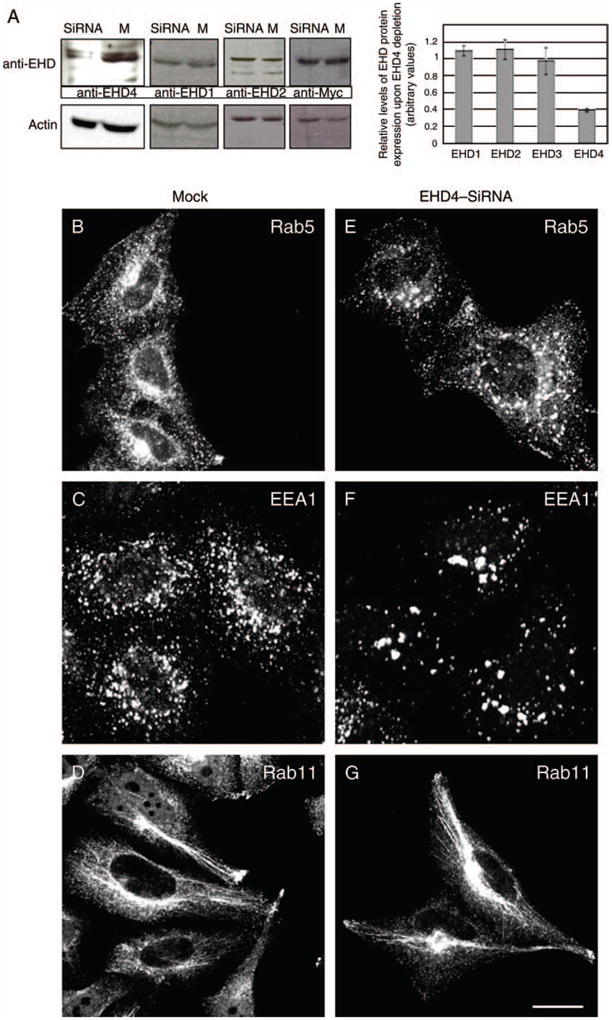
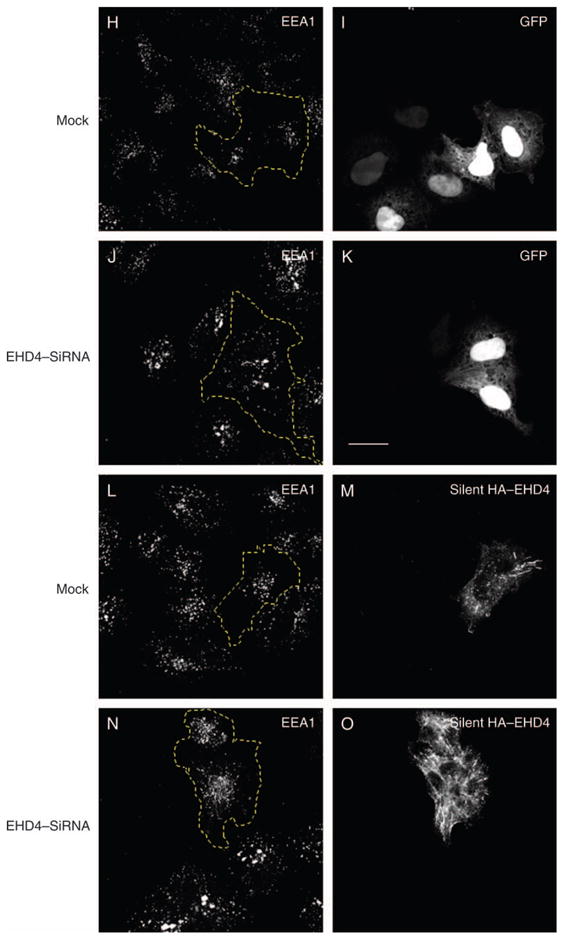
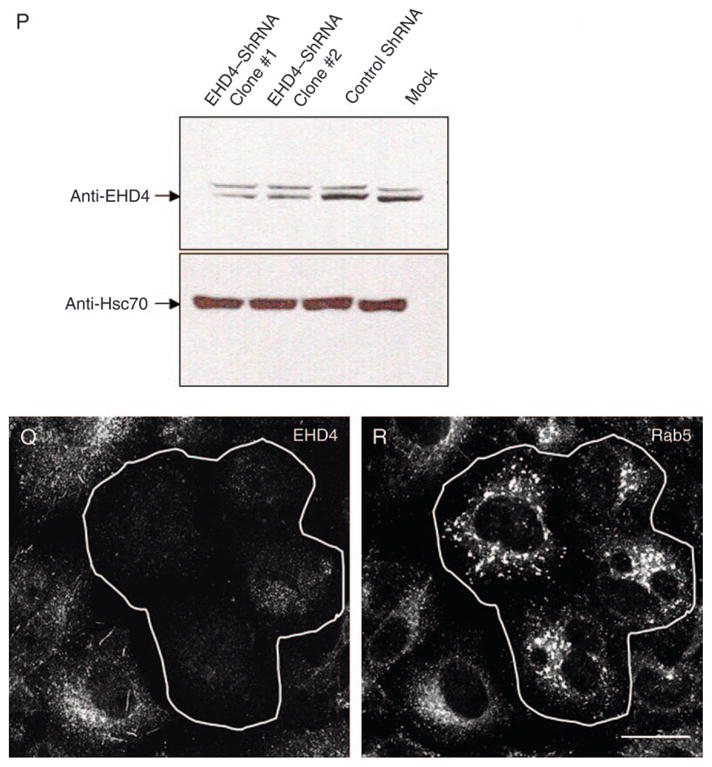
(A) HeLa cells growing on 35-mm plates were either mock-treated (denoted as M) or treated with siRNA for EHD4 (denoted as siRNA) and harvested after 72 h. Cells were then lysed and proteins separated by 8% SDS–PAGE and subjected to immunoblotting with affinity-purified anti-EHD4 antibodies, monoclonal anti-actin antibody to validate equal protein loading (bottom panel) or antibodies directed against EHD1 or EHD2. Because of difficulties in detecting endogenous EHD3 in HeLa cells, mock- and EHD4–siRNA-treated cells were transfected with GFP–Myc–EHD3 and analyzed as above, except that immunoblotting was performed with anti-Myc. These experiments were repeated independently four times and quantified by densitometry analysis. The bar graph denotes the average level of EHD expression upon EHD4–siRNA treatment compared with mock-treated levels (normalized to a value of 1). Error bars represent standard error. (B–G) HeLa cells on coverslips were mock treated (B–D) or treated with siRNA for EHD4 (E–G). After 72 h of treatment with siRNA, cells were fixed and stained for endogenous Rab5 (B and E), EEA1 (C and F) and rabbit polyclonal antibody directed at Rab11 (D and G) followed by the appropriate secondary antibodies. (H–O) Mock-treated HeLa cells (H, I, L and M) or EHD4–siRNA-treated cells (J, K, N and O) were transfected with either GFP vector alone (H–K) or a ‘silent HA–EHD4’ construct engineered to have resistance for siRNA (L–O). Cells were fixed, permeabilized and immunostained with anti-EEA1 (H, J, L and N) or with both anti-EEA1 and anti-EHD4 (M and O), while GFP-transfected cells were imaged directly (I and K). (P) HeLa cells were either transfected with control shRNA or transfected with two vector-based shRNA clones for EHD4 and harvested after 72 h. Cells were then lysed, and proteins were separated by 8% SDS–PAGE and subjected to immunoblotting with affinity-purified anti-EHD4 antibodies (top panel) or rat monoclonal anti-Hsc70 antibody to verify equal protein loading (bottom panel). (Q and R) HeLa cells on coverslips were transfected with vector-based shRNA for EHD4. After 72 h of transfection, cells were fixed and coincubated with affinity-purified anti-EHD4 antibodies (Q) and monoclonal anti-Rab5 antibodies (R) followed by the corresponding secondary antibodies. Bar, 10 μm.
To provide additional evidence that EHD4 depletion is specifically responsible for the enlarged early endosome phenotype, we utilized an siRNA-resistant EHD4 construct engineered with a silent mutation in the region recognized by the oligonucleotide. To this end, as a control, we first expressed GFP in both mock- and EHD4–siRNA-treated cells (Figure 4H–K). As expected, upon immunostaining these cells with the early endosomal marker EEA1, the endosomes in the EHD4-depleted cells were greatly enlarged (compare 4J with 4H). Moreover, expression of the GFP protein in these cells (depicted by dashed yellow line) had no effect on the regulation of endosomal size. On the other hand, expression the siRNA-resistant silent HA–EHD4 ‘rescued’ the effect of the siRNA knock-down and caused the EEA1-positive endosomes to revert back to their normal size (Figure 4L–O, compare dashed yellow area in N with neighboring cells and cells in L). These data strongly suggest that EHD4 knock-down is indeed responsible for the enlargement of early endosomes.
To further confirm our data showing that siRNA knockdown of EHD4 caused the formation of enlarged early endosomal structures, we utilized an additional knockdown strategy based on short hairpin RNA (shRNA) (Figure 4P–R). As demonstrated by immunoblot analysis, transfection of the shRNA plasmids for EHD4 reduced EHD4 protein expression by >60% compared with either control shRNA plasmids or mock treatment with only the transfection reagent (Figure 4P). Indeed, upon transfection of the EHD4–shRNA followed by immunostaining for endogenous EHD4, ~50% of the cells had a marked decrease in the level of EHD4 expression (Figure 6Q, outlined cells). Similar to our results using siRNA knockdown (Figure 4A–G), in these cells, Rab5 showed a dramatically altered subcellular distribution by coalescing into very large perinuclear structures away from the cell periphery (Figure 4R, outlined cells). Collectively, these findings demonstrate that EHD4 controls early endosome morphology and suggest that EHD4 may be involved in the regulation of trafficking at the early endosome.
Figure 6. EHD4 depletion causes retention of internalized MHCI in enlarged early endosomes and impedes its recycling.
