

Abstract
Heat shock protein (hsp) 90 inhibitors promote proteasomal degradation of pro-growth and pro-survival hsp90 client proteins, including CDK4, c-RAF and AKT, and induce apoptosis of human lymphoma cells. The pan-histone deacetylase inhibitor vorinostat has also been shown to induce growth arrest and apoptosis of lymphoma cells. Here, we determined the effects of the more soluble, orally bio-available, geldanamycin analogue 17-NN-dimethyl ethylenediamine geldanamycin (DMAG, Kosan Biosciences Inc) and/or vorinostat in cultured and primary human MCL cells. While vorinostat induced accumulation in the G1 phase, treatment with DMAG arrested MCL cells in the G2/M phase of the cell cycle. Both agents dose-dependently induced apoptosis of MCL cells. Vorinostat also induced hyperacetylation of hsp90 and disrupted the association of hsp90 with its co-chaperones p23 and cdc37, as well as with its client proteins CDK4 and c-RAF. Treatment of MCL cells with vorinostat or 17-DMAG was associated with the induction of p21 and p27, as well as with depletion of c-Myc, c-RAF, AKT and CDK4. Compared to treatment with either agent alone, co-treatment with DMAG and vorinostat markedly attenuated the levels of cyclin D1 and CDK4, as well as of c-Myc, c-RAF and AKT. Combined treatment with DMAG and vorinostat synergistically induced apoptosis of the cultured MCL cells, as well as induced more apoptosis of primary MCL cells than either agent alone. Therefore, these findings support the rationale to determine the in vivo efficacy of co-treatment with vorinostat and DMAG against human MCL cells.
Introduction
Mantle cell lymphoma (MCL) is a relatively aggressive subtype of B-cell non-Hodgkin's lymphomas (NHL) that comprises approximately 6% of human B-cell Non-Hodgkins Lymphoma (NHL).1-3 MCL cells are characterized by deregulated expression of cyclin D1, which is an important regulator of the G1 phase of the cell cycle. 2,3 In virtually all cases of MCL, cyclin D1 overexpression is due to the CCND1-IgH translocation resulting from the chromosomal translocation t(11;14)(q13;q32).2-4 Cyclin D1 expression may be further augmented in MCL cells by enhanced activity of NFκB and AP1.5 In addition to cyclin D1 upregulation, cell cycle may also be deregulated in MCL cells by genomic amplification of the cyclin-dependent kinase (CDK)-4, deletions of the CDK inhibitor p16INK4a, overexpression of BMI-1, a transcriptional repressor of the p16INK4a locus, or inability of the truncated cyclin D1 mRNA transcript in MCL to be down-regulated by miRNA-16−1.2,6,7,8 Specific genotypes, epigenetic alterations and gene expression signatures have further elucidated MCL subtypes, explained the disparate clinical profiles and outcomes of patients, and have suggested potential therapeutic targets in MCL.2,9-12 These include CDK4, AKT and MYC.2,9-12 Recently, expressions of ZAP-70, p-AKT, short cyclin D1 variant, cyclin D2 or cyclin D3 have all been correlated with a biologically aggressive behavior in MCL.3,9,13-15
Among the stress-inducible molecular chaperones, hsp90 is a highly conserved, homo-dimeric, ATP-dependent, abundantly expressed protein in eukaryotic cells, including MCL cells.16-18 Hsp90 forms the core of a super chaperone machine. It is required for the maintenance of native and functionally active conformation of important signaling protein kinases and nuclear hormone receptors, which are collectively known as the hsp90 client proteins.16-18 ATP binding to the hydrophobic N-terminus pocket also alters hsp90 conformation, such that it promotes the interaction of hsp90 with a set of co-chaperones including p23 and p50cdc37 (when the client protein is a signaling protein kinase) forming the super chaperone machine, which stabilizes the metastable signaling client proteins.19,20 Conversely, ATP hydrolysis due to the intrinsic ATPase activity of hsp90 creates the ADP-bound conformation of hsp90.16,17 This directs the misfolded client protein to a covalent linkage with polyubiquitin through the activity of an E3 ubiquitin ligase, mediating client protein polyubiquitylation and subsequent degradation by the 26S proteasome.16,17 Among the growing list of hsp90 client proteins are the MCL relevant signaling proteins c-RAF, AKT, ZAP-70 and IKKα, as well as the cell cycle regulatory proteins e.g., CDK4, p21, CHK1.16,17,21-25 Ansamycin antibiotic geldanamycin analogues (GAs), e.g., 17-AAG and the more soluble orally bio-available 17-DMAG, bind to the ATP/ADP binding pocket of hsp90, inhibiting the nucleotide binding and the chaperone function of hsp90. This is known to cause misfolding, polyubiquitylation and degradation of the hsp90 client proteins, including the MCL-relevant client proteins, by the 20S proteosome. 16,17 Consistent with this, treatment with hsp90 inhibitor was shown to induce cell cycle arrest and apoptosis of MCL cells.26
Among a variety of structurally diverse pan-histone deacetylase (HDAC) inhibitors (HDIs) are the hydroxamic analogue (HA) HDIs, e.g., vorinostat LAQ824 and LBH589.27,28 Vorinostat is the only approved HA-HDI, currently used for the treatment of cutaneous T cell lymphoma.29 Treatment with HA-HDI has been shown to increase the levels of p21 and p27, as well as increase the levels of pro-apoptotic proteins, e.g., Bax, Bak and Bim.27,28,30 HA-HDI treatment was also shown to attenuate the levels of antiapoptotic proteins, e.g., Bcl-xL, XIAP, survivin, AKT and c-RAF.27,30-32 These effects have been correlated with the ability of HA-HDIs to induce cell cycle growth arrest and apoptosis of human leukemia and non-Hodgkin's lymphoma (NHL) cells.27,29-33 Recently, HDAC6 (a class IIB, predominantly cytosolic and microtubule associated HDAC) was shown to bind and deacetylate hsp90, and HA-HDI treatment inhibited HDAC6, resulting in hyperacetylation of hsp90 and α-tubulin.34-36 HA-HDI-induced hyperacetylation of hsp90 led to inhibition of ATP binding and chaperone association of hsp90 with its client proteins, including AKT and c-RAF.34 Hyperacetylation of hsp90 was also shown to increase GA binding to hsp90 and inhibition of its chaperone function.32,37 Consistent with this, co-treatment with an HA-HDI enhanced GA-mediated depletion of hsp90 client proteins and activity against cultured and primary human leukemia cells.32 Based on these observations, in the present studies, we determined the effects of vorinostat and/or DMAG on the levels of MCL-relevant hsp90 client proteins, as well as on the growth and survival of cultured and primary human MCL cells.
