

Abstract
Doxorubicin (DOX) is a widely used antitumor drug, but its application is limited due to its cardiotoxic side effects. Hsp20 has been recently shown to protect cardiomyocytes against apoptosis, induced by ischemia/reperfusion injury or by prolonged β-agonist stimulation. However, it is not clear whether Hsp20 would exert similar protective effects against DOX-induced cardiac injury. Actually, DOX-treatment was associated with down-regulation of Hsp20 in the heart. To elucidate the role of Hsp20 in DOX-triggered cardiac toxicity, Hsp20 was first overexpressed ex vivo by adenovirus-mediated gene delivery. Increased Hsp20 levels conferred higher resistance to DOX-induced cell death, compared to GFP-control. Furthermore, cardiac-specific overexpression of Hsp20 in vivo significantly ameliorated acute DOX-triggered cardiomyocyte apoptosis and animal mortality. Hsp20-transgenic mice also showed improved cardiac function and prolonged survival after chronic administration of DOX. The mechanisms underlying these beneficial effects were associated with preserved Akt phosphorylation/activity and attenuation of DOX-induced oxidative stress. Co-immunoprecipitation studies revealed an interaction between Hsp20 and phosphorylated Akt. Accordingly, BAD phosphorylation was preserved and cleaved caspase-3 was decreased in DOX-treated Hsp20-TG hearts, consistent with the Hsp20's anti-apoptotic effects. Parallel ex vivo experiments showed that either infection with a dominant-negative Akt adenovirus or pre-incubation of cardiomyocytes with the PI3-kinase inhibitors significantly attenuated the protective effects of Hsp20. Taken together, our findings indicate that overexpression of Hsp20 inhibits DOX-triggered cardiac injury, and these beneficial effects appear to be dependent on Akt activation. Thus, Hsp20 may constitute a new therapeutic target in ameliorating the cardiotoxic effects of DOX-treatment in cancer patients.
Keywords: apoptosis, cardiomyopathy, doxorubicin, heat-shock protein, Akt
Introduction
Doxorubicin (DOX), an anthracycline antibiotic, is a highly effective chemotherapeutic agent used in the treatment of solid and hematopoietic tumors; however, a major limiting factor for the clinical use of DOX is its cumulative, irreversible cardiac toxicity.1-3 In fact, multiple intravenous DOX treatments over a period of several months result in the development of cardiomyopathy and congestive heart failure (CHF) in humans.3 The precise cellular mechanisms responsible for this chronic cardiotoxicity of DOX remain enigmatic, but the antitumor activity of DOX is likely to be distinct from the mechanism of its cardiotoxicity.4, 5 Accumulating evidence indicates that DOX-induced cardiomyopathy is mainly caused by increased oxidant production, which eventually leads to the apoptotic loss of cardiomyocytes.2, 3, 6 Therefore, we speculated that suppression of apoptosis may largely rescue DOX-triggered cardiotoxicity.
Heat shock proteins (Hsps) play important roles in cellular stress-resistance and development of tolerance as an adaptive response after exposure to various stimuli.7 It has been found that thermal preconditioning effectively protected cardiomyocytes against DOX-induced apoptosis; 8, 9 whereas this protective effect was attenuated by knockdown of Hsp70 by antisense mRNA in cardiomyocytes, 8 suggesting that heat shock proteins may regulate DOX-triggered cardiac injury. Other studies further demonstrated that elevated Hsp27 and Hsp70 levels were associated with DOX resistance in tumor cells.10 What is more, it has been suggested that pharmacologically or physiologically induced Hsp60 and Hsp70 overexpression is involved in the cardioprotection against DOX.11, 12 However, DOX-induced expression alterations of Hsps in the heart are currently not clear. Furthermore, it is still questioned whether heart-specific overexpression of a heat shock protein could protect against DOX-induced cardiotoxicity. Recently, Hsp20 has been shown to protect hearts against cardiac myocyte apoptosis, induced by ischemia/reperfusion injury in vivo,13 and elevated Hsp20 in the cardiomyocytes renders protection against isoproterenol-triggered apoptosis in vitro and in vivo.14, 15 However, the potential benefits of Hsp20 action on DOX-induced cardiac injury and its underlying mechanism(s) are largely unknown.
Therefore, on the basis of our previous findings, we postulated that increased Hsp20 levels would alleviate DOX-triggered cardiotoxicity. Our results demonstrate that Hsp20 could protect against DOX-induced cardiomyocyte death in vitro and in vivo, and further prolong the mouse survival after either acute or chronic administration of DOX. Importantly, the mechanism underlying the cardioprotective effects of Hsp20 against DOX toxicity involves: 1) interaction of Hsp20 with phosphorylated Akt, which protects Akt against dephosphorylation by phosphatase PP1; and 2) attenuation of DOX-triggered oxidative stress.
Materials and Methods
An expanded Materials and Methods section is available in the online data supplement at http://circres.ahajournals.org.
Animal Preparation
Generation of cardiac-specific overexpressed Hsp20 mice has been previously described.13 Male wild-type (WT) and transgenic (TG) mice, inbred on a FVB/N background, were studied at 8 to 10 weeks. DOX was administered by intraperitoneal (ip) injection at one dose of 20 mg/kg or a weekly dose of 3 - 4mg/kg to a cumulative amount of 20 mg/kg. Control mice received injections of saline to a comparable volume. All procedures were in accordance with institutional guidelines for animal research.
Additional Methods
An expanded Materials and Methods section containing details regarding drug treatment in vitro; generation of plasmid pcDNA3-Hsp20 and adenovirus vectors: Ad. Hsp20 and Ad.dnAkt; cardiomyocyte isolation and cell culture; cell viability and apoptosis assay; cardiac contraction measurements and cardiotoxicity assay; western blot analysis; co-immunoprecipitation and in vitro Akt kinase activity assay; oxidative stress and ROS assay is available in the online data supplement.
Results
Effects of DOX on the Expression of Major Hsps in the Murine Heart
To assess the regulatory roles of cardiac Hsps in response to DOX treatment, we examined the expression profiles of the six major Hsps (Hsp90, Hsp70, Hsp60, Hsp27, αB-crystallin and Hsp20) at different time points, following DOX ip injection. Western blots and quantitative results (Figure 1) indicated that alterations in the levels of Hsp70 and Hsp60 exhibited a biphasic response: a) they greatly increased at 1 hour after DOX injection, and returned to basal levels by 2 hours; b) they increased again at 4-12 hours, and significantly decreased below basal levels at 2-3 days after DOX treatment. There was no significant change in Hsp90 expression, while DOX increased Hsp27 content by 2 fold at 30 min, and this increase was maintained up to 3 days. The levels of αB-crystallin were transiently increased at 30 min to 1 hour, and then returned to basal. While a similar transient increase was also observed with Hsp20 expression, its levels decreased by 40% at 2-3 days after DOX injection. These results demonstrate that the expression pattern of various Hsps is altered in the heart upon DOX insult, suggesting that these Hsps may be involved in DOX-induced cardiomyopathy via different mechanism(s).