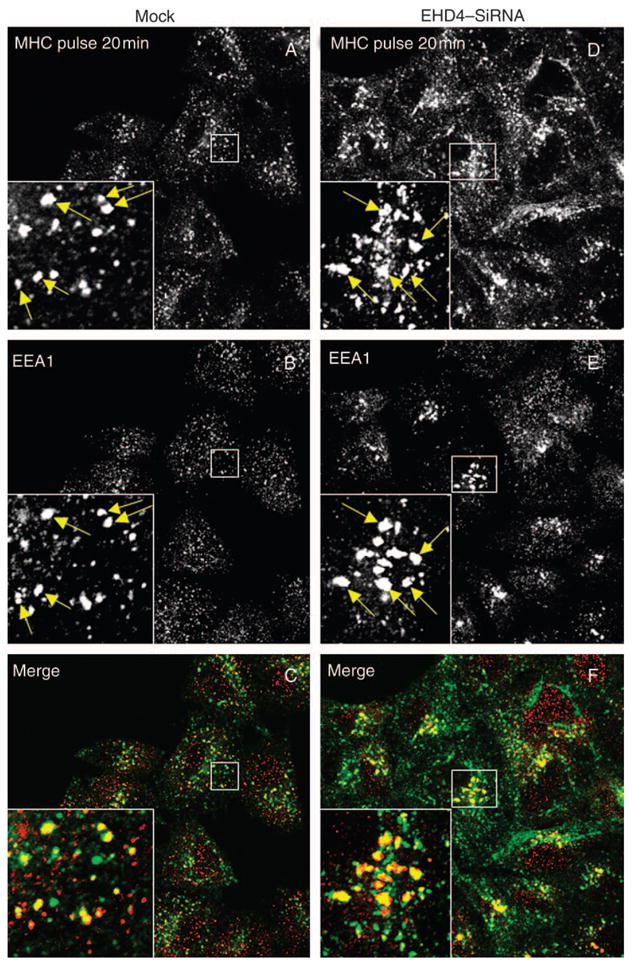
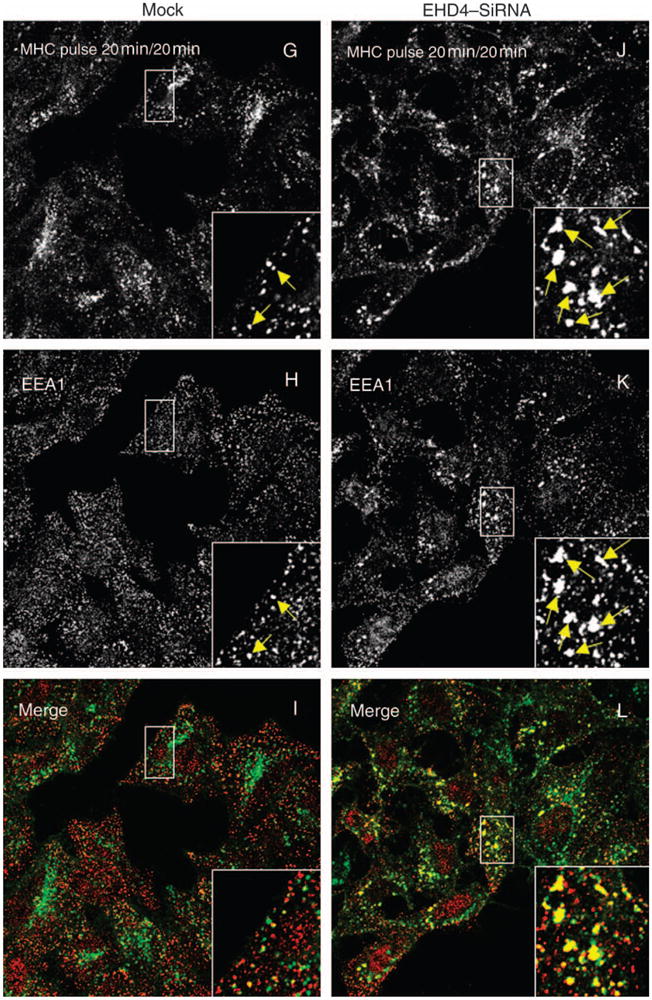
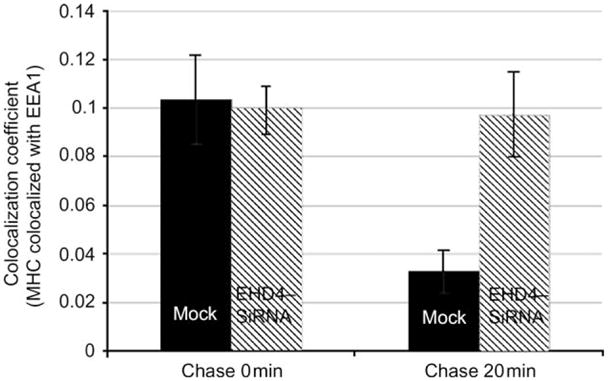
(A–L) HeLa cells on coverslips were mock treated (AC and G–I) or treated with siRNA for EHD4 for 72 h (D–F and J–L). The cells were then continuously pulsed with anti-MHCI monoclonal antibody for 20 min. After brief acid washing to remove surface-bound MHCI antibody, cells were either directly fixed (A–F) or ‘chased’ in complete media for 20 min (G–L) at 37°C prior to fixation. Cells were then permeabilized and incubated with rabbit antibodies directed against EEA1 followed by Alexa Fluor 488-conjugated anti-mouse antibodies and Alexa Fluor 568-conjugated anti-rabbit antibodies. Insets (A–L) depict higher magnifications of the boxed areas, and arrows show vesicles positive for both EEA1 and MHCI. Bar, 10 μm. (M) LSM 5 PASCAL software was used to calculate the numbers of pixels representing MHCI that colocalize with EEA1 in the above experiment. Bars depict standard error, n =8.
We next aimed to determine whether the enlarged early endosomal structures generated in the absence of EHD4 were accessible to endocytic cargo. HeLa cells on cover-slips were either mock treated (Figure 5A,B) or treated with EHD4–siRNA (Figure 5D,E) for 72 h and then subjected to brief ‘pulse–chase’ experiments with fluorochrome-labeled transferrin [transferrin–Alexa Fluor 568 (Tf-568)]. As demonstrated, after a short 3-min pulse with Tf-568, both mock-treated and EHD4–siRNA-treated cells exhibited a similar subcellular distribution pattern of the internalized transferrin (Figure 5A,D). For the most part, the transferrin was observed in very small vesicles, including pre-endosomal and early endosomal structures and at this time-point still dispersed quite uniformly through the cell (Figure 5A,D). Although some concentration in the perinuclear region was beginning to be evident in mock-treated cells (Figure 5A), this coalescence at the ERC was particularly evident only following a 5-min ‘chase’ (Figure 5B). On the other hand, in the EHD4–siRNA-treated cells, the Tf-568 was observed in enlarged structures (correlating with those seen earlier for Rab5 and EEA1), and importantly, it remained well dispersed throughout the periphery of the cells and did not concentrate at the ERC (Figure 5E). Indeed, upon EHD4 depletion, even following a 2 h continuous uptake of transferrin, this cargo did not accumulate at the ERC (Figure 5, compare F with C) but remained primarily in the periphery, often in enlarged structures. However, the rate of transferrin recycling to the plasma membrane upon EHD4 depletion remained similar to mock-treated cells (Figure 5G), suggesting that direct recycling to the plasma membrane from early endosomes may compensate for the impaired transport to the ERC. These data indicate that the enlarged early endosomes generated in the absence of EHD4 are indeed accessible to endocytic cargo and impede normal transport downstream through this pathway.
Figure 5. Loss of EHD4 causes accumulation of internalized transferrin in enlarged early endosomes and impairs its trafficking to the ERC.
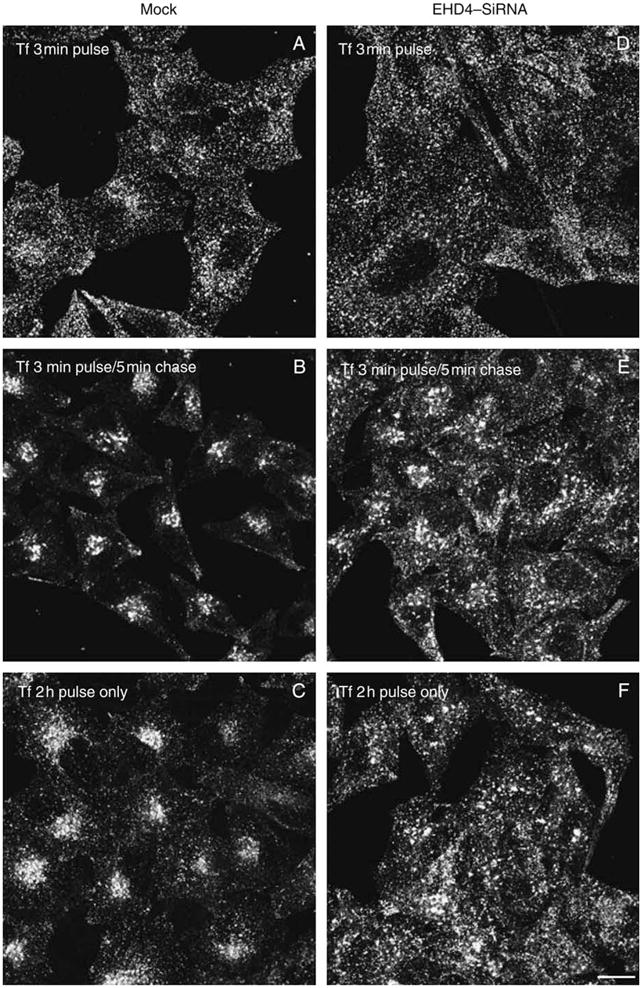
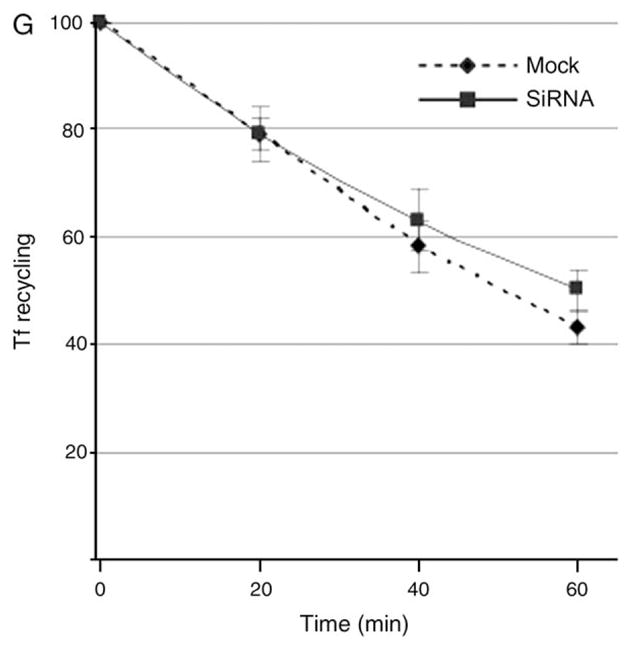
(A–F) HeLa cells on coverslips were mock treated (A–C) or treated with siRNA for EHD4 (D–F). After 72 h of treatment with siRNA, starved cells were pulsed with Tf-568 for 3 min and fixed (A and D), pulsed for 3 min and ‘chased’ in complete media for 5 min at 37°C prior to fixation (B and E) or pulsed continuously for 2 h (C and F). Bar, 10 μm. (G) HeLa cells on plates were mock treated or treated with siRNA for EHD4. After 72 h of treatment with siRNA, starved cells were either pulsed with Tf-568 for 20 min and fixed or ‘chased’ in complete media for the indicated times prior to fixation. Flow cytometry analysis was then performed to measure remaining Tf-568 levels for 10 000 cells at the indicated times. Error bars indicate standard error from three independent experiments.
