

Summary
DNA interstrand crosslinks (ICLs) are the most toxic lesions induced by chemotherapeutic agents such as Mitomycin C and Cisplatin. By covalently linking both DNA strands, ICLs prevent DNA melting, transcription, and replication. Studies on ICL signaling and repair have been limited because these drugs generate additional DNA lesions that trigger checkpoint signaling. Here, we monitor sensing, signaling from and repairing of a single, site-specific ICL in cell-free extract derived from Xenopus eggs and in mammalian cells. Notably, we demonstrate that ICLs trigger a checkpoint response independently of origin-initiated DNA replication and uncoupling of DNA polymerase and DNA helicase. The Fanconi anemia pathway acts upstream of RPA-ATR-Chk1 to generate the ICL signal. The system also repairs ICLs in a reaction that involves extensive, error-free, DNA synthesis. Repair occurs by both origin-dependent and origin-independent mechanisms. Our data suggest that cell sensitivity to crosslinking agents results from both checkpoint and DNA repair defects.
Introduction
DNA interstrand crosslinks (ICLs) are highly toxic lesions that covalently link both strands of DNA and distort the DNA helix. By preventing DNA melting, ICLs physically block DNA replication, recombination and RNA transcription. Failure to repair ICL lesions before DNA replication may induce DNA breaks, chromosomal rearrangements and cell death. ICLs are generated by chemotherapeutic agents including cisplatin (CDDP), mitomycin C (MMC), and nitrogen mustards, and by endogenous products of lipid peroxidation (Dronkert and Kanaar, 2001).
Pathways of ICL repair have mostly been inferred from the sensitivities of DNA repair-defective cell lines to crosslinking agents. However, crosslinking agents generate primarily intrastrand crosslinks and monoadducts; interstrand crosslinks (ICLs) represent only a small fraction (1-10%) of total lesions (Dronkert and Kanaar, 2001). This has made it difficult to analyze the repair of ICLs unambiguously. Current evidence suggests that ICL repair can take place during and outside of S-phase (Akkari et al., 2000; McHugh and Sarkar, 2006), and multiple repair pathways are involved in the repair of a single ICL (Dronkert and Kanaar, 2001; Niedernhofer et al., 2005). The mechanism of sensing/recognition of ICL lesions may require stalling of the replication/transcription machinery and/or structure-specific DNA binding proteins that recognize the ICL-induced distortion (Noll et al., 2006). An initial step in ICL repair is the generation of incisions in the proximity of the ICL and unhooking of the adduct attributed to nuclease complexes – ERCC1/XPF and Mus81-Eme1 (Hanada et al., 2006; Kuraoka et al., 2000). Steps implicating translesion DNA synthesis have been proposed in both origin-dependent and -independent ICL repair (McHugh and Sarkar, 2006; Raschle et al., 2008). Collapse of a replication fork at an ICL has been shown to generate DNA double-strand breaks (DSBs) (Niedernhofer et al., 2004) and these DSBs could conceivably participate in repair of the lesion. Cell lines with mutations in proteins essential for homologous recombination (HR) are hypersensitive to crosslinking agents, particularly during S-phase (McHugh et al., 2000; Nojima et al., 2005). Despite its potential importance, checkpoint signaling induced specifically by ICLs has never been investigated.
Fanconi anemia (FA) cells are also hypersensitive to crosslinking agents, suggesting a role for the FA pathway in ICL recognition and/or repair (Grompe and D'Andrea, 2001). FA cells are defective in the initial incision step in ICL repair, implicating the FA pathway as an early responder to ICL damage (Kumaresan et al., 2007). Indeed, the FA pathway can be activated by small synthetic DNA structures, further supporting a role for the FA pathway in damage recognition (Sobeck et al., 2007). The FA pathway has also been shown to function in an S-phase checkpoint (Centurion et al., 2000; Pichierri et al., 2002) and the assembly of Rev1 nuclear foci in response to ICLs (Mirchandani et al., 2008).
To investigate signaling pathways triggered by ICLs, we employed Xenopus cell-free extracts that support DNA replication, repair, and checkpoint signaling (Shechter et al., 2004; Walter et al., 1998). Using a DNA substrate harboring a single site-specific ICL, we establish that these extracts support signaling from an ICL and repair of the lesion. Notably, we demonstrate that ICLs trigger a checkpoint response independent of origin-initiated DNA replication and uncoupling of DNA polymerase and MCM helicase. We show that the FA pathway acts upstream of RPA-ATR-Chk1 to generate the ICL signal. This system repairs ICLs in a reaction that involves extensive DNA synthesis. Consistent with evidence from other systems, repair occurs by both origin-dependent and origin-independent mechanisms.
Results
Sensing and processing of a single ICL in cell-free extracts
To evaluate signaling from and repair of an ICL in the absence of other DNA lesions, we generated a circular plasmid template containing a single site-specific ICL, synthesized as previously described (Dooley et al., 2001) (Fig. 1A). Two guanines on opposing strands within a GpC sequence (CDDP target), were connected by a 3-carbon linker. The resultant ICL causes helix distortion similar to that of a MMC-induced ICL (Dooley et al., 2003). ICL-containing oligos were ligated to pBS plasmid and purified free of nicked and linear DNA. Since DNA polymerases cannot amplify DNA across an ICL, we used PCR to assess template purity. We did not detect a PCR product using primers flanking the ICL (“X”, Fig. 1B) as compared to primers flanking a control region on the plasmid (“C”, Fig. 1B) and quantitative real-time PCR (qRT-PCR) showed that our substrate was 99.98% pure (Fig. 1C – ratio of “X”:“C” products).
Figure 1. Replication of ICLs in Xenopus cell-free extract.
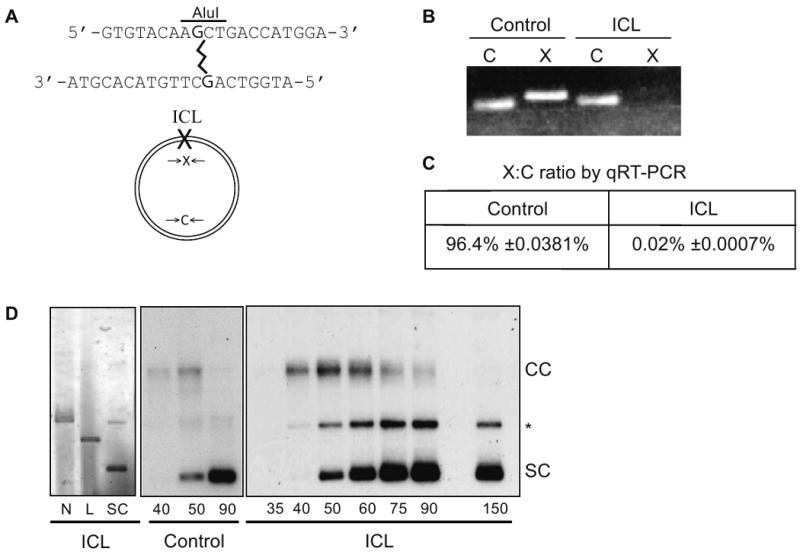
(A) Schematic representation of site-specific ICL and X and C primers. (B) PCR products of control and ICL plasmids with X and C primers. (C) qRT-PCR quantification of ratio of products across X and C primers (X:C ratio) for control and ICL plasmids. Mean ± SEM. (D) Electrophoretic analysis of nicked (N), linear (L), and supercoiled (SC) ICL plasmid (left panel). Electrophoretic analysis of replication and repair of control (center panel) and ICL plasmids (right panel) in the Xenopus HSS/NPE system in the presence of 32P-αdCTP (CC: concatemer, SC: supercoiled, * replication/repair intermediates).
