

Summary
Although the human cytomegalovirus-coded protein, pUL38, prevents apoptosis, its mode of action has remained unknown. Using an epitope-tagged pUL38 expressed from its normal context in the viral genome, we employed a rapid one-step immunoaffinity purification to isolate interacting proteins, which were then identified by mass spectrometry. TSC2, a constituent of the tuberous sclerosis tumor suppressor protein complex (TSC1/2), was one of the cellular proteins identified in the analysis. Antibodies to TSC1 and TSC2 were used to confirm their interactions with pUL38 after infection with wild-type virus. TSC1/2 normally integrates stress signals and regulates the mammalian target of rapamycin complex 1 (mTORC1), a protein complex that responds to stress by limiting cell size and growth. TSC1/2 failed to regulate mTORC1 in the presence of the viral protein, demonstrating that pUL38 supports virus replication at least in part by blocking normal cellular responses to stress.
Introduction
Human cytomegalovirus (HCMV) is a member of the β-herpesvirus family. Infections in healthy children and adults are generally asymptomatic, but the virus causes life-threatening disease in immunologically immature or compromised individuals (reviewed in (Mocarski et al., 2007)). Congenital HCMV infection is the leading viral cause of birth defects, and neonates can suffer serious complications following infection. HCMV is a major complication in immunosuppressed individuals, with a significant contribution to morbidity and mortality in allogeneic transplant recipients and AIDS patients.
The HCMV genome contains >200 open reading frames, although many have not been demonstrated to encode proteins (Murphy et al., 2003a; Murphy et al., 2003b). Upon infection of a permissive cell, HCMV expresses its genes in a regulated cascade; immediate-early genes are expressed first, followed by early and then late genes. The UL38 transcription unit is first expressed during the early phase of infection (reviewed in (Mocarski et al., 2007). A mutant virus lacking pUL38 induces apoptosis after infection, producing reduced levels of viral progeny (Terhune et al., 2007). The mechanism by which pUL38 blocks apoptosis and facilitates HCMV growth is unknown. A BLAST search of pUL38 reveals no sequence homology to cellular proteins, and more sophisticated searches for functional homologies also failed to provide compelling hints to its mode of action (Novotny et al., 2001; Rigoutsos et al., 2003).
To probe the role of pUL38, we screened for proteins that interact with it. We used a mutant virus, expressing an epitope-tagged pUL38 protein from its normal context in the viral genome, coupled with a rapid one-step immunoaffinity purification and mass spectrometry to identify interacting proteins. This combination of proteomics and genetics identified multiple viral and cellular proteins likely to interact with pUL38, one of which is TSC2, also known as tuberin.
TSC2 and TSC1 (hamartin), interact to form the tuberous sclerosis protein complex (TSC1/2), and mutations in either subunit are linked to the development of tuberous sclerosis, a recessive disorder that is characterized by tumors in multiple organs (reviewed in (Crino et al., 2006)). TSC1/2 is regulated by multiple signaling pathways (reviewed in (Kwiatkowski and Manning, 2005)). Growth factors activate Akt and RSK1, which phosphorylate TSC2 and block its activity. Stress activates AMP kinase (AMPK), which phosphorylates TSC2 and activates it. When the TSC1/2 complex is activated, TSC2 functions as a GTPase-activating protein for Rheb, a GTP-binding protein that activates the mammalian target of rapamycin complex 1 (mTORC1). mTORC1 is comprised of at least three subunits, mTOR serine-threonine kinase, raptor and gβL, and it regulates cell growth in response to growth factors and nutrient availability. mTORC1 controls cell growth by modulating multiple processes, including protein synthesis, ribosome biogenesis and autophagy (reviewed in (Sarbassov et al., 2005a)). Thus, TSC1/2 interprets signals from multiple inputs, and, when activated, it is a negative regulator of mTORC1 and thereby inhibits cellular growth.
We confirmed the interaction of pUL38 with the tumor suppressor protein complex, and demonstrated that the viral protein antagonizes the ability of TSC1/2 to negatively regulate mTORC1. Thus, pUL38 blocks a growth regulatory pathway to facilitate viral replication.
Results
Identification of candidate pUL38 interacting partners
To identify cellular and viral proteins that interact with pUL38 in the context of infection, we created a viral mutant, BADinUL38TAP, which expresses from its normal location on the viral genome the pUL38 protein fused to immunoglobulin-binding domains of protein A and a calmodulin-binding peptide (TAP) at its carboxyl terminus, pUL38TAP (Fig. 1A, left). Mutations disrupting the UL38 ORF result in attenuated virus replication with a rapid onset of apoptosis (Terhune et al., 2007). BADinUL38TAP replicated to wild-type levels (Fig. 1A, right), and pUL38TAP displayed a similar localization as observed for untagged pUL38 when assayed by immunofluorescence (Fig. 1B). Tagged pUL38 (from BADinUL38TAP) and untagged pUL38 (from BADwt) were found in both the cytoplasm and nucleus at 24 h after infection. The pUL38 TAP fusion does not disrupt the localization or an essential role of pUL38 in HCMV-infected fibroblasts.
Figure 1.
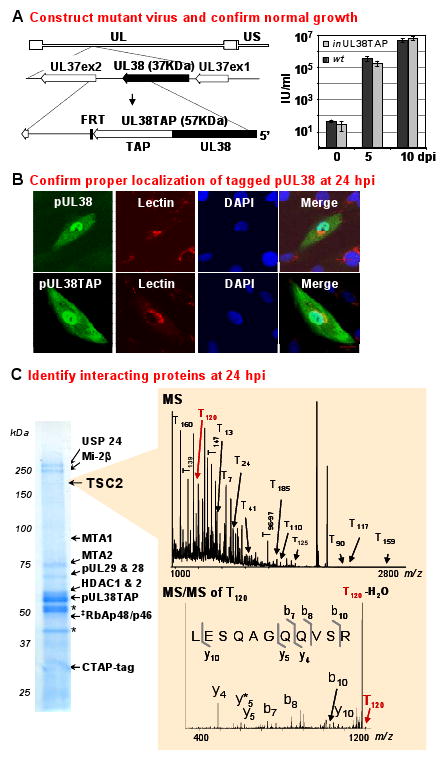
Process for identification of pUL38 binding partners during HCMV infection.
(A) Construct mutant virus with tagged protein and confirm its normal growth. The top line of the diagram to the left locates the UL38 ORF within the unique long (UL) domain of the viral genome; the second line shows the location of the UL38 ORF between UL37 exons; the third line illustrates the fusion of a TAP tag to the C-terminus of pUL38 and the position of a FLP recombinase target (FRT) sequence introduced with the TAP sequence in BADinUL38TAP. Arrowheads mark the C-termini of ORFs. Yields (IU, infectious units determined by assay for HCMV IE1 protein fluorescence at 48 hpi) were determined for BADwt and BADinUL38TAP at 0 (immediately after virus adsorption), 5 and 10 days post infection (dpi) of fibroblasts (0.01 pfu/cell).
(B) Confirm proper localization of the tagged protein at 24 h post infection (hpi). pUL38 and pUL38TAP localizations (green) were determined after infection with BADwt or BADinUL38TAP, respectively. For wild-type pUL38, anti-pUL38 antibody was used for immunofluorescence, while IgG was used to visualize pUL38TAP. Lectin HPA (red) stains the Golgi and DAPI (blue) stains nuclear DNA. Uninfected cells surround infected cells.
(C) Identify interacting proteins at 24 hpi. pUL38TAP-interacting proteins were isolated by immunoaffinity purification, separated by gel electrophoresis and identified by sequential MS and MS/MS analysis. The positions of key captured proteins as well as pUL38TAP and free TAP tag (CTAP) are indicated. *, non-specific contaminants. ‡, because of the similarity of the sequences of RbAp48 and RBAp46, either or both may be present.