Figure 1.
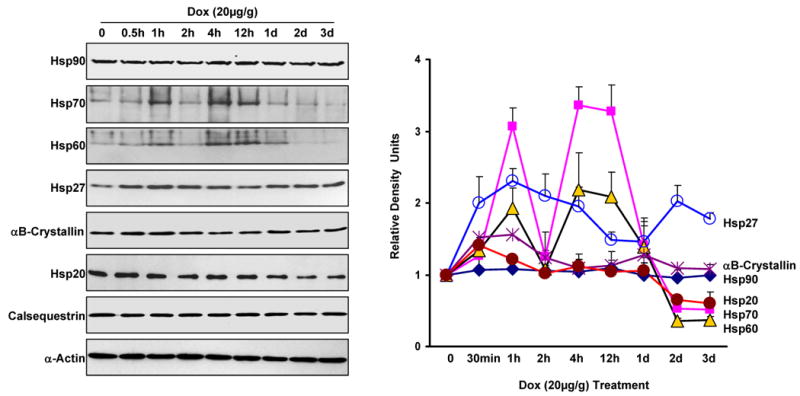
Time course of major Hsps' expression in the mouse heart after i.p. administration of doxorubicin (20 mg/kg). At the indicated intervals, mouse hearts were excised and homogenized to assess major Hsps' expression by Western blot analysis (Left panel). The right panel shows the quantitative results from the Western-blots. Calsequestrin and α-Actin as internal controls (n=4 hearts for each time point).
DOX-Induced Cardiomyocyte Death and Apoptosis is Suppressed by Increased Hsp20 Levels
Several studies have shown that some Hsps can act as negative regulators of DOX-triggered apoptotic and necrotic cell death, such as Hsp10 and Hsp60, which have been found to modulate DOX-induced mitochondrial apoptosis signaling in neonatal cardiomyocytes.16 However, the underlying mechanisms are still remained to be clarified. To investigate whether increased Hsp20 levels could have an inhibitory effect on DOX induced cardiomyocyte death, we first infected H9c2 cells with Ad.Hsp20 or Ad.GFP for 24 h and subsequently subjected them to DOX treatment. After 24 h of treatment with DOX at 0.5μM, 40% of the Ad.GFP-infected cells were not viable as observed by phase-contrast microscopy (Figure 2A), and measured by the MTS assay (Figure 2B). However, DOX-induced cell death was markedly diminished in the Ad.Hsp20-infected group (Figure 2A and B). The cytoprotective effect of Hsp20 was also observed in cells treated with different concentrations of DOX or infected with different concentrations of Ad.Hsp20, which was dependent on the level of Hsp20 overexpression (Figure 2C and D), suggesting that Hsp20 acts directly on cardiomyocytes to inhibit DOX-induced cell death.
Figure 2.
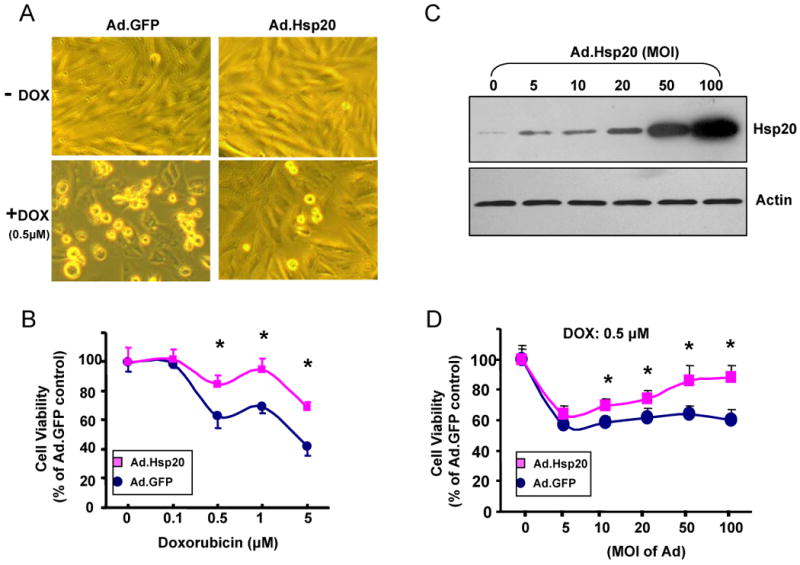
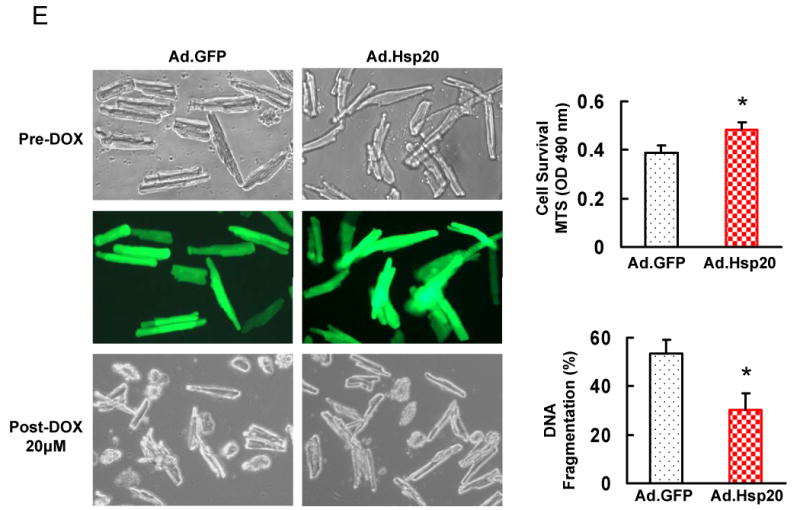
Effect of increased Hsp20 expression on cardiaomyocyte survival after doxorubicin treatment. (A) Photomicrographs of Ad.Hsp20-infected or Ad.GFP-infected H9c2 cells (50 MOI) were taken in the presence or absence of doxorubicin (0.5 μM) for 24 h. (B) Ad.Hsp20-infected H9c2 cells showed significantly higher cell viability than Ad.GFP-infected cells after treatment with doxorubicin at 0.5, 1 or 5 μM for 24 h. (C) Western-blots showed increased Hsp20 expression upon increased multiplicity of infection (MOI) of adenoviral vector. (D) Protection of H9c2 cells against doxorubicin was shown to be dependent on the Hsp20 expression levels. (E) There was no morphological change between the empty vector Ad.GFP- and Ad.Hsp20-infected myocytes, and more than 95% adult rat cardiomyocytes were infected by recombinant adenovirus vector after 24 h, as indicated by GFP fluorescence (Magnification 200×). Hsp20-overexpressed cardiomyocytes were resistant to doxorubicin-induced cell death, as evidenced by decreased number of round-shaped cardiomyocytes (Magnification 100×), and MTS analysis, as well as DNA fragmentation assay, determined by cell-death-detection ELISA kit (*P < 0.05 versus control Ad.GFP). Similar results were observed in three additional, independent experiments.
