

Abstract
Inflammation in general and proteinases generated as a result are likely mediators of early secondary pathogenesis after spinal cord injury. We report that matrix metalloproteinase-9 (MMP-9) plays an important role in blood–spinal cord barrier dysfunction, inflammation, and locomotor recovery. MMP-9 was present in the meninges and neurons of the uninjured cord. MMP-9 increased rapidly after a moderate contusion spinal cord injury, reaching a maximum at 24 hr, becoming markedly reduced by 72 hr, and not detectable at 7 d after injury. It was seen in glia, macrophages, neutrophils, and vascular elements in the injured spinal cord at 24 hr after injury. The natural tissue inhibitors of MMPs were unchanged over this time course. MMP-9-null mice exhibited significantly less disruption of the blood–spinal cord barrier, attenuation of neutrophil infiltration, and significant locomotor recovery compared with wild-type mice. Similar findings were observed in mice treated with a hydroxamic acid MMP inhibitor from 3 hr to 3 d after injury, compared with the vehicle controls. Moreover, the area of residual white matter at the lesion epicenter was significantly greater in the inhibitor-treated group. This study provides evidence that MMP-9 plays a key role in abnormal vascular permeability and inflammation within the first 3 d after spinal cord injury, and that blockade of MMPs during this critical period attenuates these vascular events and leads to improved locomotor recovery. Our findings suggest that early inhibition of MMPs may be an efficacious strategy for the spinal cord-injured patient.
Keywords: blood–spinal cord barrier, inflammation, locomotor recovery, matrix metalloproteinase-9, proteinases, spinal cord injury
Each year, there are ∼10,000 spinal cord injuries that result in permanent disabilities. There is considerable evidence that functional recovery after spinal cord injury is not simply a consequence of the initial mechanical destruction of tissue but is also attributed to the evolution of complex secondary events that contribute to early as well as delayed cell injury. Proteinases and, in particular, matrix metalloproteinases (MMPs) are likely mediators of early secondary vascular pathogenesis after spinal cord injury.
MMPs are a family of extracellular zinc- and calcium-dependent endopeptidases (Birkedal-Hansen et al., 1993) that degrade the extracellular matrix and other extracellular proteins (Sternlicht, 1999, 2001). MMPs are essential for remodeling of the extracellular matrix, tissue morphogenesis, and wound healing (Werb, 1997). However, excessive proteolytic activity of MMPs can be detrimental, leading to numerous pathologic conditions, including disruption of the blood–brain barrier (Rosenberg et al., 1994, 1995, 1998; Rosenberg and Navratil, 1997; Mun-Bryce and Rosenberg, 1998b; Yong et al., 2001) and inflammation (Mun-Bryce and Rosenberg, 1998b). MMP-9 degrades gelatin (denatured collagens); collagen IV, V, and XI; elastin; vitronectin; myelin basic protein; and other substrates (Vu and Werb, 1998). This protease is predominantly expressed by inflammatory cells, including macrophages, lymphocytes, and neutrophils, as well as endothelial cells (Mainardi et al., 1984; Hibbs et al., 1987; Murphy et al., 1989;Wilhelm et al., 1989). Recent studies suggest that MMP-9 inactivates α1-antitrypsin, the primary physiologic inhibitor of leukocyte elastase, a step that is central to leukocyte migration (Liu et al., 1998). In the CNS, there is a low constitutive expression of MMP-9 in microglia, astrocytes, and hippocampal neurons, and it can be induced in astrocytes, microglia/macrophages, and hippocampal cells (Backstrom et al., 1996; Cuzner et al., 1996; Gottschall and Deb, 1996; Liu et al., 1998; Yong et al., 2001).
We have focused on the role of MMP-9 because of its established link to disruption of the blood–brain barrier, inflammation, and tissue injury. MMP-9 has been implicated in abnormal vascular permeability (Rosenberg et al., 1994, 1995; Mun-Bryce and Rosenberg, 1998b) associated with either hemorrhagic injury (Rosenberg et al., 1994) or inflammation (Mun-Bryce and Rosenberg, 1998b). Thus, abnormal increases in MMP-9 in both inflammatory cells and endothelial cells may collectively impair barrier function by degrading the vascular basement membrane. There is also evidence that MMP-9 increases in ischemic brain injury (Rosenberg et al., 1996b; Romanic et al., 1998), and that administration of a monoclonal antibody to MMP-9 reduces the hemispheric infarct size (Romanic et al., 1998). Most recently, it has been shown that methylprednisolone, the only therapeutic agent approved by the Food and Drug Administration, suppresses the expression of MMP-9 after spinal cord injury (Xu et al., 2001). Together, these data serve as a basis for our hypothesis that MMP-9 contributes to disruption of the blood–spinal cord barrier after spinal cord injury, and that modulation of MMP-9 after spinal cord injury stabilizes the barrier, limits inflammation, and promotes locomotor recovery.
MATERIALS AND METHODS
Generation of experimental models
Surgical procedures. All procedures were performed according to protocols approved by the University of California Committee on Research (San Francisco, CA). MMP-9-null and wild-type littermates were generated as described previously (Vu et al., 1998) and bred on an FVBn background. The wild-type mice were obtained from the negative littermates of the back-crosses into the FVBn background. The MMP-9-null mouse has a mild developmental delay in bone formation (Vu et al., 1998). However, by 6 weeks of age, these animals have an axial skeleton indistinguishable from the wild-type mice. These mice have a normal life span, and there are no phenotypic differences between the MMP-9-null and wild-type mice. All studies described below were conducted in a blinded manner.
Adult, male mice (4–6 months of age) were anesthetized with 2.5% Avertin (0.02 ml/gm body weight, i.p.) and maintained at 37°C throughout the experiment by using a warming blanket placed under the animal. A contusive injury was performed based on modifications of procedures originally described by Kuhn and Wrathall (1998). Briefly, using aseptic techniques, the spinous process and laminas of T8 were removed, and a circular region of dura, ∼2.4 mm in diameter, was exposed. After stabilization of the vertebral column, a 2 gm weight was dropped 5 cm onto the exposed dura. After injury, the overlying skin was closed with wound clips. Postoperative care included manual expression of each animal's bladder until recovery of reflex emptying.
Inhibitor studies. Wild-type mice were subjected to spinal cord injury as described in the previous section. All mice were treated with either GM6001 (AMS Scientific, Inc., Concord, CA; 100 mg/kg in 4% methylcellulose, i.p.), a general inhibitor of MMPs, or vehicle (4% methylcellulose, i.p.) at 3 hr after injury. Animals were treated every 12 hr (100 mg/kg in 4% methylcellulose, i.p.) for the first 3 d after injury.
Zymography
Gelatin zymography. Samples of spinal cord, prepared from the epicenter, were quick-frozen at −80°C. Each sample was weighed and homogenized (1:4 w/v) in lysis buffer containing 50 mm Tris-HCl, pH 8.0, 150 mmNaCl, 1% NP-40, 0.5% deoxycholate, and 0.1% SDS. Soluble and insoluble extracts were separated by centrifugation and stored at −20°C. Equal amounts of the supernatant were analyzed by gel zymography as described previously (Herron et al., 1986) on 10% SDS-polyacrylamide gels, copolymerized with substrate (1 mg/ml gelatin). The proteins were renatured by incubation in 2.5% Triton X-100 and then incubated in substrate buffer (50 mm Tris-HCl, pH 8.5, 5 mmCaCl2) for 24–36 hr at 37°C to enable the MMP-9 and other gelatinases to cleave the gelatin. After rinsing in water, each gel was stained with Coomassie blue for 4 hr and destained in 50% methanol. Negative staining is indicative of the location of active protease bands. After exposure to SDS during gel separation, proenzymes, present in tissue extracts, are activated without proteolytic cleavage. To inhibit MMP proteolytic activities, substrate gels were incubated in substrate buffer with 4 mm1,10-phenanthroline (Sigma, St. Louis, MO) as described previously (Adler et al., 1990). This control ensured that the measured activity corresponded to matrix metalloproteinase activity. The identities of MMPs were based on their molecular weights.
Reverse zymography. Reverse zymography was used to identify physiologic inhibitors, the tissue inhibitors of metalloproteinases (TIMP-1 and TIMP-2). The gel was prepared as described above, with the exception that gelatinases were also added to the SDS-gelatin gel. The gelatinases degrade the gel except in those regions in which there is inhibitory activity. As a result, TIMP-1 activity is visualized in Coomassie blue-stained and destained gels as dark blue bands.
In situ zymography. In situ zymography was used to detect and localize enzyme activity in tissue sections (Oh et al., 1999). The uninjured and injured (24 hr after injury) spinal cords were quickly removed without fixation and frozen at −80°C. Sections (16 μm) were cut on a cryostat and incubated in 0.05m Tris-HCl, 0.15 m NaCl, 5 mm CaCl2, and 0.2 mm NaN3, pH 7.6, containing 40 μg of FITC-labeled gelatin (Molecular Probes, Eugene, OR), at 37°C for 1 hr. The gelatin with a fluorescent tag remains caged (does not fluoresce) until the gelatin is cleaved by gelatinase activity, such as MMP-9 (or MMP-2). This method detects regionally specific gelatinolytic activity but does not distinguish between gelatinases. Reaction product was visualized by fluorescence microscopy.