At 24 h post infection with BADinUL38TAP, pUL38TAP and associated proteins were isolated from cell extracts. By capturing complexes from cells infected with the HCMV variant, it was possible to identify pUL38-interacting proteins under conditions where pUL38 is expressed with proper kinetics and at normal levels. Isolations were performed via the protein A tag, using a rapid one-step immunoaffinity purification on magnetic beads coated with IgG (Cristea et al., 2006; Cristea et al., 2005). pUL38TAP was efficiently captured with little of the fusion protein remaining in the insoluble fraction (data not shown). Isolated viral and host proteins were resolved by electrophoresis, stained with Coomassie blue, and identified by mass spectrometry. The major protein bands evident in the polyacrylamide gel and illustrative data from sequential MALDI QqTOF MS and MALDI IT MS/MS analysis for one of the identified cell proteins, TSC2, are displayed in figure 1C. The full set of identified proteins is presented in table S1.
Two HCMV proteins were identified in the capture experiment (Table S1). The first was pUL52, and the second migrated at ∼70 kDa (Fig. 1C). The latter contained amino acid segments from the adjacent UL29 and UL28 ORFs. NetGene2 (www.cbs.dtu.dk/services/netgene2, (Brunak et al., 1991)) predicted a splice donor/acceptor motif that would generate a mRNA encoding a UL29/28 protein of 701 amino acids, consistent with its electrophoretic migration. The product of UL52 is essential, while the UL29 and 28 ORFs augment HCMV replication in fibroblasts (Yu et al., 2003). However, their functions are unknown and we have not investigated the consequences of their predicted interactions with pUL38.
Numerous cellular proteins were identified in the capture experiment (Fig. 1C and Table S1). Although their capture suggests that pUL38 might influence multiple cell functions, we have so far confirmed only one of these interactions. The list, therefore, comprises potential pUL38-interacting proteins. It is intriguing that six subunits of the nucleosome remodeling and histone deacetylation (NuRD) complex were among the proteins captured by pUL38TAP: Mi-2β, MTA1 and 2, HDAC1 and 2, and RbAp48/46 (Fig. 1C). The NuRD complex includes histone deacetylases and chromatin-remodeling ATPases, which repress transcription (Bowen et al., 2004). It is possible that pUL38 antagonizes NuRD to optimize expression of the viral genome. The HCMV immediate-early 1 (Nevels et al., 2004) and immediate-early 2 (Park et al., 2007) proteins also block histone deacetylase function, and other herpesviruses (e.g. (Gu et al., 2005)) attack repressive chromatin-modifying complexes as well.
We focused on the predicted interaction of pUL38 with TSC2, a component of the TSC1/2 tumor suppressor protein complex. TSC1/2 regulates mTORC1, which is deregulated by HCMV infection (Kudchodkar et al., 2004). This led to the hypothesis that pUL38 binds to TSC1/2 and antagonizes its ability to regulate mTORC1.
Characterization of the pUL38 interaction with TSC2 and TSC1
To confirm the putative pUL38-TSC2 interaction, we reversed the capture process used in the pUL38TAP immunoaffinity purification, and used antibodies specific for cellular proteins to test for co-immunoprecipitation of pUL38 from wild-type virus-infected cell extracts. A TSC2-specific antibody co-precipitated pUL38 from infected cells, but not mock-infected cells (Fig. 2A, top panel). No pUL38 was detected after immunoprecipitation from the infected cell extract with pre-immune IgG, and the use of wild-type virus ruled out a non-specific interaction of TSC2 with the TAP component of pUL38TAP. The thickness of the pUL38 band detected in figure 2A suggested that multiple species might be present, so the analysis was repeated using a higher resolution electrophoresis protocol (Fig. 2B). Three pUL38-specific bands were evident, corresponding to proteins of approximately 33, 35 and 37 kDa. All three isoforms are found in cells expressing only pUL38 (Fig. 4A), indicating that the three species are specific to the UL38 ORF. We do not yet know the origin of the three species, but note that there are three in-frame AUG codons that could code for proteins this size, and there is precedent in HCMV for use of multiple in-frame starts within an ORF (Stamminger et al., 2002). Antibody to TSC2 preferentially co-precipitated the pUL38 37 kDa isoform and to a lesser extent the 35 kDa species (Fig. 2B).
Figure 2.
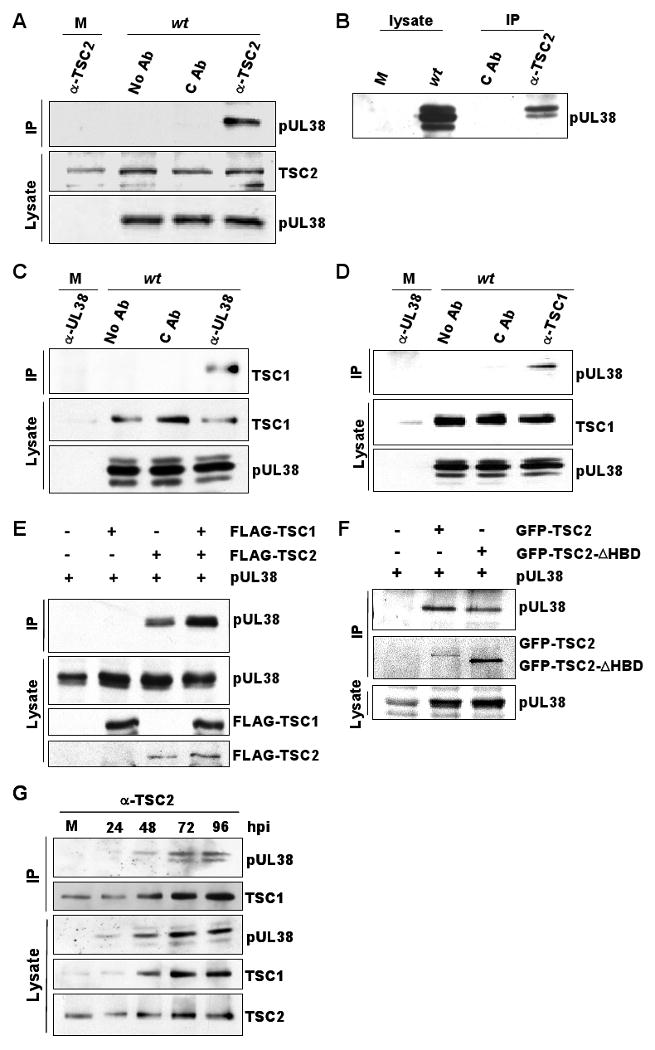
Confirmation of the interaction between pUL38 and the TSC1/2 protein complex. Fibroblasts were mock-infected (M), infected with BADwt (wt) (3 pfu/cell) or 293T cells were transfected with indicated expression vectors. Results are representative of two independent experiments.
(A) pUL38 co-precipitates with TSC2. Cell lysates were prepared at 48 h post mock or BADwt infection, subjected to immunoprecipitation (IP) using rabbit polyclonal antibody to TSC2 (α-TSC2), control preimmune rabbit IgG (C Ab), or beads with no antibody (No Ab), and precipitated proteins were analyzed by Western blot (WB) using the indicated antibodies. As controls, the levels of TSC2 and pUL38 were monitored by Western blot assay.
(B) Two pUL38 isoforms co-precipitate with TSC2. To improve the separation of pUL38 isoforms present at 48 h post infection, electrophoresis was performed using a larger format polyacrylamide gel. Left lanes, Western blots (WB) were preformed on mock- and infected lysates using antibody to pUL38; right lanes, Western blots were performed after immunoprecipitation (IP) from lysates using α-TSC2 or preimmune rabbit IgG (C Ab).
