

Abstract
The epidermal growth factor receptor (EGFR) is a transmembrane tyrosine kinase receptor involved in the proliferation and survival of cancer cells. EGFR is the first molecular target against which monoclonal antibodies (mAb) have been developed for cancer therapy. Here we review the mechanisms underlying the effects of EGFR-specific mAb in cancer therapy. The efficacy of EGFR-specific mAb in cancer occurs thanks to inhibition of EGFR-generated signalling; furthermore, the effects of antibodies on the immune system seem to play an important role in determining the overall anti-tumour response. In this review, attention is focused on cetuximab and panitumumab, two mAb introduced recently into clinical practice for treatment of metastatic colorectal and head and neck cancer which target the external part of EGFR.
Keywords: cancer, cetuximab, epidermal growth factor receptor, monoclonal antibodies, panitumumab
Epidermal growth factor receptor
Epidermal growth factor receptor (EGFR) is a cell membrane growth factor receptor characterized by tyrosine kinase activity that plays a crucial role in the control of key cellular transduction pathways in both normal and cancerous cells. EGFR is over-expressed in a variety of human tumours, including head and neck, breast, lung, colorectal, prostate, kidney, pancreas, ovary, brain and bladder cancer [1–3].
The 170 kDa protein function depends either on the formation of EGFR – EGFR homodimers or heterodimers – that comprise the three members of the EGFR [human epidermal receptor 1 (HER1)] family of growth factor receptors (HER2, HER3 and HER4) following binding of an EGFR-selective ligand. The activating ligands include the epidermal growth factor (EGF), transforming growth factor-α (TGF-α), amphiregulin or neuregulin. The binding EGFR/ligand results in conformational changes that allow the activation of EGFR tyrosine kinase and the phosphorylation of specific tyrosine residues within the EGFR intracellular carboxyl- terminal domain. Phosphorylated tyrosine residues serve as docking sites for several signalling proteins finally stimulating cell proliferation, loss of differentiation, invasion, angiogenesis and blocking of apoptosis. Within a few hours of activation, receptors are internalized into cytoplasm, where they are either degraded or recycled back to the membrane.
EGFR homodimers undergo degradation, whereas EGFR and HER2 heterodimerization is associated with recycling upon endocytosis, which enhances mitogenic signalling. Homodimers are weaker effectors compared with heterodimers: EGFR and HER2 is the most common heterodimer; HER2:3 plus neuregulin is the most potent combination; HER2 decelerates the internalization of HER1; HER1 requires ligand binding before dimerization, while HER2 does not require a ligand to dimerize and is often expressed at a 100-fold higher concentration compared with HER1. The complex signalling network generated by triggering EGFR includes the ras- and mitogen-activated protein kinase (MAPK) pathway that leads to cell proliferation, the phosphatidylinositol-3 kinase (PI3K) and protein kinase B (Akt) pathway driving cell cycle progression and cell survival [4]. There is also evidence that EGFR can translocate to the nucleus, where it acts as a transcription factor (Fig. 1) [5–7].
Fig. 1.
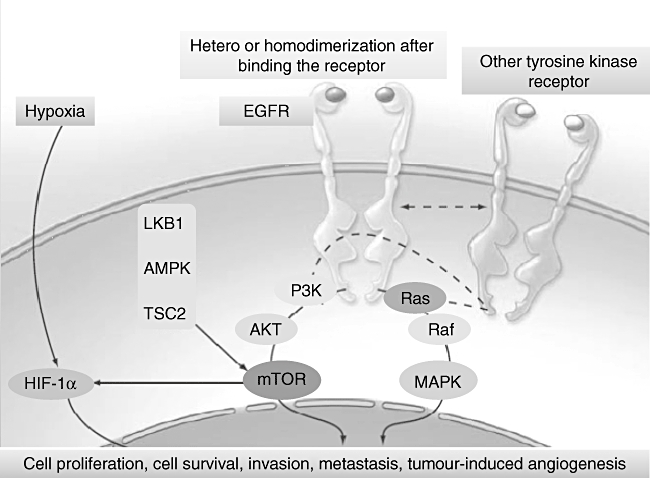
Signal transduction pathway mediated by epidermal growth factor receptor (EGFR). The interaction with ligand that occurs in the extracellular portion of the EGFR family induces the formation of a functionally active EGFR–EGFR homodimer or of an EGFR–human epidermal receptor 2 (HER2), EGFR–HER3 or EGFR–HER4 heterodimer. As explained in the text, this results in conformational changes that allow the EGFR tyrosine kinase to be activated with the phosphorylation of specific tyrosine residues within the EGFR intracellular carboxyl-terminal domain. Phosphorylated tyrosine residues trigger a complex programme of intracellular signals to the cytoplasm and then to the nucleus that stimulates cell proliferation, loss of differentiation, invasion and angiogenesis, and blocks the apoptosis.
Inhibition of the target
Two pharmacological approaches have been used successfully to inhibit EGFR functions in cancer treatment: neutralizing monoclonal antibodies and small-molecule tyrosine kinase inhibitors.
Anti-EGFR monoclonal antibodies bind to the extracellular domain of EGFR in its inactive state; they compete for receptor binding by occluding the ligand-binding region, and thereby block ligand-induced EGFR tyrosine kinase activation [8,9]. Small-molecule EGFR tyrosine kinase inhibitors compete reversibly with Adenosine 5′ triphosphate to bind to the intracellular catalytic domain of EGFR tyrosine kinase and, thus, inhibit EGFR auto-phosphorylation and downstream signalling. In addition, various small-molecule EGFR tyrosine kinase inhibitors can block different growth factor receptor tyrosine kinases, including other members of the EGFR family, or the vascular endothelial growth factor receptor. Anti-EGFR monoclonal antibodies recognize EGFR exclusively and are therefore highly selective to this receptor. Nevertheless, an intrinsic or acquired resistance to the EGFR inhibitor that limits the use of these drugs in cancer therapy has been evidenced. This could be related to constitutive activation of downstream mediators or over-expression of other tyrosine kinase receptors [10]. For example, the persistent activation of downstream signalling steps such as MAPK and PI3K/Akt could promote cell proliferation, survival, differentiation and motility [11], as illustrated in a study in which the resistant phenotype unaffected by the treatment with C225 (monoclonal antibody anti-EGFR) is due to the intrinsic activity of those pathways [12].