Immunocytochemistry and histochemistry
Immunocytochemistry. At a designated time point, animals were deeply anesthetized and perfused with 4% paraformaldehyde in 0.1 m PBS, pH 7.4. The spinal cord was removed, rinsed in PBS, either prepared for embedding in paraffin or cryoprotected in sucrose (20% in PBS), and frozen. For paraffin embedding, tissue was dehydrated through graded alcohols and xylene and embedded in paraffin. Sections, 5–10 μm in thickness, were cut using a Leica (Deerfield, IL) 2135 microtome and deparaffinized. Frozen sections (10–15 μm) were cut on a cryostat.
Conventional immunocytochemistry was performed on either deparaffinized or cryostat sections. A 1:200 dilution was used for rabbit anti-mouse MMP-9 (Behrendtsen et al., 1992), a 1:500–1000 dilution was used for porcine anti-mouse glial fibrillary acidic protein (Sigma), a 1:200 dilution was used for rabbit anti-mouse platelet endothelial cell adhesion molecule-1 (PECAM-1; BD-PharMingen, San Diego, CA), and a 1:5 dilution was used for the macrophage-specific rat anti-mouse F4/80 (a gift from S. Gordon, University of Oxford, Oxford, UK) (Austyn and Gordon, 1981). These antibodies were prepared in blocking solution consisting of 1% sheep serum and, unless otherwise specified, blocking agents and secondary antibodies were provided by the Tyramide Signal Amplification Direct and Indirect Kits (Molecular Probes), according to the manufacturer's instructions. Incubation of primary antibodies occurred overnight at 4°C, with the exception of anti-GFAP, which was incubated for 30 min at room temperature. Secondary antibodies were used at a 1:500 dilution. Final detection of the signal used either a fluorescent or a biotinylated tyramide derivative and visualized using a peroxidase substrate. The anti-GFAP was conjugated to Cy3 (Sigma) and directly visualized. Immunocytochemical controls consisted of omission of the primary antibody. All images were digitally captured on a Leica microscope equipped with a CCD camera (SPOT software, model 1.3.0; Diagnostic Instruments, Inc., Sterling Heights, MI) and imaged using PhotoShop 6.0 (Adobe Systems, San Jose, CA).
Histochemistry. Neutrophils were identified in the injured cord at 42 d after injury by means of a chloroacetate esterase stain (naphthol AS-D chloroacetate esterase kit; Sigma). The protocol was as described by the manufacturer with several exceptions. All cords were fixed by intravascular perfusion with 4% paraformaldehyde. These fixed sections were then incubated for 20 min in the esterase stain. Serial sections, 14 μm in thickness, were obtained from a 1 cm length of cord, centered over the region of maximal damage. Every fourth section was mounted on slides for evaluation. One section, exhibiting maximal neutrophil infiltration, was selected from a 1 mm length of cord, centered over the impact site. In addition, one section 0.8 mm rostral to the lesion epicenter and another section 0.8 mm caudal to the lesion epicenter were selected. The number of neutrophils within each of these sections was determined from digital images, captured with a CCD camera at 40× magnification, as described above. Contrast for each image was optimized using Adobe PhotoShop 6.0. Only darkly stained structures, >38 μm2 (7 μm in diameter), were counted. For the rostral and caudal sections, the numbers of neutrophils within the entire cross section were determined and summed. For the epicenter, neutrophils were quantified within specific regions of the cross section. Those neutrophils were counted if they resided in rectangular boxes, centrally positioned in the right and left dorsal horns (231 × 126 μm2), dorsal columns (173 × 96 μm2), right and left lateral white matter (2211 × 384 μm2), pericentral gray matter (369 × 173 μm2), and right and left ventral white matter (373 × 164 μm2). Values were expressed at a ratio of the summed number of neutrophils in each of the rostral and caudal segments divided by the number of neutrophils at the epicenter.
The area of residual white matter at the epicenter was determined in vehicle- and drug-treated wild-type mice at 42 d after injury. We selected residual white matter for analysis because we have shown previously that it is the best single measurement for characterizing the degree of injury in the contused spinal cord and is predictive of motor recovery (Noble and Wrathall, 1985). Serial cross sections from the lesion epicenter, prepared from animals that had been killed at 42 d after injury, were stained for myelin using Luxol fast blue. Stained cross sections, prepared from a 5 mm segment centered over the impact site, were selected for analysis. The section that exhibited the greatest loss of white matter, as demonstrated by Luxol fast blue, was selected for subsequent analysis. These sections were captured with a Spotcam camera mounted on a Nikon (Tokyo, Japan) Optiphot and analyzed using PhotoShop 6.0. In each captured image, a histogram was generated that resulted in two distinct peaks, corresponding to areas of Luxol fast blue staining and background staining. Each image was then adjusted such that only pixels related to Luxol fast blue were visible. These pixels were quantified and expressed per unit area.
Barrier permeability studies
Permeability to horseradish peroxidase. Barrier permeability to horseradish peroxidase (HRP) was evaluated in spinal cord-injured wild-type and MMP-9-null mice and mice treated with either GM6001 or vehicle. Mice were anesthetized and administered HRP (type II; 75 mg/kg in 0.4 ml of 0.9% saline, i.p.) 10 min before killing at 24 hr after injury. The mice were perfused with fixative, as described for the immunocytochemistry. After removal of the spinal cord, the tissue was rinsed in buffer, cryoprotected, and frozen. A 1 cm length of cord, centered over the impact site, was selected for serial sectioning. Cross sections, 20 μm in thickness, were cut using a cryostat. Sections were dehydrated in graded alcohols, cleared, and mounted on slides. Reaction product was visualized using 3,3-diaminobenzidine tetrachloride as the chromogen. Every 50th section was selected for semiquantitative analysis. Each cross section was subdivided into 11 regions, corresponding to the dorsal columns, pericentral gray matter and ventral white matter, and right and left regions of each of the following: (1) pericentral lateral white matter, (2) peripheral lateral white matter, (3) dorsal horns, and (4) ventral horns. The extent of extravasation was evaluated on a three point, graded scale as follows: 1, light staining that is limited to discrete patches; 2, light staining throughout the region and/or darkly stained patches; and 3, dark staining throughout the region. A maximal score for any given section was 33.
Permeability to luciferase. MMP-9-null and wild-type mice and mice treated with either vehicle or GM6001 were reanesthetized at 24 hr after injury and injected intravenously with a 1:1 solution consisting of recombinant luciferase (“Quantilum,” 1 mg/ml in luciferase storage buffer; Promega, Madison, WI) and PBS/BSA (2.7 mm potassium chloride, 1.5 mm potassium phosphate, 8.1 mm sodium phosphate, 0.8% sodium chloride, and 0.001% bovine serum albumin). Animals were then placed on a warming blanket to maintain body temperature. Each animal was killed 30 min after injection of luciferase, and the spinal cord was quickly removed. A 3 mm length of cord, centered over the impact site, was homogenized in a 1:50 dilution by weight of 1× cell lysis buffer (25 mm Tris, pH 7.8, 2 mmtrans-1,2-diaminocyclohexaneN,N,N′,N′-tetra-acetate monohydrate, 2 mm dithiothreitol, 10% glycerol, and 1% Triton X-100). Lysates were centrifuged (12,000 rpm for 8 min) to remove cellular debris, and the supernatants were brought to a final dilution of 1:5000. Enzyme activities, measured in 10 μl aliquots of the dilution, were based on luminescence using a luciferase assay kit (Promega) and a luminometer (TD-20/20; Turner Designs, Sunnyvale, CA) with a 10 sec measuring time. Values were expressed as the ratio of activity within the injured segment relative to an internal control (cervical segment).
Functional assessment
Locomotor recovery was assessed using an open-field testing paradigm, the Basso–Beattie–Bresnahan (BBB) Locomotor Rating Scale, which is based on a 21 point scale originally developed in the spinal cord-injured rat (Basso et al., 1995). This scale assesses 10 distinct categories that range from limb movement to tail position and involve detailed observations of joint movement, stepping, and coordination. Uninjured animals exhibit a locomotor score of 21, whereas animals that exhibit complete hindlimb paralysis are scored as a 0. Animals that are moderately injured typically show recovery over time and exhibit a locomotor score of between 10 and 11 by ∼6 weeks after injury (Basso et al., 1995, 1996). Spinal cord-injured animals were tested on days 1 and 3 after injury and weekly thereafter for 6 weeks. Each animal was tested within an enclosed arena of clear acrylic (53 × 108 × 5.5 cm) that was supported over a mirror. Positioning of the limbs and locomotion were then observed by either directly or indirectly (via the mirror) viewing the animal.
Quantitative analysis
The mean values for luciferase permeability, neutrophil infiltration, locomotor recovery, and residual white matter for each control and experimental group are reported ± SDs. MMP-9-nulls and drug-treated groups were compared with their respective controls (wild types and vehicle-treated mice) using unpaired Student'st tests. Statistical significance was defined atp ≤ 0.05.