(C) TSC1 co-precipitates with pUL38. Extracts were prepared at 48 h after mock or BADwt infection, and immunoprecipitations were performed using antibody to pUL38 (α-pUL38), a non-specific monoclonal antibody (C Ab) or beads with no antibody (No Ab).
(D) pUL38 co-precipitates with TSC1. Extracts were prepared at 48 h after mock or BADwt infection, and immunoprecipitations were performed using antibody to TSC1 (α-TSC1), a non-specific monoclonal antibody (C Ab) or beads with no antibody (No Ab).
(E) pUL38 co-precipitates with FLAG-TSC2 but not FLAG-TSC1. Cells were transfected with vectors expressing the indicated proteins, extracts were prepared 48 h later and immunoprecipitations were performed by using FLAG epitope-specific antibody.
(F) pUL38 co-precipitates with EGFP-TSC2ΔHBD. Cells were transfected with vectors expressing the indicated proteins, extracts were prepared 48 h later and immunoprecipitations were performed by using GFP-specific antibody.
(G) TSC1 and TSC2 co-immunoprecipitate with pUL38. Lysates were prepared at indicated times after mock or BADwt infection (h post infection, hpi), immunoprecipitated using antibody to TSC2, and assayed by Western blot using antibodies to the indicated proteins. Lysates were also assayed directly by western blot.
Figure 4.
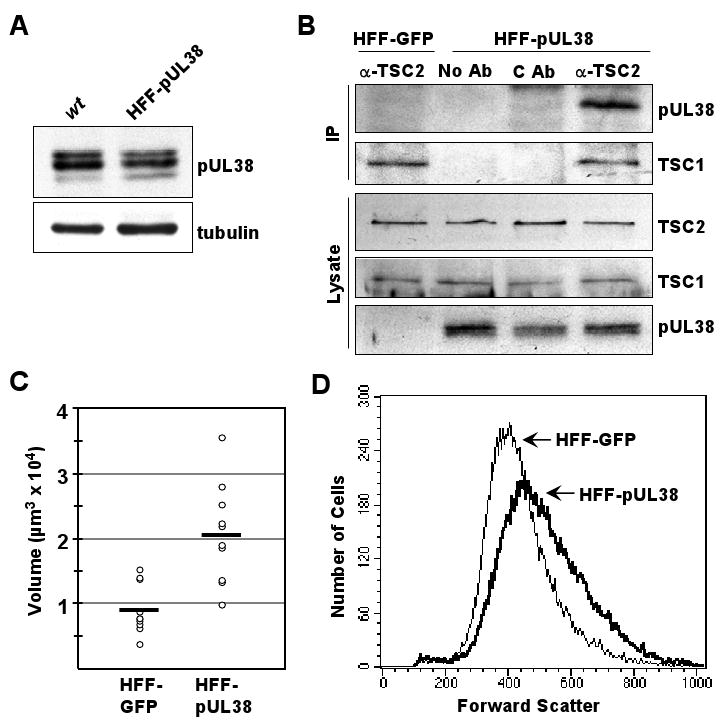
Fibroblasts expressing pUL38 are larger than normal fibroblasts.
(A) Three isoforms of pUL38 are expressed in the absence of infection. Extracts of HFF-pUL38 cells and fibroblasts at 24 h post infection with BADwt were assayed by Western blot using antibody to pUL38. α-tubulin was monitored as a loading control.
(B) pUL38 interacts with TSC2 in the absence of infection. Extracts were prepared from HFF-pUL38 or control HFF-GFP cells, immunoprecipitations (IP) were performed using rabbit antibody to TSC2 (α-TSC2) or control preimmune rabbit antibody (C Ab), and western blot (WB) assays utilized the indicated antibodies. The results are representative of two independent experiments.
(C) Assay of cell volume by quantification of fluorescent intensity in HFF-GFP versus HFF-pUL38 cells. Cells were loaded with calcein green AM, complete z-stacks (0.3 μm slices) were collected for individual, fluorescent cells, and volumes were calculated. 10 well-isolated cells of each type were analyzed, and bars mark average cell volumes.
(D) Assay of relative cell volume by forward scatter of HFF-GFP versus HFF-pUL38 cells. Forward scatter of ∼5×105 cells per sample was measured by flow cytometry.
TSC2 interacts with TSC1 to form the tumor suppressor protein complex, TSC1/2. To determine whether pUL38 also interacts, directly or indirectly, with TSC1, the same set of lysates examined in figure 2A were subjected to immunoprecipitation with antibody to pUL38 (Fig. 2C, top panel). pUL38-specific immune complexes isolated from BADwt-infected cells contained TSC1 protein, and this interaction was found to be specific using the same criteria outlined above for TSC2. A similar experiment demonstrated that antibody to TSC1 can co-precipitate pUL38 (Fig. 2D).
To determine whether pUL38 can interact with each of the TSC1/2 subunits independently, 293T cells were transfected with a pUL38 expression vector plus constructs encoding FLAG-tagged TSC1 and/or FLAG-tagged-TSC2. pUL38 was co-precipitated with tagged TSC2 but not TSC1 (Fig. 2E), arguing that the viral protein does not interact with free TSC1. To further probe the interaction of the viral protein with TSC2, cells were transfected with the pUL38 expression vector plus a vector encoding GFP-tagged TSC2 or a GFP-tagged derivative of TSC2 lacking the TSC1 interaction domain (Goncharova et al., 2004). The deleted TSC2 was, as expected, smaller than the wild-type protein, and immunoprecipitation of the TSC2 variant with GFP-specific antibody co-precipitated pUL38 (Fig. 2F). We conclude that pUL38 interacts with the tumor suppressor complex primarily through its TSC2 subunit. A direct interaction with TSC2, but not TSC1, might explain the failure to detect TSC1 in our analysis of pUL38TAP-interacting proteins by mass spectrometry (Table S1). Perhaps the TSC1/2 complex is disrupted during the one-step isolation method.
Since pUL38 can interact with a TSC2 variant lacking a TSC1 binding domain, it is likely that pUL38 does not disrupt the TSC1/2 complex. To verify this prediction, we tested whether normal levels of the TSC1/2 complex were maintained in infected cells (Fig. 2G). Cell lysates were prepared after mock or BADwt infection, subjected to immunoprecipitation with antibody to TSC2, and co-precipitated TSC1 was monitored by western blot assay. TSC1 was present in TSC2 immune precipitates at each time assayed after infection. In fact, more TSC1 was found associated with TSC2 at 72 and 96 hpi, consistent with the increase observed in the total amount of TSC1; in contrast, the level of TSC2 remained relatively constant after infection. In a recent high throughput analysis, TSC1 protein was shown to increase by a factor of 3.1 after HCMV infection (Stanton et al., 2007).
To further investigate the interaction of pUL38 with the TSC1/2 complex, we performed immunofluorescent analysis. Visual inspection of the fluorescent images suggested that TSC1 and TSC2 exhibited substantial co-localization within uninfected and infected cells (Fig. 3), and quantitative measurement of the images (Costes et al., 2004) confirmed the co-localization, demonstrating that Pearson's correlation (r) for co-localization, was even greater in infected (r = 0.79) than uninfected cells (r = 0.65). This difference was consistently observed in multiple images (data not shown). Perfect co-localization was evident when a TSC1 fluorescent image was compared to itself (r = 1.0) and little was evident when cytoplasmic virus-coded pUL99 protein was compared to DAPI-stained DNA (r = 0.06). We infer that pUL38 does not significantly disrupt the normal association of TSC1 and TSC2.
Figure 3.