Xenopus cell-free extracts support chromatin assembly and DNA replication of plasmid templates. We could thus assess the role of replication on ICL recognition and repair. The ICL-containing plasmid or a control plasmid of identical sequence was first incubated in cytosolic extract (HSS) to allow pre-replicative complex (pre-RC) assembly, followed by addition of nucleoplasmic extract (NPE) to initiate S-phase in the presence of radiolabeled nucleotides (Walter et al., 1998). We monitored labeled DNA products following gel electrophoresis. The control plasmid yielded a 5.4 kb replication intermediate (CC) which resolved rapidly to a 2.7 kb product (SC) (Fig. 1D, middle panel). The ICL plasmid also generated a 5.4 kb product which slowly resolved to a 2.7 kb form (Fig. 1D, right panel). In the case of ICL plasmid, we propose that the 5.4 kb form, which migrated slower than nicked and linear plasmid (Fig. 1D, left panel), corresponds to fully replicated DNA concatemers still harboring an unrepaired ICL. The appearance of a concatemer intermediate was recently demonstrated for a similar ICL-containing DNA template in Xenopus extracts (Raschle et al., 2008). The 2.7 kb form co-migrates with the supercoiled plasmid and corresponds to replicated and/or repaired circular molecules. We observed products of intermediate mobility (* in Fig. 1D); which are, presumably, replication/repair intermediates, such as linear and relaxed circular molecules. There was some variability in the kinetics of appearance and resolution of the 5.4 kb intermediate depending on the NPE preparation (Fig. S1).
ICLs are repaired via origin-dependent and origin-independent mechanisms
To confirm that repaired DNA molecules, free of ICLs, were generated in these extracts, we monitored the appearance of PCR products across the ICL site. Using PCR and qRT-PCR, we detected increasing amounts of “X” product as lesions were repaired (Fig. 2A, B). Repair efficiency assessed by qRT-PCR was 20% after 3 hours (Fig. 2B). Since PCR can amplify a fully repaired product as well as a partially repaired intermediate with only one repaired strand (Table S1, intermediates B and C), we used AluI (Fig. 1A) to determine the fraction of accurately repaired double-stranded DNA. We found that AluI digestion reduced the yield of “X” product by almost 73% indicating that most of the DNA was repaired on both strands. The remaining 27% could represent single-stranded intermediates, abortive repair products, or imprecisely repaired products (Table S2).
Figure 2. Replication initiation-dependent and -independent repair of ICLs.
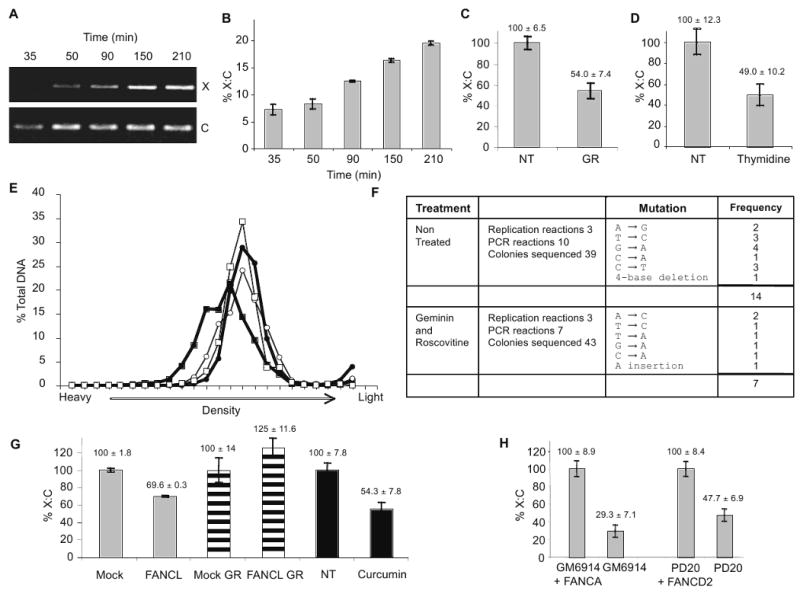
(A) PCR products using X and C primers on ICL-plasmid incubated in HSS/NPE for times indicated. (B) qRT-PCR quantification of X:C ratio for ICL-plasmid incubated in HSS/NPE for times indicated. (C) qRT-PCR quantification of X:C ratio of ICL-plasmid incubated for 75 min in non-treated (NT) extracts or extracts treated 50 ng/μL geminin and 500 μM roscovitine (GR). (D) HeLa cells were arrested by double thymidine block and transfected with ICL plasmid then released (NT) or maintained in thymidine block (Thymidine). 24 hr post transfection plasmid DNA was recovered for qRT-PCR quantification of X:C ratio. (E) Control and ICL plasmids were incubated in HSS/NPE for 0 min (○ Control, □ ICL) or 120 min (● Control, ■ ICL) in the presence of 500 mM BrdUTP and geminin/roscovitine. DNA was separated on a 1.72 g/mL CsCl gradient and analyzed by qRT-PCR using C primers for total DNA content. (F) ICL plasmid was replicated in HSS/NPE for 150 min in the absence or presence of geminin/roscovitine and amplified with A and B primers. PCR products were cloned and sequenced. (G) qRT-PCR quantification of X:C ratio in mock- and FANCL-depleted HSS/NPE (grey bars), in mock- and FANCL-depleted HSS/NPE with geminin and roscovitine (striped bars), or in non-treated (NT) or curcumin-treated (curcumin) HSS/NPE (black bars) for 90 min. Values normalized to the mock or NT controls. (H) PD20, PD20 + wtFANCD2, GM6914, GM6914 + wtFANCA were transfected with ICL-plasmid. 24 hr post transfection plasmid DNA was recovered and subjected to qRT-PCR quantification of X:C ratio.
Indirect evidence suggests that ICL repair in vertebrates occurs primarily during S phase (Akkari et al., 2000), whereas ICL repair in bacteria and yeast can occur in the absence of an origin-initiated replication (Dronkert and Kanaar, 2001). Mammalian cell extracts that do not support DNA replication can process ICLs (Li et al., 1999; Mu et al., 2000). To assess the contribution of DNA replication to ICL repair, we monitored repair in the presence of geminin, which sequesters Cdt1 and prevents MCM loading, and roscovitine, a cyclin-dependent kinase inhibitor (Wang et al., 2008). The efficiency of ICL repair in geminin/roscovitine-treated extracts was 54% that of untreated extracts (Fig. 2C). Inhibition of origin-initiated DNA replication did not significantly affect the fraction of lesions repaired on both strands (Table S2). Similar findings were obtained in human cells transfected with an ICL-containing plasmid. The repair efficiency in HeLa cells arrested and kept in G1 with thymidine, as confirmed by FACS analysis (Fig. S2A), was 49% that of exponentially growing cells (Fig. 2D and Fig. S2B).
The extent of DNA synthesis during ICL repair has not been described. To this end, we separated DNA molecules on CsCl gradients following incorporation of BrdUTP. To monitor exclusively DNA repair, origin-dependent DNA replication was inhibited by geminin and roscovitine. The positions of unsubstituted DNA and hemi-substituted DNA were determined using control circular plasmid DNA, before and after semi-conservative replication, respectively (Fig. S3), allowing us to correlate the extent of DNA synthesis with plasmid position in the gradient. As expected, the control plasmid did not change in density following incubation in extract (Fig. 2E, open squares). In contrast, most ICL-DNA migrated at a significantly higher density than unsubstituted DNA (Fig. 2E, filled squares). The increase in density corresponds to approximately 11% substitution with BrdUTP (300 nucleotides for a 2.7 kb plasmid) (Fig. 2E and S3). This level of DNA synthesis is incompatible with canonical NER processing, which is associated with synthesis of about 30 nucleotides (Friedberg et al., 1995). Thus, the majority of ICL-DNA was recognized and processed, even though only 20% of crosslinks were fully repaired. Some potential repair intermediates are shown in Table S1. These findings indicate a unique mode of repair that involves extensive repair DNA synthesis. It further suggests an efficient first step, possibly required for ICL tolerance, which involves DNA synthesis across the unhooked ICL followed by a subsequent step to complete repair (Bergstralh and Sekelsky, 2008).