RESULTS
MMP-9 activity increases after spinal cord injury
We first analyzed MMP activity in the spinal cord in wild-type mice after injury by gelatin zymography (Fig.1A). Animals were subjected to contusive spinal cord injury and killed at 24 hr, 72 hr, and 1 week after injury. Samples were also taken from uninjured (laminectomized) wild-type mice and MMP-9-null mice 24 hr after injury. In all animals, a 4 mm segment of cord corresponding to the center of the injury was homogenized and subject to gelatin zymography. In the uninjured mouse, no gelatinase activity was detected. In the wild-type injured mouse, MMP-9 activity was strongest at 24 hr after injury, with bands corresponding to the MMP-9-active form, the inactive zymogen, and MMP-9/TIMP-1 complexes. This activity was reduced by 72 hr, and only the inactive zymogen was present by 1 week after injury. The inactive form of MMP-2 (gelatinase A) appeared in all injured samples. The MMP-9-null mouse did not have the MMP-9 bands, as would be expected, but did not show a compensatory increase in MMP-2. A general inhibitor of metalloproteinases, 1,10-phenanthroline, completely blocked the gelatinolytic activity, thus confirming that these bands were caused by metalloproteinases (Fig. 1A).
Fig. 1.
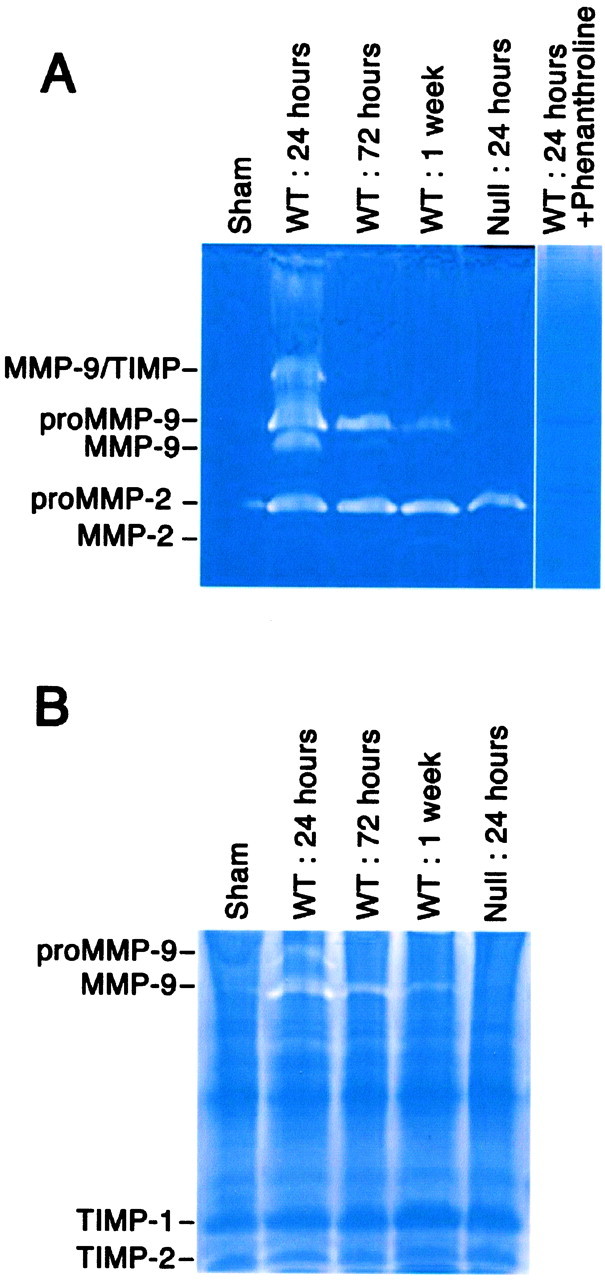
Time course of MMP-9 activity increases after spinal cord injury. A 3 mm length of spinal cord, centered over the impact site, was flash-frozen and homogenized. Soluble fractions were analyzed by gelatin zymography (A) or by reverse gelatin zymography (B). A, MMP-9 activity increases acutely after spinal cord injury and decreases by 1 week after injury. Note that the absence of MMP-9 activity in the null mouse does not result in a compensatory increase in MMP-2 activity in the injured spinal cord. 1,10-phenanthroline, a general inhibitor of metalloproteinases, completely blocks the inactive and active forms, thus confirming the specificity of these molecules. The positions of migration of active and zymogen forms of MMP-9 and MMP-2 and the MMP-9/TIMP-1 complexes determined from standards are marked.B, TIMP activity, seen by reverse zymography, is unchanged after spinal cord injury. The positions of migration of TIMP-1 and TMP-2 determined from standards are marked, as well as the migration of proMMP-9 and active MMP-9. WT, Wild type.
MMP-9 is present in uninjured meninges and motoneurons and in blood vessels, macrophages, and astrocytes in injured spinal cord
MMP-9 was localized in the meninges and in ventral horn motoneurons in the uninjured spinal cord of wild-type mice (Fig.2). There was no evidence for expression of MMP-9 in other neurons or in glia. At 24 hr after injury, there was a similar pattern of expression of MMP-9 in segments of spinal cord that were at least one segment removed from the injury (Fig. 2). However, at the epicenter (the region of maximal damage) and in the immediately adjacent tissue, there was pronounced induction of MMP-9 that was associated with blood vessels, as well as cells that were identified as macrophages and astrocytes by double immunolabeling (Figs. 2, 3).
Fig. 2.
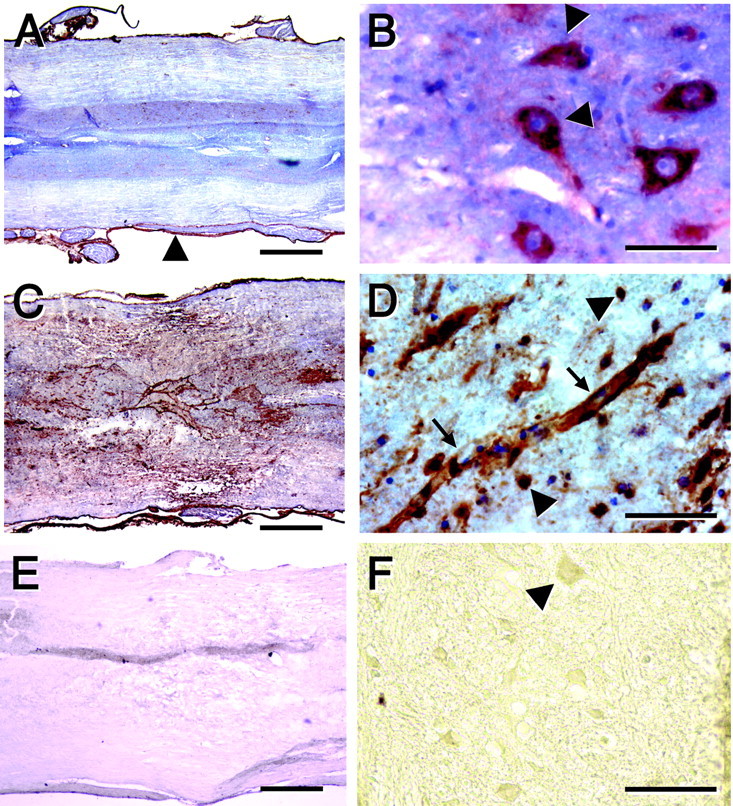
Immunolocalization of MMP-9 in the uninjured spinal cord and at 24 hr after injury. A, B, Uninjured spinal cord. C–F, Injured spinal cord.A–D, Anti-MMP-9 in the wild-type mouse. E, F, Anti-MMP-9 in the MMP-9-null mouse. MMP-9, visualized by HRP immunohistochemistry, is localized in meninges (A,arrowhead) and ventral horn motoneurons (B, arrowheads). After spinal cord injury there is prominent expression of MMP-9 at the lesioned epicenter (C). At higher magnification, MMP-9 is localized within vascular structures (D, arrows), as well as in round cells bearing no processes (D,arrowheads). There is no staining within the epicenter (E) or motoneurons in the adjacent penumbral zone (F, arrowhead) in the MMP-9-null animal. Scale bars: A, C, E, 500 μm; B, D, 50 μm; F, 100 μm.
Fig. 3.

Immunolocalization of MMP-9 at 24–72 hr after injury. Based on double immunofluorescence, MMP-9 (A, B, E) is localized in blood vessels (C, PECAM immunolocalization), macrophages (D, F4/80 immunolocalization), and astrocytes (F, glial fibrillary acidic protein immunolocalization). Controls had no immunofluorescence (data not shown). Scale bars: A–D, 50 μm; E, F, 100 μm.
In situ zymography of gelatinolytic activity shows increased activity after injury
According to in situ zymography on sections of uninjured spinal cord in the wild-type animals, gelatinolytic activity was primarily restricted to the meninges (Fig.4). The traumatized spinal cord at 24 hr after contusion injury exhibited several distinct changes in gelatinolytic activity. There was increased activity associated within the meninges bordering the impact site and abundant activity within the epicenter (Fig. 4). Gelatinase activity was associated with a variety of cell areas, including blood vessels (Fig. 4). It is noteworthy that gelatinase activity was detected in the spinal cord of the MMP-9-null animal after injury (Fig. 4). This is not surprising, because it is known that other members of the MMP family can contribute to gelatinolytic activity (Yong et al., 2001).