Moreover, the increase of angiogenesis caused by up-regulation of the vascular endothelial growth factor in human cancer cells by EGF and TGF-α could promote resistance to EGFR inhibition [13].
To date, two anti-EGFR monoclonal antibodies, panitumumab and cetuximab, are currently in widespread use in cancer treatment.
Cetuximab
Cetuximab (C225, Erbitux™) is an immunoglobulin (Ig) G1 human–murine chimeric counterpart of the murine monoclonal antibody M225. It binds to the EGFR with a 2-log higher affinity compared with the natural ligands TGF-α and EGF [14]. Binding of cetuximab to the EGFR promotes receptor internalization and subsequent degradation without receptor phosphorylation and activation [15]. This results in receptor down-regulation, reducing the availability of EGFR on the cell surface and preventing activation of EGFR-associated, downstream signalling pathways. Cetuximab also binds to the mutant receptor EGFRvIII, inducing internalization of 50% of antibody-receptor complexes after 3 h, and an 80% reduction in phosphorylated EGFRvIII.
Binding of cetuximab to EGFR inhibits the progression of the cell cycle at the G0/G1 boundary, increases expression of the cell cycle regulator p27KIP1 and induces apoptosis by increasing expression of pro-apoptotic proteins (e.g. Bax and caspase-3, caspase-8 and caspase-9) [16] or by inactivation of anti-apoptotic proteins (e.g. Bcl-2) inducing decreased expression or phosphorylation [17]. Cetuximab has also been reported to inhibit the production of pro-angiogenic factors such as vascular endothelial growth factor, interleukin-8 and the basic fibroblast growth factor; inhibition of these factors is associated with a decrease in new blood vessel formation and the development of distant metastases in orthotopic cancer models [18].
Clinical overview of cetuximab
The first cetuximab Phases I and I/II trials demonstrated the safety of cetuximab alone or in combination with cytotoxic chemotherapy used as treatments for patients with metastatic squamous cell carcinoma of the head and neck, colorectal cancer and non-small-cell lung cancer [19–22].
A multi-centre, randomized Phase II trial examined the combination of cetuximab and irinotecan (n = 218) compared to cetuximab alone (n = 111) in patients with EGFR-positive irinotecan-refractory metastatic colon cancer. The response rate (RR) of the combination was significantly higher, 23% versus 11% with cetuximab alone (P = 0·007), and the disease control rates were 56–32%, respectively. The time to progression was also significantly greater for the combination arm (4·1 versus 1·5 months, P < 0·001), and the median survival time was 8·6 months in the cetuximab arm (P = 0·48); survival in the two arms was not statistically significant. The presence of cutaneous rash correlated significantly with response, as the RR was higher in patients presenting rash compared with rash-free patients (25·8% versus 6·3%, P = 0·005) [23]. These findings were also confirmed by the MABEL (Monoclonal Antibody Erbitux in a European Pre-License study) study, which assessed definitively the efficacy and safety of cetuximab in the treatment of metastatic colorectal cancer (mCRC) [24].
Based on these results, cetuximab was approved for use in patients with EGFR-expressing mCRC refractory to irinotecan-based chemotherapy, in combination with irinotecan (for irinotecan-refractory patients) or as monotherapy (for irinotecan-intolerant patients).
The impact of cetuximab plus irinotecan in second-line metastatic colorectal EGFR-expressing cancer patients was examined in a multi-national Phase III trial known as the EPIC (Erbitux Plus Irinotecan for Metastatic Colorectal Cancer) study. The results reveal that treatment with cetuximab improved the progression-free survival (PFS), RR and health-related quality of life [25]. Moreover, cetuximab demonstrated activity in patients with colorectal cancer in whom other treatments have failed, improving the PFS, overall survival and quality of life over best supportive care (BSC) [26].
The role of cetuximab was also investigated in first-line treatment of mCRC. Phase II studies indicate that cetuximab combined with both irinotecan- and oxaliplatin-based chemotherapies is active with a 10–20% absolute increase in RR [27–30]. Recently, a multi-centre, randomized, Phase III study evaluated the combination of cetuximab with a standard chemotherapeutic regimen of fluorouracil, leucovorin and irinotecan (Folfiri) in previously untreated mCRC. Cetuximab plus Folfiri increased RR significantly and prolonged PFS (Table 1) [31,32].
Table 1.
Up-front therapy with cetuximab and oxaliplatin or irinotecan (intention to treat population).
| Authors | Patients (n) | Regimen | Response rate % | PFS (months) |
|---|---|---|---|---|
| Tabernero 2007 [27] | 43 | Folfox-4 | 72 | 10·2 |
| Bokemeyer 2007 [28] | 337 | Folfox-4 versus Folfox-4 + cmab | 35·7 45·6 | 7·2 |
| Borner 2006 [71] | 74 | Xelox versus Xelox + cmab | 33 53 | – |
| Folprecht 2006 [29]Van Cutsem 2007 [31] | 21 1217 | Irinotecan/5FU/FA + cmab Folfiri versus Folfiri + cmab | 67 38·7 46·9 | 9·9 8 8·9 |
| Tabernero 2006 [30] | 62 | Folfiri + cmab every 2 weeks (escalating dose) | 42 | 7·2 |
Cmab, cetuximab; PFS, progression-free survival; FU/FA, fluorouracil and leucovorin; Folfiri, fluorouracil, leucovorin and irinotecan; Folfox, oxaliplatin, leucovorin and fluorouracil.