Materials and Methods
Reagents and Antibodies
DMAG was kindly provided by Kosan Biosciences Inc., while vorinostat was a gift from Merck and Co. Inc.. Rat monoclonal anti-hsp90 and rabbit polyclonal anti-hsp70 antibody were purchased from StressGen (Ann Arbor, MI). Monoclonal anti-acetyl α-tubulin and anti-β-actin antibody was purchased from Sigma Aldrich (St. Louis, MO). Monoclonal anti-acetyl lysine, anti-Akt and anti-cyclin D1 antibodies were purchased from Cell Signaling Technology (Beverly, MA). Anti-CDK4, cdc37, c-Myc antibodies were purchased from Santa Cruz Biotechnology, Inc., (Santa Cruz, CA). Anti-p27 antibody was purchased from BD Pharmingen, (San Diego, CA), anti-p21 from Thermo Scientific, (Fremont CA), anti-c-RAF from BD Transduction laboratories (New jersey, USA), and anti-p23 was procured from Alexis biochemicals (San Francisco, CA). Other reagents and antibodies were purchased from sources, as previously described.30-32,34,37
Cell Culture
Cultured human MCL Z138, JeKo-1 and MO2058 cells were maintained in RPMI medium containing 10% FBS. Cultured medium was changed every 3 days and cells were maintained at a density of 250,000 cells/mL. Logarithmically growing cells were exposed to the designated concentrations and exposure interval of the drugs. Following these treatments, cells were pelleted and washed free of the drug(s), prior to the performance of the studies described below.
Primary MCL and Normal CD34+ Hemopoietic Progenitor Cells
Primary MCL cells were obtained with informed consent as part of a clinical protocol approved by the Institutional Review Board of the Medical College of Georgia. As previously described, bone marrow and/or peripheral blood samples were collected in heparinized tubes, or were obtained from leukophoresis units, and mononuclear cells were separated using Lymphoprep (Axis-Shield, Oslo, Norway), washed once with complete RPMI-1640 media, re-suspended in complete RPMI-1640 and counted to determine the number of cells isolated prior to their use in the various experiments.30 The purity of the MCL cell was confirmed to be 80% or better by morphologic evaluation of cytospun cell preparations stained with Wright stain. Banked, de-linked, and de-identified donor peripheral blood CD34+ mononuclear cells procured for recipients who had since died were purified by immunomagnetic beads conjugated with anti-CD34 antibody prior to use in the cell viability assay (StemCell Technologies).30
Cell Lysis and Protein Quantitation
Untreated or drug-treated cells were centrifuged and the cell pellets were re-suspended in lysis buffer, then centrifuged and protein concentrations determined, as previously described.30-32
Western Blot analyses
Western analyses of c-RAF-1, AKT, hsp90, hsp70 and β-actin were performed using specific anti-sera or monoclonal antibodies as described previously.30-32 Horizontal scanning densitometry was performed on Western blots by using acquisition into Adobe PhotoShop (Adobe Systems Inc., San Jose, CA) and analysis by the NIH Image Program (U.S. National Institutes of Health, Bethesda, MD).30-32,34,37 The expression of β-actin was used as a loading control.
Immunoprecipitation of hsp90 and immunoblot analyses of hsp90 client proteins
Following designated treatments, cells were lysed with the lysis buffer as described above. Two hundred micrograms of cell lysate was mixed gently with 2 μg of rat monoclonal anti-hsp90 or 2 μg of mouse monoclonal anti-cdc37, anti-p23, anti-CDK4, anti-AKT or anti-c-RAF antibody and incubated at 4°C for 1−2 hours on a rotator. Pre-washed protein-G beads were added to the lysate-antibody mixture and incubated overnight at 4°C on a rotator. The immunoprecipitates were washed 4 times with lysis buffer and eluted from the agarose beads by boiling with 6X SDS sample buffer before SDS-PAGE and Western blot analysis, as previously described.30-32
Assessment of apoptosis and synergism
Untreated or drug-treated cells were fixed with 70% ethanol then stained with annexin V and propidium iodide (PI). The percentage of apoptotic cells were determined by flow cytometry as previously described.32 To analyze synergism between vorinostat and DMAG in inducing apoptosis, cells were treated with vorinostat and DMAG at a constant ratio for 48 hours. The percentage of apoptotic cells was determined by flow cytometry as previously described.30 Synergism defined as a more than expected additive effect was assessed using the median dose effect of Chou-Talalay method and the combination index (CI) for each drug combination was obtained using the commercially available software Calcusyn (Biosoft, Ferguson, MO).38 CI values of less than 1.0 represent synergism of the two drugs in the combination.