To further confirm that Hsp20 acts as a survival factor in cardiomyocytes, ventricular myocytes were isolated from adult rat hearts, and infected with Ad.GFP or Ad.Hsp20. The infection efficiency reached more than 95% after 24 h (Figure 2E). These infected-cells were subsequently subjected to 20 μM DOX treatment for 24 h. Both MTS and DNA fragmentation assays showed that ectopic overexpression of Hsp20 protected adult rat cardiomyocytes against DOX-triggered cell death and apoptosis (Figure 2E).
Decreased DOX-Induced Cardiomyocyte Injury and Apoptosis in Hsp20-TG Hearts
To determine whether Hsp20 overexpression protects cardiomyocytes against DOX-induced apoptosis in vivo, TG mice overexpressing Hsp20 under the control of the cardiac-specific α-MHC promoter were analyzed. TG hearts showed a ten-fold increase in total (transgenic and endogenous) Hsp20 content, compared with WT mouse hearts.13 Eight-week-old WT and TG mice were subjected to one intraperitoneal (ip) injection of 20 mg/kg of DOX or saline as a control. At 4 days after the injection of DOX, the hearts of DOX-treated WT mice (Figure 3A), but not those of DOX-treated TG mice (Figure 3B), displayed focal cytoplasmic vacuolization, a hallmark of cell injury, which is consistent with previous reports.17, 18 The percentage of TUNEL-positive nuclei in the DOX-treated WT hearts was 2.5-fold higher than in DOX-treated TG hearts (Figure 3C). Accordingly, DNA fragmentation, measured by an ELISA method, was reduced by 30% in DOX-treated TG hearts, compared with controls (Figure 3D). These results indicate that overexpression of Hsp20 in vivo significantly decreased DOX-induced cardiac injury and apoptosis.
Figure 3.
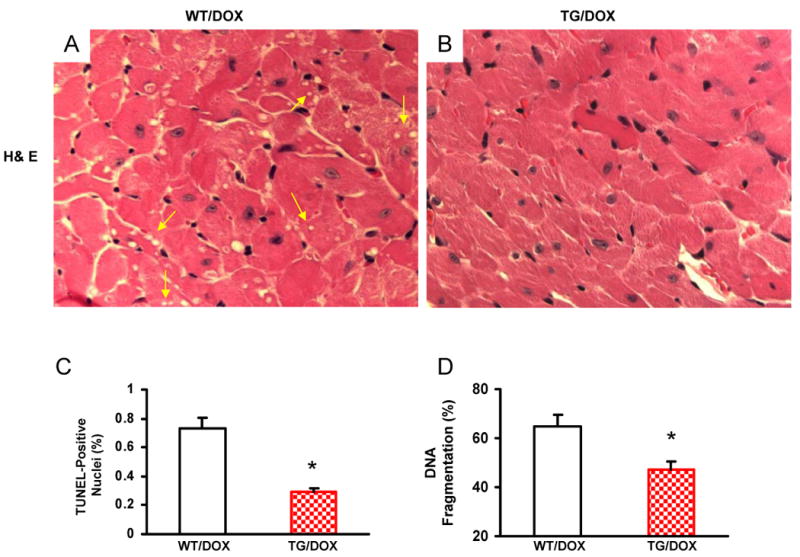
Cardiac-specific overexpression of Hsp20 attenuates cardiomyocyte death and apoptosis after acute administration of DOX. Hematoxylin and eosin (H& E) staining of mouse ventricle [A: wild-type (WT); B: Hsp20 transgenic (TG)] on day 4 after DOX treatment. Arrows indicate representative vacuolization. Quantitative results of TUNEL staining are shown in (C). n=5, with 2 sections from each heart. *P<0.01, compared with DOX-treated WT hearts. (D) DNA fragmentation assay, 200 μg heart homogenate was determined by cell-death-detection ELISA kit (*P < 0.05 versus DOX-treated WT hearts, n=6).
Preserved Cardiac Function in Hsp20-TG Mice after Acute DOX Administration
Recent studies have shown that very low levels of myocyte apoptosis (23 myocytes per 105 nuclei of animal hearts; 80-250 myocytes per 105 nuclei of human hearts) significantly affect cardiac function and survival.19 Given the marked disparity of cell injury and apoptosis in DOX-treated WT and Hsp20-TG hearts, we examined contractile function and animal survival in a separate series of experiments. 4 days after the injection of 20 mg/kg of DOX or saline, WT and TG mouse hearts were subjected to functional measurement by the Langendorff mode. As shown in Figure 4 A-D, DOX treatment caused a significant decrease in the rates of contraction (+dP/dt) and relaxation (−dP/dt) in WT hearts, accompanied by a significantly increased left ventricular end-diastolic pressure (LVEDP) and reduced left ventricular developed pressure (LVDP). However, these functional parameters showed no significant difference in TG hearts (Figure 4A-D). In addition, while 6 out of 11 WT mice died within 12 days after treatment with DOX, most TG mice (12 out of 14) survived for up to 12 days after treatment (Figure 4E, P<0.05, vs WT). These findings indicate that overexpression of Hsp20 in the heart reduced the extent of DOX-induced acute heart failure and prolonged survival.
Figure 4.

Overexpression of Hsp20 in vivo improves cardiac function after acute administration of DOX (20 mg/kg), as determined by the rates of contraction (+dP/dt) and relaxation (−dP/dt) (A and B), the left ventricular end-diastolic pressure (LVEDP) (C) and the left ventricular developed pressure (LVDP) (D). Hsp20 overexpression also prolongs the animal survival after acute treatment of DOX (E). (*P<0.05; compared with vehicle or WT group, n=5 for function measurement, and n=11-14 for animal survival observation).
Improved Cardiac Function and Survival in Hsp20-TG Mice after Chronic DOX Administration
Since patients typically receive lower levels of DOX given over many weeks, 2, 3 we further investigated whether cardiac-specific overexpression of Hsp20 could improve myocardial function and prolong survival in a chronic model of DOX-induced cardiotoxicity. To develop such a chronic model, an initial DOX dose of 4 mg/kg was selected and mice were injected weekly for 5 weeks. The survival curve of the treated groups indicated that there was no significant difference between WTs and TGs; although all the treated WT mice died, whereas 3 out of 9 treated TG mice survived during the 10 weeks follow-up after the last DOX injection (Figure 5A). However, in an additional chronic model, mice received 3 mg/kg weekly for 6 weeks, and we observed that all the treated TG mice survived during the 7 weeks follow-up after the last DOX administration; whereas 5 out of 12 treated WT mice died (Figure 5B, P<0.05 vs WT). To determine the alteration of myocardial function in this chronic model, one week after the last administration of DOX (3 mg/kg weekly for 6 weeks), we measured myocardial contractility by the Langendorff preparation to avoid the in vivo confounding effects, such as loading conditions and neurohormonal factors. As shown in Figure 5C, the rates of contraction (+dP/dt) and relaxation (−dP/dt) were significantly improved, accompanied by a reduced LVEDP and an increased LVDP in DOX-treated TG hearts, compared with DOX-treated WT hearts.