Cetuximab was also approved in 2006 for the treatment of head and neck cancer as a single agent in patients with advanced platinum resistant disease and in combination with radiotherapy for treatment of locally advanced disease.
A Phase III randomized trial of cetuximab in advanced head and neck cancer was carried out. In this study, a sample of 424 patients was divided into those treated with radiation therapy alone and those receiving supplementary treatment with cetuximab. At a median follow-up of 54 months, it was observed that the rate of survival was almost double in patients receiving cetuximab compared with patients receiving radiation therapy alone (49 versus 29 months; P = 0·03). Statistically significant increases in loco-regional control and PFS were also reported for the group receiving cetuximab. This is the first study to demonstrate a statistically significant survival benefit rate for patients treated with curative intent using an anti-EGFR antibody [33].
In addition, cetuximab plays a crucial role in the treatment of platinum-resistant squamous-cell carcinoma of head and neck cancer alone or in combination with chemotherapy (first-line setting) [34].
Panitumumab
Panitumumab (Vectibix, Amgen, Thousand Oaks, CA, USA) is a fully human IgG2 targeting the extracellular domains of EGFR monoclonal antibody. Developed by Abgenix's XenoMouse technology, which creates antibodies that do not contain murine proteins, it offers effective high affinity therapy (Kd = 5 × 10−11 M) with a minimum rate of allergic reactions or anaphylaxis [35].
Well tolerated – its main toxic effect is dermatological – it has never reached grade 4 in clinical trials. Because of its structure (fully human antibody), infusion-related reactions are minimal (only one of 148 patients experienced grade 3 reaction while one of 463 patients discontinued treatment due to grade 2 hypersensitivity reaction). Panitumumab could be administered weekly, fortnightly or every 3 weeks without visible change in pharmacokinetic parameters [36].
Clinical overview of panitumumab
Panitumumab has been evaluated in clinical trials both as monotherapy and in combination with other agents for the treatment of various types, including colorectal and kidney cancer [37].
The Phase I study demonstrated that pharmacokinetic exposure showed similarities at 2·5 mg/kg per week, 6 mg/kg every 2 weeks and 9 mg/kg every 3 weeks. Grades 3- or 4-related adverse events were noted in 10% of patients, with grade 3 skin-related effects being the most frequent (7% of patients). No maximum tolerated dose was reached, and no human anti-human antibody formation or infusion-related reactions were observed [38].
In the Phase II studies, panitumumab in monotherapy and in combination with chemotherapy for the treatment of chemo-refractory colorectal cancer was active and well tolerated. A relationship between skin rash severity and survival was noted as being similar to that observed with the use of cetuximab. Moreover, its efficacy appears to be similar in patients with both low and negative EGFR levels [39–42].
A large Phase III multi-centre pivotal trial randomized patients with oxaliplatin and irinotecan-refractory EGFR-expressing mCRC between BSC and BSC plus panitumumab at a dose of 6 mg/m2 every 2 weeks. The aim of this study was to show the significant difference in PFS. Panitumumab showed a 46% decrease in tumour progression rate compared with that of BSC alone (hazard ratio: 0·54; 95% confidence interval: 0·44, 0·66; P < 0·000000001, stratified log-rank test). The subset analyses demonstrated the consistent treatment effect of panitumumab in all subgroups of patients. The RR was significantly higher in the panitumumab arm, with an 8% partial RR and 28% achieving stable disease, compared with no partial responses and 10% stable disease with BSC alone. The time to response was 8 weeks, and the median duration of the response was 17 weeks. Panitumumab also showed activity in cross-over study patients, with 10% achieving partial response and 32% stable disease. No difference was observed in overall survival (hazard ratio: 1·00; 95% confidence interval: 0·82–1·22) due probably to the high rate of cross-over population to panitumumab.
Skin reactions of any grade occurred in 90% of patients receiving panitumumab, and in 9% of those receiving BSC; the incidence of grades 3–4 skin-related adverse reactions was 14% compared with 0% in patients not treated with panitumumab. The incidence of skin toxicity in the panitumumab group was dose-related; however, no correlation was observed between dose and severity [43].
A further analysis of biomarkers was conducted to determine whether the effect of panitumumab monotherapy on PFS differed in patients with tumours characterized by mutant compared with wild-type (WT) K-ras.
Amado et al. first demonstrated that the response to panitumumab monotherapy and the improvement in PFS was limited only to patients with WT K-ras. No patient harbouring a K-ras mutation (46%) responded to panitumumab [44].
These findings led to registration by regulatory agencies worldwide as a monotherapy for third-line treatment of colorectal cancer that is refractory to fluoropyrimidines, oxaliplatin or irinotecan. Moreover, in 2007, panitumumab was approved by the European Medicines Agency for use in patients with colorectal cancer carrying a normal, WT K-ras gene.
The role of panitumumab in combination with anti-angiogenic drugs has also been explored in a randomized Phase III study (panitumumab advanced colorectal cancer evaluation). In this trial patients with mCRC were assigned randomly for first-line treatment within each chemotherapy cohort (823 patients oxaliplatin- and 230 irinotecan-based) to bevacizumab and chemotherapy with or without panitumumab 6 mg/kg every 2 weeks. The primary end-point was PFS within the oxaliplatin cohort. The results of the study were negative, as the combination of panitumumab with bevacizumab and chemotherapy resulted in a decrease of PFS and in excessive toxicity, particularly diarrhoea, infections and pulmonary embolism. The results were consistent in both the oxaliplatin and irinotecan cohorts. Moreover, as demonstrated previously, the triple combination did not provide additional benefit in the K-ras WT population treated with panitumumab [45].