To assess the accuracy of repair, a set of PCR-amplified products from independent repair assays were cloned and sequenced around the ICL. 82 PCR products of 265 nucleotides each (total of 21,730 nucleotides) contained 21 errors (Fig. 2F). Taq polymerase has an error rate of 1/9,000 nucleotides; therefore, 2 of the 21 errors could arise during PCR amplification. Our data indicates that ICL repair is associated with incorporation of approximately 300 nucleotides (Fig. 2E). Therefore, on average, most of the 265 nucleotides surrounding the ICL site will be synthesized de novo during repair. Our error rate of 19 ÷ (265 × 82) = 87×10-5, is thus consistent with the involvement of translesion synthesis polymerases with low error rate such as pol ζ (McCulloch and Kunkel, 2008). This is in contrast to the repair of psoralen ICLs where error rates are 10- to 100-fold higher (Cipak et al., 2006; Lu et al., 2005).
The accuracy of ICL repair in geminin/roscovitine-treated extracts (61×10-5) was similar to that observed in replication-competent extracts (Fig. 2F), again consistent with the fidelity of translesion synthesis polymerases (McCulloch and Kunkel, 2008).
The hypersensitivity of FA cells to crosslinking agents led us to ask whether FA proteins play a role in ICL repair. We immunodepleted FANCL, the catalytic E3 ligase component of the FA core complex, from extracts. As we have previously shown (Wang et al., 2008), FANCL depletion prevented FANCD2 monoubiquitylation, indicated by the absence of FANCD2L (Fig. 4D), and FANCD2 and FANCA recruitment to DNA (Fig. 4E, only FANCD2L binds DNA, (Sobeck et al., 2006; Wang et al., 2008)). FANCL depletion delayed the appearance of DNA concatemers and their resolution into circular forms but did not affect the mobility of the DNA replication intermediates (Fig. S4). Using qRT-PCR, we determined that FANCL depletion decreased ICL repair in extracts by 30% (Fig. 2G). A similar decrease in ICL repair was observed in extracts treated with curcumin, a small molecule inhibitor of the FA pathway (Chirnomas et al., 2006) (Fig. 2G). Interestingly, FANCL depletion did not inhibit ICL repair in extracts treated with geminin and roscovitine (Fig. 2G). This indicates that the FA pathway participates in ICL repair primarily when a replication fork is available.
Figure 4. ICL-checkpoint requires the FA pathway upstream of Chk1 activation.
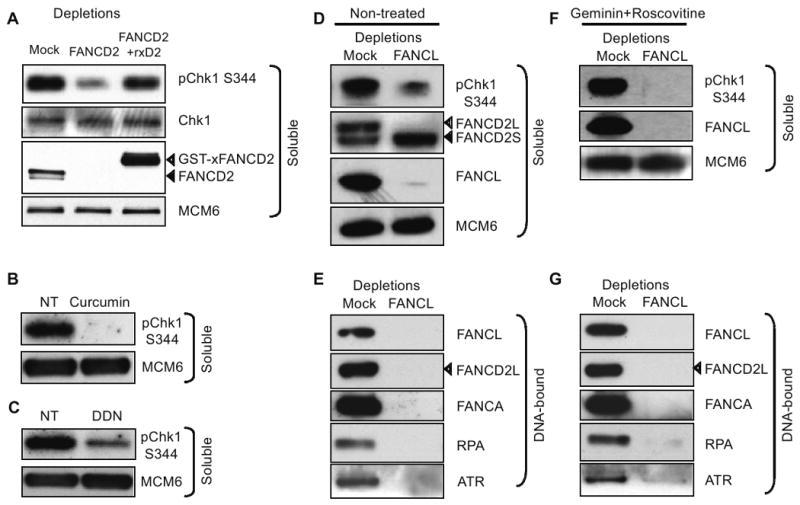
(A) Western blot analysis of mock- FANCD2-, and FANCD2-depleted HSS/NPE extracts supplemented with wt GST-xFANCD2 HSS/NPE extracts following incubation with ICL plasmid. Soluble extract was probed with indicated antibodies. (B) HSS/NPE extracts were incubated with ICL plasmid in buffer (NT) or 100 μM Curcumin. Soluble extract was probed with indicated antibodies. (C) HSS/NPE extracts were incubated with ICL plasmid in buffer (NT) or 5 mM DDN. Soluble extract was probed with indicated antibodies. (D) Western blot analysis of mock- and FANCL-depleted HSS/NPE extracts incubated with ICL plasmid. Soluble extract was probed with indicated antibodies. (E) Western blot analysis of mock- and FANCL-depleted HSS/NPE extracts incubated with ICL plasmid. DNA-bound proteins were probed with indicated antibodies. (F) Western blot analysis of mock- and FANCL-depleted HSS/NPE extracts in the presence of geminin and roscovitine incubated with ICL plasmid. Soluble extract was probed with indicated antibodies. (G) Western blot analysis of mock- and FANCL-depleted HSS/NPE extracts in the presence of geminin and roscovitine incubated with ICL plasmid. DNA-bound proteins were probed with indicated antibodies. For soluble extracts, MCM6 was used as a loading control and for DNA-bound proteins were normalized by qRT-PCR quantification of DNA for each sample (Table S3). ICL-plasmid was incubated in extracts for 90 min.
To determine the fraction of fully repaired double-stranded DNA in presence of geminin and roscovitine in FANCL-depleted extracts we used AluI digestion (Fig. 1A). We found that a significant fraction (52%) of lesions was repaired on both strands (Table S2). Furthermore, we monitored DNA synthesis specifically associated with repair in extracts treated with curcumin or depleted of FANCL in presence of geminin/roscovitine. We found reduced, but significant, nucleotide synthesis, consistent with ICL repair (Fig. S5),
Next, we asked whether the FA pathway participates in ICL repair in human cells. We used cells from FA-A and FA-D2 patients and their isogenic counterparts complemented with vectors encoding for wild type FANCA and FANCD2, respectively. ICL repair in exponentially growing cells decreased by 70.7% in FANCA-deficient cells and by 52.3% in FANCD2-deficient cells, as compared with their complemented counterparts (Fig. 2H).
We conclude that the FA pathway, as assessed by inactivation of FANCL, FANCA or FANCD2 or treatment with curcumin, is required for origin-dependent ICL repair. However, since FA pathway inactivation did not completely abrogate ICL repair, the sensitivity of FA cells to crosslinking agents could be due in part to a repair defect as well as defect(s) in sensing or signaling from ICLs (see below).
Nucleotide excision repair (NER), base excision repair (BER), DNA mismatch repair (MMR) and DNA double-strand break (DSB) repair (Wold, 1997) all generate single-strand DNA intermediates that require the single-strand DNA binding protein RPA. Because RPA is essential for cell viability, its role in ICL repair could not be evaluated directly. We found that RPA depletion significantly decreased ICL repair in both geminin/roscovitine-treated and untreated extracts and was partially rescued by addition of recombinant human RPA (rhRPA) (Fig. S6). These data indicate that RPA-dependent processes participate in ICL repair in the presence and absence of origin-initiated replication. Note that RPA depletion was more inhibitory in the latter case (5-fold reduction vs. 2-fold), indicating that origin-initiated replication-independent and FA-independent ICL repair depends primarily on RPA.
ICLs trigger a FA-dependent checkpoint that inhibits DNA replication
DNA crosslinking agents primarily produce intrastrand crosslinks that block DNA polymerases but allow DNA unwinding by MCM helicases, triggering an ATR-dependent checkpoint (Pichierri and Rosselli, 2004). We show below, for the first time, that a site-specific ICL generates a checkpoint signal. We monitored replication of the ICL-containing plasmid in the presence of caffeine, an inhibitor of checkpoint kinases. Consistent with ICL activation of an ATR/ATM checkpoint, replication of the ICL-plasmid was strongly stimulated by caffeine (Fig. 3A). Next, we monitored replication of an intact plasmid in extracts pre-incubated with a control or an ICL plasmid. The ICL-containing plasmid generated a checkpoint signal that inhibited replication of the intact template in trans (Fig. 3B).
Figure 3. ICLs activate a FA- and ATR-Chk1-dependent checkpoint.