Figure 5.

Overexpression of Hsp20 in vivo prolongs the animal survival (A: 4mg/kg/week for 5 weeks; B, 3 mg/kg/week for 6 weeks) and improves cardiac function (C) after chronic administration of DOX. Cardiac function was measured by ex vivo Langendorff preparations at 1 week after last i.p injection of DOX (3 mg/kg weekly for 6 times) (*P<0.05; compared with WT group, n=9-12 for animal survival observation, and n=4-5 for functional measurement).
Akt-BAD Signaling Pathway is Preserved in DOX–Treated Hsp20-TG Hearts
To elucidate the possible mechanism(s) of Hsp20 cardioprotection against DOX, we first assessed other major Hsps' expression levels in acute DOX-treated TG hearts and controls. Western blots showed that overexpression of Hsp20 did not alter the expression levels of other major Hsps (Hsp90, Hsp70, Hsp60, Hsp27, and αB-crystallin) either in DOX-treated or saline-treated hearts, compared with WT hearts (Online Figure I), suggesting that Hsp20 overexpression does not result in compensatory adaptation by other Hsps, and clearly indicates that the sole increase in Hsp20 levels can lead to protection against DOX-induced cardiotoxicity.
A number of studies have shown that two classes of cell growth and survival/death signaling pathways, the PI3K/Akt and MAPKs pathways, play important roles in regulating cell survival. 20, 21 Our results showed that there were no differences in activation of MAPK signaling between WT hearts and TGs after acute DOX administration (Online Figure II). However, DOX-treated WT hearts revealed significantly decreased activation of Akt (generally referred to as Akt1 hereafter), as determined by immunoblotting for phosphorylation of either Thr308 or Ser473. Importantly, phosphorylation of Akt was preserved in DOX-treated TG hearts (Figure 6A), which was further supported by in vitro kinase activity assays, using GSK-3α/β as a substrate (Figure 6B). Surprisingly, we found that this immuno-complex of pSer 473-Akt contained Hsp20 (Figure 6B). Furthermore, reciprocal co-immunoprecipitation with an Hsp20 antibody showed an increased association between Hsp20 and phosphorylated Akt in DOX-treated Hsp20-TG hearts (Figure 6C). To further examine whether Hsp20 interacts with non-phosphorylated isoform of Akt, we co-infected H9c2 cells with a nonphosphorylatable form of Akt1-AAA (Ad.dnAkt) and Ad.Hsp20. While Ad.Hsp20/Ad.dnAkt-cell lysates revealed a significant increase in total Akt level (the most is exogenous non-phosphorylated Akt1), immunoprecipitation experiments with the Hsp20 antibody showed that Hsp20-associated Akt levels were similar to those in the Ad.GFP control (Figure 6D), suggesting that Hsp20 may selectively interact with phosphorylated Akt. In addition, co-immunoprecipitation results indicated that Hsp20 was associated with not only Akt1, but also Akt2 (Online Figure III) in the murine heart.
Figure 6.
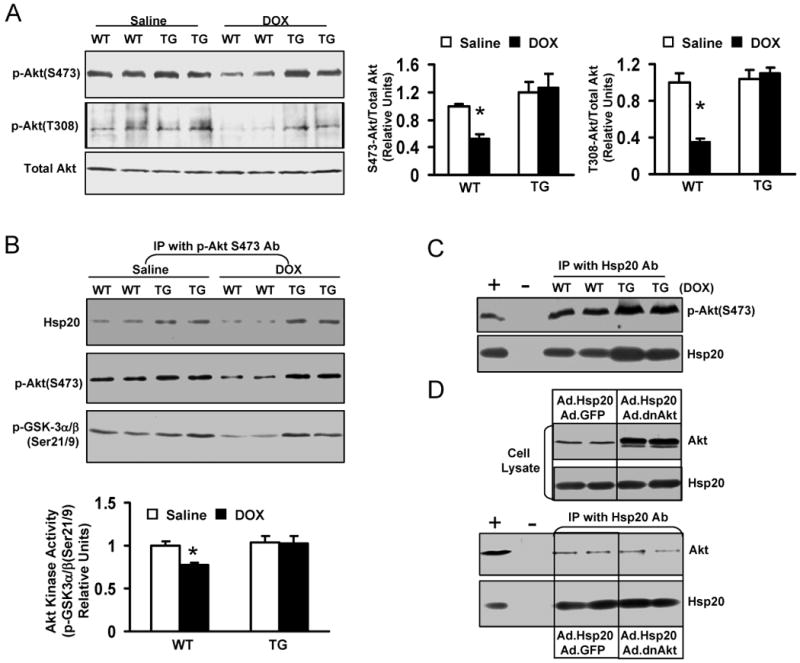
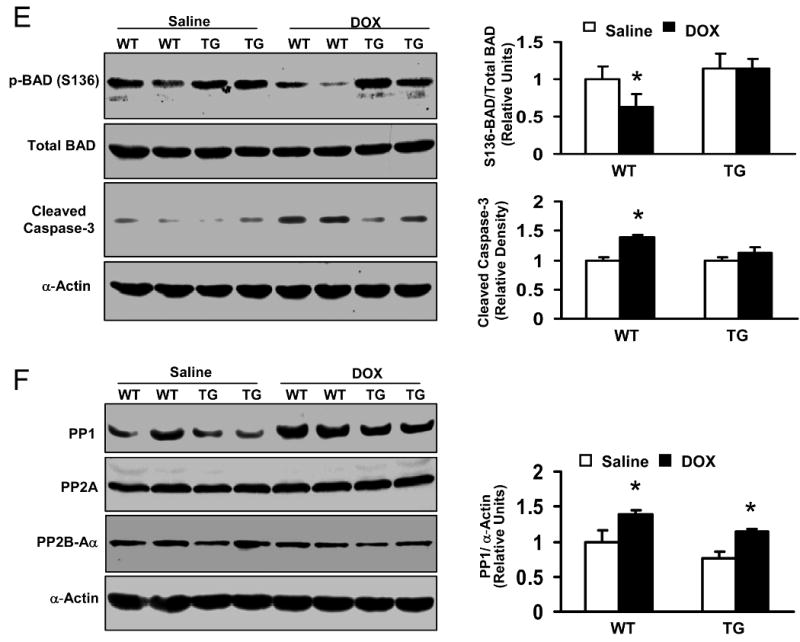
Effects of Hsp20 on the expression of Akt-BAD-caspase3 signaling cascades in the DOX-treated hearts. Hearts were excised on day 4 after acute administration of DOX (20 mg/kg), homogenized with lysis buffer, and subjected to Western-blotting analysis. (A) The phosphorylation levels of Akt at either S473 or T308 site were significantly decreased in WT hearts after DOX treatment, while there were no alterations in Hsp20-TG hearts. (B) In vitro Akt kinase activity assay by using GST-fused GSK-3α/β crosstide corresponding to residues surrounding GSK-3α/β (Ser21/9) as a substrate. This immunoplex was also probed with Ser473-Akt or Hsp20 antibody. The same amount of heart homogenate (1mg/200μl) was immunoprecipited with Ser473-Akt antibody. (C) Reciprocal co-immunoprecipitation with Hsp20 antibody revealed an increased association of Hsp20 with Ser473-Akt in DOX-treated TG hearts. Preimmunoprecipitated WT heart homogenate was used as positive control (+), and immunoprecipitate without Hsp20 antibody (added only protein G PLUS agarose) was used as negative control (-). (D) H9c2 cells were co-infected with a non-phosphorylatable Akt (Ad.dnAkt) and Ad.Hsp20, and 48 hours later, cells were collected and added NP40 lysis buffer. The same amount of cell lysates was subjected to SDS-PAGE or immunoprecipitated with Hsp20 antibody. Preimmunoprecipitated H9c2 cell lysate was used as positive control (+), and immunoprecipitate without Hsp20 antibody was used as negative control (-). (E) 150 μg heart homogenate was immuno-blotted with p-BAD (S136) or BAD antibody, or the cleaved caspase-3 antibody. (F) 100 μg heart homogenate was immuno-blotted with PP1, PP2A, or PP2B-Aα antibody. (n=4 hearts for each group, *P<0.01, compared with saline controls).