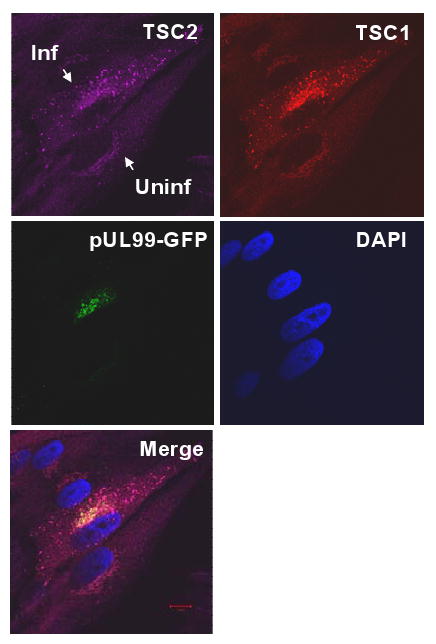
Co-localization of TSC1 and TSC2 in infected fibroblasts. Immunofluorescent analysis at 24 h post infection with BADinUL99GFP, a variant that contains a GFP tag at the C-terminus of the UL99 ORF. The field contains both an uninfected (Uninf) and infected cell (Inf) identified by GFP expression. Antibodies were employed to identify TSC2 (purple) and TSC1 (red), and DNA was stained with DAPI.
pUL38 blocks TSC1/2 function, antagonizing its regulation of mTORC1
Since TSC1/2 normally inhibits the mTORC1 kinase under stress conditions, limiting cell size and mass (Sarbassov et al., 2005a), we tested whether pUL38 can release this constraint. Fibroblasts were generated expressing pUL38 (HFF-pUL38) or GFP (HFF-GFP). Initially, we compared pUL38 expression in HFF-pUL38 cells to that in fibroblasts at 48 h after infection with BADwt (Fig. 4A). Western blot analysis demonstrated that similar amounts of pUL38 and the same variety and relative proportions of pUL38 subspecies were produced in cells expressing the protein as in infected cells. Further, antibody to TSC2 co-immunoprecipitated pUL38 from extracts of HFF-pUL38 cells (Fig. 4B, top panel), demonstrating that no additional virus-coded protein is needed for the interaction. TSC2-specific antibody also co-immunoprecipitated TSC1 (Fig. 4B, second panel from top), consistent with our interpretation that the TSC1/2 complex remains intact in the presence of pUL38. Western blot assays demonstrated that extracts of HFF-pUL38 and HFF-GFP cells contained very similar amounts of TSC1 and TSC2 (Fig. 4B, bottom panels). The failure to modulate TSC1 levels in the presence of pUL38 suggests that one or more additional virus-coded functions is needed for the induction observed in infected cells (Fig. 2).
Cell volumes were assayed by measurement of calcein green AM fluorescence in sequential 0.3 μm optical sections through cells, and the average volume calculated for HFF-pUL38 cells was about twice that of HFF-GFP cells (Fig. 4C). Further, measurement of forward scatter by flow cytometry confirmed that HFF-pUL38 cells are larger than HFF-GFP cells (Fig. 4D). Thus, pUL38-expressing cells were larger than control cells, consistent with the inhibition of TSC1/2 (Fingar et al., 2002).
As noted above, TSC1/2 inhibits the activity of mTORC1 in response to stress. TSC2 is activated when serum is withdrawn from cells, because the loss of growth factors inhibits PI3K-Akt and ERK1/2-RSK1 signaling, which normally block TSC1/2 activity; TSC2 is also activated by the withdrawal of nutrients, because energy deprivation activates AMPK, which then activates TSC1/2 (Fig. 7) (Kwiatkowski and Manning, 2005). When activated, mTORC1 phosphorylates the ribosomal protein S6 kinase (p70 S6 kinase) and eukaryotic initiation factor 4E binding protein 1 (4E-BP1) (Sarbassov et al., 2005a). To further evaluate the ability of pUL38 to antagonize TSC1/2, we monitored the activity of mTORC1 in control or HFF-pUL38 cells after nutrient stress. Initially, the phosphorylation of rpS6 at S235/236, a target of the mTORC1-activated p70 S6 kinase, was assayed by using antibodies that recognized total or phosphorylated rpS6 (Fig. 5A). Maintenance in medium lacking growth factors for 12 h induced a modest decrease in total rpS6 in both cell types, and HFF-pUL38 cells accumulated ∼2.5-fold more phosphorylated rpS6 than HFF-GFP cells. Incubation in PBS (no growth factors, amino acids or sugars) for 1 h after the initial 12 h period in medium lacking serum resulted in a dramatic reduction in the amount of phosphorylated rpS6 in HFF-GFP cells. In contrast, HFF-UL38 cells contained near wild-type levels of phosphorylated rpS6 after 1 h in PBS, and the phosphoprotein was still detected, albeit at a reduced level, after 2 h (Fig. 5A). Continued phosphorylation of rpS6 in HFF-pUL38 cells after maintenance in PBS was dependent on rapamycin-sensitive mTORC1 activity (Fig. 5B). We extended the analysis to direct targets of mTORC1, and found that phosporylation of both p70 S6 kinase at T389 and 4E-BP1 at T37/46 was more resistant to a 2h incubation in PBS, following 12 h in serum-free medium, in cells expressing pUL38 as compared to control cells (Fig. 5B). Thus, pUL38 alone is sufficient to maintain mTORC1 signaling under stress-inducing conditions.
Figure 7.

HCMV influences multiple cellular pathways that communicate with mTORC1 through TSC1/2. Activities that are known to be enhanced or inhibited by HCMV infection are indicated by green or red boxes, respectively. Gray boxes mark activities not known to be modified by HCMV. The “core” TSC1/2-mTOR pathway is rendered in larger, bold print: TSC1/2, tuberous sclerosis complex; Rheb-GTP, ras homologue enriched in brain protein; mTORC1, mammalian target of rapamycin complex 1; S6K, p70 S6 kinase; 4E-BP1, eukaryotic initiation factor 4E binding protein 1; rpS6, ribosomal protein S6. Some additional activities that impact the core pathway: ATM, ataxia-telangiectasia mutated protein; p53, p53 tumor suppressor protein; LKB1, Peutz-Jeghers syndrome protein; ERK1/2, extracellular signal-regulated kinase 1 and 2; RSK1, p90 ribosomal S6 kinase 1; IRS1, insulin receptor substrate protein; PI3K, phosphoinositide 3-kinase; Akt, protein kinase B; AMPK, AMP kinase.
Figure 5.
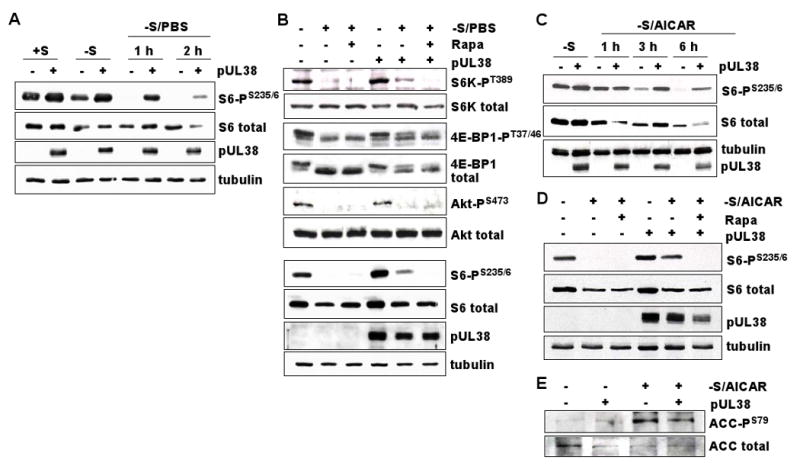
HCMV pUL38 is sufficient to prevent inhibition of the mTORC1 kinase by stress. pUL38 and tubulin were monitored as controls, and results are representative of 2 or 3 independent experiments.