Because EHD4 has been implicated in the Arf6 clathrin-independent endocytic pathway (49), we attempted to determine whether EHD4 regulates the trafficking of clathrin-independent cargo through the early endosome. To address this question, mock- and siRNA-treated cells were allowed to uptake antibodies against MHCI proteins (30) for 20 min. As demonstrated, a significant portion of MHCI was detected in early endosomal structures containing EEA1 (Figure 6A–C, see arrows and insets). As expected, early endosomes were enlarged upon depletion of EHD4, and these structures were accessible to MHCI (Figure 6D–F, see arrows and insets). In mock-treated cells following an additional 20 min of ‘chase’, MHCI was predominantly in a perinuclear region corresponding to the ERC, with little MHCI remaining colocalized with EEA1 (Figure 6G–I). However, depletion of EHD4 caused retention of MHCI in the early endosomal compartment where it remained highly colocalized with EEA1 (Figure 6J–L, see arrows in insets). These results were quantified and consistently indicated that twofold to threefold more MHCI was retained in EEA1-positive early endosomes at this time-point (Figure 6M). Indeed, even after 40 min of chase, the EHD4-depleted cells displayed considerable retention of MHCI in the early endosome compared with mock-treated cells (unpublished data). These data indicate that trafficking of both clathrin-dependent and clathrin-independent cargo through the early endosome is regulated by EHD4.
Early endosomes are also a nexus for the sorting of proteins to the late endosomal degradatory pathway. We therefore proposed to determine whether loss of EHD4 also impairs the trafficking of cargo that traverses this route. To this aim, mock- and EHD4–siRNA-treated cells were pulsed for 15 min with DiI-low-density lipoprotein (LDL), chased for 40 min and analyzed to determine the extent to which LDL was localized to EEA1-containing early endosomes. At this time-point in mock-treated cells, the vast majority of LDL molecules had already exited the early endosomes and no longer displayed colocalization with EEA1 (Figure 7A–C). On the other hand, in EHD4–siRNA-treated cells, a significant portion of LDL could still be observed colocalizing with EEA1 (Figure 7D–F, quantified in G). These data imply that EHD4 regulates exit from the early endosome toward both the recycling and the degradative pathways.
Figure 7. Loss of EHD4 causes a delay in LDL transport to the late endocytic pathway.

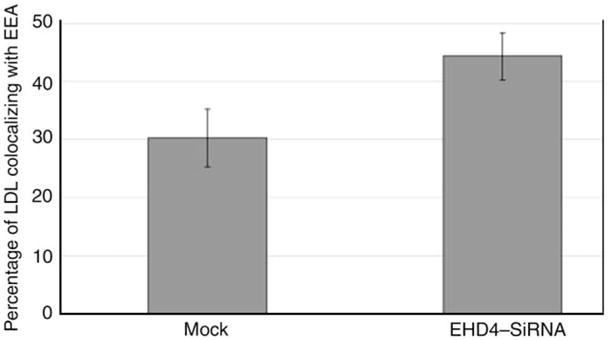
(A–F) He-La cells on coverslips were mock treated (A–C) or treated with EHD4–siRNA (D–F). After 72 h of treatment with siRNA, cells were starved in media containing fatty-acid-free serum for 1 h followed by a brief pulse of 15 min with DiI-labeled LDL and ‘chased’ in complete media for 40 min at 37°C prior to fixation. Cells were then permeabilized and incubated with mouse antibodies directed against EEA1 followed by Alexa Fluor 488-conjugated anti-mouse antibodies. Insets (A–F) depict higher magnifications of the boxed areas. Bar, 10 μm. (G) Quantitative analysis of LDL colocalized with EEA1. Error bars show standard error, n = 8.
Enlarged early endosomes have been observed previously when either wild-type Rab5 (50) or a GTP-locked Rab5 mutant was transfected into cells (51). We therefore hypothesized that the enlarged early endosomes in EHD4 knock-down cells may contain enhanced levels of GTP–Rab5 associated with their membranes. To test this hypothesis, we utilized three different methods. First, we took advantage of a plasmid containing a yellow fluorescent protein (YFP)/Rab5-binding domain of Rabaptin-5 fusion protein (YFP–R5BD) that binds specifically to GTP–Rab5 (52). Mock- and EHD4–siRNA-treated cells were transfected with YFP–R5BD, fixed and examined by confocal microscopy (Figure 8A–F). In mock-treated cells, endogenous Rab5 showed a typical early endosomal distribution pattern and was localized throughout the cell to small- and medium-sized vesicles (Figure 8A). The YFP–R5BD in these cells was mostly cytosolic as previously discussed (52), suggesting that relatively little GTP–Rab5 was localized to endosomal membranes (Figure 8A–C). In EHD4–siRNA-treated cells, as we have shown, the Rab5 localized to much larger structures (Figure 8D, see inset). YFP–R5BD was recruited from the cytosol to these structures, indicating that they were enriched in GTP–Rab5 (Figure 8D–F, see insets). Second, endogenous full-length Rabaptin-5 was also recruited to these enlarged endosomal membranes in EHD4-depleted cells (Figure 8G, H). Third, to obtain additional evidence supporting these findings that depletion of EHD4 leads to activation of Rab5, HeLa cells were mock treated or treated with both EHD4–siRNA and shRNA (to maximize the level of knock-down), followed by homogenization and separation to membrane and cytosolic fractions and then examined by immunoblotting with anti-Rab5 antibodies. To verify that membrane proteins are exclusively in the pellet fraction, immunoblotting was first performed with anti-transferrin receptor (TfR) (Figure 8I, right panel). As demonstrated in a representative experiment, cells depleted for EHD4 displayed more Rab5 in the membrane pellet, indicating an increase in the membrane-bound active Rab5–GTP (Figure 8I). In addition, a modest decrease in cytosolic Rab5 was also observed in EHD4-depleted cells (Figure 8I). To quantitatively evaluate the level of Rab5 activation, the membrane-to-cytosol ratios were calculated for mock- and EHD4–siRNA-treated cells from three independent experiments (Figure 8J). When mock ratios were normalized (to a value of 1 to permit fair comparison between separate experiments), the increase in membrane-to-cytosolic Rab5 in EHD4-depleted cells was consistently twofold to fivefold higher (Figure 8J, depicts a mean of fourfold increase in the ratio). Collectively, these data favor a model by which loss of EHD4 induces activation of Rab5.
Figure 8. Loss of EHD4 results in increased levels of active Rab5.
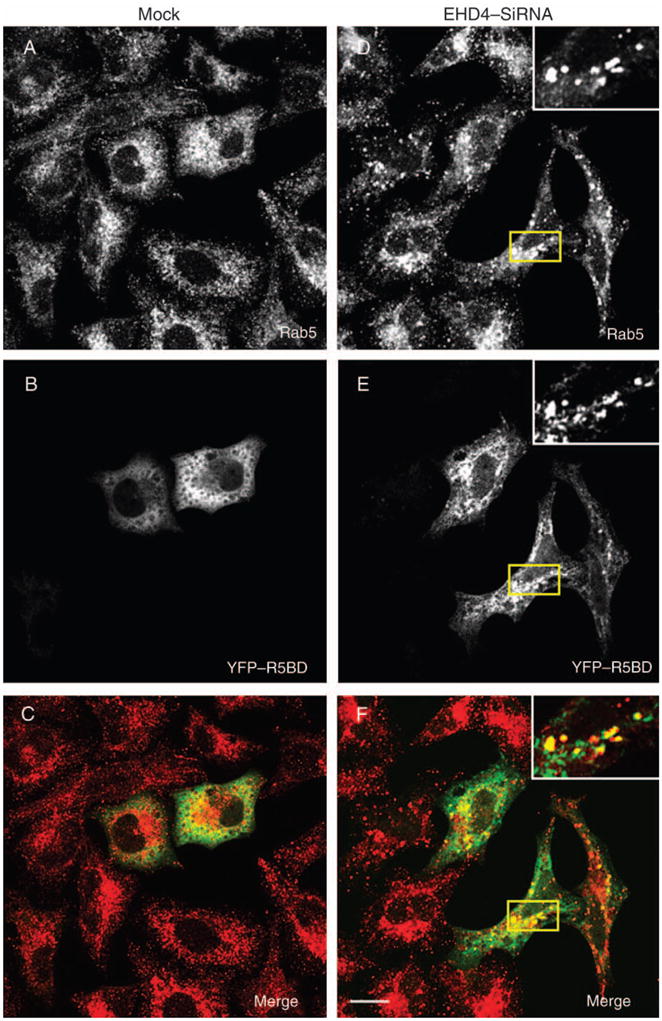

HeLa cells on coverslips were mock treated (A–C and G) or treated with siRNA for EHD4 (D–F and H) for 72 h. In the final 24 h, both mock- and siRNA-treated cells were either transfected with the YFP–Rab5-binding domain of Rabaptin-5 (R5BD, A–F) or left untransfected (G and H). Cells were fixed and incubated with mouse monoclonal antibodies directed at Rab5 (A and D) or endogenous Rabaptin-5 (G and H) followed by secondary Alexa Fluor 568-conjugated goat anti-mouse antibody. Bars, 10 μm. (I) Mock-treated HeLa cells and HeLa cells treated together with EHD4–siRNA and EHD4–shRNA were homogenized in ice-cold homogenization buffer, and post-nuclear supernatants were separated to membrane and cytosol fractions by ultracentrifugation. The samples were resolved by 10% SDS–PAGE, transferred to nitrocellulose and immunoblotted with either anti-Rab5 or anti-TfR antibodies (as indicated) followed by anti-mouse–HRP-conjugated antibodies. Enhanced chemiluminescence was used for detection. (J) Densitometric analysis was performed on three independent experiments. Briefly, the ratios of membrane-associated Rab5 to cytosolic Rab5 was calculated for mock-treated and EHD4–siRNA-treated cells in each experiment. Note that the actual numerical values for membrane/cytosol distribution varied with each experiment and for siRNA versus mock:EHD4 depletion produced membrane-to-cytosol values of 82.7, 17.1 and 5.2, whereas mock depletion yielded values of 13.4, 6.4 and 1.5. However, as these values are arbitrary in nature (depending on film exposure times, etc.), by comparing knock-down-to-control ratios in each individual experiment, it was possible to normalize mock values at 1 (see dotted line in bar graph) and represent a mean fold increase observed upon EHD4–siRNA knock-down for all three experiments (including standard error bars).