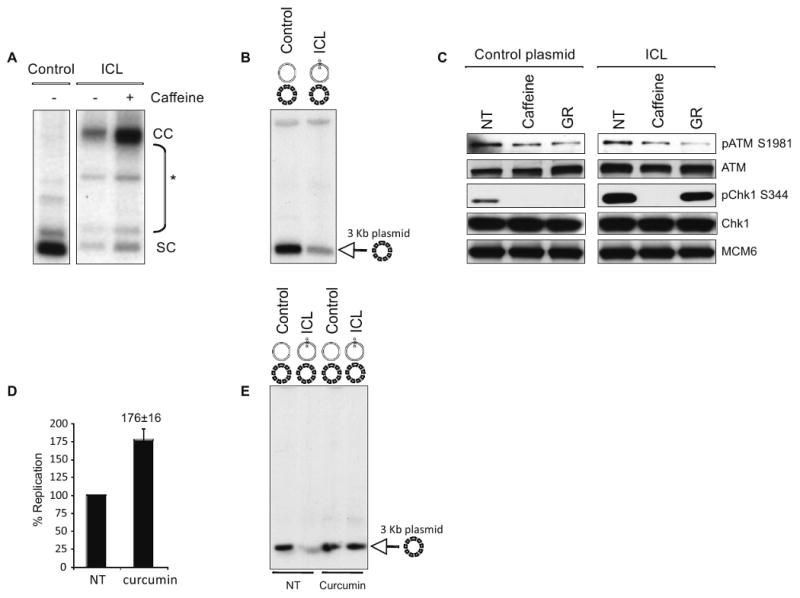
(A) Electrophoretic analysis of control or ICL plasmid with or without 5 mM caffeine incubated in HSS/NPE for 120 min, in the presence of 32P-αdCTP. (B) 2.7 kb control or ICL plasmids were incubated in HSS/NPE for 90 min followed by the addition of a 3 kb plasmid (dashed) and 32P-αdCTP for 20 min. DNA was analyzed on agarose gel. (C) HSS/NPE extracts in the presence of buffer (NT), 5 mM caffeine (Caffeine), or geminin and roscovitine (GR) were incubated with control (left panel) or ICL plasmid (right panel) for 90 min. Western blot analysis of soluble extracts was performed with the indicated antibodies. (D) ICL plasmid was incubated in HSS/NPE with or without 100 μM curcumin for 90 min, in the presence of 32P-αdCTP. Nucleotide incorporation was quantified using phosphoimager and SEM is shown for 4 independent experiments. (E) 2.7 kb control or ICL plasmids were incubated in HSS/NPE with buffer (NT), 100 μM curcumin or 5 mM caffeine for 90 min, followed by the addition of a 3 kb plasmid (dashed) and 32P-αdCTP for 20 min. DNA was analyzed on agarose gel.
To distinguish between ATR and ATM activation, we monitored Chk1 phosphorylation at S344. Chk1 modification was strongly stimulated by the ICL-plasmid and was abrogated by caffeine (Fig. 3C). This indicates that ICLs, in the absence of other DNA lesions, activate ATR-dependent signaling. In contrast, the ICL-plasmid did not increase ATM phosphorylation at S1981 (Fig. 3C). Because the replicative DNA helicase cannot unwind DNA past an ICL, ICL- and UV-induced checkpoints must be mechanistically different (Byun et al., 2005). Notably, ICL-induced Chk1 phosphorylation was resistant to geminin and roscovitine, and therefore did not require origin-initiated DNA replication (Fig. 3C, right panel). This again underscores the clear distinction between the ICL and canonical DNA replication checkpoints.
Because inactivation of the FA pathway did not completely abrogate ICL repair, we tested the contribution of the FA pathway to the ICL-induced checkpoint. We monitored replication of the ICL-containing plasmid in the presence of curcumin (Fig. 3D). Curcumin stimulated replication of the ICL plasmid, consistent with the idea that the ICL-containing DNA activated a FA pathway-dependent checkpoint. Moreover, curcumin abrogated the checkpoint generated by the ICL-containing plasmid that inhibits replication of an intact template in trans (Fig 3E).
ICL-checkpoint requires the FA pathway upstream of Chk1 activation
In contrast to the partial dependence of ICL repair on the FA pathway (Fig. 2G and 2H), FANCD2 immunodepletion (Fig. 4A, top row) eliminated almost all ICL-induced Chk1 phosphorylation. ICL-dependent Chk1 phosphorylation was also abrogated in extracts treated with curcumin (Fig. 4B) and significantly reduced in extracts treated with DDN, another FA pathway inhibitor (Landais et al., 2009) (Fig. 4C). FANCD2 depletion (Fig. 4A, second row) did not affect the levels of total Chk1. Notably, Chk1 phosphorylation in FANCD2-depleted extracts was rescued by addition of recombinant GST-xFANCD2 protein expressed in baculovirus-infected cells. This confirms that ICL-induced signaling was dependent on the FA pathway. Similarly, FANCL immunodepletion abrogated ICL-dependent Chk1 phosphorylation (Fig. 4D). Strikingly, ICL-dependent Chk1 phosphorylation was also inhibited in FANCL-depleted extracts treated with geminin and roscovitine (Fig. 4F). This indicates that FANCL functions upstream of Chk1 in the ICL-induced checkpoint pathway, whether or not origin activity is blocked. This was confirmed by treating extracts with curcumin in presence of geminin and roscovitine, which reduced Chk1 phosphorylation (Fig. S7A). In contrast, ICL-dependent Chk1 phosphorylation was not affected in FANCD2-depleted extracts treated with geminin and roscovitine (Fig. S7B). FANCL depletion did not reduce total Chk1 protein levels (Fig. S8). FANCL depletion did not inhibit DNA replication of a control plasmid or aberrantly trigger Chk1 signaling (Fig. S8). Together, this data suggests that repair of an ICL does not require checkpoint signaling from the ICL.
Since ATR-dependent Chk1 phosphorylation is activated by ssDNA-RPA intermediates (Costanzo et al., 2003; Zou and Elledge, 2003), we asked if the FA pathway functions upstream of these proteins. FANCL depletion strongly reduced RPA and ATR recruitment to ICL-plasmids (Fig. 4E, 4G, Fig. S9). We confirmed that loss of RPA and ATR recruitment did not result from the co-depletion of these proteins with FANCL (Fig. S10). These data indicate that the FA pathway lies upstream of the RPA-ATR-Chk1 pathway in ICL signaling.
These data predict ICL-containing DNA will not activate Chk1 in cells from patients with Fanconi anemia. We therefore monitored ICL-induced Chk1 phosphorylation in human FA-A cells deficient for FANCA and their isogenic counterparts complemented with vector expressing wild type FANCA. FA-A cells (GM6914) show elevated levels of phosphorylated Chk1 in untransfected cells (data not shown) and in cells transfected with a control plasmid (Fig. S11A). Constitutive Chk1 activation might reflect genomic instability associated with mutation in the FA pathway. As predicted, Chk1 phosphorylation did not increase in FA-A cells upon transfection with the ICL plasmid. In contrast, background Chk1 phosphorylation, in complemented FA cells was reduced, but significantly increased upon transfection with ICL plasmid (Fig. S11B). Chk1 activation in response to the ICL-plasmid in FANCA-proficient cells was 3-fold that of FA-A cells (Fig. S11B).
Mechanisms of ICL-induced checkpoint
The FA pathway also functions downstream of checkpoint signaling (Pichierri and Rosselli, 2004) and binding of FA proteins to chromatin during unperturbed DNA replication requires RPA (Wang et al., 2008). To determine the role of RPA in ICL-induced signaling, FA pathway activation and Chk1 phosphorylation was examined in RPA-depleted extracts in the presence or absence of geminin and roscovitine. RPA depletion reduced FANCD2 activation (Fig. 5A) and FA protein loading onto ICL-containing DNA (Fig. 5B), suggesting that positive feedback between the FA pathway and RPA is required for robust checkpoint signaling. Furthermore, ICL-induced Chk1 phosphorylation was clearly RPA-dependent in the presence of geminin and Roscovitine (Fig. 5A). Note that RPA depletion inhibits DNA synthesis even if origin activation is permitted. We then asked how ATR depletion affected the ICL-activated checkpoint. We recently demonstrated a structural role for ATR in FA protein recruitment during DNA replication (Wang et al., 2008). This is confirmed in Fig. 5D, where ATR depletion significantly reduced FA protein loading to the ICL-plasmid and abrogated ICL-induced Chk1 phosphorylation (Fig. 5C). RPA loading was also reduced in the absence of ATR, which could be the consequence of reduced FA protein binding to chromatin. These data are consistent with previous studies showing recruitment of ATR-ATRIP to RPA-bound ssDNA and subsequent activation of Chk1 (Costanzo et al., 2003; Zou and Elledge, 2003).