(A) Subconfluent HFF-pUL38 (+pUL38) or control HFF-GFP cells (-pUL38) were cultured with (+S) or without serum (-S) for 12 h. Some −S cultures were switched to PBS for 1 h or 2 h, after which cells were harvested, lysates prepared, and protein was analyzed by Western blot assay using antibodies specific for rpS6 phosphorylated at S235/236 and total rpS6.
(B) Subconfluent +pUL38 and −pUL38 cells were maintained in serum-free medium for 12 h, and the medium was replaced with PBS for 2 h in the presence or absence of Rapamycin (Rapa, 20 nM). Proteins were assayed by Western blot using antibodies specific for indicated phosphorylated proteins or that recognize proteins irrespective of phosphorylation state.
(C) Subconfluent +pUL38 and −pUL38 cells were maintained in serum-free medium (-S) for 12 h, then AICAR (AMPK activator, 5mM) was added to a portion of the cultures and cells were harvested 1, 3 or 6 h later. Assays were as described in panel A.
(D) Subconfluent +pUL38 and −pUL38 cells were maintained in serum-free medium for 12 h, then AICAR with or without rapamycin was added to a portion of the cultures and cells were harvested 6 h later. Assays were as described in panel A.
(E) Subconfluent +pUL38 and −pUL38 cells were maintained in serum-free medium for 12 h, then AICAR with or without rapamycin was added to a portion of the cultures and cells were harvested 6 h later. Proteins were assayed by Western blot using antibodies specific for acetyl CoA carboxylase (ACC) phosphorylated at S79 and total ACC.
Phosphorylation of 4E-BP1 T37/46 was more resistant to rapamycin in pUL38-expressing as compared to control cells (Fig. 5B). The mechanistic basis for this observation is not clear, but it has been noted that HCMV-infected cells contain a rapamycin-resistant raptor-containing activity (raptor is a constituent of the normally rapamycin-sensitive mTORC1 complex) that can mediate hyperphosphorylation of 4E-BP1 (Kudchodkar et al., 2006). Nevertheless, rapamycin treatment at the start of infection delays the production of virus progeny by about 12 h and reduces the final yield of virus by a factor of 5-50 in the presence or absence of serum (Kudchodkar et al., 2004).
Limiting nutrients induce an increase in the AMP/ATP ratio in the cell. AMP binds to and allows activation of AMPK, which can phosphorylate and activate TSC2 with subsequent inhibition of mTORC1 activity. Thus, AMPK negatively regulates mTORC1 through TSC1/2. AMPK also can be activated by the cell-permeable AMP analog AICAR. AICAR treatment decreases mTORC1 activity and induces cell growth arrest (Corton et al., 1995). We tested the ability of pUL38 to block the inhibition of mTORC1 activity by AMPK, using AICAR to stimulate AMPK activity. Serum-starved HFF-GFP control cells or HFF-pUL38 cells were treated with AICAR, and mTORC1 activity assessed by measuring rpS6 phosphorylation at S235/236. Phosphorylated rpS6 was markedly reduced in HFF-GFP as compared to HFF-pUL38 cells after 3 h or 6 h of drug treatment (Fig. 5C). The 6 h AICAR treatment was repeated in the presence or absence of rapamycin (Fig. 5D, top two panels). AICAR-induced phosphorylation of rpS6 was sensitive to the inhibitor, confirming that pUL38 preserved the activity of rapamycin-sensitive mTORC1. The concentration of AICAR used in these experiments induced phosphorylation at S79 of a known AMPK target, acetyl-CoA carboxylase (Fig. 5E), demonstrating that AMPK was activated by the drug. Further, the level of phosphorylated acetyl-CoA carboxylase was not influenced by the presence of pUL38, arguing that pUL38 does not act at AMPK or upstream of AMPK to influence mTORC1 function. We conclude that pUL38 blocks the negative regulation of mTORC1 by AMPK by inhibiting TSC1/2 function.
A second mTOR-containing complex, mTORC2 phosphorylates Akt at S473 (Hresko and Mueckler, 2005; Sarbassov et al., 2005b). This modification contributes to activation of Akt for efficient phosphorylation of some but not all of its targets (Guertin et al., 2006; Jacinto et al., 2006). Akt S473 phosphorylation is impaired in TSC1/2-deficient cells (Yang et al., 2006), suggesting that the tumor suppressor regulates mTORC2, at least under some circumstances. Accordingly, we tested whether pUL38 influences Akt S473 phosphorylation as a consequence of its inhibitory interaction with TSC1/2. Although, as observed previously (Jacinto et al., 2006), starvation in PBS blocked Akt S473 phosphorylation, there was no difference in the level of Akt S473 phosphorylation in HFF-pUL38 as compared to control HFF-GFP cells maintained in medium containing serum (Fig. 5B, middle panels). We find no evidence for an effect of pUL38 on mTORC2.
The effect of pUL38 on p70 S6 kinase and 4E-BP1 phosphorylation in response to stress was confirmed within HCMV-infected cells (Fig. 6A). Fibroblasts were maintained in medium lacking serum overnight, and then infected with BADwt (pUL38+) or BADdlUL38 (pUL38-). After virus adsorption, the inoculum was removed and replaced with serum-free medium, and the phosphorylation status of mTORC1 targets was measured at 48 hpi. The level of phosphorylated p70 S6 kinase and 4E-BP1 was substantially reduced in cells infected with the pUL38-deficient virus. The phosphorylation of rpS6 was monitored as measure of p70 S6 kinase activity, and its phosphorylation was also substantially reduced in cells infected with pUL38-deficient virus as compared to cells infected with wild-type HCMV at each time tested (Fig. 6B).
Figure 6.
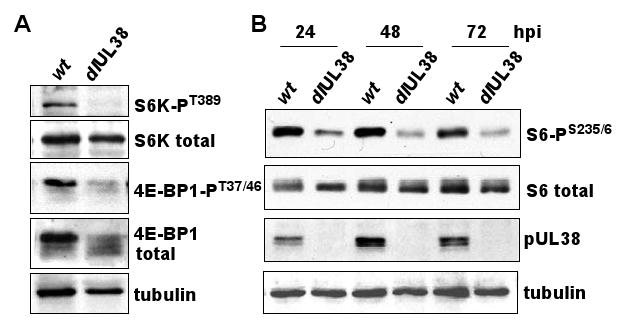
Phosphorylation of rpS6 in response to stress is decreased following infection with a pUL38-deficient virus. As controls, pUL38 and tubulin were monitored. The results are representative of two independent experiments.
(A) Fibroblasts were maintained for 12 h in serum-free medium, infected with BADwt (1 pfu/cell) or with BADdlUL38 at an equivalent number of genomes per cell, and re-fed with serum-free medium. At 48 h post infection, cells were harvested, lysates prepared and analyzed by Western blot using antibodies specific for indicated phosphorylated proteins or that recognize proteins irrespective of phosphorylation state.
(B) Fibroblasts were treated as in panel (A), and, at the indicated h post infection (hpi), cells were harvested and analyzed by Western blot using antibodies specific for total rpS6 and rpS6 phosphorylated at ser235/236.
No phosphorylated p70 S6 kinase T389 was detected at 48 hpi with the pUL38-deficient virus (Fig. 6A), whereas residual phosphorylated rpS6 S235/236 was evident at 48 and 72 hpi (Fig. 6B). Earlier work has documented rapamycin-resistant phosphorylation of rpS6 in HCMV-infected cells in the absence of detectable phosphorylated p70 S6 kinase T389, and this led to speculation that the virus might induce an mTORC1-independent kinase activity that mediates the residual rpS6 S235/236 phosphorylation (Kudchodkar et al., 2004). Perhaps we see evidence of this kinase activity in our experiment; alternatively, the pUL38-deficient virus-infected cells might contain a small amount of active p70 S6K that we have failed to detect.