Fig. 4.
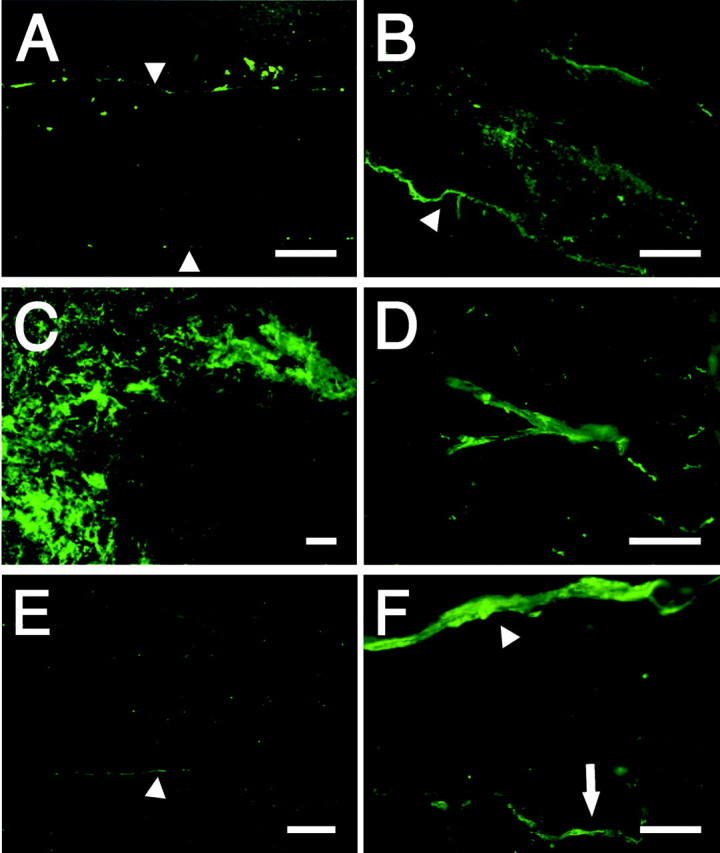
Localization of gelatinolytic activity in situ after spinal cord injury. Unfixed spinal cords from mice (uninjured or at 24 hr after injury) were frozen, and cryosections were prepared for in situ gelatin zymography as described in Materials and Methods. Fluorescence is indicative of gelatinolytic activity. In the uninjured wild-type spinal cord, small amounts of gelatinase activity are identified in the meninges (A, arrowheads). After spinal cord injury, gelatinase activity is prominent in the meninges (B, arrowhead) as well as within the epicenter (C). The gelatinase activity within the epicenter is localized at least in part to blood vessels (D). In the MMP-9-null, injured mouse (E, F), gelatinase activity is not as robust in the epicenter (E). Activity still appears in the meninges (E, F, arrowheads) and blood vessels (F, arrow). Scale bars: A, B, 500 μm; C, 100 μm; D, 50 μm.
Lack of MMPs blunts the blood–spinal cord barrier breakdown after spinal cord injury
We have shown previously that spinal cord injury results in prominent breakdown of the blood–spinal cord barrier to endogenous proteins, as well as to HRP (Noble and Wrathall, 1989a). To determine whether increased MMP-9 activity after spinal cord injury contributes to this abnormal permeability, we performed two types of experiments. In the first experiment, we compared the blood–spinal cord barrier to HRP 24 hr after spinal cord injury in wild-type mice with that for MMP-9-null mice and wild-type mice that were treated with the MMP inhibitor GM6001. The lesion epicenter in the wild-type and null mice was characterized by prominent intraparenchymal hemorrhage (Fig.5). The magnitude of hemorrhage in animals was quite variable. This finding is consistent with other studies, which have demonstrated that the extent of intraparenchymal hemorrhage does not correlate with injury severity after graded spinal cord contusion injuries, nor is it a good predictor of functional outcome (Noble and Wrathall, 1989b). Red blood cells appeared as dark brown, because of the peroxidatic-like activity of hemoglobin, and were distributed in a “spoke-like” pattern that radiated outward from the center of the cord. Importantly, there was a distinct difference in the pattern of barrier permeability to HRP in the wild-type mice compared with the MMP-9-null mice and mice treated with GM6001 (Figs.5, 6). HRP, which appeared as a diffuse, light brown reaction product, was maximally expressed in the lesion epicenter of the wild type. In contrast, there were only light patches of HRP reaction product in the epicenter of the MMP-9-null and drug-treated animals (Fig. 5). Although barrier permeability was most dramatic at the lesion epicenter in all animals, segments rostral and caudal to the lesion of the wild-type mice also exhibited abnormal permeability. In contrast, this axial distribution of abnormal barrier permeability was not observed in the MMP-9-null or GM6001-treated animals (Fig. 6).
Fig. 5.
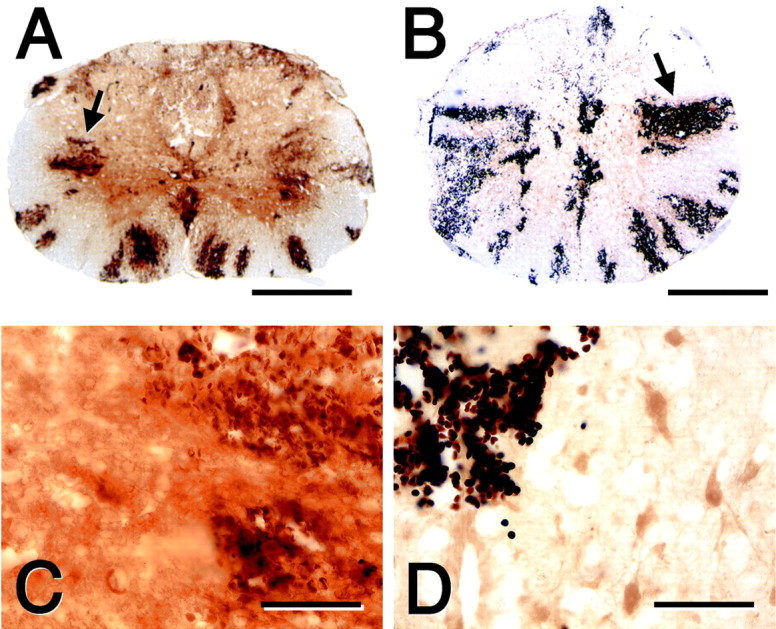
Blood–spinal cord barrier disruption to HRP at 24 hr after injury in the wild-type (A, C) and the MMP-9-null (B, D) mice. The lateral white matter is characterized by radial spokes of intraparenchymal hemorrhage (A, B, arrows). HRP, appearing as adark brown diffuse reaction product, is more pronounced in the wild-type (C) compared with the MMP-9-null (D) spinal cord. Scale bars: A, B, 500 μm; C, D, 100 μm.
Fig. 6.

Pattern of blood–spinal cord barrier disruption to HRP at 24 hr after injury in MMP-9 wild-type (WT) and null mice and in mice treated with GM6001. The relative intensity of staining for HRP, scaled from 1 to 3, was determined in 18 serial sections centered over the impact site. Within each cross section, 11 regions of the spinal cord, indicated in the schematic, were evaluated. The maximal score, indicative of intense HRP reactivity for any given section, was scored 33. The most pronounced staining for HRP occurred at the epicenter in all animals. There was a marked increase in permeability to HRP in the wild-type mice compared with either the MMP nulls or the wild-type mice treated with GM6001. DC, Dorsal column; DH, dorsal horn; L, peripheral lateral white matter;M, pericentral lateral white matter; V, ventral white matter; VH, ventral horn.
In the second type of experiment, barrier breakdown to the protein luciferase was quantified within the homogenates prepared from the lesion epicenter of spinal cord-injured MMP-9-null and wild-type mice and wild-type mice that were treated with either vehicle or GM6001 (Fig. 7). Similar to the anatomical studies described in the HRP study, abnormal barrier permeability was significantly attenuated in the MMP-9-null mice compared with the wild types and in the drug-treated compared with the vehicle-treated animals.
Fig. 7.

Effect of blocking MMPs on permeability to luciferase after spinal cord injury. Abnormal permeability to luciferase was quantified in the epicenter at 24 hr after injury in MMP-9-null (n = 6) and wild-type (WT) (n = 5) littermates and in mice treated with vehicle (n = 4) or GM6001 (begun at 3 hr after injury; n = 6). There is a significant reduction in barrier permeability in the MMP-9-null compared with the wild-type littermates (*p = 0.02) and in drug-treated compared with vehicle controls (*p = 0.04). Values are means ± SD.
Blocking MMPs decreases neutrophil infiltration
Acute inflammation is a normal response to injury. We found that 70–74% of all neutrophils within the lesion epicenter of the vehicle- and drug-treated animals resided in the white matter. Moreover, we observed that there were fewer neutrophils infiltrating within the lesion epicenter of MMP-9-null compared with the wild-type mice at 24 hr after injury (Fig. 8). When neutrophil infiltration was quantified in spinal cord-injured mice that were treated with either vehicle or the MMP inhibitor GM6001 (Fig. 8), we observed a significant reduction in the numbers of neutrophils in drug-treated compared with vehicle-treated animals. These data suggest that improved locomotor recovery in the drug-treated group may be attributable to decreased white matter damage by neutrophils.