Figure 5. Mechanisms of ICL-induced checkpoint.
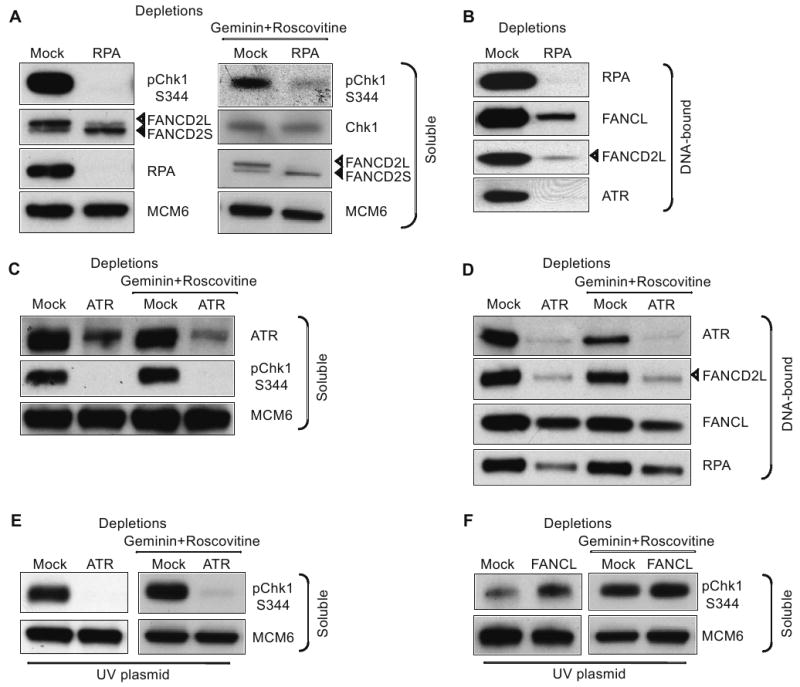
(A) Western blot analysis of mock- and RPA-depleted HSS/NPE extracts incubated with ICL plasmid. Soluble extract was probed with indicated antibodies. (B) Western blot analysis of mock- and RPA-depleted HSS/NPE extracts incubated with ICL plasmid. DNA-bound proteins were probed with indicated antibodies. (C) Western blot analysis of mock- and ATR-depleted HSS/NPE extracts incubated with ICL plasmid. Soluble extract was probed with indicated antibodies. (D) Western blot analysis of mock- and ATR-depleted HSS/NPE extracts incubated with ICL plasmid. DNA-bound proteins were probed with indicated antibodies. (E) Western blot analysis of mock- and ATR-depleted HSS/NPE extracts incubated with plasmids irradiated with 500 mJ/cm2 UVB. (F) As in E, with mock- and FANCL-depleted extracts. For soluble extracts, MCM6 was used as a loading control and DNA-bound proteins were normalized by qRT-PCR quantification of DNA for each sample. Extracts were incubated with or without geminin and roscovitine as indicated. ICL or UV plasmids were incubated in extracts for 90 min.
UV lesions trigger a DNA replication checkpoint following uncoupling of MCM helicase and stalled replicative DNA polymerase (Byun et al., 2005). This uncoupling generates RPA-coated ssDNA that activates the ATR-Chk1 pathway. We monitored Chk1 phosphorylation in ATR-depleted extracts. As anticipated, we observed that ATR was required for signaling from both UV and ICL lesions, establishing that these pathways converge at the level of ATR (Fig. 5C and 5E). However, in contrast to the ICL lesion, FANCL depletion did not inhibit signaling from the UV lesion (compare Fig. 5F with Figs. 4D and 4F). Similar results were obtained in the absence of initiation of DNA replication (Fig. 5E and 5F). This demonstrates that ICL and UV signaling pathways are mechanistically distinct; checkpoint signaling from UV lesions is independent of the FA pathway, consistent with the normal sensitivity of FA cells to UV irradiation (Kalb et al., 2004).
Discussion
DNA interstrand crosslinks pose a difficult challenge to cells since they create barriers to both DNA and RNA polymerases. Because they inhibit these essential biological processes, it has been proposed that cellular sensitivity to crosslinking agents might correlate exclusively with ICL repair (Dronkert and Kanaar, 2001). In contrast to this notion, we find that a plasmid harboring a single, site-specific ICL activates a robust checkpoint in the presence or absence of origin-initiated DNA replication. This indicates that signaling from an ICL can occur outside of S-phase. It also suggests that defects in signaling from an ICL could play a previously unsuspected role in sensitivity or acquired resistance to crosslinking agents. Supporting this idea, we find that inactivation of the FA pathway, which is thought to inhibit ICL repair, in fact has a stronger impact on checkpoint signaling than on removal of the lesion (Figures 2G, 4A, 4D and 4F).
Our data indicate that repair of a defined ICL in Xenopus extracts or in mammalian cells does not rely exclusively on origin-initiated DNA replication. Indeed, we observe both origin-dependent and – independent ICL repair and checkpoint activation. Although this finding contrasts with prevalent models that link ICL repair with replication fork progression (Niedernhofer et al., 2005; Patel and Joenje, 2007; Wang, 2007), several studies strongly suggest that ICL repair can operate outside of S-phase (Henriques and Moustacchi, 1980; Sarkar et al., 2006).
A recent study in Xenopus egg extracts addresses the steps of ICL repair (Raschle et al., 2008). These authors report that repair of a nitrogen mustard ICL is entirely blocked by p27Kip, an inhibitor of origin firing. In contrast, our study of an ICL similar in structure to an MMC crosslink reveals geminin and roscovitine-resistant repair. The difference between this report and that of Raschle et al. might be explained in part by the different substrates. NMR studies indicate that the crosslink used in our experiments distorts and destabilizes the DNA helix (Dooley et al., 2003). In contrast, the nitrogen mustard crosslink they use in p27Kip-treated extracts is not thought to generate significant DNA distortion (Raschle et al., 2008). Recognition of an ICL in the absence of an incoming DNA replication fork could be influenced by the structural modifications in the DNA molecule associated with the lesion (Smeaton et al., 2008). Notably, distortion of the DNA molecule by platinum derivatives has been proposed to be critical to their efficacy; cisplatin and its trans isomer (trans-diamminedichloroplatinum) induce distinct patterns of DNA lesions. Cisplatin, which is more toxic than the trans isomer, generates more distorting lesions than the latter (Sherman and Lippard, 1987).
Repair of ICLs during or outside of S-phase might involve distinct DNA polymerases. Indeed, homologous recombination (HR) has been proposed to participate in ICL repair in S-phase and G2 when homologous sequences are present, and not at other points in the cell cycle. However, we find that the error rates within the newly synthesized DNA surrounding the repaired ICL are similar whether or not origin-initiated DNA replication is blocked, consistent with the involvement of the same DNA polymerase(s) in the repair process. Studies that have addressed the fidelity of psoralen ICL repair in cell-free extracts (Cipak et al., 2006; Lu et al., 2005) report error rates 10- to 100-fold higher than that which we observe in our system (61 ×10-5 − 80 × 10-5). These error rates are consistent with DNA Pol ζ - promoted replication (McCulloch and Kunkel, 2008). Raschle et al. also suggests that Pol ζ might operate in ICL repair during DNA replication; Pol ζ has also been implicated in ICL repair in G1 in yeast cells (McHugh and Sarkar, 2006). However, a recent report of ICL repair involving a high rate of incorporation errors suggest that a less accurate DNA polymerase such as Pol κ, might also participate in this reaction in mammalian cells (Minko et al., 2008). We showed above that ICL repair is associated with extensive incorporation of deoxynucleotides, consistent with long-patch DNA synthesis.