Predictive biomarkers
The early biomarker, developed in mCRC to correlate with the activity of anti-EGFR antibodies, was confined to merely expressing the target.
Chung et al. demonstrated that colorectal cancer patients with EGFR-negative tumours have the potential to respond to cetuximab-based therapies, registering a 25% objective RR. Consequently, the presence of the target (EGFR) does not ensure the response to anti-EGFR inhibitors. Furthermore, EGFR analysis by current immunohistochemistry techniques does not seem to have predictive value for the selection or the exclusion of patients for cetuximab, therefore EGFR immunohistochemistry is not warranted currently [46].
In addition, not even the gene copy number for EGFR determined by fluorescence in situ hybridization on tumour samples was associated significantly with clinical response to this targeted therapy [47].
Several studies have been carried out to define a subgroup of patients with potentially differential responses to anti-EGFR antibody therapy, and these show that benefits are confined to the subgroup with WT K-ras tumours.
K-ras is a guanosine triphosphate-binding protein with a critical role in cellular growth and survival pathways [48]. It is mutated in approximately 30–50% of colorectal cancer and results in a constitutive activation of the MAPK pathway and in lack of response with EGFR inhibitors [49–53].
Published reports so far have investigated the role of K-ras as a selection marker for EGFR inhibitor treatment on tumour samples from uncontrolled studies and include therapy with EGFR inhibitors alone or in combination with chemotherapy. In this respect, the relative effect with antibody treatment on the outcome of K-ras, as a predictive marker, is not so clear. In, the study published by Amado et al., comparing panitumumab monotherapy with BSC, no clinical benefit to panitumumab at all in patients with the K-ras mutation was evidenced in any clinical end-point, thus confirming the role of the K-ras mutant as a negative predictor of response (Table 2) [44].
Table 2.
Phase III study of panitumumab versus best supportive care (BSC).
| Panitumumab (n = 231) | ITT population BSC (n = 232) | K-ras WT panitumumab (n = 124) | BSC (n = 119) | |
|---|---|---|---|---|
| RR, n (%) | 22 (10) | 0 (0) | 21 (17) | 0 (0) |
| mPFS (m) | 1·9 | 1·7 | 2·9 | 1·7 |
| PFS HR | 0·54 (0·44–0·66) | 0·45 (0·34–0·59) | ||
| Amado et al. 2008 [44] |
ITT, intention to treat; RR, response rate; PFS, progression-free survival; HR, hazard ratio; WT, wild-type.
Analyses of K-ras status and response to cetuximab have provided similar results.
The retrospective correlative analysis of patients enrolled in the CO.17 trial performed by Karapetis et al. show that the benefit of cetuximab treatment was confined to patients who had a tumour with no K-ras mutations, with few or no effects in the presence of a K-ras mutation (Table 3) [54].
Table 3.
Phase III study of cetuximab versus best supportive care (BSC).
| Cetuximab (n = 287) | ITT population BSC (n = 285) | K-ras WT cetuximab (n = 124) | BSC (n = 119) | |
|---|---|---|---|---|
| RR, n (%) | – | – | 13 (13) | 0 (0) |
| mPFS (m) | 1·9 | 1·8 | 3·8 | 1·9 |
| PFS HR | 0·68 (0·57–0·80) | 0·40 (0·30–0·54) | ||
| mOS (m) | 6·1 | 4·6 | 9·5 | 4·8 |
| OS HR | 0·77 (0·64–0·92) | 0·55 (0·41–0·74) | ||
| Karapetis et al. 2008 [54] |
ITT, intention to treat; PFS, progression-free survival; RR, response rate; HR, hazard ratio; OS, overall survival; WT, wild-type.
Moreover, in patients with mCRC treated with first-line infused fluorouracil, folinic acid and oxaliplatin with or without cetuximab the improved RR and PFS associated with cetuximab was confined to those patients having a K-ras WT tumour [55–57].
Recently, another breakthrough has been achieved focusing on the role of v-raf murine sarcoma viral oncogene homologue B1 (BRAF) as a potential biomarker for anti-EGFR antibody treatment. Di Nicolantonio et al. showed that in the presence of BRAF mutations (BRAF V600E allele) there was no response to either cetuximab or panitumumab [58].
Immunological mechanisms
In recent years, it has been shown that the anti-tumoral effects of mAb may be due to their ability to act on the immune system [59]. In general terms, the use of mouse chimeric antibodies may elicit immune responses specifically for the mouse portion of the molecule leading to a destruction of the antibody; in some cases, however, also to the destruction of targeted tumoral cells [60].In addition, several reports have described, both in vitro and in vivo, how these antibodies are able to elicit antibody-dependent cellular cytotoxicity (ADCC), complement-mediated cytotoxicity, or both. These effector responses are due to the binding of the Fc portion of antibodies to the Fc receptors expressed on the surface of different cell types. This binding leads to a wide array of effects, from uptake to killing. It should be noted that macrophages, dendritic cells, neutrophils, eosinophils, B cells, mast cells, natural killer (NK) cells, platelets and Langerhans cells express Fc receptors capable of discriminating different Ig classes. The effects of ligation of the Fc portion of the antibody with the Fc receptors on the cells depend upon the specificity of the Fc receptors for a given Ig class and on the cell types. For instance, cetuximab is an IgG1, therefore it is able to bind several Fc receptors: FcγRI (CD64), FcγRII-A (CD32), FcγRII-B1 (CD32), FcγRII-B2 (CD32) and FcγRIII (CD16) [61]. For this reason, cetuximab is able to mediate ADCC induced by NK activity through binding to FcγRIII, but it is also able to engage Fc receptors on the surface of other cells such as eosinophils, mast cells, dendritic cells, B cells and other cell types [62]. Therefore, an obvious scenario can be envisaged in which the overall effects of these antibodies are also due to complex mechanisms other than those of ADCC and complement-mediated cytotoxicity. Recently, another intriguing mechanism has been highlighted that can help to exploit the potential effect of mAb in solid cancer. Upon recognition by antibodies of ligands on target cells, the components of this immune complex (antibody and ligand) can be removed (shaved) from these cells and internalized by cells expressing Fcγ receptors [63–66].