HCMV pUL38 is necessary (Fig. 6) and sufficient (Fig. 5) to de-regulate mTORC1.
Discussion
We used a rapid, one-step purification method (Cristea et al., 2005) to capture epitope-tagged pUL38 expressed from the HCMV genome, and employed mass spectrometry to identify multiple viral and cell proteins that co-purified with it (Fig. 1C and Table S1). The interaction with TSC1/2 was confirmed by using antibodies to the cellular partners to co-precipitate pUL38 from extracts of cells infected with wild-type HCMV (Fig. 2).
We focused our functional analysis on the interaction of pUL38 with TSC2. Ectopic expression of pUL38 in fibroblasts increased their size (Fig.4C and 4D). Increased cell size is a hallmark of tumors formed in tuberous sclerosis complex and results from constitutive mTORC1 signaling (Fingar et al., 2002; Tee et al., 2003). Increased size is also a hallmark of HCMV-infected cells (Gandhi and Khanna, 2004). The interaction of pUL38 with the tumor suppressor protein complex blocked its ability to regulate mTORC1 in response to stress in cells expressing the viral protein outside the context of infection (Fig. 5) and in virus-infected cells (Fig. 6). Limiting growth factors and nutrients activate TSC1/2, which blocks mTORC1. In control fibroblasts this stress blocked phosphorylation of two direct mTORC1 targets, p70 S6K T389 and 4E-BP1 T37/46, as well as rpS6 S235/236, which is phosphorylated by activated p70 S6K (Fig. 5A and B). In contrast, rapamycin-sensitive mTORC1 remained significantly more active in pUL38-expressing cells subjected to stress (Fig. 5A and B). Further, AICAR-mediated stimulation of AMPK, which activates TSC1/2 and inhibits mTORC1, did not block phosphorylation of rpS6 in the presence of pUL38 (Fig. 5C).
The physical interaction of pUL38 with TSC1/2 and the pUL38-mediated block to stress-induced inhibition of mTORC1 activity support the conclusion that the viral protein antagonizes the ability of TSC1/2 to regulate mTORC1 activity. This conclusion is consistent with earlier work showing that HCMV induces mTORC1 activity (Kudchodkar et al., 2004, 2006), and blocks the effect of AMPK on mTORC1 function (Kudchodkar et al., 2007). The relevance of mTORC1 function to HCMV pathogenesis is underscored by multiple observations: inhibition of mTORC1 by rapamycin antagonizes HCMV replication in cultured cells (Kudchodkar et al., 2004); shRNA depletion of the Raptor mTORC1 subunit inhibits virus growth (Kudchodkar et al., 2006); and, importantly, rapamycin protects against reactivation of HCMV in patients who have undergone allogeneic hematopoietic stem cell transplantation (Marty et al., 2007).
How does pUL38 block TSC1/2 activity? The human papillomavirus type 16 E6 protein directs the degradation of TSC2 (Lu et al., 2004), and Kaposi's sarcoma-associated herpesvirus vGPCR causes phosphorylation and inactivation of TSC2 (Sodhi et al., 2006). Further, expression of Epstein-Barr virus LMP2 protein correlates with Akt activation and hyperphosphorylation of the mTORC1 target 4E-BP1 (Moody et al., 2005), suggesting that this oncoprotein might also target TSC1/2. HCMV pUL38 does not reduce the level of TSC1 or TSC2 (Fig. 2), it does not contain motifs predictive of intrinsic kinase activity, nor is there evidence for disruption of the TSC1/2 complex (Fig. 2G and 3). Perhaps the interaction of pUL38 with TSC2 (Fig. 2E and F) blocks an activating phosphorylation of TSC2 or facilitates an inhibitory phosphorylation. Alternatively, pUL38 could displace a component from the complex that we have not monitored, direct an antagonistic cellular protein to the complex or interfere directly with the GAP activity of TSC2.
We have established that pUL38 alone is sufficient to antagonize the regulation of mTORC1 by TSC1/2. Additional inputs likely cooperate with pUL38 to modulate the TSC1/2-mTORC1 response pathway in infected cells (Fig. 7). HCMV activates phosphatidylinositol 3-kinase (PI3K) and its downstream targets, including Akt (Johnson et al., 2001). This activation is mediated at least in part by the HCMV immediate-early 1 and 2 proteins, which can induce PI3K-dependent phosphorylation of Akt (T308) outside the context of infection (Yu and Alwine, 2002). This phosphorylation activates Akt, which can phosphorylate and inactivate TSC2 (Kwiatkowski and Manning, 2005). Similarly, RSK1 kinase is active in HCMV-infected cells (Rodems and Spector, 1998), and it also can phosphorylate and inactivate TSC2 (Kwiatkowski and Manning, 2005). In addition, HCMV blocks upstream elements of the DNA damage response, which normally activates TSC1/2. ATM kinase, which propagates the DNA damage response (Kastan and Bartek, 2004), is mislocalized within HCMV-infected cells (Gaspar and Shenk, 2006), and p53, which activates AMPK in response to DNA damage (Feng et al., 2005), is inactivated after infection (Casavant et al., 2006).
The ability of pUL38 to inhibit TSC1/2 and maintain active mTORC1 leads to predictions of additional consequences within infected cells. mTORC1 activates p70 S6 kinase, which favors the translation of mRNAs containing a 5′ TOP motifs (Jefferies et al., 1997). By maintaining active p70 S6 kinase, pUL38 might alter translational specificity within infected cells. In a similar vein, active p70 S6 kinase down regulates insulin receptor substrates 1 and 2 (Harrington et al., 2004; Shah et al., 2004), so pUL38 might induce insulin resistance in HCMV-infected cells. Autophagy is regulated by mTORC1. Inhibition of mTORC1 by rapamycin (Shintani and Klionsky, 2004) or by activation of AMPK (Meley et al., 2006) can increase autophagy, so maintenance of active mTORC1 by pUL38 might inhibit autophagy. Finally, pUL38 might influence cell cycle status. The cyclin-dependent kinase inhibitor, p27, is a Cip/Kip family member, and it contributes to the maintenance of cells in G0 (Coats et al., 1996; Zhang et al., 2000). TSC2 binds to p27, stabilizing it (Rosner and Hengstschlager, 2004), and inhibition of TSC2 expression can induce quiescent fibroblasts to enter the G1 phase of the cell cycle (Soucek et al., 1997). HCMV induces G0 cells to enter G1, and three viral proteins, pUL82 (Kalejta et al., 2003), immediate-early 1 (Castillo et al., 2000) and immediate early 2 (Murphy et al., 2000), contribute to the induction. Given the ability of pUL38 to block TSC1/2 function, it is possible that it also helps to move quiescent cells into the cycle, providing an environment conducive to viral DNA replication.
Experimental Procedures
Cells, viruses, and reagents
Human foreskin fibroblasts (passages 5-10) and 293T cells were cultured in medium containing 10% newborn calf serum (NCS). To produce fibroblasts expressing pUL38 (HFF-pUL38), the UL38 open reading frame was amplified by PCR and cloned into pRetro-EBNA to make pRetroUL38. pRetroUL38 was transfected into the Phoenix Ampho packaging cells (Kinsella and Nolan, 1996) to generate retrovirus (RetroUL38), which was then used to infect fibroblasts. Control fibroblasts expressing green fluorescent protein (HFF-GFP) were produced by infection with RetroGFP (Silva et al., 2005). >90% of cells expressed pUL38 or GFP.
BADwt is produced from a clone of the HCMV AD169 strain, pAD/Cre (Yu et al., 2002). BADdlUL38 and BADinUL38TAP are derivatives of BADwt that lack the UL38 coding region (Terhune et al., 2007) or contain a TAP tag fused to the C-terminus of the UL38 ORF (Supplemental Data).