We previously reported that FA proteins load on chromatin in a DNA replication-dependent manner and may travel with the replication fork (Sobeck et al., 2006; Wang et al., 2008). In this way, FA proteins would be poised to repair ICL lesions encountered by the replication fork. Here, we demonstrate that FA proteins FANCL, FANCA, and FANCD2 are recruited to an ICL-containing plasmid, even when origin-initiated DNA replication is inhibited. Furthermore, we show that in the absence of origin firing, FA core proteins are critical for signaling upstream of the RPA-ATR-Chk1 checkpoint pathway. This suggests that FA proteins could directly sense DNA ICLs, possibly via the distortion that these lesions create in the DNA helix. In support of this idea, FA proteins FANCD2, FANCM, and FANCJ are known to bind to specific DNA structures mimicking replication forks, 3′-flaps, and G quadraduplexes (Ciccia et al., 2007; Sobeck et al., 2007; Wu et al., 2008). Following recognition of ICLs by FA proteins, ssDNA-RPA intermediates are generated and checkpoint signaling is activated. Two FA proteins FANCJ (BRIP1/BACH1) and FANCM (Hef) have helicase/translocase activity (Wang, 2007). It is conceivable that in the absence of origin-initiated DNA synthesis, FANCJ and/or FANCM unwind DNA in the area of the ICL, leading to RPA recruitment and activation of ATR-Chk1 checkpoint signaling. Alternatively, checkpoint signaling could be triggered by ssDNA-RPA intermediates generated by exonuclease activity coupled to lesion excision. Importantly, we have uncovered distinct roles for FA core proteins and FANCD2 in checkpoint signaling activated during S-phase or in the absence of DNA replication initiation. Whereas FA core complex is required for checkpoint activation under both conditions, FANCD2 is dispensable for checkpoint activation in presence of geminin and roscovitine. This is reminiscent of the requirement for FANCL but not for FANCD2 in activating the G2/M checkpoint following re-replication (Zhu and Dutta, 2006). It further suggests that ICLs trigger mechanistically distinct checkpoints in S-phase and in the absence of DNA replication.
Here, we show that a single DNA interstrand crosslink activates a robust replication checkpoint downstream of the FA pathway. Although this checkpoint involves RPA-ATR-Chk1 checkpoint signaling, it is distinct from UV-induced checkpoints that depend on the functional uncoupling of the replication polymerase and the MCM helicase. Disruption of the FA pathway is associated with cancer susceptibility and increased resistance to cisplatin (Jacquemont and Taniguchi, 2007). It will be important to determine whether abrogation of the ICL-induced checkpoint in FA-deficient tumors underlies their cisplatin sensitivity. In addition, specifically targeting this checkpoint function may restore cisplatin sensitivity in tumors that have acquired cisplatin resistance.
Experimental Procedures
Plasmid preparation
A plasmid containing a single defined ICL mimicking MMC-induced crosslinks was generated as follows: A linker sequence containing a PflMI restriction site was inserted between SacI and NotI sites in the multiple cloning site of pBS KS- (STRATAGENE) (5′ CACCCATGGAATGGACGC 3′ 5′ GGCCGCGTCCATTCCATGGGTGAGCT 3′) to generate pBS-PflMI. This plasmid was digested with PflMI and DraIII restriction enzymes and purified from agarose gel using QIAGEN gel extraction kit and ligated to the crosslinked oligonucleotide duplex (5′ GTGTACAAG*CTGACCATGGA 3′ 5′ ATGGTCAG*CTTGTACACGTA 3′, crosslink between G*s) or a control duplex without the crosslink using T4 DNA ligase (NEB). The crosslinked duplex was synthesized by incubation of the oligonucleotides containing 2-fluorohypoxanthine and purification by HPLC and capillary gel electrophoresis, followed by treatment with 5% acetic acid and purification by PAGE and desalting (Dooley et al., 2001; Dooley et al., 2003). Following ligation, non-circular and nicked molecules were eliminated using ExonucleaseIT (BAYOU biolabs Cat # X-101) and purified using a PCR purification kit (QIAGEN). qRT-PCR using primers flanking the ICL confirmed the presence of ICL in the final plasmid with 99.98% purity.
pBS was irradiated with 500 mJ/cm2 UVB. This amount of UVB corresponds to an average of 3 lesions per plasmid (Perdiz et al., 2000).
PCR, Real-Time PCR (qRT-PCR) and primer sequences
Using Taq polymerase (Roche) and the LightCycler-FastStart DNA MasterPlus SYBR Green kit (Roche), PCR and qRT-PCR detection of the crosslink site was performed with 5′-AGATAGGGTTGAGTGTTGTTC-3′ and 5′-GCGTCCATTCCATGGTC-3′ primers (X primers). The same sample was also amplified across a control region using 5′-TGGCAGCACTGCATAATTCT-3′ and 5′-TCTCAACAGCGGTAAGATCC-3′ primers (C primers). PCR conditions: 35 cycles of 94°C for 30 s, 62°C for 45 s, and 72°C for 1 min with a 10 min extension at 72 °C. qRT-PCR conditions: 45 cycles of 95°C for 10 s, 62°C for 4 s, and 72°C for 9 s. The X:C ratio was calculated by dividing the quantity amplified using the X primers by the quantity amplified using the C primers and normalizing the ratio against a control plasmid. The area flanking the ICL was cloned using PCR primers A and B: 5′-AGATAGGTTGAGTGTTGTTC and 5′-AGCTATGACCATGATTACGC, amplifying 265 bases, into PGEM-T Easy vector (Promega) and white colonies were selected after 16 hr incubation. DNA from clones was sequenced using T7 primer. Where error bars are present, results were expressed as mean ± SEM.
Replication reactions in the Xenopus system, immunodepletions, and DNA binding
X. laevis were handled in accordance to guidelines provided by the Institutional Animal Care and Use Committee at Columbia University, protocol AA5192. Cell-free extracts (LSS, HSS and NPE) were prepared from unfertilized Xenopus eggs as described (Shechter et al., 2004). All reactions using HSS/NPE were incubated in HSS for 30 min and NPE was added for remainder of the time indicated. When using 32P-αdCTP, nucleotide was added after the initial 30 min incubation in HSS. Immunodepletions were performed using anti-FANCL, FANCD2, RPA, or ATR antibodies and rabbit IgG (Sigma) coupled to Protein A-Sepharose CL-4B beads (Amersham Biosciences). Two to four rounds of depletions were performed by incubating extracts with bead-bound antibodies at 4°C for 30 min per round.
For DNA binding assays, extracts were incubated as described and each reaction was diluted with chromatin isolation buffer (CIB) (100 mM KCl, 2.5 MgCl2, 50 mM Hepes pH 7.8) supplemented with 0.125% Triton X-100. DNA-bound fractions were isolated through a 30% sucrose solution in CIB at 8000 × g for 30 min at 4°C. The amount of DNA loaded per well was normalized by qRT-PCR quantification. Calculated DNA concentrations were derived using the second derivative maximum analysis method from the LightCycler3 software. The amounts loaded per sample were calculated to normalize the total DNA loaded per lane in each experiment (Table S3). Samples were run on SDS-PAGE and analyzed by Western blotting. Equal protein loading was confirmed using ORC1 as loading control (Fig. S12).
Preparation of recombinant proteins
Recombinant human RPA protein was expressed and purified as described (Henricksen et al., 1994). Recombinant GST-xFANCD2 protein was expressed and purified from baculovirus-infected sf9 cells using GST-agarose beads (Molecular Probes) as described (Sobeck et al., 2006).