Assessment of percentage non-viable cells
Following designated treatments, primary MCL or normal CD34+ bone marrow progenitor cells were stained with trypan blue (Sigma, St. Louis, MO). The numbers of non-viable cells were determined by counting the cells that showed trypan blue uptake in a hemocytometer, and reported as percentage of untreated control cells.
Cell cycle analysis
Cultured MCL cells were exposed to the indicated concentrations of drugs for 24 to 48 hours and the cell cycle analysis was performed following PI staining as described previously.30,32
Statistical Analyses
Data were expressed as mean ± SEM. Comparisons used student's t test or ANOVA, as appropriate. P values of < 0.05 were assigned significance.
Results
Vorinostat and DMAG induce cell cycle growth arrest and apoptosis in MCL cells
Based on the previous reports that vorinostat induces apoptosis in human leukemia and lymphoma cells and 17-AAG exerts anti-MCL activity, we first determined the cell cycle growth inhibitory and apoptotic effects of vorinostat and DMAG on the cultured MCL JeKo-1 and MO2058 cells.26,30,39 Logarithmically growing cells were exposed to clinically relevant concentrations of vorinostat (0.5 to 2.0 μM) or DMAG (0.1 to 0.5 μM) for 48 hours, and the percentage of apoptotic cells was assessed by flow cytometry (Figure 1A & IB). Our results show that both vorinostat and DMAG dose-dependently induced apoptosis of MCL cells. Treatment of MCL cells with vorinostat for 24 hours caused the accumulation of MCL cells in the G1 phase of the cell cycle, while DMAG treatment increased the percentage of cells in both the G1 and G2/M phases of the cell cycle (Table 1).
Figure 1. Treatment with vorinostat or 17-DMAG dose-dependently induces apoptosis of the MCL MO2058, Z138 and JeKo-1 cells.
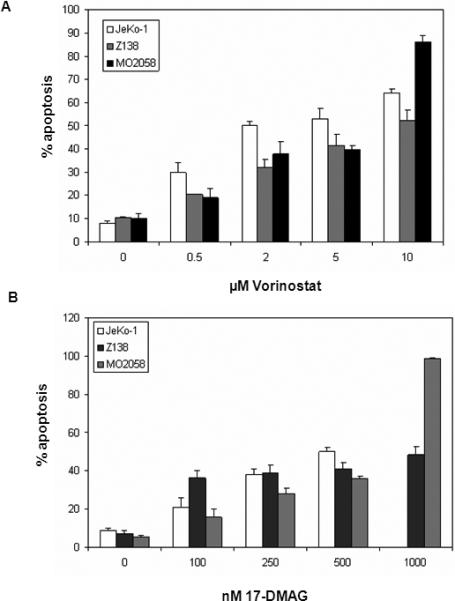
Cells were treated with indicated concentrations of vorinostat (A) or 17-DMAG (B) for 48 hours. Following this, the percentage of annexin-V-stained apoptotic cells was determined by flow cytometry. Values represented as bar graphs are the mean of 3 experiments plus or minus the standard error (SE)
Table 1. Vorinostat or 17-DMAG treatments cause increased accumulation of MCL cells MO2058 in G1 and G2/M phases of cell cycle in a dose dependent manner.
MO2058 cells were treated with different doses of vorinostat or 17-DMAG for 24 hours. Following these treatments, the percentage of cells in the various phases of the cell cycle was determined after staining with propidium iodide and performing flow cytometry. Values represent the means ± SE of three experiments.
| MO2058 Cells | |||
|---|---|---|---|
| G1 |
S |
G2/M |
|
| vorinostat (24 hours) | |||
| 0 μM | 18.3 ± 1.6 | 63.4 ± 5.3 | 18.3 ± 5.1 |
| 0.5 μM | 29.2 ± 4.1 | 58.1 ± 5.9 | 12.7 ± 2.2 |
| 2.0 μM | 48.0 ± 3.7 | 34.3 ± 3.7 | 17.7 ± 7.4 |
| DMAG (24 hrs) | |||
| 0 μM | 18.3 ± 1.6 | 63.4 ± 5.3 | 18.7 ± 5.1 |
| 0.1 μM | 27.9 ± 6.5 | 55.6 ± 2.3 | 16.5 ± 8.0 |
| 0.25 μM | 37.1 ± 6.1 | 36.7 ± 2.0 | 26.2 ± 7.9 |
| 0.5 μM | 26.3 ± 4.8 | 38.4 ± 2.9 | 36.4 ± 4.0 |
Vorinostat and DMAG disrupt chaperone function of hsp90 and deplete hsp90 client proteins in MCL cells
Consistent with previous reports and the cell cycle growth arrest described above, treatment with vorinostat and DMAG resulted in a dose-dependent up-regulation of cell cycle inhibitors such as p21 and p27 (Figure 2A). Additionally, without significantly affecting the levels of hsp90, treatment with vorinostat and DMAG abrogated the chaperone function of hsp90, which was associated with induction of hsp70 levels in MCL cells (Figure 2A).40 Immunoprecipitation of hsp90 followed by immunoblot analysis by anti-acetyl lysine antibody demonstrated that vorinostat treatment also induced hyperacetylation of hsp90 (Figure 2B). Disruption of hsp90 chaperone function by vorinostat was also characterized by attenuation of the chaperone association between hsp90 and the co-chaperones p23 and cdc37 (Figure 2B).16,17 Importantly, short exposure to vorinostat also disrupted the chaperone association between hsp90 and its MCL-relevant client proteins CDK4 and c-RAF (Figure 2C). Treatment with vorinostat and DMAG for 24 hours also depleted the levels of the hsp90 client proteins c-RAF, CDK4 and AKT, in JeKo-1 and MO2058 cells (Figure 3A and 3B). We observed a dose dependent increase in Histone H3 acetylation following vorinostat treatment in both MO2058 and JeKo-1 cells. While treatment with vorinostat also depleted cyclin D1 and c-MYC levels in the two cell types, DMAG depleted only cyclin D1 in JeKo-1 and c-MYC levels in MO2058 cells (Figure 3A and 3B). Collectively, these observations suggest that both vorinostat and DMAG disrupt hsp90 chaperone function resulting in the depletion of key pro-growth and pro-survival proteins in the MCL cells.