Endogenous EHD4 interacts with EHD1
Several studies have recently demonstrated that EHD1 and its closest paralog, EHD3 (86% identical), are capable of oligomerizing through their central coiled-coil domains (25,42) and that loss of oligomerization leads to the protein becoming cytosolic. Because EHD4 also contains a predicted central coiled coil, we next determined whether this protein oligomerizes with the other EHD paralogs. Using a selective two-hybrid binding assay, we showed that EHD4 robustly binds to EHD1 and EHD3 (Figure 9A). However, while binding of EHD4 to EHD2 and EHD4 homo-oligomerization was also observed, it appears to be significantly weaker than binding of EHD4 to either EHD1 or EHD3 (Figure 9A). To confirm these observations in mammalian cells by coimmunoprecipitations, we transfected HeLa cells with HA–EHD4 and with GFP–Myc–EHD1, GFP–Myc–EHD2 or GFP-Myc-EHD3 (Figure 9B). As shown, expression levels of GFP–Myc–EHD1, GFP–Myc–EHD2 and GFP–Myc–EHD3 were relatively similar, and HA–EHD4 was coexpressed in each of these cells (Figure 9B, cell lysate panel). Despite similar expression levels, significantly more EHD1 was coimmunoprecipitated with EHD4 compared with EHD2 (Figure 9B, upper left panel). EHD3 was also immunoprecipitated at higher levels than EHD2 (Figure 9B). Although EHD4 could also homo-oligomerize, EHD4/EHD4 interactions identified by coimmunoprecipitation appeared to be weaker than those between EHD4 and either EHD1 or EHD3 (Figure 9C). This association of EHD4 with all three paralogs and its homo-oligomerization, as shown in Figure 9B,C, were quantified and are presented in Figure 9E (for calculation of values, see legend). To ascertain whether EHD4 hetero-oligomerizes with EHD1 through coiled-coil interactions, we cotransfected HA–EHD4 together with Myc–EHD1 V203P, a point mutant that we have previously shown to have a disrupted coiled coil (25). As demonstrated, this mutation significantly reduced EHD4/EHD1 interactions to <5% of the level of interactions between the two wild-type proteins (Figure 9D). These data show that EHD4 interacts with its EHD paralogs in a hierarchical manner through coiled-coil interactions.
Figure 9. Preferential binding of EHD4 with EHD1 and EHD3.

(A) The Saccharomyces cerevisiae yeast strain AH109 was cotransformed with the following GAL4-binding domain (GAL4bd) fusion constructs: GAL4bd–p53 (control), GAL4bd–EHD4, GAL4bd–EHD3, GAL4bd–EHD2 and GAL4bd–EHD1 together with the following GAL4 transcription activation (GAL4ad) fusion products: GAL4ad–EHD4 and GAL4ad–SV40 Large T-Antigen (SV40LT-Ag, control). Cotransformants were assayed for their growth on non-selective (+HIS) and selective (−HIS) media. (B–D) HeLa cells were cotransfected with HA–EHD4 and GFP–Myc–EHD1, GFP–Myc–EHD2 or GFP–Myc–EHD3 (B), GFP–EHD4 (C) and Myc–EHD1 V203P (D). After 24 h, cells were lysed and subjected to immunoprecipitations with anti-HA antibody-conjugated agarose beads. Immunoprecipitates and total cell lysates (as indicated) were resolved by 8% SDS–PAGE, transferred to nitrocellulose and immunoblotted with anti-Myc antibodies (B and D, top panels), anti-HA antibodies (B–D, bottom panels) followed by detection with anti-mouse–HRP-conjugated antibodies (B–D) or with biotinylated anti-GFP antibodies followed by streptavidin–HRP (C, top panel). Enhanced chemiluminescence was used for detection. (E) Quantitative analysis of the coimmunoprecipitation displayed in (B) and (C) is shown; intensity of protein bands on the immunoblots was calculated by multiplying the mean of luminosity by number of pixels as measured by ADOBE PHOTOSHOP software. Given the relatively similar levels of EHD proteins coexpressed with EHD4 in the cotransfected cells, the relative level of interaction was calculated by dividing the intensity of a given precipitated EHD protein by the intensity of the directly immunoprecipitated HA–EHD4 from a specific sample. Error bars show standard error, n = 3. IP, immunoprecipitation.
Despite increasing evidence that EHD proteins oligomerize and that oligomerization is important for biological function, to date, no studies have addressed the interaction between these endogenous proteins. Our coimmunoprecipitation studies with overexpressed EHD proteins showed that EHD4 interacted preferably with EHD1 and EHD3; however, for the purposes of endogenous coimmunoprecipitations, our anti-EHD3 antibodies are unsuitable. Accordingly, we aimed to determine whether endogenous EHD4 and endogenous EHD1 interact in HeLa cells (Figure 10). To this aim, untransfected HeLa cells were lysed and the proteins were separated by SDS–PAGE. The non-reducing conditions utilized here allowed us to distance the non-reduced immunoglobulin G (~155 kDa) from the EHD proteins (EHDs, ~60 kDa). Immunoblotting with either anti-EHD1 (Figure 10A, left panel) or anti-EHD4 (Figure 10A, right panel) showed the position of these EHD proteins on the gel, with EHD4 migrating at a slightly higher apparent molecular weight. Anti-EHD1 antibodies successfully immunoprecipitated EHD1 (Figure 10A, third lane), whereas pre-immune serum did not precipitate protein that was recognized by anti-EHD1 antibodies (Figure 10A, second lane), demonstrating the specificity of the anti-EHD1 antibodies for EHD1 immunoprecipitations. Moreover, when control immunoprecipitations were carried out with either EHD1 pre-immune serum or protein A–Sepharose beads, no EHD4 was detected by immunoblotting (Figure 10A, second panel from right). However, when lysates were immunoprecipitated with anti-EHD1 antibodies and immunoblotted with anti-EHD4, a band of ~64 kDa was detected (Figure 10A, fourth lane). A band of identical apparent molecular weight was observed when immunoprecipitations were carried out directly with anti-EHD4 and immunoblotted with the same antibody (Figure 10A, fifth lane). While the anti-EHD4 was generated against an entirely unique C-terminal amino acid sequence present only in EHD4, it was not possible to obtain useful antibodies against EHD1 that did not partially overlap with the sequence of EHD4. Although immunoblotting clearly showed a high preference of the EHD1 peptide antibodies for the 60-kDa EHD1 band (as opposed to the ~64-kDa EHD4 band), it was nevertheless necessary to rule out the possibility that EHD4 might be immunoprecipitated directly by the anti-EHD1 antibody. To this aim, we immunoprecipitated EHD1 from mock-treated cells as well as cells depleted for EHD1 with EHD1–siRNA (Figure 10B). As shown, EHD1–siRNA-treated cells showed a significant reduction (>90%) in the levels of EHD1 when assessed by immunoprecipitation and immunoblotting analysis (Figure 10B, top panel). Moreover, when EHD1 was depleted from the cells with siRNA, there was almost a complete loss of the coimmunoprecipitated EHD4 (which is often but not always observed as a doublet band), indicating that EHD4 precipitates as a result of its association with EHD1 and not because of direct binding with the anti-EHD1 antibody (Figure 10B, bottom panel). These data demonstrate that endogenous EHD4 and EHD1 interact in cells.
Figure 10. Endogenous EHD4 and EHD1 are present together in a complex.
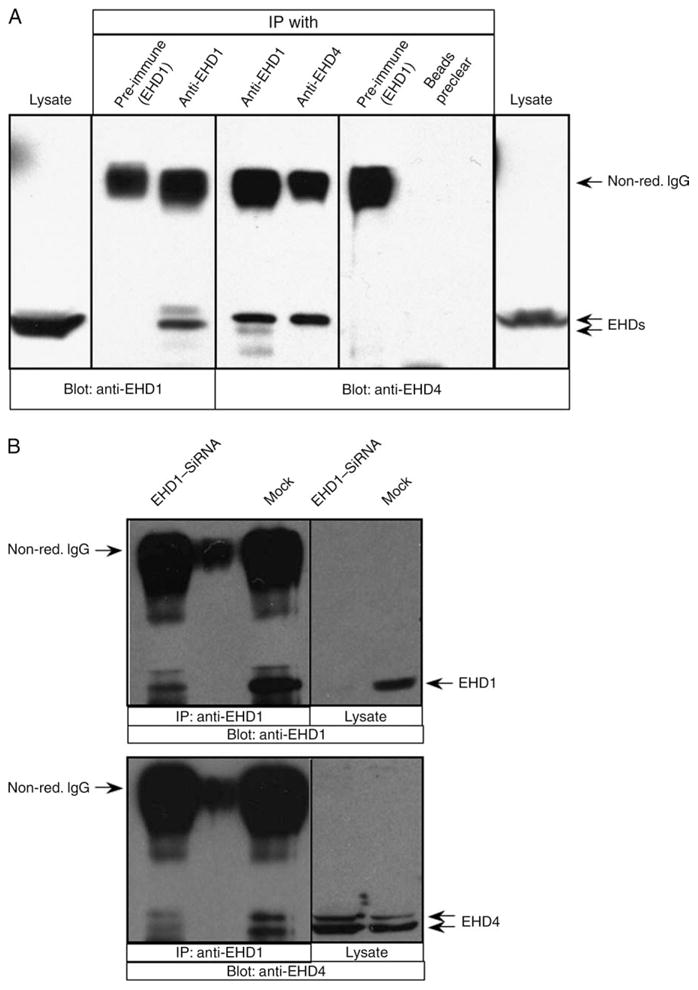
Detergent lysates were prepared from untransfected HeLa cells (A) or HeLa cells either mock treated or treated with EHD1–siRNA (B). The lysates were subjected to immunoprecipitation with affinity-purified polyclonal rabbit anti-EHD1 antibodies, affinity-purified polyclonal rabbit anti-EHD4 antibodies or pre-immune serum (control) or protein A–Sepharose beads preclear (control). Immunoprecipitates and total lysates (as indicated) were resolved by non-reducing 8% SDS–PAGE, transferred to nitrocellulose and immunoblotted with either anti-EHD4 or anti-EHD1 antibodies (as indicated) followedbyanti-rabbit–HRP-conjugated antibodies. Enhanced chemiluminescence was used for detection. Positions of the non-reduced immunoglobulin G (non-red. IgG) and EHD proteins are indicated with arrows. IP, immunoprecipitation.