Cell lines, transfection method, cell cycle synchronization, and BRDU labeling
HeLa, PD20, PD20 complemented with human wild-type FANCD2, GM6914 and GM6914 complemented with human wild-type FANCA (Duckworth-Rysiecki et al., 1986; Taniguchi et al., 2002) cells were grown in Dulbecco's Modified Eagle Medium (DMEM) + 15 % FCS at 37°C with 5% CO2. Transfection – HeLa and FA cells were transfected with 50 ng of ICL plasmid (for repair assays) and a carrier plasmid using lipofectamine LTX and plus reagents in Opti-MEM reduced serum medium as instructed by the manufacture (Invitrogen). FA cells were transfected with 500 ng of ICL plasmid for checkpoint activation experiments. 24 hr after transfection, cells were harvested and DNA was extracted using QIAGEN Miniprep modified protocol. HeLa cells were synchronized at the G1/S transition by a double-thymidine block. Exponentially growing cells were incubated for 14-16 hr in complete medium with 2 mM thymidine (Sigma), released in fresh medium for 9 hr and incubated a second time with 2 mM thymidine for 14-16 hr. At this point, >99% of cells were in G1/S as assessed by FACS. For BrdU pulse labeling, cells were incubated in the presence of 10 μM Bromodeoxyuridine (BrdU, Sigma) for 30 min and fixed. Synchronized HeLa cells were trypsinized, resuspended in PBS and fixed in 70% ethanol. Fixed cells were stained with a solution of 1 mM sodium citrate, 500 mg/ml propidium iodide, and 100 mg/ml RNAseA in PBS. Cells were analyzed using FACScalibur analyzer and CellQuest Software (BD Biosciences).
Cell lysates were prepared using CSK buffer (10 mM PIPES, ph 6.8; 250 mM NaCl; 300 mM sucrose; 3 mM MgCl2; 1 mM EGTA; 0.5% Triton X-100).
Density substitution
ICL plasmid was replicated for 2.5 hr in the presence of BrdUTP in HSS/NPE., and then treated with proteinase K. The DNA was precipitated, mixed with 1.72 g/ml CsCl in TE, and centrifuged at 36,000 rpm (Sorvall rotor VTi65.1) for 20 hr at 20°C. 500 μL fractions were collected and DNA was precipitated for qRT-PCR.
Antibodies and Western Blotting
For Western Blot analysis, the commercial antibodies used were the following: Phospho-ATM (Ser1981; Rockland Immunochemicals); Phospho-Chk1 (Ser 345, Cell Signaling); β-Actin (Sigma). Antibodies against xATM and xMCM6, were previously described (Robertson et al., 1999; Ying and Gautier, 2005). Anti-RPA p70 antibodies were a gift from P Jackson, anti-xChk1 antibodies were a gift from H Lindsay, and anti-ATR antibodies were a gift from V Costanzo. xFANCD2 and xFANCA antibodies were generated as described by Sobeck et al. (Sobeck et al., 2006). xFANCL-GST fusion protein was made by inserting the xFANCL sequence into the pDONR201 vector of the Gateway Cloning System (Invitrogen) and recombination reactions to produce the expression vector pDEST GST xFANCL. Recombinant GST-xFANCL protein was purified with Glutathione-Sepharose A beads (GE) and both bead-bound GST-FANCL and denatured GST-FANCL were used to immunize rabbits to generate anti-FANCL antibodies.
Chemicals
32P-αdCTP (10 Ci/μL) was purchased from GE Healthcare. Where indicated, caffeine (Sigma #58-08-2, 5 mM in 10 mM PIPES at pH 8.0), geminin (50 ng/μL), curcumin (Sigma #458-37-7, 100 μM in EtOH), roscovitine (Sigma # R7772, 500 μM in DMSO), 2,3-Dichloro-5,8-dihydroxy-1,4-naphthoquinone (DDN, Calbiochem #287805, 5 mM in DMSO) were added when assembling the reactions.
Supplementary Material
Acknowledgments
We thank Dr. R. Baer for critical reading of the manuscript; Dr. M. Wold for the RPA construct; Dr. M. Hoatlin for FANCA, FANCD2 antibodies and for FANCD2 baculovirus; Dr. V. Costanzo for ATR antibodies; Dr. P. Jackson for RPA antibodies; Dr. T. Huang for FA cells; Dr. C. Ying and Dr. S. Peterson for experimental assistance. This work was supported by Breast Cancer NYSDOH grant C020907 to M.B-Y and NIH grants CA92245 and GM077495 to J.G.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akkari YM, Bateman RL, Reifsteck CA, Olson SB, Grompe M. DNA replication is required To elicit cellular responses to psoralen-induced DNA interstrand cross-links. Molecular and cellular biology. 2000;20:8283–8289. doi: 10.1128/mcb.20.21.8283-8289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstralh DT, Sekelsky J. Interstrand crosslink repair: can XPF-ERCC1 be let off the hook? Trends Genet. 2008;24:70–76. doi: 10.1016/j.tig.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centurion SA, Kuo HR, Lambert WC. Damage-resistant DNA synthesis in Fanconi anemia cells treated with a DNA cross-linking agent. Exp Cell Res. 2000;260:216–221. doi: 10.1006/excr.2000.4995. [DOI] [PubMed] [Google Scholar]
- Chirnomas D, Taniguchi T, de la Vega M, Vaidya AP, Vasserman M, Hartman AR, Kennedy R, Foster R, Mahoney J, Seiden MV, D'Andrea AD. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol Cancer Ther. 2006;5:952–961. doi: 10.1158/1535-7163.MCT-05-0493. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, Meetei AR, Laghmani el H, Joenje H, McDonald N, de Winter JP, et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Molecular cell. 2007;25:331–343. doi: 10.1016/j.molcel.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Cipak L, Watanabe N, Bessho T. The role of BRCA2 in replication-coupled DNA interstrand cross-link repair in vitro. Nature structural & molecular biology. 2006;13:729–733. doi: 10.1038/nsmb1120. [DOI] [PubMed] [Google Scholar]
- Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell. 2003;11:203–213. doi: 10.1016/s1097-2765(02)00799-2. [DOI] [PubMed] [Google Scholar]
- Dooley PA, Tsarouhtsis D, Korbel GA, Nechev LV, Shearer J, Zegar IS, Harris CM, Stone MP, Harris TM. Structural studies of an oligodeoxynucleotide containing a trimethylene interstrand cross-link in a 5′-(CpG) motif: model of a malondialdehyde cross-link. J Am Chem Soc. 2001;123:1730–1739. doi: 10.1021/ja003163w. [DOI] [PubMed] [Google Scholar]
- Dooley PA, Zhang M, Korbel GA, Nechev LV, Harris CM, Stone MP, Harris TM. NMR determination of the conformation of a trimethylene interstrand cross-link in an oligodeoxynucleotide duplex containing a 5′-d(GpC) motif. Journal of the American Chemical Society. 2003;125:62–72. doi: 10.1021/ja0207798. [DOI] [PubMed] [Google Scholar]
- Dronkert ML, Kanaar R. Repair of DNA interstrand cross-links. Mutat Res. 2001;486:217–247. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- Duckworth-Rysiecki G, Toji L, Ng J, Clarke C, Buchwald M. Characterization of a simian virus 40-transformed Fanconi anemia fibroblast cell line. Mutation research. 1986;166:207–214. doi: 10.1016/0167-8817(86)90019-2. [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W. DNA repair and mutagenesis. Washington, D.C.: ASM Press; 1995. [Google Scholar]
- Grompe M, D'Andrea A. Fanconi anemia and DNA repair. Hum Mol Genet. 2001;10:2253–2259. doi: 10.1093/hmg/10.20.2253. [DOI] [PubMed] [Google Scholar]
- Hanada K, Budzowska M, Modesti M, Maas A, Wyman C, Essers J, Kanaar R. The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. The EMBO journal. 2006;25:4921–4932. doi: 10.1038/sj.emboj.7601344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henricksen LA, Umbricht CB, Wold MS. Recombinant replication protein A: expression, complex formation, and functional characterization. The Journal of biological chemistry. 1994;269:11121–11132. [PubMed] [Google Scholar]