Figure 2. Treatment with vorinostat induces hsp90 acetylation and results in decreased association of hsp90 with the cochpaerones p23 and cdc37.

A) MO2058 cells were exposed to vorinostat and DMAG for 24 hours and the resulting cell lysates were immunoblotted for hsp70, hsp90, p27 and p21. B) JeKo-1 cells were exposed to indicted concentration of vorinostat and hsp90 was immunoprecipitated from the cell lysates. The immunoprecipitates were resolved on 10% SDS PAGE and hsp90 acetylation was detected using anti-acetylated lysine antibody. Binding of co-chaperones to hsp90 was detected by immunoblotting for p23 and cdc37. Total hsp90 immunopreciptated was detected by immunoblotting for hsp90. C) To detect binding of client proteins to hsp90 following vorinostat treatment, JeKo-1 cells were treated with indicated concentration of vorinostat and hsp90 was immunoprecipitated from the resulting cell lysates. CDK4 and c-RAF bound to hsp90 was detected by immunoblotting.
Figure 3. Treatment with vorinostat and 17-DMAG attenuates the intracellular levels of c-Myc, c-RAF, AKT and CDK4 in a dose-dependent manner.
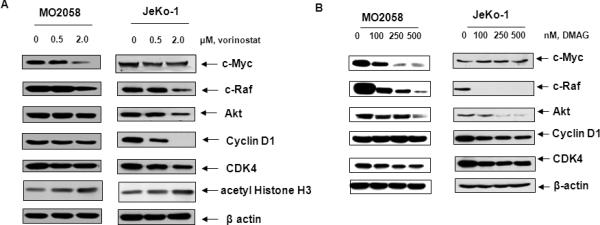
A & B) Cell lysates from JeKo-1 and Mo2058 cells exposed to vorinostat and 17-DMAG were immunoblotted for c-Myc, c-Raf, AKT, cyclin D1, CDK4 and acetylated histone H3. The levels of β-actin served as the loading control.
Co-treatment with vorinostat and 17-DMAG synergistically induces apoptosis in MCL cells
Next, we determined the apoptotic effects of the combination of vorinostat and DMAG in Z138, JeKo-1 and MO2058 cells. As shown in Figure 4A and 4B, co-treatment with vorinostat significantly enhanced the apoptosis induced by DMAG. Furthermore, combined treatment with vorinostat and DMAG synergistically induced apoptosis in MO2058, JeKo-1 and Z138 cells, as indicated by the combination indices registering below 1.0 (Figure 4C). We also determined the effect of combined treatment with DMAG and vorinostat on the levels of c-MYC and cyclin D1, as well as on the MCL relevant hsp90 client proteins CDK4, AKT and c-RAF. Figure 5A demonstrates that co-treatment with DMAG and vorinostat caused more depletion of c-MYC and cyclin D1 than treatment with either agent alone in both MO2058 and JeKo-1 cells (Figure 5A and 5B). Additionally, the combined treatment was superior to either agent alone in attenuating the levels of AKT, c-RAF and CDK4 MO2058 and JeKo-1 cells (Figure 5A and 5B). Notably, these findings suggest that increased sensitivity of MCL cells to simultaneous HDAC and hsp90 inhibition could be at least partly due to marked abrogation of pro-growth and pro-survival hsp90 client proteins and the concomitant up-regulation of the cell cycle inhibitors p21 and p27.
Figure 4. Co-treatment with vorinostat and 17-DMAG induces more apoptosis and exerts synergistic cytotoxic effects in MCL cells.

A) MO2058 cells were treated with the indicated concentrations of vorinostat and/or 17-DMAG for 48 hours. Following this, the percentage of annexin-V-stained apoptotic cells was determined by flow cytometry. B) JeKo-1 cells were treated with the indicated concentrations of vorinostat and/or 17-DMAG for 48 hours. Following this treatment the percent of nonviable cells was determined by the trypan blue exclusion method. The values represent the means ± SE of three experiments C) MO2058, JeKo-1 and Z138 cells were exposed to varying concentrations of vorinostat (between 500 and 2500 nM) and 17-DMAG (between 100 and 500 nM) at a fixed ratio (5:1) for 48 hours, and apoptosis was measured by annexin V/propidium iodide staining. Combination index values for each fraction affected were determined using a Calcusyn; Biosoft. Combination index values <1.0 correspond to synergistic interactions.
Figure 5. Co-treatment with vorinostat and 17-DMAG causes up-regulation of cell cycle inhibitors and enhanced depletion of hsp90 client proteins compared to either treatment alone.