The mechanism has been called trogocytosis, or nibbling. Trogocytosis triggers a cascade of complex events, depending upon the ligand size and IgG class involved, which affect the immune response in different ways, sometimes even depressing the immune response or the efficacy of the binding antibody/target due to antigen shedding in blood [67]. It has also been shown to be effective when IgG2 antibodies, such as panitumumab, are used (Fig. 2) [68].
Fig. 2.
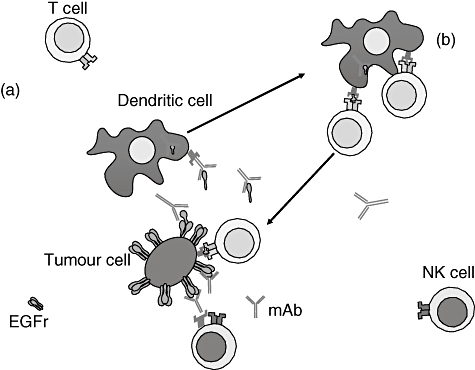
Effects of monoclonal antibody (mAb) therapies on the immune system. (a) The mAb bind the epidermal growth factor receptor (EGFR) expressed on the surface of the tumour cell; this binding activates antibody dependent cellular cytotoxicity by engaging Fc receptors on the surface of natural killer (NK) cells. Some T cells may also be activated by specific recognition of the FAb portion of the mAb. Finally, the binding mAb/EGFR can allow, by trogocytosis, the internalization of part of EGFR by antigen-presenting cells as the dendritic cells. (b) Dendritic cells present peptides derived from the internalized portion of EGFR to T cells, priming an immune response more efficient in recognition of EGFR peptides associated with major histocompatibility complex expressed by tumour cells. As reported in the text, Fc receptors are expressed on the surface of many cells (mast cells, eosinophils, etc.) that can secrete a variety of different cytokines and chemokines, thus amplifying the overall immune response induced by mAb.
Finally, it should be remembered that the ability to interfere with the signalling initiated by EGFR affects many other receptor systems, for instance chemokine and cytokine receptors Toll-like receptors, that are critical for immune responses [69]. Another interesting perspective worthy of note is that activation of the immunonological mechanisms described above corresponds with the observation that increased efficacy in patients treated with EGFR mAb is correlated with the presence of cutaneous rash. Indeed, the links between complement activation, ADCC and cutaneous rash are well known to clinical immunologists.
On the basis of such premises, it can be suggested that the beneficial effects of anti-EGFR antibodies do not depend only upon their ability to interfere with the signalling generated by the receptor and this could explain, at least in part, the different response observed in treated patients. Furthermore, a better understanding of the interactions between the mAb used as therapy for solid tumours and the immune system could be critical in designing new approaches for immunotherapy in cancer.
Conclusion
The use of anti-EGFR monoclonal antibodies is diffuse in cancer therapy; however, clinical responses have been observed in only 15% of patients treated. Moreover, the use of these drugs has contributed only a modest overall survival benefit in comparison to commonly practised BSC. Although these results could be considered interesting, it should be taken into account that in several countries the analysis of cost-effectiveness is now proportionally extremely limiting, given the economic crisis worldwide and its effects on the budgets of national health systems. As a result, these data would not be sufficient to influence or avoid a progressive reduction in the use of mAb therapy in clinical practice.
The challenge for the near future is to identify biomarkers that are capable of predicting and targeting eligible patients for anti-EGFR antibody treatment on a one-to-one scale. On the basis of reported trials and current debate [70], complex efforts are needed that take into consideration not only the mechanistic effect of blocking EGFR, but also the further exploiting of mAb effects on the immune system. Clearly, this is no simple task and, although there is now an impressive quantity of data in the field, we are still far from having a thorough understanding of the scenario as a whole. In this respect, the consideration that EGFR-related signalling is involved in many steps of immune responses provides an inkling of just how complex is the road ahead. However, in the long term the final destination will be well worth all our painstaking and strenuous efforts.
Disclosure
The authors declare no conflict of interest.