Functional relationship between EHD4 and EHD1
To probe the relationship between EHD4 and EHD1, HeLa cells were either mock treated or treated with EHD4–siRNA, and the effect of EHD4 depletion on the subcellular distribution of EHD1 was examined. As expected, in mock-treated cells, endogenous EHD1 localized to an array of punctae and tubular membranes that displayed minimal overlap with endogenous Rab5 (Figure 11A–C). As described earlier, EHD4 depletion led to Rab5 activation and localization to enlarged early endosomes (Figure 11D). However, the pattern of EHD1 was greatly altered, the tubular EHD1 structures were no longer observed and EHD1 now appeared to colocalize significantly with Rab5 on the enlarged early endosomal membranes (Figure 11D–F). In a reciprocal experiment, the subcellular distribution of EHD4 was examined in mock-treated and EHD1–siRNA-treated cells (Figure 11G–L). In mock-treated cells, Rab5 localized to perinuclear and peripheral punctate endosomes, whereas EHD4 localized peripherally to punctae and tubular endosomes, with some colocalization in evidence (Figure 11G–I). However, in EHD1-depleted cells, both Rab5 and EHD4 localize primarily to the perinuclear ERC region, are largely absent from the periphery and show a strong colocalization with each other (Figure 11J–L). These data imply that EHD4/EHD1 interactions are important to maintain proper subcellular distribution of EHD and other endocytic regulatory proteins.
Figure 11. The interaction between EHD4 and EHD1 is required for the normal subcellular distribution of both proteins.
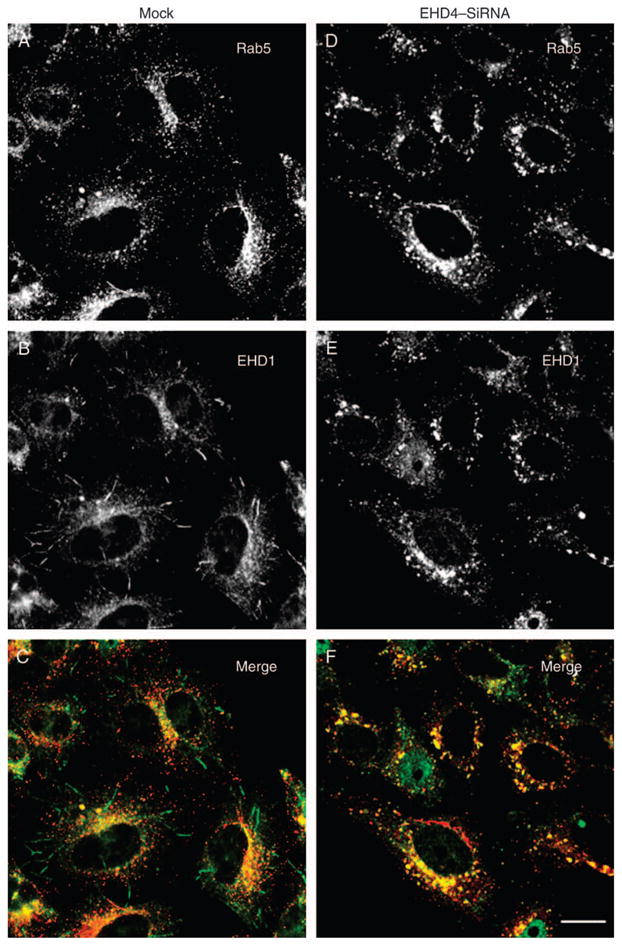
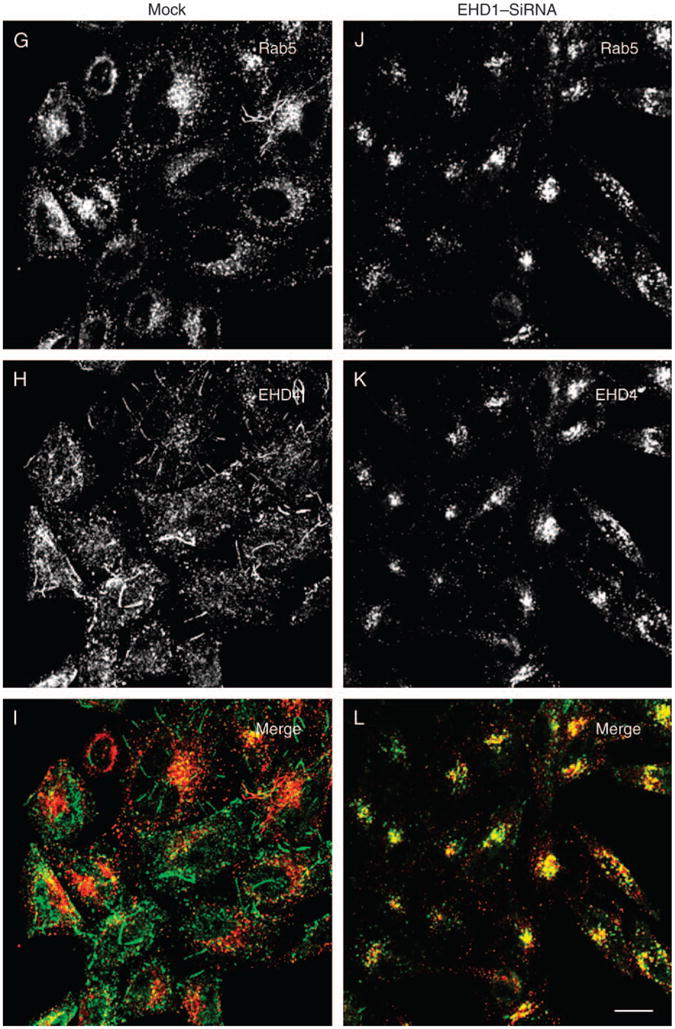
(A–F) HeLa cells on coverslips were mock treated (A–C) or treated with siRNA for EHD4 (D–F). After 72 h of treatment with EHD4–siRNA, cells were fixed and coincubated with antibodies directed at Rab5 (A and D) and affinity-purified polyclonal rabbit anti-EHD1 antibodies (B and E) followed by the corresponding secondary antibodies. (G–L) HeLa cells on coverslips were mock treated (G–I) or treated with siRNA for EHD1 (J–L). After 72 h of treatment with EHD1–siRNA, cells were fixed and coincubated with antibodies directed at Rab5 (G and J) and affinity-purified polyclonal rabbit anti-EHD4 antibodies (H and K) followed by the corresponding secondary antibodies. Bar, 10 μm.
Discussion
The four C-terminal human EHD proteins are highly homologous, with the best-characterized EHD1 sharing 70–86% identity with its paralogs. Accordingly, one of the primary hurdles in analyzing the unique function of each individual C-terminal EHD proteins has been the difficulty in generating specific antibodies for each of these proteins. Indeed, while the functional role of EHD1 has been well characterized, the exact roles of the other EHD proteins remain more obscure. In particular, the function of EHD4 has remained somewhat controversial. While EHD4 was originally described as a component of the extracellular matrix (47), more recent studies have proposed endocytic roles. However, there is currently no clear consensus for the endocytic step(s) that EHD4 might regulate. Studies on PC12 rat adrenal pheochromocytoma cells suggest a role for EHD4 in the early stages of the endocytic pathway, regulating the internalization of nerve growth factor receptor (48). More recently, two studies that have concurrently addressed the respective roles of all four EHD proteins have proposed disparate roles for EHD4. The first study, by Plomann and colleagues (45), suggests that EHD4 is involved in highly specialized transport processes that do not involve trafficking through endosomes. A paper by Band and colleagues provides evidence that EHD4 regulates a transport step at the early endosome (46). Our studies, which take advantage of a highly specific antibody generated against a peptide found exclusively in EHD4, coupled with siRNA and shRNA knock-downs, functional assays and biochemical studies, promote a role for EHD4 in controlling the exit of receptors and internalized cargo from early endosomes and suggest that EHD4 plays a role in regulating Rab5 activation.
Several lines of evidence support a role for EHD4 at the early endosome. First of all, both transfected and endogenous EHD4 are distributed on punctate peripheral structures that partially colocalize with the early endosomal membrane proteins Rab5 and EEA1. EHD4 also partially colocalizes with Arf6, Rab4 and Sorting Nexins 1 and 2. In addition, endogenous EHD4 colocalizes with internalized transferrin. Moreover, the loss of EHD4 expression, induced by either siRNA or shRNA, leads to the accumulation of internalized transferrin on aberrantly enlarged early endosomes.
The role emerging for EHD4 in transport at the early endosome differs from the established role of EHD1, which shares 74% identity. EHD1 controls recycling of receptors and membranes to the plasma membrane from the ERC where it emanates in a series of long tubular membrane structures. Although EHD4 is found primarily associated with punctate structures in the cell periphery, akin to EHD1, it also localizes to elongated tubular membranes. While it is not possible to concurrently immunostain for both endogenous EHD1 and EHD4 because the peptide-specific antibodies are both polyclonal, cotransfection experiments suggest a modest degree of overlap on tubular membranes (data not shown).
Recent studies indicate that C-terminal EHD proteins interact to form homo- and hetero-oligomers through their coiled-coil domains (24–26,42). Such EHD homo- and/or hetero-oligomerization appears to be important for EHD localization and function because mutations that are predicted to interfere with coiled-coil formation cause EHD proteins to lose their ability to associate with membranes (24–26,42). It has also been demonstrated that EHD4 is capable of interacting with EHD1 and its paralogs (46), suggesting co-operation and partial functional overlap between EHD4 and EHD1. However, because overexpression of pairs of EHD proteins tends to impact their subcellular localizations, it is difficult to extrapolate the functional significance from such studies. Until now, because of the inherent difficulties in obtaining specific antibodies for coimmunoprecipitation experiments, it was unknown whether endogenous EHD proteins form a complex in vivo. Our evidence indicates that while the majority of EHD4 is on peripheral early endosomes and EHD1 primarily resides at the tubular recycling compartment, EHD4 and EHD1 do interact transiently, and this association may be critical for transport of cargo along the early endosome/ERC axis.
The physiological significance of the EHD4 interaction with EHD1 is supported by the effect of knocking down either EHD protein on its EHD paralog. Loss of EHD4 expression ablates the formation of endogenous EHD1 tubules and alters its subcellular localization, causing its accumulation in peripheral Rab5-containing early endosomes (Figure 11A–F). Similarly, depletion of EHD1 causes a dramatic change in EHD4 localization, with tubular EHD4-containing structures no longer in evidence and EHD4 accumulating at the perinuclear endocytic recycling region (Figure 11G–L). While the significance of these findings remains to be studied, these data hint at the possibility of an ‘EHD cycle’ between the early endosomes and the recycling compartment.