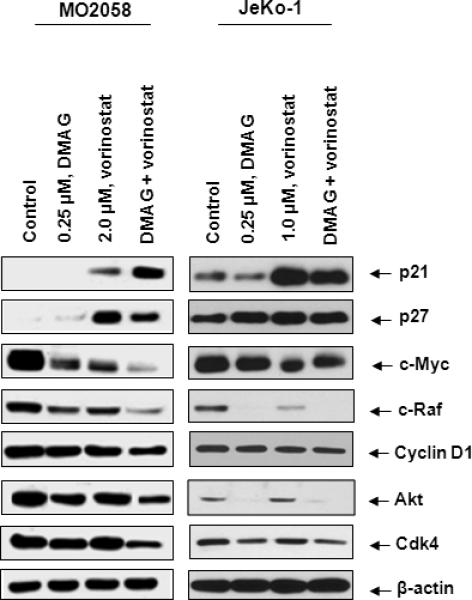
A) MO2058 and JeKo-1 cells were treated with vorinostat and/or 17-DMAG for 24 hours. Western blot analyses of p21, p27, c-Myc, cRaf, cyclin D1, AKT and CDK4 were performed on the cell lysates. The levels of β-actin served as the loading control.
Co-treatment with SAHA And 17-DMAG affects primary MCL cell survival with little or no effect on normal CD34 positive cells
Finally, we also tested the effect of co-treatment with vorinostat and DMAG in primary MCL versus normal CD34+ hemopoitic progenitor cells. Our data indicate that treatment with vorinostat and DMAG mediated loss of cell viability in primary MCL cells (Figure 6A). Additionally, co-treatment with vorinostat significantly enhanced DMAG mediated loss of viability of primary MCL cells (Figure 6A). In contrast, normal CD34+ hematopoietic progenitor cells were relatively less sensitive than primary MCL cells to vorinostat and DMAG, and co-treatment with vorinostat did not significantly increase DMAG mediated loss of cell viability of normal CD34+ hematopoietic progenitor cells (Figure 6B).
Figure 6. Treatment with vorinostat and/or 17-DMAG induces cell death of primary lymphoma cells but not CD34+ hematopoietic progenitor cells.
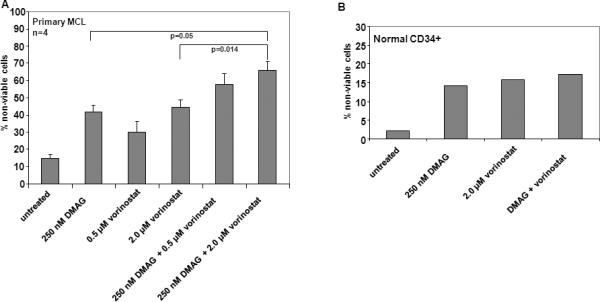
A) Primary lymphoma cells were treated with the indicated concentrations of vorinostat and or 17-DMAG for 48 hours and the percentage of nonviable cells was determined by Trypan blue exclusion method. The values represent an average of four individual patient samples. B) CD34+ normal bone marrow hematopoietic progenitor cells were treated with the indicated concentration (nM) of vorinostat and/or 17-DMAG for 48 hours. Following this the percentage of non-viable cells was determined by Trypan blue exclusion method. The values represent an average of two independent experiments performed in duplicates.
Discussion
Several recent studies have documented clinical responses and benefit secondary to a variety of novel agents in the treatment of MCL. These include the mTOR kinase inhibitor temsirolimus, the proteasome inhibitor bortezomib, the immunomodulatory agent lenalidomde and the inhibitor of cyclin-dependent kinases flavipiridol.41-44 However, none of these therapies induce long term remission, and patients ultimately succumb to the disease. Because of this poor clinical outcome, there is a critical need to develop more effective combination therapies for MCL. Both an hsp90 inhibitor and a pan-HDAC inhibitor has individually shown activity against MCL cells.26,45 Based on this, here, we determined the growth inhibitory and apoptotic activity of treatment with the pan-HDAC inhibitor vorinostat and/or the hsp90 inhibitor DMAG against human MCL cells. Our findings demonstrate that individually both vorinostat and DMAG induce cell cycle growth arrest and apoptosis of cultured human MCL cells. Moreover, co-treatment with vorinostat and DMAG synergistically induce apoptosis of human MCL cells.
Several combination regimens include combination therapies with bortezomib and rituximab and cyclophosphamide which caused significantly enhanced apoptosis in MCL cell lines and primary cells.49 Combination of SAHA with bortezomib was recently shown to exert synergistic cytotoxic effect in MCL cell lines with an increase in the production of ROS, enhanced caspase 3, 8 and 9 activation and inhibition of NFκB activity.50 The combination of the mTOR inhibitor RAD001 with SAHA, bortezomib, or vincristine showed synergistic anti-MCL activity while Temsirolimus induced autophagy and in combination with SAHA resulted in enhanced cytotoxicity in MCL cell lines.51,52
Treatment with Vorinostat induced cell cycle G1 accumulation of MCL cells. This was associated with and possibly due to vorinostat mediated induction of p21 and p27, as well as depletion of cyclin D1, CDK4 and c-MYC. Since cyclinD1 and c-MYC collaborate in abrogating RB function and lymphomagenesis, depletion of cyclin D1 and c-MYC could contribute to the anti-MCL activity of vorinostat plus DMAG.46,47 In MCL cells, vorinostat mediated hyperacetylation of hsp90 due to inhibition of HDAC6 is consistent with reports of a similar effect in human leukemia cells.34 Hyperacetylation of hsp90 has been shown to be associated with inhibition of chaperone association of hsp90 with its client proteins and increased binding of GA to hsp90.32,34 Decreased chaperone function of hsp90 would explain vorinostat mediated attenuation of the levels of the pro-growth and pro-survival hsp90 client proteins, e.g., CDK4, AKT and c-RAF, as well as in upregulation of hsp70.16,17 Increased binding of GA to hyperacetylated hsp90 may be responsible for greater disruption of chaperone function of hsp90, as has been previously described, which may explain greater depletion of the MCL relevant, pro-growth and pro-survival hsp90 client proteins in vorinostat and DMAG treated MCL cells.32,37 Although cyclin D1 has not been shown to bind directly to