Given the marked disparity of Akt activity in DOX-treated hearts, we examined its downstream targets to assess their potential role in DOX-induced cardiotoxicity. Akt has been shown to promote cell survival via its ability to phosphorylate BAD at Ser136.22, 23 Accordingly, pS136-BAD was markedly reduced in DOX-treated WT hearts, but not in DOX-treated TG hearts (Figure 6E). However, another critical downstream element of the Akt cell survival pathway, GSK-3β phosphorylation (Ser 21), was unaltered in either WT or TG hearts after DOX treatment (data not shown). These results suggest that pBAD alteration could be attributed to the anti-apoptotic effects of Hsp20. Also, an active form of caspase 3, cleaved caspase-3, was 1.8-fold higher in WT than TG upon DOX treatment (Figure 6E). Taken together, these findings indicate that Hsp20 interacts with phosphorylated Akt, and maintains its phosphorylated state, leading to preserved Bad phosphorylation and inhibition of caspase-3 cleavage, which consequently results in attenuation of DOX-triggered cardiomyocyte apoptosis.
It has been documented that inactivation/dephosphorylation of Akt can be regulated by Ser/Thr phosphatases (PP1 and PP2).23-25 As such, we next examined whether the levels of phosphatase (PP1, PP2A, or PP2B) expression were altered in the DOX-treated hearts. As shown in Figure 6F, protein phosphatase 1 (PP1) was significantly upregulated in both DOX-treated WT and TG hearts, while PP2a and PP2B were not altered in either condition, suggesting that the Hsp20-p-Akt complex may block the action of PP1 on Akt dephosphorylation.
Maintenance of Akt Activation Is Required in Protection of Hsp20 against DOX-Induced Cardiomyocyte Death
To further confirm our in vivo findings on involvement of phosphorylated Akt in the Hsp20 cardioprotection against DOX, we inhibited activation of Akt and MAPKs or their upstream kinases prior to DOX addition in either Ad.GFP- or Ad.Hsp20-infected adult rat cardiomyocytes. Quantitation of cell survival with MTS revealed that pre-treatment with either the p38 inhibitor SB203580, MEK1/2 inhibitor PD98059, or JNK inhibitor had no apparent effect on protection of Hsp20 against DOX-induced cell death (Figure 7A). However, upon addition of the PI3K inhibitors Ly29402 and wortmannin, which inhibit phosphorylation/activation of its substrate Akt, the protective action of Hsp20 was eliminated in DOX-treated cardiomyocytes (Figure 7B). Furthermore, infection with a dominant-negative form of Akt (dnAkt) also offset the protection of Hsp20 against DOX-triggered cardiomyocyte death (Figure 7C). Taken together, these results suggest that Hsp20 protects against DOX-induced cardiomyocyte death mainly via the Akt-dependent signaling pathway.
Figure 7.


Cardioprotection of Hsp20 against DOX -induced cell death is dependent on Akt signaling. (A) Pre-treated cardiomyocytes with SB203580, PD98059, or JNK inhibitor had no effect on protection of Hsp20 against DOX-induced cell death. Cardiomyocytes were infected with Ad.GFP or Ad.Hsp20 for 24 h; subsequently, SB203580, PD98059, or JNK inhibitor was added for one hour, and then these cells were incubated with DOX (20 μM) for 24 h. Cell survival was measured by MTS assay. (B) Pre-treatment of the Ad.Hsp20-infected cardiomyocytes with either Ly294002 or wortmannin totally inhibited the protective effects of Hsp20 against DOX. (C) Infection with dominant-negative Akt (Ad.dnAkt) eliminated the protection provided by overexpressed Hsp20 in cardiomyocytes against DOX. *P<0.05; compared with Ad.GFP-infected group. Data shown are representative of 3 separated experiments.
Attenuation of Oxidative Stress in the Hsp20-TG Hearts Following DOX Treatment
DOX-induced cardiotoxicity has previously been suggested to be attributable mostly to cardiomyocyte damage caused by increased oxidative stress.26, 27 To determine if increased Hsp20 expression might be accompanied by the induction of antioxidant pathways, which detoxify reactive oxygen species (ROS), we next measured the activities of antioxidant enzymes and ROS levels in heart homogenates. The enzymatic activities of both superoxide dismutase (SOD) and glutathione peroxidase (GPx) were significantly reduced in DOX-treated hearts, compared with saline-treated samples. However, these decreases were less pronounced in TG hearts, relative to WTs (Figure 8A and B). Similarly, the ROS levels were markedly increased in WTs after DOX treatment, but overexpression of Hsp20 attenuated this increase (Figure 8C). Furthermore, increased Hsp20 expression could effectively suppress DCF fluorescence in DOX-stressed cardiomyocytes (H9c2) in a dose-dependent fashion (Figure 8D), consistent with its anti-apoptotic effects (Figure 2D).