Fig. 8.
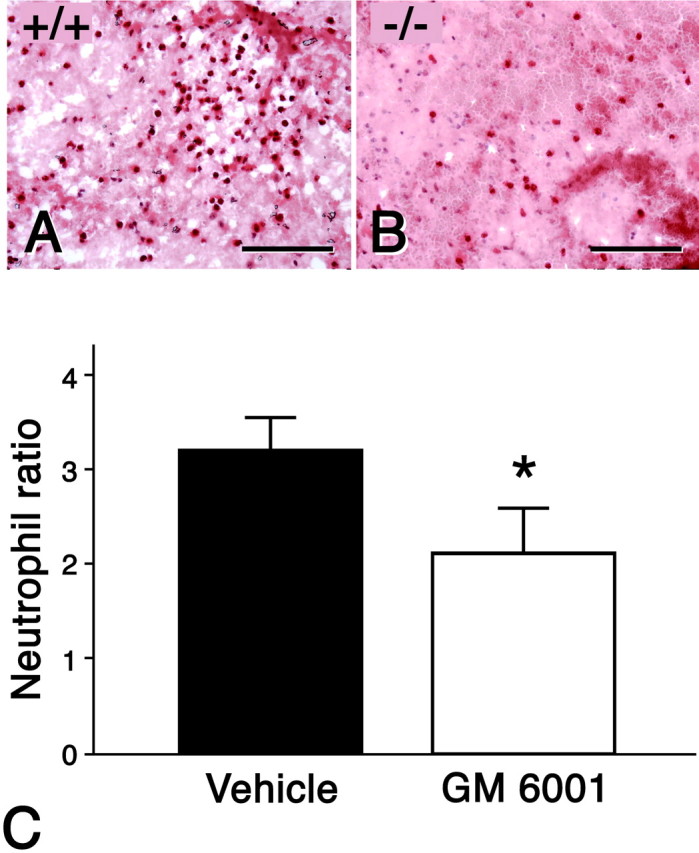
Effect of blocking MMPs on recruitment of inflammatory neutrophils into lesions after spinal cord injury. According to chloroacetate esterase staining, there appear to be greater numbers of neutrophils in the spinal cord-injured wild-type (A) compared with the MMP-9-null (B) animals at 24 hr after injury. The numbers of neutrophils were quantified within the epicenter of spinal cord-injured mice treated with either GM6001 (n = 4) or vehicle (n = 4) at 24 hr after injury (C). There is a significant reduction in the numbers of neutrophils in mice treated with GM6001 compared with vehicle controls (*p = 0.01). Values represent means ± SD. Scale bars: A, B, 100 μm.
Blocking MMPs improves locomotor recovery and attenuates histologically assessable white matter damage
We subsequently asked whether MMP activity affected locomotor recovery after spinal cord injury using an open-field testing paradigm, the BBB Locomotor Rating Scale, (Basso et al., 1995). There was significant locomotor recovery as early as 3 d after injury and weekly thereafter in MMP-9-null and in wild-type mice that were treated with the MMP inhibitor GM6001, compared with their respective controls (Fig. 9).
Fig. 9.
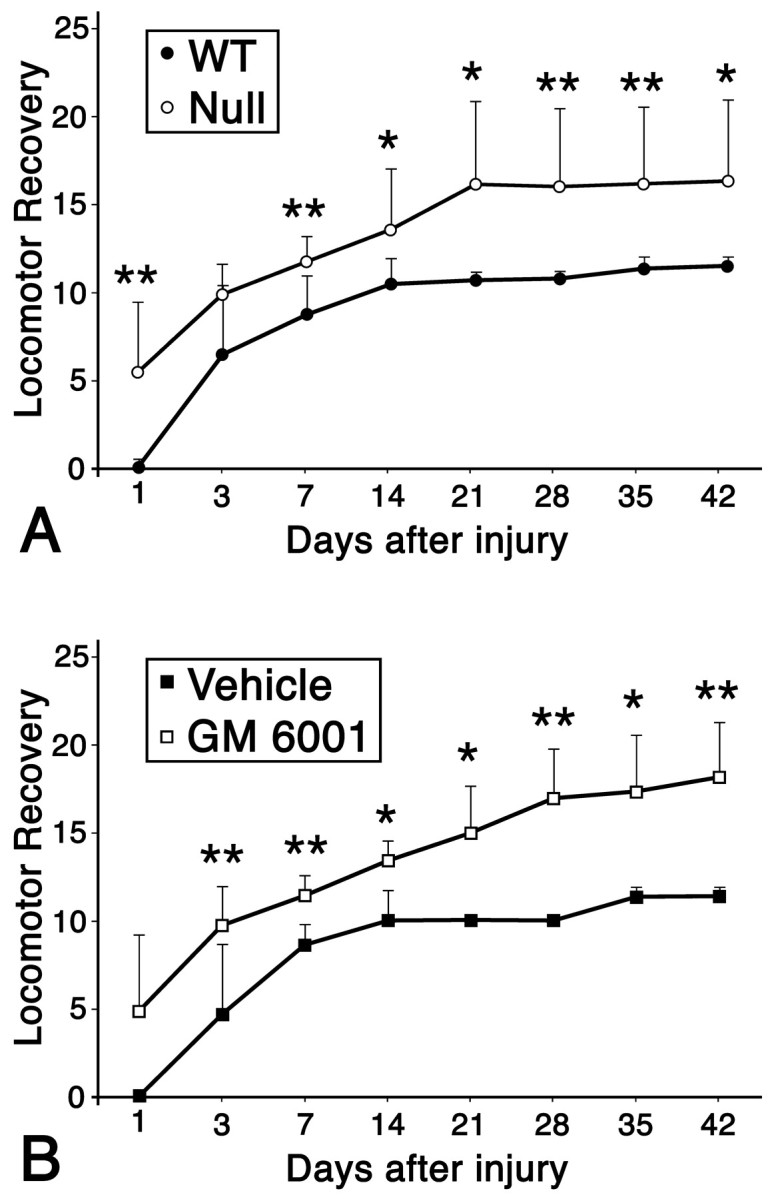
Effect of blocking MMPs on locomotor activity after spinal cord injury. Locomotor recovery was evaluated over a 6 week period, using a 21 point scale, in wild-type (WT) (n = 7) and MMP-9-null (n = 7) animals (A) and in GM6001-treated (n = 9), and vehicle-treated (n = 4) animals (B). Both the nulls and GM6001-treated animals exhibited greater locomotor recovery compared with their respective controls. Values represent means ± SD; ∗p = 0.05, ∗∗p ≤ 0.01.
When the area of residual white matter was quantified in the injured spinal cords of wild-type mice that were treated with the MMP inhibitor GM6001, there was significant preservation of white matter compared with the vehicle-treated groups (Fig.10). These data indicate that blocking MMP activity attenuates tissue damage and promotes locomotor recovery.
Fig. 10.
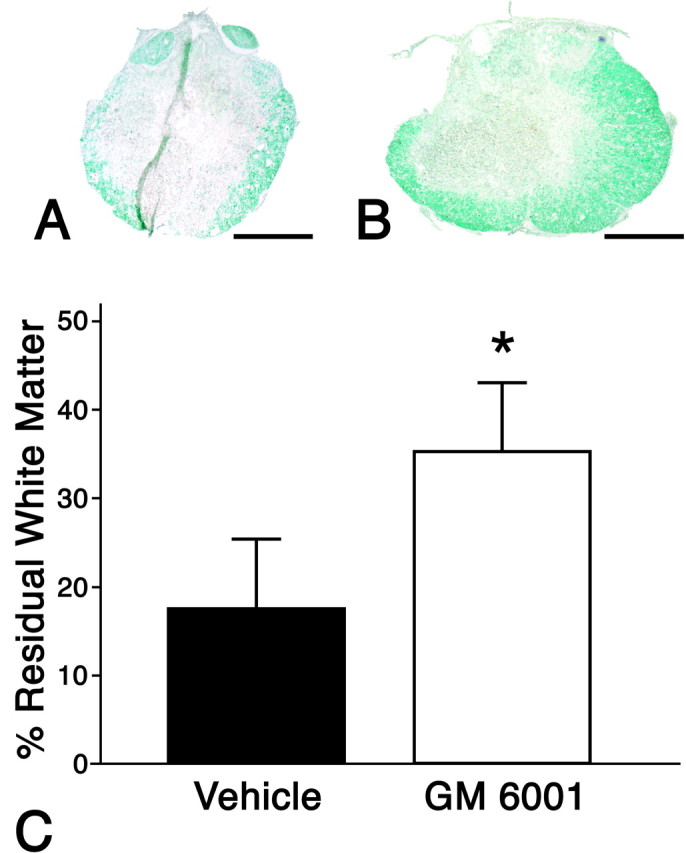
Effect of blocking MMPs on preservation of spinal cord white matter. A typical appearance of residual white matter at 42 d after injury, as identified with a Luxol fast blue stain, is shown for representative sections of GM6001-treated (A) and vehicle-treated (B) animals. There is significantly greater preservation of white matter in the GM6001-treated compared with the vehicle-treated animals (C). Values represent means ± SD; *p = 0.01.