References
- 1.Woodburn JR. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol Ther. 1999;82:241–50. doi: 10.1016/s0163-7258(98)00045-x. [DOI] [PubMed] [Google Scholar]
- 2.Yarden Y. The EGFR family and its ligands in human cancer signaling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37:S3–S8. doi: 10.1016/s0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- 3.Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19:6550–65. doi: 10.1038/sj.onc.1204082. [DOI] [PubMed] [Google Scholar]
- 4.Yarden Y, Sliwkowski MX. Untangling the ErbB signaling network. Nat Rev Mol Cell Biol. 2001;2:127–37. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 5.Lin SY, Makino K, Xia W, et al. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–8. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 6.Oksvold M, Huitfeldt H, Stang E, Madshus I. Localizing the EGF receptor. Nat Cell Biol. 2002;4:E22. doi: 10.1038/ncb0202-e22a. author reply E22. [DOI] [PubMed] [Google Scholar]
- 7.Waugh MG, Hsuan JJ. EGF receptors as transcription factors: ridiculous or sublime? Nat Cell Biol. 2001;3:E209, 11. doi: 10.1038/ncb0901-e209. [DOI] [PubMed] [Google Scholar]
- 8.Ciardiello F, Tortora G. EGFR antagonist in cancer treatment. N Engl J Med. 2008;358:1160–74. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 9.Normanno N, Bianco C, De Luca A, Maiello MR, Salomon DS. Target-based agents against ErbB receptors and their ligands: a novel approach to cancer treatment. Endocr Relat Cancer. 2003;10:1–21. doi: 10.1677/erc.0.0100001. [DOI] [PubMed] [Google Scholar]
- 10.Bianco R, Damiano V, Gelardi T, Daniele G, Ciardiello F, Tortora G. Rational combination of targeted therapies as a strategy to overcome the mechanisms of resistance to inhibitors of EGFR signaling. Curr Pharm Des. 2007;13:3358–67. doi: 10.2174/138161207782360564. [DOI] [PubMed] [Google Scholar]
- 11.Kolch W, Calder M, Gilbert D. When kinases meet mathematics: the systems biology of MAPK signalling. FEBS Lett. 2005;579:1891–5. doi: 10.1016/j.febslet.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 12.Janmaat ML, Kruyt FA, Rodriguez JA, Giaccone G. Response to epidermal growth factor receptor inhibitors in nonsmall cell lung cancer cells: limited antiproliferative effects and absence of apoptosis associated with persistent activity of extracellular signal-regulated kinase or Akt kinase pathways. Clin Cancer Res. 2003;9:2316–26. [PubMed] [Google Scholar]
- 13.Gille J, Swerlick RA, Caughman SW. Transforming growth factor-alpha-induced transcriptional activation of the vascular permeability factor (VPF/VEGF) gene requires AP-2- dependent DNA binding and transactivation. EMBO J. 1997;16:750–9. doi: 10.1093/emboj/16.4.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim ES, Khuri FR, Herbst RS. Epidermal growth factor receptor biology (IMC-C225) Curr Opin Oncol. 2001;13:506–13. doi: 10.1097/00001622-200111000-00014. [DOI] [PubMed] [Google Scholar]
- 15.Hadari YR, Doody JF, Wang Y, et al. The IgG1 monoclonal antibody cetuximab induces degradation of the epidermal growth factor receptor. J Clin Oncol. 2004;22 abstract 234. [Google Scholar]
- 16.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA. 2001;98:11598–603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gingras AC, Kennedy SG, O'Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt (PKB) signaling pathway. Genes Dev. 1998;12:502–13. doi: 10.1101/gad.12.4.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perrotte P, Matsumoto T, Inoue K, et al. Anti-epidermal growth factor receptor antibody C225 inhibits angiogenesis in human transitional cell carcinoma growing orthotopically in nude mice. Clin Cancer Res. 1999;5:257–65. [PubMed] [Google Scholar]
- 19.Thienelt CD, Bunn PA, Jr, Hanna N, et al. A multi-centered phase I/II study of cetuximab in combination with paclitaxel and carboplatin in untreated patients with stage IV non-small cell lung cancer. J Clin Oncol. 2005;23:8786–93. doi: 10.1200/JCO.2005.03.1997. [DOI] [PubMed] [Google Scholar]
- 20.Robert F, Blumenschein G, Dicke K, Tseng J, Saleh MN, Needle M. Phase Ib/Ia study of anti-epidermal growth factor receptor antibody, cetuximab, in combination with gemcitabine/carboplatin in patients with advanced stage IV non-small cell lung cancer. Proc Am Soc Clin Oncol. 2003;22:643. abstract 2587. [Google Scholar]
- 21.Saltz LB, Meropol NJ, Loehrer PJ, Sr, Needle MN, Kopit J, Mayer RJ. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22:1201–8. doi: 10.1200/JCO.2004.10.182. [DOI] [PubMed] [Google Scholar]
- 22.Vega-Villegas E, Awada R, Mesia L, et al. A phase I study of cetuximab in combination with cisplatin or carboplatin and 5-FU in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. Proc Am Assoc Cancer Res. 2003;22:2020. [Google Scholar]
- 23.Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–45. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 24.Wilke H, Glyne-Jones R, Thaler J, et al. Cetuximab plus irinotecan in heavily preatreated metastatic colorectal cancer progression on irinotecan. MABEL study. J Clin Oncol. 2008;26:5335–43. doi: 10.1200/JCO.2008.16.3758. [DOI] [PubMed] [Google Scholar]
- 25.Sobrero AF, Maurel J, Fehrenbacher L. EPIC: phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:2311–9. doi: 10.1200/JCO.2007.13.1193. [DOI] [PubMed] [Google Scholar]
- 26.Jonker DJ, O'Callaghan CJ, Karapetis CS, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–8. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- 27.Tabernero J, Van Cutsem E, Díaz-Rubio E, et al. Phase II trial of cetuximab in combination with fluorouracil, leucovorin, and oxaliplatin in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2007;25:5225–32. doi: 10.1200/JCO.2007.13.2183. [DOI] [PubMed] [Google Scholar]
- 28.Bokemeyer I, Bondarenko A, Makhson H, et al. FOLFOX-4 cetuximab plus 5-FU/FA/oxaliplatin (FOLFOX-4) versus in the first-line treatment of metastatic colorectal cancer (mCRC): OPUS, a randomized phase II study. J Clin Oncol. 2007;25 abstract 4035. [Google Scholar]
- 29.Folprecht G, Lutz MP, Schöffski P, et al. Cetuximab and irinotecan/5-fluorouracil/folinic acid is a safe combination for the first-line treatment of patients with epidermal growth factor receptor expressing metastatic colorectal carcinoma. Ann Oncol. 2006;17:450–56. doi: 10.1093/annonc/mdj084. [DOI] [PubMed] [Google Scholar]
- 30.Tabernero J, Cervantes A, Martinelli E, et al. Optimal dose of cetuximab (C) given every 2 weeks (q2w): a phase I pharmacokinetic (PK) and pharmacodynamic (PD) study of weekly (q1w) and q2w schedules in patients (pts) with metastatic colorectal cancer (mCRC) J Clin Oncol. 2006;24:1425. abstract 3085. [Google Scholar]
- 31.Van Cutsem E, Nowacki M, Lang I, et al. Randomized phase III study of irinotecan and 5-FU/FA with or without cetuximab in the first-line treatment of patients with metastatic colorectal cancer (mCRC): the CRYSTAL trial. J Clin Oncol. 2007;25 abstract 4000. [Google Scholar]
- 32.Van Cutsem E, Peeters M, Siena S, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy – refractory metastatic colorectal cancer. J Clin Oncol. 2007;25:1658–64. doi: 10.1200/JCO.2006.08.1620. [DOI] [PubMed] [Google Scholar]
- 33.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–78. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 34.Vermorken JB, Mesia R, Vega E, et al. Cetuximab extends survival of patients or recurrent or metastatic SCCHN when added to first line platinum-based therapy results of a randomized phase III (EXTREME) study. Late-breaking abstract presented at the 43rd American Society of Clinical Oncology Annual Meeting, Chicago, 1–5 June 2007.