Figure 8. Hsp20 overexpression attenuates DOX-triggered oxidative stress.


A: superoxide dismutase (SOD); B: glutathione peroxidase (GPx); and (C) amount of ROS, determined by DCF fluorescence intensity. The specificity of DCF signal was confirmed by the use of SOD (Online Figure IV). (n=5 hearts, *P<0.01, # P<0.05, compared to saline controls, respectively). (D) The amount of ROS was decreased in pcDNA3-Hsp20-transfected H9c2 cells in a dose-dependent fashion. *P<0.05; compared with pcDNA3-cells. Data shown are representative of 3 separated experiments. (E) Proposed scheme for protection of Hsp20 against DOX-triggered cardiotoxicity. Treatment of cardiomyocytes with DOX triggers oxidative stress, evidenced by reduction of endogenous antioxidants and increase of ROS levels, which leads to upregulation of phosphatase PP1, dephosphorylation of Akt and its downstream molecular BAD, which subsequently activates caspase-3, resulting in cardiac apoptosis and dysfunction. Overexpression of Hsp20 attenuates DOX-induced cardiac oxidative stress. Furthermore, Hsp20 interacts with p-Akt, preventing its dephosphorylation, which subsequently maintains BAD phosphorylation, and inhibits activation of caspase-3, resulting in attenuation of DOX-mediated cardiac injury.
Discussion
It is well accepted that cardiomyocyte apoptosis could be a fundamental part of the myocardial process that initiates or aggravates heart failure.19, 28 For example, conditional overexpression of active caspase-8 demonstrated that very low levels of myocyte apoptosis are sufficient to cause lethal, dilated cardiomyopathy.19 In this report, we provide the first evidence that overexpression of Hsp20 in vivo and in vitro protects against DOX-induced cardiomyocyte apoptosis and necrosis, resulting in prevention of cardiac dysfunction and improvement of animal survival. These data suggest that Hsp20 may play a positive regulatory role in the treatment of DOX-induced cardiomyopathy. Of interest, for the fist time, we demonstrate that Hsp20 interacts with phosphorylated Akt, and prevents DOX-triggered Akt dephosphorylation by phosphatase PP1, suggesting that increased association of Hsp20-p-Akt in the heart can suppress DOX-mediated activation of the PP1-Akt-BAD-caspase-3 pathway, and hence inhibit the occurrence of apoptosis (Figure 8E).
Previous studies indicate that DOX -induced cardiotoxicity is associated with decreased levels of Akt phosphorylation.29, 30 There is a increasing list of cadioprotective reagents, such as neuregulin-1β, erythropoietin, leukemia inhibitory factor (LIF) and insulin-like growth factor-1, which attenuate DOX-induced cardiotoxicity via an Akt-dependent signaling pathway. 31-34 Actually, activation of Akt is necessary for cell survival and resistance to DOX-induced apoptosis, which may occur by phosphorylation of the Bcl-x inhibitor BAD at Ser-136. However, it was not clear why DOX treatment causes dephosphorylation of Akt, which consequently results in cardiac injury. In the present study, we report that administration of DOX upregulated the expression of PP1 in the heart, which dephosphoyrlated Akt and its downstream, S136-BAD. Furthermore, Hsp20 binds with phosphorylated Akt, which may block the action of PP1 on dephosphorylation of Akt. This is supported by decreased association of Hsp20-p-Akt in DOX-treated WT hearts and no alteration in DOX-treated TGs, compared with the saline-groups. While PP2A has been identified as a mediator of Akt dephosphorylation in fibroblasts, smooth muscle, and osteosarcoma,35-37 PP1 is also reported to associate with and dephosphorylate Akt in breast cancer cells,24 consistent with our present data in cardiomyocytes. Indeed, the expression levels of either PP2A or PP2B were similar between DOX-treated hearts and saline controls. These differences may reflect cell/tissue-specific significance of phosphatase activities.
At present, it is not entirely clear how chronic treatment of DOX leads to upregulation of phosphatase PP1 in the heart. Several studies have considered oxidative stress as a major mediator of DOX-induced cardiac gene dysregulation.29, 31, 38 While it is reasonable that DOX inhibits cardiac gene expression by inhibition of DNA replication/transcription or by elevated degradation of protein, it should be noted that certain genes encoding proteins, besides the current PP1, have also been found upregulated in a mouse model of chronic DOX-cardiotoxicity.38 We also noticed that PP1 levels were downregulated in Hsp20-TG hearts under both basal conditions and DOX-treatment, and the possible mechanism is currently under investigation.
Hsps, as molecular chaperones, have been extensively investigated on protection against ischemia/reperfusion injury and other stress stimuli; however, their possible protective effects on DOX-induced cardiotoxicity and underlying mechanisms are less well studied. Recently, one study has shown that increased Hsp27 levels in cardiomyocytes were correlated with more resistance to DOX-triggered cell death, and accordingly, DOX-induced cardiac dysfunction and animal mortality were also significantly decreased.39 However, these observations could not exclude protective roles of other Hsps, because overexpression of Hsp27 in the heart upregulated both Hsp32 and Hsp70 expression in response to DOX treatment.39 Our present study clearly shows that overexpression of Hsp20 did not alter the expression of other Hsps under either basal conditions or after DOX treatment (Online Figure I). Therefore, it is not likely that the other Hsps may play some compensatory roles in the cardioprotective effects exerted by Hsp20. Furthermore, although other Hsps, such as Hsp90 and Hsp27, can also interact with and modulate Akt activation in the apoptotic cascade,40, 41 the present study indicates that the alterations in expression levels of these Hsps were similar in both Hsp20-TGs and WTs treated with DOX. Nevertheless, protective roles, similar to that of Hsp20 against DOX-triggered cardiotoxicity, may be also played by other Hsps in vivo, and future studies may clarify the specificity of this response to Hsp20 using inducible or conditional knockout models. Furthermore, it remains unclear how protein phosphatase 1 (PP1) affects Akt activity and how DOX treatment upregulates PP1 expression in the murine hearts.
In conclusion, the present work demonstrates that overexpression of Hsp20 in the heart attenuates DOX-induced cardiac injury. The mechanism underlying its protection, at least partially, depends on: 1) maintenance of Akt signaling cascades (Akt-BAD-caspase3); and 2) attenuation of DOX triggered oxidative stress, leading to inhibition of DOX-induced cardiomyocyte death and apoptosis (Figure 8E). Consequently, myocardial function and animal survival are improved. Thus, targeted therapy to increase Hsp20 expression in the heart may hold promise in suppressing DOX-triggered cardiac toxicity.
Supplementary Material
Acknowledgments
Sources of Funding: This study was supported by NIH grant HL-087861 and AHA SDG grant 0730076N (Dr. G-C Fan).