DISCUSSION
We report that spinal cord-injured animals with a null mutation in MMP-9 exhibit reduced infiltration of neutrophils, stabilization of the blood–spinal cord barrier, and significant locomotor recovery compared with wild-type littermates. Moreover, similar observations were noted after pharmacological inhibition of MMPs, beginning 3 hr after injury over a period of 3 d, a timeframe coinciding with prominent disruption of the barrier and infiltration of neutrophils. Together, these exciting findings suggest that acute inhibition of MMPs may have efficacy as a therapeutic strategy for the treatment of human spinal cord injury.
Overview of MMP functions
MMPs are important for extracellular matrix remodeling and are integral to morphogenesis, inflammation, cancer, and wound healing (Sternlicht, 2001; Yong et al., 2001). MMPs degrade components of the extracellular matrix, including fibrillar and nonfibrillar collagens, fibronectin, laminin, glycoproteins, and nonmatrix substrates, including serine proteinase inhibitors (Vu and Werb, 1998; Vu et al., 1998). MMP-9 is capable of degrading gelatin, collagens, elastin, vitronectin, and the major components of the basal lamina comprising the blood–brain barrier (Mun-Bryce and Rosenberg, 1998a).
MMP-9 in the intact and injured spinal cord
MMP-9 expression was restricted to motoneurons and the meninges in the uninjured spinal cord. By 24 hr after injury, immunoreactive MMP-9 was expressed in vascular structures, astrocytes, neutrophils, and microglia/macrophages.
The change in expression of MMP-9 in the injured cord at 24 hr after injury corresponded to its prominent activation, as demonstrated with gelatin zymography. MMP-9 is regulated by several mechanisms: (1) transcriptional control, (2) secretion as an inactive zymogen subject to proteolytic activation, and (3) inhibition by its endogenous inhibitor, TIMP-1 (Vu and Werb, 1998; Sternlicht, 1999). Proteolysis by MMPs in normal or pathological states depends on the balance of proteinase to inhibitor (Sternlicht, 1999). In the present study, TIMP-1 was unchanged during the period of maximal activity of MMP-9. This finding and the results from in situ zymography, which shows sites of net active protease, suggest active proteolysis by MMP-9 in the acutely injured spinal cord and establish the basis for defining the contribution of this protease to secondary damage.
MMPs and inflammation after spinal cord injury
Neutrophils infiltrate the traumatized cord within the first several days after injury (Dusart and Schwab, 1994; Taoka et al., 1997;Carlson et al., 1998). We observed reduced numbers of neutrophils in the injured spinal cord at 24 hr after injury in MMP-depleted mice, a finding consistent with the role of MMP-9 in the transmigration of neutrophils from the vascular compartment (Pluznik et al., 1992;Delclaux et al., 1996).
Our findings likewise implicate neutrophils in impaired locomotor recovery after spinal cord injury. We demonstrate decreased infiltration of neutrophils and improved locomotor recovery in animals treated over the first 3 d after injury, a period coinciding with maximal infiltration of neutrophils into the traumatized spinal cord. Although controversial (Bartholdi and Schwab, 1995), neutrophils have been implicated in secondary pathogenesis after spinal cord injury (Taoka et al., 1997).
Neutrophils damage tissue by generating reactive oxygen species as well as proteases, including MMPs (Weiss, 1989). MMP-9 is characterized by a broad substrate specificity that includes extracellular matrix proteins as well as nonmatrix proteins such as α1-proteinase inhibitor, the primary inhibitor of neutrophil elastase (Liu et al., 2000). MMP-9 promotes tissue damage either directly by disrupting structural proteins or indirectly by inactivating proteins such as α1-proteinase inhibitor (Banda et al., 1980; Sires et al., 1994; Liu et al., 2000). Its involvement with α1-proteinase inhibitor is of particular interest because neutrophil elastase degrades the perivascular extracellular matrix, and its inhibition attenuates intraparenchymal hemorrhage and improves neurologic recovery after spinal cord injury (Armao et al., 1997; Taoka et al., 1998). Because α1-proteinase inhibitor–elastase complexes are chemotactic for neutrophils, inhibiting MMP-9 may also diminish additional recruitment of neutrophils.
The recruitment of neutrophils requires sequential appearance of several molecularly distinct chemoattractants (Liu et al., 2000; Chen et al., 2001). We found that administration of an MMP inhibitor was highly effective in blocking neutrophil recruitment and tissue damage when administered hours after the injury. MMP-9 is also prominently expressed during macrophage infiltration after peripheral nerve injury. This suggests that MMPs in general, and MMP-9 in particular, play a significant role in the sustained phases of inflammatory cell recruitment.
MMPs and the blood–spinal cord barrier
Increased activity of MMP-9 at 24 hr after injury coincided with prominent disruption of the blood–spinal cord barrier. Furthermore, this abnormal permeability was significantly reduced in MMP-depleted animals. MMP-9 has been implicated in blood–brain barrier disruption associated with inflammatory demyelinating disorders (Gijbels et al., 1994; Rosenberg et al., 1994, 1996b; Lim et al., 1996; Maeda and Sobel, 1996; Anthony et al., 1997), hemorrhagic brain injury, intracerebral administration of cytokines, and cerebral ischemia (Rosenberg et al., 1996a, 1998; Rosenberg and Navratil, 1997; Mun-Bryce and Rosenberg, 1998b; Fujimura et al., 1999). Moreover, there is evidence that inhibition of MMPs blocks barrier disruption (Rosenberg and Navratil, 1997; Mun-Bryce and Rosenberg, 1998b).
MMP-9 and other MMPs may influence the integrity of the blood–spinal cord barrier by degrading the basal lamina and/or tight junctions of endothelial cells. Leukocytes use MMPs during transmigration (Pluznik et al., 1992;Yong et al., 2001). MMPs degrade cadherins and other cell–cell communication molecules (Sternlicht, 2001). The recruitment of leukocytes triggers signal transduction cascades leading to junctional disorganization and abnormal vascular permeability (Bolton et al., 1998). The basal lamina surrounding endothelial cells plays a critical role in maintaining the integrity of the barrier in part by providing structural support to the endothelial cell (Kalaria, 2000; Farkas and Luiten, 2001). MMPs released from degranulating leukocytes may therefore compromise blood–brain barrier function and promote vasogenic edema.
MMPs and recovery of locomotor function
A critical finding of these studies is that MMP-9-null mice exhibited significant locomotor recovery compared with their wild-type controls after spinal cord injury. This neuroprotection may be attributed to the early involvement of MMPs in secondary pathogenesis. This hypothesis is based on our observation that animals treated with an MMP inhibitor within the first 3 d after injury likewise exhibited similar significant improvement in locomotor recovery.
Our studies suggest that the protection afforded by acute intervention with an MMP inhibitor targets early vascular responses associated with both disruption of the blood–spinal cord barrier and inflammation. Disruption of the barrier after spinal cord injury results in the influx of inflammatory cells and indiscriminate extravasation of molecules, including plasma proteins (Noble and Wrathall, 1989;Popovich et al., 1996). This abnormal permeability exposes the spinal cord to the toxic effects of inflammatory cells, as well as to amino acids such as glutamate and glycine, which, when present at high concentrations, can be toxic to cells (Schlosshauer, 1993).
We demonstrate significant neuroprotection of white matter in animals treated with the MMP inhibitor compared with the vehicle controls. Such protection may in part account for the improved locomotor recovery. Similar findings of neuroprotection have been reported after cerebral infarction, produced by systemic blockade of MMP-9 with a neutralizing antibody (Romanic et al., 1998). In this study, administration of the MMP inhibitor was initiated at 3 hr after injury and was maintained for 3 d after injury. The delay in treatment for 3 hr was selected for its potential relevance to the spinal cord-injured patient. Moreover, the limited duration of treatment, from 3 hr to 3 d, more precisely defined the contribution of MMPs to early pathogenesis and the extent to which this acute inhibition would influence white matter pathology and locomotor recovery. We found that the MMP inhibitor not only stabilized the barrier but also reduced the infiltration of neutrophils. Thus, neuroprotection and the resulting improvement in locomotion may be caused by decreased acute secondary damage.
It is also possible that MMP-9 produced by macrophages damages myelinated axons that were originally spared by the initial injury. Macrophages are integral to delayed demyelination of those populations of axons that have withstood the traumatic insult (Blight, 1985, 1994). Macrophages are in close proximity to the myelin sheaths of axons and infiltrate the myelin lamellas of axons that appear normal (Gledhill et al., 1973; Griffiths and McCulloch, 1983; Blight, 1985). This relationship is significant, because both macrophages and microglia secrete MMP-9 and other MMPs.