- 35.Mendez MJ, Green LL, Corvalan JR, et al. Functional transplant of megabase human immunoglobulin loci recapitulates human antibody response in mice. Nat Genet. 1997;15:146–56. doi: 10.1038/ng0297-146. [DOI] [PubMed] [Google Scholar]
- 36.Mitchell EP, Rose L, Ramirez M, Jacobs M. Dermatological toxicities of panitumumab in the treatment of patients with metastatic colorectal cancer from two clinical studies. Annual Meeting Proceedings. Gastrointest Cancers Symp. 2007 abstract 449. [Google Scholar]
- 37.Rowinsky EK, Schwartz GH, Gollob JA, et al. Safety, pharmacokinetics, and activity of ABX-EGF, a fully human anti-epidermal growth factor receptor monoclonal antibody in patients with metastatic renal cell cancer. J Clin Oncol. 2004;22:3003–15. doi: 10.1200/JCO.2004.11.061. [DOI] [PubMed] [Google Scholar]
- 38.Weiner L, Belldegrun A, Rowinsky E, et al. Update results from a dose and schedule study of panitumumab (ABX-EGF) monotherapy, in patients with advanced solid malignancies. J Clin Oncol. 2005;23:16S. doi: 10.1158/1078-0432.CCR-07-1509. abstract 3059. [DOI] [PubMed] [Google Scholar]
- 39.Berlin J, Neubauer M, Swanson P, et al. Panitumumab antitumor activity in patients (pts) with metastatic colorectal cancer (mCRC) expressing > 10% epidermal growth factor receptor (EGFR) J Clin Oncol. 2006;24(suppl.18S):3548. ASCO Annual Meeting Proceedings Part I. [Google Scholar]
- 40.Berlin J, Posey J, Tchekmedyian S, et al. Panitumumab with irinotecan/leucovorin/5-fluorouracil for first-line treatment of metastatic colorectal cancer. Clin Colorectal Cancer. 2007;6:427–32. doi: 10.3816/CCC.2007.n.011. [DOI] [PubMed] [Google Scholar]
- 41.Hecht JR, Mitchell E, Baranda J, et al. Panitumumab antitumor activity in patients (pts) with metastatic colorectal cancer (mCRC) expressing low (1–9%) or negative (1%) levels of epidermal growth factor receptor (EGFR) J Clin Oncol. 2006;24(suppl.18S):3547. ASCO Annual Meeting Proceedings Part I. [Google Scholar]
- 42.Malik I, Hecht JR, Patnaik A, et al. Safety and efficacy of panitumumab monotherapy in patients with metastatic colorectal cancer (mCRC) J Clin Oncol. 2005;23:251s. abstract 3520. [Google Scholar]
- 43.Van Cutsem E, Peeters M, Siena S, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol. 2007;25:1658–64. doi: 10.1200/JCO.2006.08.1620. [DOI] [PubMed] [Google Scholar]
- 44.Amado RG, Wolf M, Peeters M, et al. Wild-type K-ras is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–31. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 45.Hecht JR, Mitchell E, Chidiac T, et al. A randomized phase III of chemotherapy, bevacizumab, and panitumumab a compared with chemotherapy and bevacizumab alone for metastatic colorectal cancer. J Clin Oncol. 2009;27:672–80. doi: 10.1200/JCO.2008.19.8135. [DOI] [PubMed] [Google Scholar]
- 46.Chung KY, Shia J, Kemeny NE, et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol. 2005;23:1803–10. doi: 10.1200/JCO.2005.08.037. [DOI] [PubMed] [Google Scholar]
- 47.Moroni M, Veronese S, Benvenuti S, et al. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to anti-EGFR treatment in colorectal cancer: a cohort study. Lancet Oncol. 2005;6:279–86. doi: 10.1016/S1470-2045(05)70102-9. [DOI] [PubMed] [Google Scholar]
- 48.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–65. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 49.Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res. 2007;67:2643–8. doi: 10.1158/0008-5472.CAN-06-4158. [DOI] [PubMed] [Google Scholar]
- 50.Di Fiore F, Blanchard F, Charbonnier F, et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by cetuximab plus chemotherapy. Br J Cancer. 2007;96:1166–9. doi: 10.1038/sj.bjc.6603685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Esteller M, Gonzalez S, Risques RA, et al. K-ras and p16 aberrations confer poor prognosis in human colorectal cancer. J Clin Oncol. 2001;19:299–304. doi: 10.1200/JCO.2001.19.2.299. [DOI] [PubMed] [Google Scholar]
- 52.Lievre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–5. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 53.De Roock W, Piessevaux H, De Schutter J, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008;19:508–15. doi: 10.1093/annonc/mdm496. epub ahead of print on 12 November 2007. [DOI] [PubMed] [Google Scholar]