Footnotes
Disclosures: None
References
- 1.Ganey PE, Carter LS, Mueller RA, Thurman RG. Doxorubicin toxicity in perfused rat heart. Decreased cell death at low oxygen tension. Circ Res. 1991;68:1610–1613. doi: 10.1161/01.res.68.6.1610. [DOI] [PubMed] [Google Scholar]
- 2.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 3.Safra T, Muggia F, Jeffers S, Tsao-Wei DD, Groshen S, Lyass O, Henderson R, Berry G, Gabizon A. Pegylated liposomal doxorubicin (doxil): reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Ann Oncol. 2000;11:1029–1033. doi: 10.1023/a:1008365716693. [DOI] [PubMed] [Google Scholar]
- 4.Kluza J, Marchetti P, Gallego MA, Lancel S, Fournier C, Loyens A, Beauvillain JC, Bailly C. Mitochondrial proliferation during apoptosis induced by anticancer agents: effects of doxorubicin and mitoxantrone on cancer and cardiac cells. Oncogene. 2004;23:7018–7030. doi: 10.1038/sj.onc.1207936. [DOI] [PubMed] [Google Scholar]
- 5.Wang S, Konorev EA, Kotamraju S, Joseph J, Kalivendi S, Kalyanaraman B. Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms. J Biol Chem. 2004;279:25535–25543. doi: 10.1074/jbc.M400944200. [DOI] [PubMed] [Google Scholar]
- 6.Li T, Singal PK. Adriamycin-induced early changes in myocardial antioxidant enzymes and their modulation by probucol. Circulation. 2000;102:2105–2110. doi: 10.1161/01.cir.102.17.2105. [DOI] [PubMed] [Google Scholar]
- 7.Benjamin IJ, McMillan DR. Stress (heat shock) proteins: molecular chaperones in cardiovascular biology and disease. Circ Res. 1998;83:117–132. doi: 10.1161/01.res.83.2.117. [DOI] [PubMed] [Google Scholar]
- 8.Ito H, Shimojo T, Fujisaki H, Tamamori M, Ishiyama S, Adachi S, Abe S, Marumo F, Hiroe M. Thermal preconditioning protects rat cardiac muscle cells from doxorubicin-induced apoptosis. Life Sci. 1999;64:755–761. doi: 10.1016/s0024-3205(98)00617-1. [DOI] [PubMed] [Google Scholar]
- 9.Venkatakrishnan CD, Tewari AK, Moldovan L, Cardounel AJ, Zweier JL, Kuppusamy P, Ilangovan G. Heat shock protects cardiac cells from doxorubicin-induced toxicity by activating p38 MAPK and phosphorylation of small heat shock protein 27. Am J Physiol Heart Circ Physiol. 2006;291:H2680–2691. doi: 10.1152/ajpheart.00395.2006. [DOI] [PubMed] [Google Scholar]
- 10.Ciocca DR, Rozados VR, Cuello Carrion FD, Gervasoni SI, Matar P, Scharovsky OG. Hsp25 and Hsp70 in rodent tumors treated with doxorubicin and lovastatin. Cell Stress Chaperones. 2003;8:26–36. doi: 10.1379/1466-1268(2003)8<26:hahirt>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demidenko ZN, Vivo C, Halicka HD, Li CJ, Bhalla K, Broude EV, Blagosklonny MV. Pharmacological induction of Hsp70 protects apoptosis-prone cells from doxorubicin: comparison with caspase-inhibitor- and cycle-arrest-mediated cytoprotection. Cell Death Differ. 2006;13:1434–1441. doi: 10.1038/sj.cdd.4401812. [DOI] [PubMed] [Google Scholar]
- 12.Chicco AJ, Schneider CM, Hayward R. Exercise training attenuates acute doxorubicin-induced cardiac dysfunction. J Cardiovasc Pharmacol. 2006;47:182–189. doi: 10.1097/01.fjc.0000199682.43448.2d. [DOI] [PubMed] [Google Scholar]
- 13.Fan GC, Ren X, Qian J, Yuan Q, Nicolaou P, Wang Y, Jones WK, Chu G, Kranias EG. Novel cardioprotective role of a small heat-shock protein, Hsp20, against ischemia/reperfusion injury. Circulation. 2005;111:1792–1799. doi: 10.1161/01.CIR.0000160851.41872.C6. [DOI] [PubMed] [Google Scholar]
- 14.Fan GC, Chu G, Mitton B, Song Q, Yuan Q, Kranias EG. Small heat-shock protein Hsp20 phosphorylation inhibits beta-agonist-induced cardiac apoptosis. Circ Res. 2004;94:1474–1482. doi: 10.1161/01.RES.0000129179.66631.00. [DOI] [PubMed] [Google Scholar]
- 15.Fan GC, Yuan Q, Song G, Wang Y, Chen G, Qian J, Zhou X, Lee YJ, Ashraf M, Kranias EG. Small heat-shock protein Hsp20 attenuates beta-agonist-mediated cardiac remodeling through apoptosis signal-regulating kinase 1. Circ Res. 2006;99:1233–1242. doi: 10.1161/01.RES.0000251074.19348.af. [DOI] [PubMed] [Google Scholar]
- 16.Shan YX, Liu TJ, Su HF, Samsamshariat A, Mestril R, Wang PH. Hsp10 and Hsp60 modulate Bcl-2 family and mitochondria apoptosis signaling induced by doxorubicin in cardiac muscle cells. J Mol Cell Cardiol. 2003;35:1135–1143. doi: 10.1016/s0022-2828(03)00229-3. [DOI] [PubMed] [Google Scholar]
- 17.Kizaki K, Ito R, Okada M, Yoshioka K, Uchide T, Temma K, Mutoh K, Uechi M, Hara Y. Enhanced gene expression of myocardial matrix metalloproteinases 2 and 9 after acute treatment with doxorubicin in mice. Pharmacol Res. 2006;53:341–336. doi: 10.1016/j.phrs.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 18.Toko H, Oka T, Zou Y, Sakamoto M, Mizukami M, Sano M, Yamamoto R, Sugaya T, Komuro I. Angiotensin II type 1a receptor mediates doxorubicin-induced cardiomyopathy. Hypertens Res. 2002;25:597–603. doi: 10.1291/hypres.25.597. [DOI] [PubMed] [Google Scholar]
- 19.Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest. 2003;111:1497–1504. doi: 10.1172/JCI17664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu W, Lee WL, Wu YY, Chen D, Liu TJ, Jang A, Sharma PM, Wang PH. Expression of constitutively active phosphatidylinositol 3-kinase inhibits activation of caspase 3 and apoptosis of cardiac muscle cells. J Biol Chem. 2000;275:40113–40119. doi: 10.1074/jbc.M004108200. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Y, You H, Yang Y, Wei L, Zhang X, Yao L, Fan D, Yu Q. Distinctive regulation and function of PI 3K/Akt and MAPKs in doxorubicin-induced apoptosis of human lung adenocarcinoma cells. J Cell Biochem. 2004;91:621–632. doi: 10.1002/jcb.10751. [DOI] [PubMed] [Google Scholar]