There is evidence that MMP-9 contributes to demyelination. There is widespread expression of MMP-9 in macrophages and reactive astrocytes in certain demyelinating diseases (Cuzner et al., 1996; Maeda and Sobel, 1996). Moreover, MMP expression is reduced in those regions exhibiting inactive lesions (Maeda and Sobel, 1996). This suggests that MMP expression correlates closely with localization of active demyelination. MMP-9 cleaves myelin basic protein (Gijbels et al., 1993) and thus may contribute to the disintegration of the myelin sheath. Future studies will be required to more specifically identify the role of contribution of MMP-9, produced by macrophages and glia, in axonal degeneration after spinal cord injury. In the shorter term, our studies suggest that MMP inhibitors administered in the first few hours after spinal cord injury could attenuate the poor outcomes attributable to secondary damage.
Footnotes
This work was supported by National Institutes of Health Grant R01 NS39278 and by a fellowship to F.D. from the Disabled Veterans of America. We thank Nino Maida and Tjoson Tjoa for their excellent technical assistance.
Correspondence should be addressed to Linda J. Noble, Department of Neurological Surgery, University of California, C224, 521 Parnassus Avenue, San Francisco, CA 94143-0520. E-mail: noblelj@itsa.ucsf.edu.
REFERENCES
- 1.Adler RR, Brenner CA, Werb Z. Expression of extracellular matrix-degrading metalloproteinases and metalloproteinase inhibitors is developmentally regulated during endoderm differentiation of embryonal carcinoma cells. Development. 1990;110:211–220. doi: 10.1242/dev.110.1.211. [DOI] [PubMed] [Google Scholar]
- 2.Anthony DC, Ferguson B, Matyzak MK, Miller KM, Esiri MM, Perry VH. Differential matrix metalloproteinase expression in cases of multiple sclerosis and stroke. Neuropathol Appl Neurobiol. 1997;23:406–415. [PubMed] [Google Scholar]
- 3.Armao D, Kornfeld M, Estrada EY, Grossetete M, Rosenberg GA. Neutral proteases and disruption of the blood-brain barrier in rat. Brain Res. 1997;767:259–264. doi: 10.1016/s0006-8993(97)00567-2. [DOI] [PubMed] [Google Scholar]
- 4.Austyn JM, Gordon S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol. 1981;11:805–815. doi: 10.1002/eji.1830111013. [DOI] [PubMed] [Google Scholar]
- 5.Backstrom JR, Lim GP, Cullen MJ, Tokes ZA. Matrix metallo-proteinase-9 (MMP-9) is synthesized in neurons of the human hippocampus and is capable of degrading the amyloid-β peptide (1–40). J Neurosci. 1996;16:7910–7919. doi: 10.1523/JNEUROSCI.16-24-07910.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banda MJ, Clark EJ, Werb Z. Limited proteolysis by macrophage elastase inactivates human alpha 1-proteinase inhibitor. J Exp Med. 1980;152:1563–1570. doi: 10.1084/jem.152.6.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartholdi D, Schwab ME. Methylprednisolone inhibits early inflammatory processes but not ischemic cell death after experimental spinal cord lesion in the rat. Brain Res. 1995;672:177–186. doi: 10.1016/0006-8993(94)01410-j. [DOI] [PubMed] [Google Scholar]
- 8.Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- 9.Basso DM, Beattie MS, Bresnahan JC, Anderson DK, Faden AI, Gruner JA, Holford TR, Hsu CY, Noble LJ, Nockels R, Perot PL, Salzman SK, Young W. MASCIS evaluation of open field locomotor scores: effects of experience and teamwork on reliability. Multicenter Animal Spinal Cord Injury Study. J Neurotrauma. 1996;13:343–359. doi: 10.1089/neu.1996.13.343. [DOI] [PubMed] [Google Scholar]
- 10.Behrendtsen O, Alexander CM, Werb Z. Metalloproteinases mediate extracellular matrix degradation by cells from mouse blastocyst outgrowths. Development. 1992;114:447–456. doi: 10.1242/dev.114.2.447. [DOI] [PubMed] [Google Scholar]
- 11.Birkedal-Hansen H, Moore WG, Bodden MK, Windsor LJ, Birkedal-Hansen B, DeCarlo A, Engler JA. Matrix metalloproteinases: a review. Crit Rev Oral Biol Med. 1993;4:197–250. doi: 10.1177/10454411930040020401. [DOI] [PubMed] [Google Scholar]
- 12.Blight AR. Delayed demyelination and macrophage invasion: a candidate for secondary cell damage in spinal cord injury. Cent Nerv Syst Trauma. 1985;2:299–315. doi: 10.1089/cns.1985.2.299. [DOI] [PubMed] [Google Scholar]
- 13.Blight AR. Effects of silica on the outcome from experimental spinal cord injury: implication of macrophages in secondary tissue damage. Neuroscience. 1994;60:263–273. doi: 10.1016/0306-4522(94)90220-8. [DOI] [PubMed] [Google Scholar]
- 14.Bolton SJ, Anthony DC, Perry VH. Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood-brain barrier breakdown in vivo. Neuroscience. 1998;86:1245–1257. doi: 10.1016/s0306-4522(98)00058-x. [DOI] [PubMed] [Google Scholar]
- 15.Carlson SL, Parrish ME, Springer JE, Doty K, Dossett L. Acute inflammatory response in spinal cord following impact injury. Exp Neurol. 1998;151:77–88. doi: 10.1006/exnr.1998.6785. [DOI] [PubMed] [Google Scholar]
- 16.Chen R, Fleming MG, Dias LA, Werb Z, Liu Z. Mast cells play a key role in neutrophil recruitment in experimental bullous pemphigoid. J Clin Invest. 2001;108:1151–1158. doi: 10.1172/JCI11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuzner ML, Gveric D, Strand C, Loughlin AJ, Paemen L, Opdenakker G, Newcombe J. The expression of tissue-type plasminogen activator, matrix metalloproteases and endogenous inhibitors in the central nervous system in multiple sclerosis: comparison of stages in lesion evolution. J Neuropathol Exp Neurol. 1996;55:1194–1204. doi: 10.1097/00005072-199612000-00002. [DOI] [PubMed] [Google Scholar]
- 18.Delclaux C, Delacourt C, D'Ortho MP, Boyer V, Lafuma C, Harf A. Role of gelatinase B and elastase in human polymorphonuclear neutrophil migration across basement membrane. Am J Respir Cell Mol Biol. 1996;14:288–295. doi: 10.1165/ajrcmb.14.3.8845180. [DOI] [PubMed] [Google Scholar]
- 19.Dusart I, Schwab ME. Secondary cell death and the inflammatory reaction after dorsal hemisection of the rat spinal cord. Eur J Neurosci. 1994;6:712–724. doi: 10.1111/j.1460-9568.1994.tb00983.x. [DOI] [PubMed] [Google Scholar]
- 20.Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer's disease. Prog Neurobiol. 2001;64:575–611. doi: 10.1016/s0301-0082(00)00068-x. [DOI] [PubMed] [Google Scholar]
- 21.Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH. Early appearance of activated matrix metallopro-teinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res. 1999;842:92–100. doi: 10.1016/s0006-8993(99)01843-0. [DOI] [PubMed] [Google Scholar]
- 22.Gijbels K, Proost P, Masure S, Carton H, Billiau A, Opdenakker G. Gelatinase B is present in the cerebrospinal fluid during experimental autoimmune encephalomyelitis and cleaves myelin basic protein. J Neurosci Res. 1993;36:432–440. doi: 10.1002/jnr.490360409. [DOI] [PubMed] [Google Scholar]
- 23.Gijbels K, Galardy RE, Steinman L. Reversal of experimental autoimmune encephalomyelitis with a hydroxamate inhibitor of matrix metalloproteases. J Clin Invest. 1994;94:2177–2182. doi: 10.1172/JCI117578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gledhill RF, Harrison BM, McDonald WI. Demyelination and remyelination after acute spinal cord compression. Exp Neurol. 1973;38:472–487. doi: 10.1016/0014-4886(73)90169-6. [DOI] [PubMed] [Google Scholar]
- 25.Gottschall PE, Deb S. Regulation of matrix metalloproteinase expressions in astrocytes, microglia and neurons. Neuroimmunomodulation. 1996;3:69–75. doi: 10.1159/000097229. [DOI] [PubMed] [Google Scholar]
- 26.Griffiths IR, McCulloch MC. Nerve fibres in spinal cord impact injuries. Part 1. Changes in the myelin sheath during the initial 5 weeks. J Neurol Sci. 1983;58:335–349. doi: 10.1016/0022-510x(83)90093-x. [DOI] [PubMed] [Google Scholar]
- 27.Herron GS, Werb Z, Dwyer K, Banda MJ. Secretion of metalloproteinases by stimulated capillary endothelial cells. I. Production of procollagenase and prostromelysin exceeds expression of proteolytic activity. J Biol Chem. 1986;261:2810–2813. [PubMed] [Google Scholar]