Unlike EHD1, whose function is dominant in controlling the morphology and function of the ERC (22,23,29,30), EHD4 appears to be intimately involved in the regulation of traffic from early endosomes. The loss of EHD4 expression had a dramatic impact on the size of early endosomes, as determined by immunostaining with the endosomal membrane markers Rab5 and EEA1. Although both EEA1 and Rab5 are classical markers for early endosomes, subtle differences could be discerned with EEA1 often localizing to aberrant early endosomal vesicles that were larger than those containing Rab5 (Figure 4B–F). One possibility for this difference is that EEA1 localizes to a partially distinct subset of early endosomes, although it was not possible to confirm this because the antibodies directed against both EEA1 and Rab5 are monoclonal and cannot be used simultaneously.
How might EHD4 regulate the endosome size? Although the precise mechanism remains an open question worthy of further study, we rationalized that the activation of Rab5 to its GTP-bound state could play a critical role in this process. Indeed, we utilized a previously characterized YFP-tagged Rab5-binding domain from Rabaptin-5 (YFP–R5BD), which binds exclusively to Rab5 in its GTP form (52), to analyze the nucleotide status of Rab5 in vivo in cells lacking EHD4 (Figure 8). Under normal conditions, there is insufficient Rab5–GTP associated with endosomes so that YFP–R5BD remains mostly in the cytosol [(52) and Figure 8B]. However, when EHD4 was depleted by siRNA, YFP–R5BD was recruited to the Rab5-containing enlarged early endosomal structures (Figure 8D–F). Full-length endogenous Rabaptin-5 was also recruited to enlarged early endosomal structures (Figure 8G,H), further supporting the role of GTP-loaded Rab5 in the formation of enlarged early endosomes in the absence of EHD4. Moreover, fractionation experiments demonstrate a modest increase in membrane-associated (GTP-bound, active) Rab5 when EHD4 is depleted from cells (Figure 8I), further supporting a role for EHD4 in Rab5 regulation.
While the mode by which EHD4 regulates Rab5 activation is not yet understood, one might speculate that loss of EHD4 on early endosomes might either accelerate the function of a GEF for Rab5 or deactivate a GAP. The conversion of Rab5 to a GTP-bound state (or its stabilization) would allow Rab5 to become anchored to the endosomal membrane where it might interact with phosphatidylinositol-3-phosphate (PI3P) kinase, facilitating the conversion of phosphatidylinositol to PI3P. As the latter phosphoinositide is enriched on early endosomal structures (53,54), this could lead to the enlargement of these organelles. Alternatively, EHD4 might lead to a ‘fission arrest’ whereby the process of budding from early endosomes toward the ERC, the Golgi or the late endocytic pathways is impaired, thus affecting early endosomal morphology.
Overall, our data provide evidence detailing a role for EHD4 at the early endosome in the regulation of endosomal and receptor transport from this organelle. These functions implicate EHD4 in both novel and partially overlapping roles with other EHD paralogs in the regulation of endocytic trafficking.
Materials and Methods
Recombinant DNA constructs
Human EHD1, EHD3 and EHD1 V203P constructs have been described previously (25,28,30). A full-length human EHD4 complementary DNA clone was purchased from GeneCopoeia and subcloned into a pCDNA 3.1(+) expression vector (Invitrogen) using a 5′ primer containing the nucleotide sequence coding for the HA epitope with BamHI and 3′ XhoI restriction sites. HA-tagged EHD4 ΔEH and HA-tagged EHD4 G68R mutants were generated utilizing the QuickChange site-directed mutagenesis kit (Stratagene) by introducing a stop codon at amino acids 429 and changing G68 to R68, respectively. Human EHD4 was also subcloned into EGFPC3 vector (Clontech) using 5′ XhoI and 3′ BamHI restriction sites with GFP at the N-terminus of the fusion protein. Two-hybrid control vectors (GAL4ad–SV40 large T-antigen and GAL4bd–p53) were obtained from BD Biosciences Clontech. pGADT7 and pGBKT7 two-hybrid vectors containing EHD1, EHD2 and EHD3 have been described previously (28), and EHD4 was subcloned into the multicloning sites of these vectors by engineering 5′ NdeI and 3′ BamHI restrictions sites. All constructs were sequence verified and are available upon request. The YFP–R5BD (Rab5-binding domain of Rabaptin-5) was a kind gift from Dr A. Sorkin (University of Colorado Health Sciences Center), the HA–Arf6 was from Dr Julie Donaldson (National Institutes of Health) and the GFP–Rab constructs were a gift from Dr Robert Lodge (Universite Laval).
Antibodies and reagents
The following antibodies were used in this study: affinity-purified rabbit polyclonal peptide antibodies directed against the C-terminus of human EHD1 (DLPPHLVPPSKRRHE), EHD2 (VERGPDEAMEDGEEGSDDEA) and EHD4 (SHRKSLPKAD) (AnaSpec), rabbit antibodies against Rab11 (US Biologicals) and EEA1 (Novus Biologicals, Inc.), mouse monoclonal antibodies directed against the Myc epitope (9E10) and HA epitope (Covance), against EEA1, Rab5, Rabaptin-5, Sorting Nexin 1 (SNX1) and Sorting Nexin 2 (SNX2) (Transduction Laboratories), against LAMP1 (Santa Cruz Biotechnology), against actin (Abcam), against anti-human HLA-A,B and C (Leinco Tech) and rat monoclonal antibody against Hsp70 (Stressgen Bioreagents). Secondary donkey anti-mouse Alexa 488 nm and goat anti-rabbit Alexa 568 nm antibodies were purchased from Invitrogen. Goat anti-mouse horseradish peroxidase (HRP) was obtained from Jackson Immuno Research Laboratories, Inc. Donkey anti-rabbit–HRP was obtained from Amersham Biosciences, and Goat anti-rat HRP was obtained from Jackson ImmunoResearch Laboratories, Inc. DiI-LDL was obtained from Biomedical Technologies. Fatty-acid-free bovine serum and protease inhibitor cocktail were obtained from Sigma.
Immunoprecipitations and immunoblotting
For immunoprecipitation experiments, cells were harvested and lysed for 15 min in buffer containing 25 mM Tris, pH 7.4, 150 mM NaCl, 0.5% Triton-X-100 (w/v), protease inhibitor cocktail (Roche Molecular Biochemicals) and 10 mM iodoacetamide. After removal of insoluble matter by centrifugation, the lysate supernatants were subjected to immunoprecipitations with goat anti-HA antibody-conjugated agarose beads (Bethyl laboratories). After 2-h incubation at 4°C, immunoprecipitates were washed four times in lysis buffer containing only 0.1% Triton-X-100 and boiled in 125 mM Tris (pH 6.8)/2% sodium dodecyl sulfate (SDS)/12% glycerol/0.02% bromophenol blue to elute the proteins. Proteins were separated by 10% SDS–PAGE, blocked in 5% non-fat milk in PBS and immunoblotted with either mouse anti-Myc (9E10) followed by mouse anti-rabbit light chain HRP (Jackson Immuno Research Laboratories, Inc.) or biotin-conjugated anti-GFP antibodies. Streptavidin–HRP was used to detect the presence of biotinylated anti-GFP antibodies by enhanced chemiluminescence. To immunoprecipitate endogenous EHD proteins, protein A–Sepharose beads prebound to pre-immune serum or to affinity-purified anti-EHD1 and anti-EHD4 antibodies were used. Immunoblotting was performed with affinity-purified anti-EHD1 and anti-EHD4 antibodies, and detection was with donkey anti-rabbit HRP. Densitometric analysis was utilized to calculate the intensity of protein bands on the immunoblots by multiplying the mean of luminosity by number of pixels as measured by ADOBE PHOTOSHOP software, subtracting the background and using actin expression levels for normalization of each lane.
Membrane fractionation
HeLa cells were harvested in 600 μL of ice-cold homogenization buffer (25 mM HEPES, 100 mM NaCl, 1 mM ethylenediaminetetraacetic acid, pH 7.4 and protease inhibitor cocktail) by 30 vertical strokes in a glass Dounce homogenizer (Thomas) at 4°C. After centrifugation at 800 × g for 10 min to remove unbroken cells and cellular debris, the supernatant was then ultracentrifuged at 108 000 ×g for 1 h at 4°C to yield a pellet of total cellular membranes and a supernatant representing the cytosolic fraction. The membrane pellet was dissolved in urea buffer (70 mM Tris–Hcl, pH 6.8, 8 M urea, 10 mM n-ethylmaleimide, 10 mM iodoacetamide, 2.5% SDS and 0.1 M DTT) by incubating at 37°C for 15 min. The entire membrane fraction and 3.5% of the cytosolic fraction were mixed with ×2 Laemmli buffer to a final concentration of ×1 and subjected to SDS–PAGE and immunoblot analysis.
Yeast two-hybrid analysis
The Saccharomyces cerevisiae strain AH109 (Clontech) was maintained on Yeast extract, Peptone, Dextrose (YPD) agar plates. Transformation was carried out by the lithium acetate procedure as described in the instructions for the MATCHMAKER two-hybrid kit (Clontech). For colony growth assays, AH109 cotransformants were streaked on plates lacking leucine and tryptophan and allowed to grow at 30°C usually for 3 days or until colonies were large enough for further assays. An average of three to four colonies was then chosen and suspended in water, equilibrated to the same optical density of 600 nm and replated on plates lacking leucine and tryptophan (+HIS) as well as plates also lacking histidine (−HIS).
Gene knock-down by RNAi and rescue using RNAi-resistant EHD4 expression
RNA interference (RNAi) duplexes (synthesized by Dharmacon) were transfected using Dharmafect (Dharmacon) as previously described (28). Calibration experiments showed that 72 h of treatment was sufficient to attain significantly decreased expression levels of both EHD1 and EHD4. The sequence used for EHD1 (base pairs 943–963) was gaaagagatgcccaatgtc, and the sequence for EHD4 (base pairs 222–241) was gaccaccttcatcagatac. Rescue of EHD4 RNAi was performed by expression of an EHD4 construct engineered with a silent mutation at base pairs 231, 234, 235 and 237, which maintain phenylalanine, isoleucine and arginine residues in these positions. This construct was transfected using FuGENE HD (Roche Applied Science) in mock-treated and EHD4–siRNA-treated cells 48 h after the start of siRNA treatment. For the shRNA-mediated knock down of EHD4, the same sequence of EHD4 as mentioned above was cloned into RNAi-Ready pSIREN-Shuttle vector (BD Biosciences) as per the manufacturer’s instructions. Negative control shRNA was provided by the manufacturer. Efficacy of EHD1 and EHD4–RNAi was confirmed by immunoblotting of transfected and control cell lysates with affinity-purified rabbit polyclonal antibodies specific for EHD1 and EHD4, respectively.