Vectors expressing FLAG-tagged TSC1 and TSC2 (pRK7-FLAG-TSC1 and pRK7-FLAG-TSC2; (Tee et al., 2002)) and EGFP-tagged TSC2 lacking the TSC1-binding domain (EGFP-TSC2-ΔHBD; (Goncharova et al., 2004)) have been described.
Rapamycin (20 nM; Cell Signaling Technology) was used to block mTORC1 function, and 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR, 5 mM; Cell Signaling Technology) was used to activate AMPK.
Mass spectrometry analysis of pUL38 interacting proteins
Fibroblasts, grown to ∼70% confluence, were mock-infected or infected at a multiplicity of 3 pfu/cell. After 24 h, cells were washed with phosphate buffered saline (PBS), and harvested by scraping. After centrifugation at 1200 × g for 10 min at 4°C, the cell pellet was weighed and resuspended (0.1 ml/g) in 20 mM HEPES, pH 7.5, containing 1.2% (w/v) polyvinylproline, 1/100 (v/v) protease inhibitor mixture (20 mg/ml PMSF + 0.4 mg/ml pepstatin A), and 1/200 (v/v) protease inhibitor cocktail (Sigma). The cells were frozen as small pellets by dropping into liquid nitrogen. Protein extraction, immunoaffinity purification, gel electrophoresis and mass spectrometric analysis (Supplemental Data) have been described (Cristea et al., 2006; Cristea et al., 2004; Cristea et al., 2005).
Protein analysis
Proteins were analyzed by immunoprecipitation, Western blot assay and immunofluorescence (Supplemental Data). Experiments utilized rabbit peptide-specific, IgG antibodies from Cell Signaling Technologies: phospho-S6 protein (S235/236) and S6 protein (#2211 and 2212), phospho-p70 S6 kinase (T389) and p70 S6 kinase (#9205 and 2708), phospho-4E-BP1 (T37/46) and 4E-BP1(#9459 and 9452), phospho-Akt (S473) and Akt (#9271 and 9272), and phospho-acetyl-coA carboxylase (S79) and acetyl-coA carboxylase (#3661 and 3662); and from Santa Cruz Biotechnology: TSC2 (sc-893) and normal rabbit IgG (sc-2027). Other experiments used mouse monoclonal antibodies: anti-FLAG (M2, Sigma Aldrich) anti-pUL38 (8D6, (Terhune et al., 2007)); anti- TSC1 (MAB5532, Upstate); and anti-tubulin (T6199, Sigma Aldrich).
Determination of cell volume
For measurement of cell volume by fluorescence, adherent cells were incubated with calcein green AM for 1 h at 37°C, z-stack images (0.3 μm slices) were collected through entire individual cells using an RS3 spinning disk confocual microscope (Perkin Elmer), and fluorescent cell volume was calculated using Velocity 4.0 software (Improvision). For determination of relative cell volume by flow cytometry, cells were removed from culture dishes by trypsinization, and forward scatter was measured within 10 min by flow cytometry (BD Biosciences FACScan). Larger cells have a greater forward scatter.
Supplementary Material
Acknowledgments
We thank E. Goncharova and V.P. Krymskaya (University of Pennsylvania) for gifts of plasmids and D. Spector (Hershey Medical School) for critical reading of the manuscript. This work was supported by grants from U.S. National Institutes of Health to B.T.C. (RR00862), B.T.C. and M.P.R. (CA89810 and RR22220), M.P.R. (GM62427), T.S. (AI54430 and CA85786), a Rockefeller University Women and Science Fellowship (CEN5300379) to I.M.C. and an American Cancer Society Postdoctoral Fellowship (PF-07073-01-MBC) to N.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bowen NJ, Fujita N, Kajita M, Wade PA. Mi-2/NuRD: multiple complexes for many purposes. Biochimica et biophysica acta. 2004;1677:52–57. doi: 10.1016/j.bbaexp.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Brunak S, Engelbrecht J, Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J Mol Biol. 1991;220:49–65. doi: 10.1016/0022-2836(91)90380-o. [DOI] [PubMed] [Google Scholar]
- Casavant NC, Luo MH, Rosenke K, Winegardner T, Zurawska A, Fortunato EA. Potential role for p53 in the permissive life cycle of human cytomegalovirus. J Virol. 2006;80:8390–8401. doi: 10.1128/JVI.00505-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo JP, Yurochko AD, Kowalik TF. Role of human cytomegalovirus immediate-early proteins in cell growth control. J Virol. 2000;74:8028–8037. doi: 10.1128/jvi.74.17.8028-8037.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coats S, Flanagan WM, Nourse J, Roberts JM. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science. 1996;272:877–880. doi: 10.1126/science.272.5263.877. [DOI] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229:558–565. doi: 10.1111/j.1432-1033.1995.tb20498.x. [DOI] [PubMed] [Google Scholar]
- Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. The New England journal of medicine. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- Cristea IM, Carroll JW, Rout MP, Rice CM, Chait BT, Macdonald MR. Tracking and elucidating alphavirus-host protein interactions. J Biol Chem. 2006 doi: 10.1074/jbc.M603980200. [DOI] [PubMed] [Google Scholar]
- Cristea IM, Gaskell SJ, Whetton AD. Proteomics techniques and their application to hematology. Blood. 2004;103:3624–3634. doi: 10.1182/blood-2003-09-3295. [DOI] [PubMed] [Google Scholar]
- Cristea IM, Williams R, Chait BT, Rout MP. Fluorescent proteins as proteomic probes. Mol Cell Proteomics. 2005;4:1933–1941. doi: 10.1074/mcp.M500227-MCP200. [DOI] [PubMed] [Google Scholar]
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16:1472–1487. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi MK, Khanna R. Human cytomegalovirus: clinical aspects, immune regulation, and emerging treatments. The Lancet infectious diseases. 2004;4:725–738. doi: 10.1016/S1473-3099(04)01202-2. [DOI] [PubMed] [Google Scholar]
- Gaspar M, Shenk T. Human cytomegalovirus inhibits a DNA damage response by mislocalizing checkpoint proteins. Proc Natl Acad Sci U S A. 2006;103:2821–2826. doi: 10.1073/pnas.0511148103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncharova E, Goncharov D, Noonan D, Krymskaya VP. TSC2 modulates actin cytoskeleton and focal adhesion through TSC1-binding domain and the Rac1 GTPase. J Cell Biol. 2004;167:1171–1182. doi: 10.1083/jcb.200405130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Liang Y, Mandel G, Roizman B. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc Natl Acad Sci U S A. 2005;102:7571–7576. doi: 10.1073/pnas.0502658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Developmental cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem. 2005;280:40406–40416. doi: 10.1074/jbc.M508361200. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Jefferies HB, Fumagalli S, Dennis PB, Reinhard C, Pearson RB, Thomas G. Rapamycin suppresses 5′TOP mRNA translation through inhibition of p70s6k. Embo J. 1997;16:3693–3704. doi: 10.1093/emboj/16.12.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RA, Wang X, Ma XL, Huong SM, Huang ES. Human cytomegalovirus up-regulates the phosphatidylinositol 3-kinase (PI3-K) pathway: inhibition of PI3-K activity inhibits viral replication and virus-induced signaling. J Virol. 2001;75:6022–6032. doi: 10.1128/JVI.75.13.6022-6032.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalejta RF, Bechtel JT, Shenk T. Human cytomegalovirus pp71 stimulates cell cycle progression by inducing the proteasome-dependent degradation of the retinoblastoma family of tumor suppressors. Mol Cell Biol. 2003;23:1885–1895. doi: 10.1128/MCB.23.6.1885-1895.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- Kinsella TM, Nolan GP. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum Gene Ther. 1996;7:1405–1413. doi: 10.1089/hum.1996.7.12-1405. [DOI] [PubMed] [Google Scholar]