- 28.Hibbs MS, Hoidal JR, Kang AH. Expression of a metalloproteinase that degrades native type V collagen and denatured collagens by cultured human alveolar macrophages. J Clin Invest. 1987;80:1644–1650. doi: 10.1172/JCI113253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalaria RN. The role of cerebral ischemia in Alzheimer's disease. Neurobiol Aging. 2000;21:321–330. doi: 10.1016/s0197-4580(00)00125-1. [DOI] [PubMed] [Google Scholar]
- 30.Kuhn PL, Wrathall JR. A mouse model of graded contusive spinal cord injury. J Neurotrauma. 1998;15:125–140. doi: 10.1089/neu.1998.15.125. [DOI] [PubMed] [Google Scholar]
- 31.Lim GP, Backstrom JR, Cullen MJ, Miller CA, Atkinson RD, Tokes ZA. Matrix metalloproteinases in the neocortex and spinal cord of amyotrophic lateral sclerosis patients. J Neurochem. 1996;67:251–259. doi: 10.1046/j.1471-4159.1996.67010251.x. [DOI] [PubMed] [Google Scholar]
- 32.Liu Z, Shipley JM, Vu TH, Zhou X, Diaz LA, Werb Z, Senior RM. Gelatinase B-deficient mice are resistant to experimental bullous pemphigoid. J Exp Med. 1998;188:475–482. doi: 10.1084/jem.188.3.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Z, Zhou X, Shapiro SD, Shipley JM, Twining SS, Diaz LA, Senior RM, Werb Z. The serpin alpha1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell. 2000;102:647–655. doi: 10.1016/s0092-8674(00)00087-8. [DOI] [PubMed] [Google Scholar]
- 34.Maeda A, Sobel RA. Matrix metalloproteinases in the normal human central nervous system, microglial nodules, and multiple sclerosis lesions. J Neuropathol Exp Neurol. 1996;55:300–309. doi: 10.1097/00005072-199603000-00005. [DOI] [PubMed] [Google Scholar]
- 35.Mainardi CL, Hibbs MS, Hasty KA, Seyer JM. Purification of a type V collagen degrading metalloproteinase from rabbit alveolar macrophages. Coll Relat Res. 1984;4:479–492. doi: 10.1016/s0174-173x(84)80014-x. [DOI] [PubMed] [Google Scholar]
- 36.Mun-Bryce S, Rosenberg GA. Matrix metalloproteinases in cerebrovascular disease. J Cereb Blood Flow Metab. 1998a;18:1163–1172. doi: 10.1097/00004647-199811000-00001. [DOI] [PubMed] [Google Scholar]
- 37.Mun-Bryce S, Rosenberg GA. Gelatinase B modulates selective opening of the blood-brain barrier during inflammation. Am J Physiol. 1998b;274:R1203–R1211. doi: 10.1152/ajpregu.1998.274.5.R1203. [DOI] [PubMed] [Google Scholar]
- 38.Murphy G, Ward R, Hembry RM, Reynolds JJ, Kuhn K, Tryggvason K. Characterization of gelatinase from pig polymorphonuclear leucocytes. A metalloproteinase resembling tumour type IV collagenase. Biochem J. 1989;258:463–472. doi: 10.1042/bj2580463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noble LJ, Wrathall JR. Spinal cord contusion in the rat: morphometric analyses of alterations in the spinal cord. Exp Neurol. 1985;88:135–149. doi: 10.1016/0014-4886(85)90119-0. [DOI] [PubMed] [Google Scholar]
- 40.Noble LJ, Wrathall JR. Distribution and time course of protein extravasation in the rat spinal cord after contusive injury. Brain Res. 1989a;482:57–66. doi: 10.1016/0006-8993(89)90542-8. [DOI] [PubMed] [Google Scholar]
- 41.Noble LJ, Wrathall JR. Correlative analyses of lesion development and functional status after graded spinal cord contusive injuries in the rat. Exp Neurol. 1989b;103:34–40. doi: 10.1016/0014-4886(89)90182-9. [DOI] [PubMed] [Google Scholar]
- 42.Oh LY, Larsen PH, Krekoski CA, Edwards DR, Donovan F, Werb Z, Yong VW. Matrix metalloproteinase-9/gelatinase B is required for process outgrowth by oligodendrocytes. J Neurosci. 1999;19:8464–8475. doi: 10.1523/JNEUROSCI.19-19-08464.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pluznik DH, Fridman R, Reich R. Correlation in the expression of type IV collagenase and the invasive and chemotactic abilities of myelomonocytic cells during differentiation into macrophages. Exp Hematol. 1992;20:57–63. [PubMed] [Google Scholar]
- 44.Popovich PG, Horner PJ, Mullin BB, Stokes BT. A quantitative spatial analysis of the blood-spinal cord barrier. I. Permeability changes after experimental spinal contusion injury. Exp Neurol. 1996;142:258–275. doi: 10.1006/exnr.1996.0196. [DOI] [PubMed] [Google Scholar]
- 45.Romanic AM, White RF, Arleth AJ, Ohlstein EH, Barone FC. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke. 1998;29:1020–1030. doi: 10.1161/01.str.29.5.1020. [DOI] [PubMed] [Google Scholar]
- 46.Rosenberg GA, Navratil M. Metalloproteinase inhibition blocks edema in intracerebral hemorrhage in the rat. Neurology. 1997;48:921–926. doi: 10.1212/wnl.48.4.921. [DOI] [PubMed] [Google Scholar]
- 47.Rosenberg GA, Dencoff JE, McGuire PG, Liotta LA, Stetler-Stevenson WG. Injury-induced 92-kilodalton gelatinase and urokinase expression in rat brain. Lab Invest. 1994;71:417–422. [PubMed] [Google Scholar]
- 48.Rosenberg GA, Estrada EY, Dencoff JE, Stetler-Stevenson WG. Tumor necrosis factor-alpha-induced gelatinase B causes delayed opening of the blood-brain barrier: an expanded therapeutic window. Brain Res. 1995;703:151–155. doi: 10.1016/0006-8993(95)01089-0. [DOI] [PubMed] [Google Scholar]
- 49.Rosenberg GA, Navratil M, Barone F, Feuerstein G. Proteolytic cascade enzymes increase in focal cerebral ischemia in rat. J Cereb Blood Flow Metab. 1996a;16:360–366. doi: 10.1097/00004647-199605000-00002. [DOI] [PubMed] [Google Scholar]
- 50.Rosenberg GA, Dencoff JE, Correa N, Jr, Reiners M, Ford CC. Effect of steroids on CSF matrix metalloproteinases in multiple sclerosis: relation to blood-brain barrier injury. Neurology. 1996b;46:1626–1632. doi: 10.1212/wnl.46.6.1626. [DOI] [PubMed] [Google Scholar]
- 51.Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–2195. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- 52.Schlosshauer B. The blood-brain barrier: morphology, molecules, and neurothelin. BioEssays. 1993;15:341–346. doi: 10.1002/bies.950150508. [DOI] [PubMed] [Google Scholar]
- 53.Sires UI, Murphy G, Baragi VM, Fliszar CJ, Welgus HG, Senior RM. Matrilysin is much more efficient than other matrix metalloproteinases in the proteolytic inactivation of alpha 1-antitrypsin. Biochem Biophys Res Commun. 1994;204:613–620. doi: 10.1006/bbrc.1994.2503. [DOI] [PubMed] [Google Scholar]
- 54.Sternlicht MDWZ. Extracellular matrix proteinases. In: Kreis T, Vale R, editors. Guidebook to the extracellular matrix, anchor and adhesion proteins. Oxford UP; New York: 1999. pp. 503–603. [Google Scholar]
- 55.Sternlicht MDWZ. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taoka Y, Okajima K, Uchiba M, Murakami K, Kushimoto S, Johno M, Naruo M, Okabe H, Takatsuki K. Role of neutrophils in spinal cord injury in the rat. Neuroscience. 1997;79:1177–1182. doi: 10.1016/s0306-4522(97)00011-0. [DOI] [PubMed] [Google Scholar]
- 57.Taoka Y, Okajima K, Murakami K, Johno M, Naruo M. Role of neutrophil elastase in compression-induced spinal cord injury in rats. Brain Res. 1998;799:264–269. doi: 10.1016/s0006-8993(98)00459-4. [DOI] [PubMed] [Google Scholar]
- 58.Vu TH, Werb Z. Gelatinase B: structure, regulation, and function. Academic; San Diego: 1998. [Google Scholar]
- 59.Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weiss SJ. Tissue destruction by neutrophils. N Engl J Med. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 61.Werb Z. ECM and cell surface proteolysis: regulating cellular ecology. Cell. 1997;91:439–442. doi: 10.1016/s0092-8674(00)80429-8. [DOI] [PubMed] [Google Scholar]
- 62. Wilhelm SM, Collier IE, Marmer BL, Eisen AZ, Grant GA, Goldberg GI. SV40-transformed human lung fibroblasts secrete a 92-kDa type IV collagenase which is identical to that secreted by normal human macrophages. J Biol Chem 264 1989. 17213 17221 [Erratum (1990) 265:22570] [PubMed] [Google Scholar]
- 63.Xu J, Kim G-M, Ahmed S, Xu J, Yan P, Xu X, Hsu CY. Glucocorticoid receptor-mediated suppression of activator protein-1 activation and matrix metalloproteinase expression after spinal cord injury. J Neurosci. 2001;21:92–97. doi: 10.1523/JNEUROSCI.21-01-00092.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yong VW, Power C, Forsyth P, Edwards DR. Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci. 2001;2:502–511. doi: 10.1038/35081571. [DOI] [PMC free article] [PubMed] [Google Scholar]