Immunofluorescence and transferrin, MHCI and LDL uptake assays
HeLa cells were grown on cover glasses, transfected with FuGENE-6 (Roche Diagnostics) and fixed with 4% (v/v) paraformaldehyde in PBS as described previously (55). Fixed cells were incubated with primary antibodies prepared in staining solution [0.2% saponin (w/v) and 0.5% (w/v) BSA in PBS] for 1 h at room temperature. After washes in PBS, the cells were incubated with the appropriate fluorochrome-conjugated secondary antibody mixture in staining solution for 30 min at room temperature. All images were acquired using a Zeiss LSM 5 Pascal confocal microscope (Carl Zeiss) by using a ×63 1.4 numerical aperture objective with appropriate filters. Transferrin uptake was studied by first starving the cells in DMEM-lacking serum (but containing 0.5% BSA) for 30 min and then applying a 3-min pulse of 16.7 μg/mL Tf-568 (Invitrogen) followed by chase in complete media for 5 min. Cells were fixed and mounted as described above for image analysis. For MHCI pulse–chase assays, cells were pulsed with anti-MHCI antibody for the indicated times, followed by a 90-second incubation in stripping buffer (0.5 M NaCl and 0.5% acetic acid, pH 3.0) to remove surface-bound MHCI antibody. Cells were either fixed directly or incubated in complete media for the indicated times to ‘chase out’ the antibody. Colocalization coefficients were calculated with LSM 5 PASCAL software. For LDL uptake, cells were first starved in media containing fatty-acid-free serum for 1 h, followed by a brief pulse with DiI-labeled LDL and ‘chased’ in complete media for 40 min prior to fixation. All images shown are representative images from experiments that have been repeated at least three times. Quantification of colocalization was performed with IMAGEJ software utilizing JACoP (http://rsb.info.nih.gov/ij/plugins/track/jacop.html) (56) to calculate Pearson’s coefficient (57). Each pairwise comparison was performed on eight sets of images acquired with the same optical settings. Bars represent the mean ± standard error calculated from eight sets of images.
Supplementary Material
Figure S1: Partial colocalization of endogenous EHD4 with Rab4 and Sorting Nexins but not late endosome/lysosome and Golgi. (A–F and M–R) Untransfected HeLa cells grown on coverslips were fixed and coincubated with affinity–purified polyclonal rabbit anti–EHD4 antibodies (A, D, M and P) and mouse antibodies directed at Lamp1 (B), GM130 (E), Sorting Nexin 1 (SNX1, N) and Sorting Nexin 2 (SNX2, Q) followed by the corresponding secondary antibodies. (G–L) HeLa cells were transiently transfected with either GFP–Rab4 (G–I) or GFP–Rab7 (J–L). After 24 h, cells were fixed and coincubated with rabbit anti–EHD4 antibodies (G and J) followed by the corresponding secondary antibodies. Insets (A–R) depict higher magnifications of the boxed areas, and arrows show vesicles positive for organelle markers with endogenous EHD4. Bars, 10 μm.
Acknowledgments
We thank Dr A. Sorkin (Univerity of Colorado) for generously providing the YFP–R5BD construct, Dr R. Lodge (Universite Laval) for kindly providing the GFP–Rab5 constructs and Dr E.C. Dell’Angelica (University of California Los Angeles) and the members of the Caplan laboratory for helpful discussions. Support for this research was provided by grants from the National Institutes of Health (1R01GM074876 to S. C. and 5P20 RR018759-04 fellowship to M. S.) and the Nebraska Department of Health (S. C.).
References
- 1.Pfeffer SR. Rab GTPases: specifying and deciphering organelle identity and function. Trends Cell Biol. 2001;11:487–491. doi: 10.1016/s0962-8924(01)02147-x. [DOI] [PubMed] [Google Scholar]
- 2.Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2:107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- 3.Grosshans BL, Ortiz D, Novick P. Rabs and their effectors: achieving specificity in membrane traffic. Proc Natl Acad Sci U S A. 2006;103:11821–11827. doi: 10.1073/pnas.0601617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gorvel JP, Chavrier P, Zerial M, Gruenberg J. Rab5 controls early endosome fusion in vitro. Cell. 1991;64:915–925. doi: 10.1016/0092-8674(91)90316-q. [DOI] [PubMed] [Google Scholar]
- 5.Stenmark H, Valencia A, Martinez O, Ullrich O, Goud B, Zerial M. Distinct structural elements of rab5 define its functional specificity. EMBO J. 1994;13:575–583. doi: 10.1002/j.1460-2075.1994.tb06295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daro E, van der Sluijs P, Galli T, Mellman I. Rab4 and cellubrevin define different early endosome populations on the pathway of transferrin receptor recycling. Proc Natl Acad Sci U S A. 1996;93:9559–9564. doi: 10.1073/pnas.93.18.9559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheff DR, Daro EA, Hull M, Mellman I. The receptor recycling pathway contains two distinct populations of early endosomes with different sorting functions. J Cell Biol. 1999;145:123–139. doi: 10.1083/jcb.145.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ren M, Xu G, Zeng J, De Lemos-Chiarandini C, Adesnik M, Sabatini DD. Hydrolysis of GTP on rab11 is required for the direct delivery of transferrin from the pericentriolar recycling compartment to the cell surface but not from sorting endosomes. Proc Natl Acad Sci U S A. 1998;95:6187–6192. doi: 10.1073/pnas.95.11.6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casanova JE, Wang X, Kumar R, Bhartur SG, Navarre J, Woodrum JE, Altschuler Y, Ray GS, Goldenring JR. Association of Rab25 and Rab11a with the apical recycling system of polarized Madin-Darby canine kidney cells. Mol Biol Cell. 1999;10:47–61. doi: 10.1091/mbc.10.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simpson JC, Griffiths G, Wessling-Resnick M, Fransen JA, Bennett H, Jones AT. A role for the small GTPase Rab21 in the early endocytic pathway. J Cell Sci. 2004;117:6297–6311. doi: 10.1242/jcs.01560. [DOI] [PubMed] [Google Scholar]
- 11.Pellinen T, Arjonen A, Vuoriluoto K, Kallio K, Fransen JA, Ivaska J. Small GTPase Rab21 regulates cell adhesion and controls endosomal traffic of {beta}1-integrins. J Cell Biol. 2006;173:767–780. doi: 10.1083/jcb.200509019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weigert R, Donaldson JG. Fluorescent microscopy-based assays to study the role of Rab22a in clathrin-independent endocytosis. Methods Enzymol. 2005;403:243–253. doi: 10.1016/S0076-6879(05)03020-X. [DOI] [PubMed] [Google Scholar]
- 13.Mesa R, Salomon C, Roggero M, Stahl PD, Mayorga LS. Rab22a affects the morphology and function of the endocytic pathway. J Cell Sci. 2001;114:4041–4049. doi: 10.1242/jcs.114.22.4041. [DOI] [PubMed] [Google Scholar]
- 14.Kauppi M, Simonsen A, Bremnes B, Vieira A, Callaghan J, Stenmark H, Olkkonen VM. The small GTPase Rab22 interacts with EEA1 and controls endosomal membrane trafficking. J Cell Sci. 2002;115:899–911. doi: 10.1242/jcs.115.5.899. [DOI] [PubMed] [Google Scholar]
- 15.Babbey CM, Ahktar N, Wang E, Chen CC, Grant BD, Dunn KW. Rab10 regulates membrane transport through early endosomes of polarized Madin-Darby canine kidney cells. Mol Biol Cell. 2006;17:3156–3175. doi: 10.1091/mbc.E05-08-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schuck S, Gerl MJ, Ang A, Manninen A, Keller P, Mellman I, Simons K. Rab10 is involved in basolateral transport in polarized Madin-Darby canine kidney cells. Traffic. 2007;8:47–60. doi: 10.1111/j.1600-0854.2006.00506.x. [DOI] [PubMed] [Google Scholar]
- 17.Chen CC, Schweinsberg PJ, Vashist S, Mareiniss DP, Lambie EJ, Grant BD. RAB-10 is required for endocytic recycling in the Caenorhabditis elegans intestine. Mol Biol Cell. 2006;17:1286–1297. doi: 10.1091/mbc.E05-08-0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Novick P, Medkova M, Dong G, Hutagalung A, Reinisch K, Grosshans B. Interactions between Rabs, tethers, SNAREs and their regulators in exocytosis. Biochem Soc Trans. 2006;34:683–686. doi: 10.1042/BST0340683. [DOI] [PubMed] [Google Scholar]
- 19.Naslavsky N, Caplan S. C-terminal EH-domain-containing proteins: consensus for a role in endocytic trafficking, EH? J Cell Sci. 2005;118:4093–4101. doi: 10.1242/jcs.02595. [DOI] [PubMed] [Google Scholar]
- 20.Mintz L, Galperin E, Pasmanik-Chor M, Tulzinsky S, Bromberg Y, Kozak CA, Joyner A, Fein A, Horowitz M. EHD1 – an EH-domain-containing protein with a specific expression pattern. Genomics. 1999;59:66–76. doi: 10.1006/geno.1999.5800. [DOI] [PubMed] [Google Scholar]
- 21.Pohl U, Smith JS, Tachibana I, Ueki K, Lee HK, Ramaswamy S, Wu Q, Mohrenweiser HW, Jenkins RB, Louis DN. EHD2, EHD3, and EHD4 encode novel members of a highly conserved family of EH domain-containing proteins. Genomics. 2000;63:255–262. doi: 10.1006/geno.1999.6087. [DOI] [PubMed] [Google Scholar]
- 22.Grant B, Zhang Y, Paupard MC, Lin SX, Hall DH, Hirsh D. Evidence that RME-1, a conserved C. elegans EH-domain protein, functions in endocytic recycling. Nat Cell Biol. 2001;3:573–579. doi: 10.1038/35078549. [DOI] [PubMed] [Google Scholar]
- 23.Lin SX, Grant B, Hirsh D, Maxfield FR. Rme-1 regulates the distribution and function of the endocytic recycling compartment in mammalian cells. Nat Cell Biol. 2001;3:567–572. doi: 10.1038/35078543. [DOI] [PubMed] [Google Scholar]