- 22.Cieslak D, Lazou A. Regulation of BAD protein by PKA, PKCdelta and phosphatases in adult rat cardiac myocytes subjected to oxidative stress. Mol Cells. 2007;24:224–231. [PubMed] [Google Scholar]
- 23.Chakir K, Daya SK, Tunin RS, Helm RH, Byrne MJ, Dimaano VL, Lardo AC, Abraham TP, Tomaselli GF, Kass DA. Reversal of global apoptosis and regional stress kinase activation by cardiac resynchronization. Circulation. 2008;117:1369–1377. doi: 10.1161/CIRCULATIONAHA.107.706291. [DOI] [PubMed] [Google Scholar]
- 24.Xu W, Yuan X, Jung YJ, Yang Y, Basso A, Rosen N, Chung EJ, Trepel J, Neckers L. The heat shock protein 90 inhibitor geldanamycin and the ErbB inhibitor ZD1839 promote rapid PP1 phosphatase-dependent inactivation of AKT in ErbB2 overexpressing breast cancer cells. Cancer Res. 2003;63:7777–7784. [PubMed] [Google Scholar]
- 25.Meier R, Thelen M, Hemmings BA. Inactivation and dephosphorylation of protein kinase Balpha (PKBalpha) promoted by hyperosmotic stress. EMBO J. 1998;17:7294–7303. doi: 10.1093/emboj/17.24.7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berthiaume JM, Wallace KB. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol Toxicol. 2007;23:15–25. doi: 10.1007/s10565-006-0140-y. [DOI] [PubMed] [Google Scholar]
- 27.Ichihara S, Yamada Y, Kawai Y, Osawa T, Furuhashi K, Duan Z, Ichihara G. Roles of oxidative stress and Akt signaling in doxorubicin cardiotoxicity. Biochem Biophys Res Commun. 2007;359:27–33. doi: 10.1016/j.bbrc.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 28.Kang PM, Izumo S. Apoptosis and heart failure: A critical review of the literature. Circ Res. 2000;86:1107–1113. doi: 10.1161/01.res.86.11.1107. [DOI] [PubMed] [Google Scholar]
- 29.Singal PK, Li T, Kumar D, Danelisen I, Iliskovic N. Adriamycin-induced heart failure: mechanism and modulation. Mol Cell Biochem. 2000;207:77–86. doi: 10.1023/a:1007094214460. [DOI] [PubMed] [Google Scholar]
- 30.Taniyama Y, Walsh K. Elevated myocardial Akt signaling ameliorates doxorubicin-induced congestive heart failure and promotes heart growth. J Mol Cell Cardiol. 2002;34:1241–1247. doi: 10.1006/jmcc.2002.2068. [DOI] [PubMed] [Google Scholar]
- 31.Timolati F, Ott D, Pentassuglia L, Giraud MN, Perriard JC, Suter TM, Zuppinger C. Neuregulin-1 beta attenuates doxorubicin-induced alterations of excitation-contraction coupling and reduces oxidative stress in adult rat cardiomyocytes. J Mol Cell Cardiol. 2006;41:845–854. doi: 10.1016/j.yjmcc.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 32.Fu P, Arcasoy MO. Erythropoietin protects cardiac myocytes against anthracycline-induced apoptosis. Biochem Biophys Res Commun. 2007;354:372–378. doi: 10.1016/j.bbrc.2007.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Negoro S, Oh H, Tone E, Kunisada K, Fujio Y, Walsh K, Kishimoto T, Yamauchi-Takihara K. Glycoprotein 130 regulates cardiac myocyte survival in doxorubicin-induced apoptosis through phosphatidylinositol 3-kinase/Akt phosphorylation and Bcl-xL/caspase-3 interaction. Circulation. 2001;103:555–561. doi: 10.1161/01.cir.103.4.555. [DOI] [PubMed] [Google Scholar]
- 34.Lai HC, Liu TJ, Ting CT, Sharma PM, Wang PH. Insulin-like growth factor-1 prevents loss of electrochemical gradient in cardiac muscle mitochondria via activation of PI 3 kinase/Akt pathway. Mol Cell Endocrinol. 2003;205:99–106. doi: 10.1016/s0303-7207(03)00200-4. [DOI] [PubMed] [Google Scholar]
- 35.Ivaska J, Nissinen L, Immonen N, Eriksson JE, Kahari VM, Heino J. Integrin α2β1 promotes activation of protein phosphatase 2A and dephosphorylation of Akt and glycogen synthase kinase 3β. Mol Cell Biol. 2002;22:1352–1359. doi: 10.1128/mcb.22.5.1352-1359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Resjo S, Goransson O, Harndahl L, Zolnierowicz S, Manganiello V, Degerman E. Protein phosphatase 2A is the main phosphatase involved in the regulation of protein kinase B in rat adipocytes. Cell Signal. 2002;14:231–238. doi: 10.1016/s0898-6568(01)00238-8. [DOI] [PubMed] [Google Scholar]
- 37.Yellaturu CR, Bhanoori M, Neeli I, Rao GN. N-Ethylmaleimide inhibits platelet-derived growth factor BB-stimulated Akt phosphorylation via activation of protein phosphatase 2A. J Biol Chem. 2002;277:40148–40155. doi: 10.1074/jbc.M206376200. [DOI] [PubMed] [Google Scholar]
- 38.Yi X, Bekeredjian R, DeFilippis NJ, Siddiquee Z, Fernandez E, Shohet RV. Transcriptional analysis of doxorubicin-induced cardiotoxicity. Am J Physiol Heart Circ Physiol. 2006;290:H1098–H1102. doi: 10.1152/ajpheart.00832.2005. [DOI] [PubMed] [Google Scholar]
- 39.Liu L, Zhang X, Qian B, Min X, Gao X, Li C, Cheng Y, Huang J. Over-expression of heat shock protein 27 attenuates doxorubicin-induced cardiac dysfunction in mice. Eur J Heart Fail. 2007;9:762–769. doi: 10.1016/j.ejheart.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 40.Solit DB, Basso AD, Olshen AB, Scher HI, Rosen N. Inhibition of heat shock protein 90 function down-regulates Akt kinase and sensitizes tumors to Taxol. Cancer Res. 2003;63:2139–2144. [PubMed] [Google Scholar]
- 41.Wu R, Kausar H, Johnson P, Montoya-Durango DE, Merchant M, Rane MJ. Hsp27 regulates Akt activation and polymorphonuclear leukocyte apoptosis by scaffolding MK2 to Akt signal complex. J Biol Chem. 2007;282:21598–21608. doi: 10.1074/jbc.M611316200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.