hsp90, treatment with the HA-HDIs trichstatin A (TSA) has been shown to induce the nuclear export of cyclin D1 and upregulate Skp2, a component of the SCF complex, that participates in the ubiquitylation and proteasomal degradation of Cyclin D1.48 Treatment with DMAG at higher concentrations (0.5 μM) caused G2-M arrest suggesting that hsp90 inhibition affects cell cycle checkpoints other than the RBCDK4/cyclin D1-dependent G1-S checkpoint. Indeed, treatment with 17-AAG has been shown to arrest cells in the G2-M phase.49 Recent studies have shown that vorinostat treatment depletes the association of hsp90 with Aurora A and Aurora B, which leads to attenuation of Aurora A and B that are pivotally involved in regulating spindle assembly, chromosome segregation and cytokinesis.30
Apart from disrupting the hsp90 chaperone machinery and depleting pro-growth and pro-survival hsp90 client proteins, it is well recognized that HA-HDIs and hsp90 inhibitors induce growth arrest and apoptosis in numerous tumor models through molecular mechanisms that lower the threshold of apoptosis either by depleting the levels of anti-apoptotic proteins Bcl-2, Bcl-xL and Mcl-1 and/or inducing the pro-apoptotic proteins Bax, Bak and Bim, or by inducing the reactive oxygen species.16,17,27,50 Furthermore, vorinostat has been demonstrated to induce not only apoptotic but also autophagic cell death, if the activity of the caspases is blocked.51 There are also other notable mechanism that may be responsible for the relatively selective anti-MCL versus normal CD34+ bone marrow progenitor cell activity of the combination of vorinostat and DMAG. Recently, both vorinostat and hsp90 inhibitors have been shown to induce unfolded protein response (UPR) and endoplasmic reticulum (ER) stress.52,53 Inhibition of HDAC6 by vorinostat also abrogates the formation of aggresome, in which unfolded proteins are protectively sequestered, thereby accentuating the UPR and causing lethal ER stress.35,36 These findings could also explain why co-treatment with vorinostat enhances DMAG-induced apoptosis of MCL cells. Collectively, these mechanisms could potentially be contributing to the synergistic activity of the combination of vorinostat and DMAG against MCL cells. Clearly, findings presented here create a strong rationale to test the in vivo efficacy of the combination against human MCL cells.
References
- 1.Andersen NS, Jensen MK, de Nully Brown P, Geisler CH. A Danish population-based analysis of 105 mantle cell lymphoma patients: incidences, clinical features, response, survival and prognostic factors. Eur J Cancer. 2002;38:401–8. doi: 10.1016/s0959-8049(01)00366-5. [DOI] [PubMed] [Google Scholar]
- 2.Bertoni F, Zucca E, Cotter FE. Molecular basis of mantle cell lymphoma. Br J Haematol. 2004;124:130–40. doi: 10.1046/j.1365-2141.2003.04761.x. [DOI] [PubMed] [Google Scholar]
- 3.Jares P, Colomer D, Campo E. Genetic and molecular pathogenesis of mantle cell lymphoma: perspectives for new target therapeutics. Nat Rev Cancer. 2007;7:750–62. doi: 10.1038/nrc2230. [DOI] [PubMed] [Google Scholar]
- 4.Caraway NP, Gu J, Lin P, Romaguera JE, Glassman A, Katz R. The utility of interphase fluorescence in situ hybridization for the detection of the translocation t(11;14)(q13;q32) in the diagnosis of mantle cell lymphoma on fine-needle aspiration specimens. Cancer. 2005;105:110–8. doi: 10.1002/cncr.20923. [DOI] [PubMed] [Google Scholar]
- 5.Pham LV, Tamayo AT, Yoshimura LC, Lo P, Ford RJ. Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J Immunol. 2003;171:88–95. doi: 10.4049/jimmunol.171.1.88. [DOI] [PubMed] [Google Scholar]
- 6.Marzec M, Kasprzycka M, Lai R, Gladden AB, Wlodarski P, Tomczak E, et al. Mantle cell lymphoma cells express predominantly cyclin D1a isoform and are highly sensitive to selective inhibition of CDK4 kinase activity. Blood. 2006;108:1744–50. doi: 10.1182/blood-2006-04-016634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hutter G, Scheubner M, Zimmermann Y, Kalla J, Katzenberger T, Hubler K, et al. Differential effect of epigenetic alterations and genomic deletions of CDK inhibitors [p16(INK4a), p15(INK4b), p14(ARF)] in mantle cell lymphoma. Genes, Chromosomes & Cancer. 2006;45:203–10. doi: 10.1002/gcc.20277. [DOI] [PubMed] [Google Scholar]
- 8.Chen RW, Bemis LT, Amato CM, Myint H, Tran H, Birks DK, et al. Truncation in CCND1 mRNA alters miR-16−1 regulation in mantle cell lymphoma. Blood. 2008;112:822–9. doi: 10.1182/blood-2008-03-142182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertoni F, Rinaldi A, Zucca E, Cavalli F. Update on the molecular biology of mantle cell lymphoma. Hematol Oncol. 2006;24:22–7. doi: 10.1002/hon.767. [DOI] [PubMed] [Google Scholar]
- 10.Staudt LM, Dave S. The biology of human lymphoid malignancies revealed by gene expression profiling. Adv Immunol. 2005;87:163–208. doi: 10.1016/S0065-2776(05)87005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kienle D, Katzenberger T, Ott G, Saupe D, Benner A, Kohlhammer H, et al. Quantitative gene expression deregulation in mantle-cell lymphoma: correlation with clinical and biologic factors. J Clin Oncol. 2007;25:2770–7. doi: 10.1200/JCO.2006.08.7999. [DOI] [PubMed] [Google Scholar]