- 24.Lee DW, Zhao X, Scarselletta S, Schweinsberg PJ, Eisenberg E, Grant BD, Greene LE. ATP binding regulates oligomerization and endosome association of RME-1 family proteins. J Biol Chem. 2005;280:280–290. doi: 10.1074/jbc.M412751200. [DOI] [PubMed] [Google Scholar]
- 25.Naslavsky N, Rahajeng J, Sharma M, Jovic M, Caplan S. Interactions between EHD proteins and Rab11-FIP2: a role for EHD3 in early endosomal transport. Mol Biol Cell. 2006;17:163–177. doi: 10.1091/mbc.E05-05-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daumke O, Lundmark R, Vallis Y, Martens S, Butler PJ, McMahon HT. Architectural and mechanistic insights into an EHD ATPase involved in membrane remodelling. Nature. 2007;449:923–927. doi: 10.1038/nature06173. [DOI] [PubMed] [Google Scholar]
- 27.Kieken F, Jovic M, Naslavsky N, Caplan S, Sorgen PL. EH domain of EHD1. J Biomol NMR. 2007;39:232–329. doi: 10.1007/s10858-007-9196-0. [DOI] [PubMed] [Google Scholar]
- 28.Naslavsky N, Boehm M, Backlund PS, Jr, Caplan S. Rabenosyn-5 and EHD1 interact and sequentially regulate protein recycling to the plasma membrane. Mol Biol Cell. 2004;15:2410–2422. doi: 10.1091/mbc.E03-10-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rapaport D, Auerbach W, Naslavsky N, Pasmanik-Chor M, Galperin E, Fein A, Caplan S, Joyner AL, Horowitz M. Recycling to the plasma membrane is delayed in EHD1 knockout mice. Traffic. 2006;7:52–60. doi: 10.1111/j.1600-0854.2005.00359.x. [DOI] [PubMed] [Google Scholar]
- 30.Caplan S, Naslavsky N, Hartnell LM, Lodge R, Polishchuk RS, Donaldson JG, Bonifacino JS. A tubular EHD1-containing compartment involved in the recycling of major histocompatibility complex class I molecules to the plasma membrane. EMBO J. 2002;21:2557–2567. doi: 10.1093/emboj/21.11.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Picciano JA, Ameen N, Grant BD, Bradbury NA. Rme-1 regulates the recycling of the cystic fibrosis transmembrane conductance regulator. Am J Physiol Cell Physiol. 2003;285:C1009–C1018. doi: 10.1152/ajpcell.00140.2003. [DOI] [PubMed] [Google Scholar]
- 32.Guilherme A, Soriano NA, Furcinitti PS, Czech MP. Role of EHD1 and EHBP1 in perinuclear sorting and insulin-regulated GLUT4 recycling in 3T3-L1 adipocytes. J Biol Chem. 2004;279:40062–40075. doi: 10.1074/jbc.M401918200. [DOI] [PubMed] [Google Scholar]
- 33.Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- 34.Jovic M, Naslavsky N, Rapaport D, Horowitz M, Caplan S. EHD1 regulates beta1 integrin endosomal transport: effects on focal adhesions, cell spreading and migration. J Cell Sci. 2007;120:802–814. doi: 10.1242/jcs.03383. [DOI] [PubMed] [Google Scholar]
- 35.Hao M, Lin SX, Karylowski OJ, Wustner D, McGraw TE, Maxfield FR. Vesicular and non-vesicular sterol transport in living cells. The endocytic recycling compartment is a major sterol storage organelle. J Biol Chem. 2002;277:609–617. doi: 10.1074/jbc.M108861200. [DOI] [PubMed] [Google Scholar]
- 36.Naslavsky N, Rahajeng J, Rapaport D, Horowitz M, Caplan S. EHD1 regulates cholesterol homeostasis and lipid droplet storage. Biochem Biophys Res Commun. 2007;357:792–799. doi: 10.1016/j.bbrc.2007.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gokool S, Tattersall D, Seaman MN. EHD1 interacts with retromer to stabilise SNX1-tubules and facilitate endosome-to-Golgi retrieval. Traffic. 2007;8:1873–1886. doi: 10.1111/j.1600-0854.2007.00652.x. [DOI] [PubMed] [Google Scholar]
- 38.Rotem-Yehudar R, Galperin E, Horowitz M. Association of insulin-like growth factor 1 receptor with EHD1 and SNAP29. J Biol Chem. 2001;276:33054–33060. doi: 10.1074/jbc.M009913200. [DOI] [PubMed] [Google Scholar]
- 39.Braun A, Pinyol R, Dahlhaus R, Koch D, Fonarev P, Grant BD, Kessels MM, Qualmann B. EHD proteins associate with syndapin I and II and such interactions play a crucial role in endosomal recycling. Mol Biol Cell. 2005;16:3642–3658. doi: 10.1091/mbc.E05-01-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu Y, Shi H, Wei S, Wong SH, Hong W. Mutually exclusive interactions of EHD1 with GS32 and syndapin II. Mol Membr Biol. 2004;21:269–277. doi: 10.1080/09687680410001716871. [DOI] [PubMed] [Google Scholar]
- 41.Shi A, Pant S, Balklava Z, Chen CC, Figueroa V, Grant BD. A novel requirement for C. elegans Alix/ALX-1 in RME-1-mediated membrane transport. Curr Biol. 2007;17:1913–1924. doi: 10.1016/j.cub.2007.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galperin E, Benjamin S, Rapaport D, Rotem-Yehudar R, Tolchinsky S, Horowitz M. EHD3: a protein that resides in recycling tubular and vesicular membrane structures and interacts with EHD1. Traffic. 2002;3:575–589. doi: 10.1034/j.1600-0854.2002.30807.x. [DOI] [PubMed] [Google Scholar]
- 43.Park SY, Ha BG, Choi GH, Ryu J, Kim B, Jung CY, Lee W. EHD2 interacts with the insulin-responsive glucose transporter (GLUT4) in rat adipocytes and may participate in insulin-induced GLUT4 recruitment. Biochemistry. 2004;43:7552–7562. doi: 10.1021/bi049970f. [DOI] [PubMed] [Google Scholar]
- 44.Guilherme A, Soriano NA, Bose S, Holik J, Bose A, Pomerleau DP, Furcinitti P, Leszyk J, Corvera S, Czech MP. EHD2 and the novel EH domain binding protein EHBP1 couple endocytosis to the actin cytoskeleton. J Biol Chem. 2004;279:10593–10605. doi: 10.1074/jbc.M307702200. [DOI] [PubMed] [Google Scholar]
- 45.Blume JJ, Halbach A, Behrendt D, Paulsson M, Plomann M. EHD proteins are associated with tubular and vesicular compartments and interact with specific phospholipids. Exp Cell Res. 2007;313:219–231. doi: 10.1016/j.yexcr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 46.George M, Ying G, Rainey MA, Solomon A, Parikh PT, Gao Q, Band V, Band H. Shared as well as distinct roles of EHD proteins revealed by biochemical and functional comparisons in mammalian cells and C. elegans. BMC Cell Biol. 2007;8:3. doi: 10.1186/1471-2121-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuo HJ, Tran NT, Clary SA, Morris NP, Glanville RW. Characterization of EHD4, an EH domain-containing protein expressed in the extracellular matrix. J Biol Chem. 2001;276:43103–43110. doi: 10.1074/jbc.M106128200. [DOI] [PubMed] [Google Scholar]
- 48.Shao Y, Akmentin W, Toledo-Aral JJ, Rosenbaum J, Valdez G, Cabot JB, Hilbush BS, Halegoua S. Pincher, a pinocytic chaperone for nerve growth factor/TrkA signaling endosomes. J Cell Biol. 2002;157:679–691. doi: 10.1083/jcb.200201063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith CA, Dho SE, Donaldson J, Tepass U, McGlade CJ. The cell fate determinant numb interacts with EHD/Rme-1 family proteins and has a role in endocytic recycling. Mol Biol Cell. 2004;15:3698–3708. doi: 10.1091/mbc.E04-01-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bucci C, Parton RG, Mather IH, Stunnenberg H, Simons K, Hoflack B, Zerial M. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell. 1992;70:715–728. doi: 10.1016/0092-8674(92)90306-w. [DOI] [PubMed] [Google Scholar]
- 51.Stenmark H, Parton RG, Steele-Mortimer O, Lutcke A, Gruenberg J, Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J. 1994;13:1287–1296. doi: 10.1002/j.1460-2075.1994.tb06381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galperin E, Sorkin A. Visualization of Rab5 activity in living cells by FRET microscopy and influence of plasma-membrane-targeted Rab5 on clathrin-dependent endocytosis. J Cell Sci. 2003;116:4799–4810. doi: 10.1242/jcs.00801. [DOI] [PubMed] [Google Scholar]
- 53.Gaullier JM, Simonsen A, D’Arrigo A, Bremnes B, Stenmark H, Aasland R. FYVE fingers bind PtdIns(3)P. Nature. 1998;394:432–433. doi: 10.1038/28767. [DOI] [PubMed] [Google Scholar]
- 54.Corvera S, D’Arrigo A, Stenmark H. Phosphoinositides in membrane traffic. Curr Opin Cell Biol. 1999;11:460–465. doi: 10.1016/S0955-0674(99)80066-0. [DOI] [PubMed] [Google Scholar]
- 55.Caplan S, Hartnell LM, Aguilar RC, Naslavsky N, Bonifacino JS. Human Vam6p promotes lysosome clustering and fusion in vivo. J Cell Biol. 2001;154:109–122. doi: 10.1083/jcb.200102142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 57.Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci. 1992;103:857–862. doi: 10.1242/jcs.103.3.857. [DOI] [PubMed] [Google Scholar]
- 58.Naslavsky N, Rahajeng J, Chenavas S, Sorgen PL, Caplan S. EHD1 and Eps15 interact with phosphatidylinositols via their Eps15 homology domains. J Biol Chem. 2007;282:16612–16622. doi: 10.1074/jbc.M609493200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Partial colocalization of endogenous EHD4 with Rab4 and Sorting Nexins but not late endosome/lysosome and Golgi. (A–F and M–R) Untransfected HeLa cells grown on coverslips were fixed and coincubated with affinity–purified polyclonal rabbit anti–EHD4 antibodies (A, D, M and P) and mouse antibodies directed at Lamp1 (B), GM130 (E), Sorting Nexin 1 (SNX1, N) and Sorting Nexin 2 (SNX2, Q) followed by the corresponding secondary antibodies. (G–L) HeLa cells were transiently transfected with either GFP–Rab4 (G–I) or GFP–Rab7 (J–L). After 24 h, cells were fixed and coincubated with rabbit anti–EHD4 antibodies (G and J) followed by the corresponding secondary antibodies. Insets (A–R) depict higher magnifications of the boxed areas, and arrows show vesicles positive for organelle markers with endogenous EHD4. Bars, 10 μm.