- Kudchodkar SB, Del Prete GQ, Maguire TG, Alwine JC. AMPK-mediated inhibition of mTOR kinase is circumvented during immediate early times of HCMV infection. J Virol. 2007 doi: 10.1128/JVI.02079-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudchodkar SB, Yu Y, Maguire TG, Alwine JC. Human cytomegalovirus infection induces rapamycin-insensitive phosphorylation of downstream effectors of mTOR kinase. J Virol. 2004;78:11030–11039. doi: 10.1128/JVI.78.20.11030-11039.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudchodkar SB, Yu Y, Maguire TG, Alwine JC. Human cytomegalovirus infection alters the substrate specificities and rapamycin sensitivities of raptor- and rictor-containing complexes. Proc Natl Acad Sci U S A. 2006;103:14182–14187. doi: 10.1073/pnas.0605825103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Manning BD. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet. 2005;14(Spec No 2):R251–258. doi: 10.1093/hmg/ddi260. [DOI] [PubMed] [Google Scholar]
- Lu Z, Hu X, Li Y, Zheng L, Zhou Y, Jiang H, Ning T, Basang Z, Zhang C, Ke Y. Human papillomavirus 16 E6 oncoprotein interferences with insulin signaling pathway by binding to tuberin. J Biol Chem. 2004;279:35664–35670. doi: 10.1074/jbc.M403385200. [DOI] [PubMed] [Google Scholar]
- Marty FM, Bryar J, Browne SK, Schwarzberg T, Ho VT, Bassett IV, Koreth J, Alyea EP, Soiffer RJ, Cutler CS, et al. Sirolimus-based graft-versus-host disease prophylaxis protects against cytomegalovirus reactivation after allogeneic hematopoietic stem cell transplantation: a cohort analysis. Blood. 2007;110:490–500. doi: 10.1182/blood-2007-01-069294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. 2006;281:34870–34879. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- Mocarski ES, Shenk T, Pass RF. Cytomegaloviruses. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia, PA: Lippincott, Williams and Wilkins; 2007. pp. 2702–2772. [Google Scholar]
- Moody CA, Scott RS, Amirghahari N, Nathan CA, Young LS, Dawson CW, Sixbey JW. Modulation of the cell growth regulator mTOR by Epstein-Barr virus-encoded LMP2A. J Virol. 2005;79:5499–5506. doi: 10.1128/JVI.79.9.5499-5506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Rigoutsos I, Shibuya T, Shenk TE. Reevaluation of human cytomegalovirus coding potential. Proc Natl Acad Sci U S A. 2003a;100:13585–13590. doi: 10.1073/pnas.1735466100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Yu D, Grimwood J, Schmutz J, Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM, Shenk TE. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc Natl Acad Sci U S A. 2003b;100:14976–14981. doi: 10.1073/pnas.2136652100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy EA, Streblow DN, Nelson JA, Stinski MF. The human cytomegalovirus IE86 protein can block cell cycle progression after inducing transition into the S phase of permissive cells. J Virol. 2000;74:7108–7118. doi: 10.1128/jvi.74.15.7108-7118.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevels M, Paulus C, Shenk T. Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc Natl Acad Sci U S A. 2004;101:17234–17239. doi: 10.1073/pnas.0407933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novotny J, Rigoutsos I, Coleman D, Shenk T. In silico structural and functional analysis of the human cytomegalovirus (HHV5) genome. J Mol Biol. 2001;310:1151–1166. doi: 10.1006/jmbi.2001.4798. [DOI] [PubMed] [Google Scholar]
- Park JJ, Kim YE, Pham HT, Kim ET, Chung YH, Ahn JH. Functional interaction of the human cytomegalovirus IE2 protein with histone deacetylase 2 in infected human fibroblasts. J Gen Virol. 2007;88:3214–3223. doi: 10.1099/vir.0.83171-0. [DOI] [PubMed] [Google Scholar]
- Rigoutsos I, Novotny J, Huynh T, Chin-Bow ST, Parida L, Platt D, Coleman D, Shenk T. In silico pattern-based analysis of the human cytomegalovirus genome. J Virol. 2003;77:4326–4344. doi: 10.1128/JVI.77.7.4326-4344.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodems SM, Spector DH. Extracellular signal-regulated kinase activity is sustained early during human cytomegalovirus infection. J Virol. 1998;72:9173–9180. doi: 10.1128/jvi.72.11.9173-9180.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosner M, Hengstschlager M. Tuberin binds p27 and negatively regulates its interaction with the SCF component Skp2. J Biol Chem. 2004;279:48707–48715. doi: 10.1074/jbc.M405528200. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Current opinion in cell biology. 2005a;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005b;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol. 2004;14:1650–1656. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MC, Schroer J, Shenk T. Human cytomegalovirus cell-to-cell spread in the absence of an essential assembly protein. Proc Natl Acad Sci U S A. 2005;102:2081–2086. doi: 10.1073/pnas.0409597102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodhi A, Chaisuparat R, Hu J, Ramsdell AK, Manning BD, Sausville EA, Sawai ET, Molinolo A, Gutkind JS, Montaner S. The TSC2/mTOR pathway drives endothelial cell transformation induced by the Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor. Cancer Cell. 2006;10:133–143. doi: 10.1016/j.ccr.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Soucek T, Pusch O, Wienecke R, DeClue JE, Hengstschlager M. Role of the tuberous sclerosis gene-2 product in cell cycle control. Loss of the tuberous sclerosis gene-2 induces quiescent cells to enter S phase. J Biol Chem. 1997;272:29301–29308. doi: 10.1074/jbc.272.46.29301. [DOI] [PubMed] [Google Scholar]
- Stamminger T, Gstaiger M, Weinzierl K, Lorz K, Winkler M, Schaffner W. Open reading frame UL26 of human cytomegalovirus encodes a novel tegument protein that contains a strong transcriptional activation domain. J Virol. 2002;76:4836–4847. doi: 10.1128/JVI.76.10.4836-4847.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton RJ, McSharry BP, Rickards CR, Wang EC, Tomasec P, Wilkinson GW. Cytomegalovirus destruction of focal adhesions revealed in a high-throughput Western blot analysis of cellular protein expression. J Virol. 2007;81:7860–7872. doi: 10.1128/JVI.02247-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci U S A. 2002;99:13571–13576. doi: 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13:1259–1268. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- Terhune S, Torigoi E, Moorman N, Silva M, Qian Z, Shenk T, Yu D. Human cytomegalovirus UL38 protein blocks apoptosis. J Virol. 2007;81:3109–3123. doi: 10.1128/JVI.02124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Inoki K, Kim E, Guan KL. TSC1/TSC2 and Rheb have different effects on TORC1 and TORC2 activity. Proc Natl Acad Sci U S A. 2006;103:6811–6816. doi: 10.1073/pnas.0602282103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Silva MC, Shenk T. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc Natl Acad Sci U S A. 2003;100:12396–12401. doi: 10.1073/pnas.1635160100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Smith GA, Enquist LW, Shenk T. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J Virol. 2002;76:2316–2328. doi: 10.1128/jvi.76.5.2316-2328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Alwine JC. Human cytomegalovirus major immediate-early proteins and simian virus 40 large T antigen can inhibit apoptosis through activation of the phosphatidylinositide 3′-OH kinase pathway and the cellular kinase Akt. J Virol. 2002;76:3731–3738. doi: 10.1128/JVI.76.8.3731-3738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wharton W, Donovan M, Coppola D, Croxton R, Cress WD, Pledger WJ. Density-dependent growth inhibition of fibroblasts ectopically expressing p27(kip1) Mol Biol Cell. 2000;11:2117–2130. doi: 10.1091/mbc.11.6.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.