- 12.O'Connor OA. Mantle cell lymphoma: identifying novel molecular targets in growth and survival pathways. Hematology / the Education Program of the American Society of Hematology. 2007;2007:270–6. doi: 10.1182/asheducation-2007.1.270. [DOI] [PubMed] [Google Scholar]
- 13.Hui D, Dabbagh L, Hanson J, Amin HM, Lai R. High ZAP-70 expression correlates with worse clinical outcome in mantle cell lymphoma. Leukemia. 2006;20:1905–8. doi: 10.1038/sj.leu.2404362. [DOI] [PubMed] [Google Scholar]
- 14.Rudelius M, Pittaluga S, Nishizuka S, Pham TH, Fend F, Jaffe ES, et al. Constitutive activation of Akt contributes to the pathogenesis and survival of mantle cell lymphoma. Blood. 2006;108:1668–76. doi: 10.1182/blood-2006-04-015586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dal Col J, Zancai P, Terrin L, Guidoboni M, Ponzoni M, Pavan A, et al. Distinct functional significance of Akt and mTOR constitutive activation in mantle cell lymphoma. Blood. 2008;111:5142–51. doi: 10.1182/blood-2007-07-103481. [DOI] [PubMed] [Google Scholar]
- 16.Whitesell L, Lindquist SL. Hsp90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–72. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 17.Pearl LH, Prodromou C, Workman P. The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem J. 2008;410:439–53. doi: 10.1042/BJ20071640. [DOI] [PubMed] [Google Scholar]
- 18.Valbuena JR, Rassidakis GZ, Lin P, Atwell C, Georgakis GV, Younes A, et al. Expression of heat-shock protein-90 in non-Hodgkin's lymphomas. Mod Pathol. 2005;18:1343–9. doi: 10.1038/modpathol.3800459. [DOI] [PubMed] [Google Scholar]
- 19.Roe SM, Ali MM, Meyer P, Vaughan CK, Panaretou B, Piper PW, et al. The Mechanism of Hsp90 regulation by the protein kinase-specific co-chaperone p50 (cdc37). Cell. 2004;116:87–98. doi: 10.1016/s0092-8674(03)01027-4. [DOI] [PubMed] [Google Scholar]
- 20.Ali MM, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, et al. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature. 2006;440:1013–7. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen G, Cao P, Goeddel DV. TNF-induced recruitment and activation of the IKK complex require cdc37 and hsp90. Mol Cell. 2002;9:401–10. doi: 10.1016/s1097-2765(02)00450-1. [DOI] [PubMed] [Google Scholar]
- 22.Castro JE, Prada CE, Loria O, Kamal A, Chen L, Burrows FJ, et al. ZAP-70 is a novel conditional heat shock protein 90 (hsp90) client: inhibition of hsp90 leads to ZAP-70 degradation, apoptosis and impaired signaling in chronic lymphocytic leukemia. Blood. 2005;106:2506–12. doi: 10.1182/blood-2005-03-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jascur T, Brickner H, Salles-Passador I, Barbier V, El Khissiin A, Smith B, et al. Regulation of p21 (WAF1/CIP1) stability by WISp39, a hsp90 binding TPR protein. Mol Cell. 2005;17:237–49. doi: 10.1016/j.molcel.2004.11.049. [DOI] [PubMed] [Google Scholar]
- 24.Arlander SJ, Eapen AK, Vroman BT, McDonald RJ, Toft DO, Karnitz LM. hsp90 inhibition depletes Chk1 and sensitizes tumor cell to replication stress. J Biol Chem. 2003;278:52572–7. doi: 10.1074/jbc.M309054200. [DOI] [PubMed] [Google Scholar]
- 25.Vaughan CK, Gohlke U, Sobott F, Good VM, Ali MM, Prodromou C, et al. Structure of an Hsp90-Cdc37-Cdk4 complex. Mol Cell. 2006;23:697–707. doi: 10.1016/j.molcel.2006.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Georgakis GV, Li Y, Younes A. The heat shock protein 90 inhibitor 17-AAG induces cell cycle arrest and apoptosis in Mantle Cell Lymphoma cell lines by depleting cyclin D1, Akt, Bid and activating caspase 9. Br J of Haematol. 2006;135:68–71. doi: 10.1111/j.1365-2141.2006.06247.x. [DOI] [PubMed] [Google Scholar]
- 27.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 28.George P, Bali P, Annavarapu S, Scuto A, Fiskus W, Guo F, et al. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against human CMLBC cells and AML cells with activating mutation of FLT-3. Blood. 2005;105:1768–76. doi: 10.1182/blood-2004-09-3413. [DOI] [PubMed] [Google Scholar]
- 29.Duvic M, Talpur R, Chiao N, Chiao J. Phase II trial of oral suberoylanilide hydroxamic acid (SAHA) for cutaneous t-cell lymphoma (CTCL) and peripheral t-cell lymphoma (PTCL). Blood. 2003;102:625. doi: 10.1182/blood-2006-06-025999. [abstract] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fiskus W, Wang Y, Joshi R, Rao R, Yang Y, Chen J, et al. Co-treatment with vorinostat enhances activity of MK-0457 (VX-680) against acute and chronic myelogenous leukemia cells. Clin Cancer Res. 2008;14:6106–15. doi: 10.1158/1078-0432.CCR-08-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bali P, Pranpat M, Swaby R, Fiskus W, Yamaguchi H, Balasis M, et al. Activity of suberoylanilide hydroxamic acid against human breast cancer cells with amplification of her-2. Clin Cancer Res. 2005;11:6382–9. doi: 10.1158/1078-0432.CCR-05-0344. [DOI] [PubMed] [Google Scholar]