- 54.Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757–65. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 55.Van Cutsem E, Lang I, D'haens G, et al. Kras status and efficacy in the first-line treatment of patients with metastic colorectal cancer (mCRC) treated with FOLFIRI with or without. The CRYSTAL experience. J Clin Oncol. 2008;26:5s. suppl; abstract 2. [Google Scholar]
- 56.Bokemeyer C, Bondarenko I, Hartmann J, et al. Kras status and efficacy of first-line treatment of patients with metastatic colorectal cancer (mCRC) with FOLFOX with or without cetuximab: the OPUS experience. J Clin Oncol. 2008;26:178s. suppl; abstract 4000. [Google Scholar]
- 57.Cervantes A, Macarulla T, Martinelli E, et al. Correlation of KRAS status (wild type [wt]versus mutant [mt]) with efficacy to first-line cetuximab in a study of cetuximab single agent followed by cetuximab + FOLFIRI in patients (pts) with metastatic colorectal cancer (mCRC) J Clin Oncol. 2008;26:2105. suppl; abstract 4129. [Google Scholar]
- 58.Di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–12. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 59.Yan L, Hsu K, Beckman RA. Antibody-based therapy for solid tumors. Cancer J. 2008;14:178–83. doi: 10.1097/PPO.0b013e318172d71a. [DOI] [PubMed] [Google Scholar]
- 60.Jakobovits A, Amado RG, Yang X, Roskos L, Schwab G. From XenoMouse technology to panitumumab, the first fully human antibody product from transgenic mice. Nat Biotechnol. 2007;25:1134–43. doi: 10.1038/nbt1337. [DOI] [PubMed] [Google Scholar]
- 61.Murphy K, Travers P, Walport M. Janeway's immunobiology. 7th edn. New York: Garland Science; 2008. The humoral immune response. [Google Scholar]
- 62.Roda JM, Joshi T, Butchar JP, et al. The activation of natural killer cell effector functions by cetuximab-coated, epidermal growth factor receptor positive tumor cells is enhanced by cytokines. Clin Cancer Res. 2007;13:6419–28. doi: 10.1158/1078-0432.CCR-07-0865. [DOI] [PubMed] [Google Scholar]
- 63.Hudrisier D, Riond J, Mazarguil H, Gairin JE, Joly E. CTLs rapidly capture membrane fragments from target cells in a TCR signaling-dependent manner. J Immunol. 2001;166:3645–9. doi: 10.4049/jimmunol.166.6.3645. [DOI] [PubMed] [Google Scholar]
- 64.Joly E, Hudrisier D. What is trogocytosis and what is its purpose? Nat Immunol. 2003;4:815. doi: 10.1038/ni0903-815. [DOI] [PubMed] [Google Scholar]
- 65.Hudrisier D, Aucher A, Puaux AL, Bordier C, Joly E. Capture of target cell membrane components via trogocytosis is triggered by a selected set of surface molecules on T or B cells. J Immunol. 2007;178:3637–47. doi: 10.4049/jimmunol.178.6.3637. [DOI] [PubMed] [Google Scholar]
- 66.Beum PV, Lindorfer MA, Taylor RP. Within peripheral blood mononuclear cells, antibody-dependent cellular cytotoxicity of rituximab-opsonized Daudi cells is promoted by NK cells and inhibited by monocytes due to shaving. J Immunol. 2008;181:2916–24. doi: 10.4049/jimmunol.181.4.2916. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Y, Pastan I. High shed antigen levels within tumors: an additional barrier to immunoconjugate therapy. Clin Cancer Res. 2008;14:7981–6. doi: 10.1158/1078-0432.CCR-08-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beum PV, Mack DA, Pawluczkowycz AW, Lindorfer MA, Taylor RP. Binding of rituximab, trastuzumab, cetuximab, or mAb T101 to cancer cells promotes trogocytosis mediated by THP-1 cells and monocytes. J Immunol. 2008;181:8120–32. doi: 10.4049/jimmunol.181.11.8120. [DOI] [PubMed] [Google Scholar]
- 69.Shepard HM, Brdlik CM, Schreiber H. Signal integration: a framework for understanding the efficacy of therapeutics targeting the human EGFR family. J Clin Invest. 2008;118:3574–81. doi: 10.1172/JCI36049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Capdevila J, Saura C, Macarulla T, Casado E, Ramos FJ, Tabernero J. Monoclonal antibodies in the treatment of advanced colorectal cancer. Eur J Surg Oncol. 2007;33(Suppl 2):S24–34. doi: 10.1016/j.ejso.2007.09.025. Epub 2007 5 November. Review. PubMed PMID: 17981431. [DOI] [PubMed] [Google Scholar]
- 71.Borner M, Mingrone W, Koeberle D. The impact of cetuximab on the capecitabine plus oxaliplatin (XELOX) combination in first-line treatment of metastatic colorectal cancer (MCC): a randomized phase II trial of the Swiss Group for Clinical Cancer Research (SAKK) J Clin Oncol. 2006;24:585. doi: 10.1093/annonc/mdn058. abstract 3551. [DOI] [PubMed] [Google Scholar]