- 32.Rao R, Fiskus W, Yang Y, Lee P, Joshi R, Fernandez P, et al. HDAC6 inhibition enhances 17-AAG--mediated abrogation of hsp90 chaperone function in human leukemia cells. Blood. 2008;112:1886–93. doi: 10.1182/blood-2008-03-143644. [DOI] [PubMed] [Google Scholar]
- 33.Sakajiri S, Kumagai T, Kawamata N, Saitoh T, Said JW, Koeffler HP. Histone deacetylase inhibitors profoundly decrease proliferation of human lymphoid cancer cell lines. Exp Hematol. 2005;33:53–61. doi: 10.1016/j.exphem.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 34.Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280:26729–34. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 35.Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–7. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 36.Boyault C, Zhang Y, Fritah S, Caron C, Gilquin B, Kwon SH, et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007;21:2172–81. doi: 10.1101/gad.436407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Y, Rao R, Shen J, Tang Y, Fiskus W, Nechtman J, et al. Role of acetylation and extracellular location of heat shock protein 90alpha in tumor cell invasion. Cancer Res. 2008;68:4833–42. doi: 10.1158/0008-5472.CAN-08-0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 39.Fiskus W, Pranpat M, Balasis M, Bali P, Estrella V, Kumaraswamy S, et al. Cotreatment with vorinostat (suberoylanilide hydroxamic acid) enhances activity of dasatinib (BMS-354825) against imatinib mesylate-sensitive or imatinib mesylate-resistant chronic myelogenous leukemia cells. Clin Cancer Res. 2006;12:5869–78. doi: 10.1158/1078-0432.CCR-06-0980. [DOI] [PubMed] [Google Scholar]
- 40.Guo F, Rocha K, Bali P, Pranpat M, Fiskus W, Boyapalle S, et al. Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. Cancer Res. 2005;65:10536–44. doi: 10.1158/0008-5472.CAN-05-1799. [DOI] [PubMed] [Google Scholar]
- 41.Witzig TE, Geyer SM, Ghobrial I, Inwards DJ, Fonseca R, Kurtin P, et al. Phase II trial of single-agent temsirolumus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol. 2005;23:5347–56. doi: 10.1200/JCO.2005.13.466. [DOI] [PubMed] [Google Scholar]
- 42.O'Connor OA, Wright J, Moskowitz C, Muzzy J, MacGregor-Cortelli B, Stubblefield M, et al. Phase II Clinical Experience with the novel proteasome inhibitor bortezomib in patients with indolent non-Hodgkin's lymphoma and mantle cell lymphoma. J Clin Oncol. 2005;23:676–84. doi: 10.1200/JCO.2005.02.050. [DOI] [PubMed] [Google Scholar]
- 43.Wiernik PH, Lossos IS, Tuscano JM, Justice G, Vose JM, Cole CE, et al. Lenalidomide monotherapy in relapsed or refractory aggressive non-Hodgkin's lymphoma. J Clin Oncol. 2008;26:4952–7. doi: 10.1200/JCO.2007.15.3429. [DOI] [PubMed] [Google Scholar]
- 44.Smith MR. Mantle cell lymphoma: advances in biology and therapy. Curr Opin Hematol. 2008;15:415–21. doi: 10.1097/MOH.0b013e328302c9c5. [DOI] [PubMed] [Google Scholar]
- 45.Heider U, Kaiser M, Sterz J, Zavrski I, Jakob C, Fleissner C, et al. Histone deacetylase inhibitors reduce VEGF production and induce growth suppression and apoptosis in human mantle cell lymphoma. Eur J Haematol. 2006;76:42–50. doi: 10.1111/j.1600-0609.2005.00546.x. [DOI] [PubMed] [Google Scholar]
- 46.Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev. 1993;7:331–42. doi: 10.1101/gad.7.3.331. [DOI] [PubMed] [Google Scholar]
- 47.Bodrug SE, Warner BJ, Bath ML, Lindeman GJ, Harris AW, Adams JM. Cyclin D1 transgene impedes lymphocyte maturation and collaborates in lymphomagenesis with the myc gene. EMBO J. 1994;13:2124–30. doi: 10.1002/j.1460-2075.1994.tb06488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alao JP, Lam EW, Ali S, Buluwela L, Bordogna W, Lockey P, et al. Histone deacetylase inhibitor trichostatin A represses estrogen receptor alpha-dependent transcription and promotes proteasomal degradation of cyclin D1 in human breast carcinoma cell lines. Clin Cancer Res. 2004;10:8094–104. doi: 10.1158/1078-0432.CCR-04-1023. [DOI] [PubMed] [Google Scholar]
- 49.Niikura Y, Ohta S, Vandenbeldt KJ, Abdulle R, McEwen BF, Kitagawa K. 17-AAG, an Hsp90 inhibitor, causes kinetochore defects: a novel mechanism by which 17-AAG inhibits cell proliferation. Oncogene. 2006;25:4133–46. doi: 10.1038/sj.onc.1209461. [DOI] [PubMed] [Google Scholar]
- 50.Bhalla KN. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J Clin Oncol. 2005;23:3971–93. doi: 10.1200/JCO.2005.16.600. [DOI] [PubMed] [Google Scholar]
- 51.Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci. 2004;101:18030–5. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci. 2005;102:8567–72. doi: 10.1073/pnas.0503221102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davenport EL, Moore HE, Dunlop AS, Sharp SY, Workman P, Morgan GJ, et al. Heat shock protein inhibition is associated with activation of the unfolded protein response pathway in myeloma plasma cells. Blood. 2007;110:2641–9. doi: 10.1182/blood-2006-11-053728. [DOI] [PubMed] [Google Scholar]