- Henriques JA, Moustacchi E. Isolation and characterization of pso mutants sensitive to photo-addition of psoralen derivatives in Saccharomyces cerevisiae. Genetics. 1980;95:273–288. doi: 10.1093/genetics/95.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont C, Taniguchi T. The Fanconi anemia pathway and ubiquitin. BMC biochemistry. 2007;8 1:S10. doi: 10.1186/1471-2091-8-S1-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalb R, Duerr M, Wagner M, Herterich S, Gross M, Digweed M, Joenje H, Hoehn H, Schindler D. Lack of sensitivity of primary Fanconi's anemia fibroblasts to UV and ionizing radiation. Radiation research. 2004;161:318–325. doi: 10.1667/rr3138. [DOI] [PubMed] [Google Scholar]
- Kumaresan KR, Sridharan DM, McMahon LW, Lambert MW. Deficiency in incisions produced by XPF at the site of a DNA interstrand cross-link in Fanconi anemia cells. Biochemistry. 2007;46:14359–14368. doi: 10.1021/bi7015958. [DOI] [PubMed] [Google Scholar]
- Kuraoka I, Kobertz WR, Ariza RR, Biggerstaff M, Essigmann JM, Wood RD. Repair of an interstrand DNA cross-link initiated by ERCC1-XPF repair/recombination nuclease. The Journal of biological chemistry. 2000;275:26632–26636. doi: 10.1074/jbc.C000337200. [DOI] [PubMed] [Google Scholar]
- Landais I, Sobeck A, Stone S, LaChapelle A, Hoatlin ME. A novel cell-free screen identifies a potent inhibitor of the Fanconi anemia pathway. Int J Cancer. 2009;124:783–792. doi: 10.1002/ijc.24039. [DOI] [PubMed] [Google Scholar]
- Li L, Peterson CA, Lu X, Wei P, Legerski RJ. Interstrand cross-links induce DNA synthesis in damaged and undamaged plasmids in mammalian cell extracts. Mol Cell Biol. 1999;19:5619–5630. doi: 10.1128/mcb.19.8.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Zhang N, Vasquez K, Barton M, Legerski R. Repair of psoralen interstrand cross-links in Xenopus laevis egg extracts is highly mutagenic. Biochemical and biophysical research communications. 2005;336:69–75. doi: 10.1016/j.bbrc.2005.08.042. [DOI] [PubMed] [Google Scholar]
- McCulloch SD, Kunkel TA. The fidelity of DNA synthesis by eukaryotic replicative and translesion synthesis polymerases. Cell research. 2008;18:148–161. doi: 10.1038/cr.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh PJ, Sarkar S. DNA interstrand cross-link repair in the cell cycle: a critical role for polymerase zeta in G1 phase. Cell cycle (Georgetown, Tex. 2006;5:1044–1047. doi: 10.4161/cc.5.10.2763. [DOI] [PubMed] [Google Scholar]
- McHugh PJ, Sones WR, Hartley JA. Repair of intermediate structures produced at DNA interstrand cross-links in Saccharomyces cerevisiae. Molecular and cellular biology. 2000;20:3425–3433. doi: 10.1128/mcb.20.10.3425-3433.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minko IG, Harbut MB, Kozekov ID, Kozekova A, Jakobs PM, Olson SB, Moses RE, Harris TM, Rizzo CJ, Lloyd RS. Role for DNA polymerase kappa in the processing of N2-N2-guanine interstrand cross-links. The Journal of biological chemistry. 2008;283:17075–17082. doi: 10.1074/jbc.M801238200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirchandani KD, McCaffrey RM, D'Andrea AD. The Fanconi anemia core complex is required for efficient point mutagenesis and Rev1 foci assembly. DNA repair. 2008;7:902–911. doi: 10.1016/j.dnarep.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu D, Bessho T, Nechev LV, Chen DJ, Harris TM, Hearst JE, Sancar A. DNA interstrand cross-links induce futile repair synthesis in mammalian cell extracts. Mol Cell Biol. 2000;20:2446–2454. doi: 10.1128/mcb.20.7.2446-2454.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernhofer LJ, Lalai AS, Hoeijmakers JH. Fanconi anemia (cross)linked to DNA repair. Cell. 2005;123:1191–1198. doi: 10.1016/j.cell.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NG, Beverloo HB, Hoeijmakers JH, Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Molecular and cellular biology. 2004;24:5776–5787. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima K, Hochegger H, Saberi A, Fukushima T, Kikuchi K, Yoshimura M, Orelli BJ, Bishop DK, Hirano S, Ohzeki M, et al. Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells. Cancer Res. 2005;65:11704–11711. doi: 10.1158/0008-5472.CAN-05-1214. [DOI] [PubMed] [Google Scholar]
- Noll DM, Mason TM, Miller PS. Formation and repair of interstrand cross-links in DNA. Chemical reviews. 2006;106:277–301. doi: 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel KJ, Joenje H. Fanconi anemia and DNA replication repair. DNA Repair (Amst) 2007;6:885–890. doi: 10.1016/j.dnarep.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Perdiz D, Grof P, Mezzina M, Nikaido O, Moustacchi E, Sage E. Distribution and repair of bipyrimidine photoproducts in solar UV-irradiated mammalian cells. Possible role of Dewar photoproducts in solar mutagenesis. The Journal of biological chemistry. 2000;275:26732–26742. doi: 10.1074/jbc.M001450200. [DOI] [PubMed] [Google Scholar]
- Pichierri P, Averbeck D, Rosselli F. DNA cross-link-dependent RAD50/MRE11/NBS1 subnuclear assembly requires the Fanconi anemia C protein. Hum Mol Genet. 2002;11:2531–2546. doi: 10.1093/hmg/11.21.2531. [DOI] [PubMed] [Google Scholar]
- Pichierri P, Rosselli F. Fanconi anemia proteins and the s phase checkpoint. Cell Cycle. 2004;3:698–700. [PubMed] [Google Scholar]
- Raschle M, Knipsheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Scharer OD, Walter JC. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008;134:969–980. doi: 10.1016/j.cell.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson K, Hensey C, Gautier J. Isolation and characterization of Xenopus ATM (X-ATM): expression, localization, and complex formation during oogenesis and early development. Oncogene. 1999;18:7070–7079. doi: 10.1038/sj.onc.1203194. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Davies AA, Ulrich HD, McHugh PJ. DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase zeta. The EMBO journal. 2006;25:1285–1294. doi: 10.1038/sj.emboj.7600993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter D, Costanzo V, Gautier J. ATR and ATM regulate the timing of DNA replication origin firing. Nature cell biology. 2004;6:648–655. doi: 10.1038/ncb1145. [DOI] [PubMed] [Google Scholar]
- Sherman SE, Lippard SJ. Structural aspects of platinum anticancer drug interactions with DNA. Chemical Reviews. 1987;87:1153–1181. [Google Scholar]
- Smeaton MB, Hlavin EM, McGregor Mason T, Noronha AM, Wilds CJ, Miller PS. Distortion-dependent unhooking of interstrand cross-links in mammalian cell extracts. Biochemistry. 2008;47:9920–9930. doi: 10.1021/bi800925e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobeck A, Stone S, Costanzo V, de Graaf B, Reuter T, de Winter J, Wallisch M, Akkari Y, Olson S, Wang W, et al. Fanconi anemia proteins are required to prevent accumulation of replication-associated DNA double-strand breaks. Molecular and cellular biology. 2006;26:425–437. doi: 10.1128/MCB.26.2.425-437.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobeck A, Stone S, Hoatlin ME. DNA structure-induced recruitment and activation of the Fanconi anemia pathway protein FANCD2. Molecular and cellular biology. 2007;27:4283–4292. doi: 10.1128/MCB.02196-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D'Andrea AD. S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood. 2002;100:2414–2420. doi: 10.1182/blood-2002-01-0278. [DOI] [PubMed] [Google Scholar]
- Walter J, Sun L, Newport J. Regulated chromosomal DNA replication in the absence of a nucleus. Mol Cell. 1998;1:519–529. doi: 10.1016/s1097-2765(00)80052-0. [DOI] [PubMed] [Google Scholar]
- Wang LC, Stone S, Hoatlin ME, Gautier J. Fanconi anemia proteins stabilize replication forks. DNA repair. 2008 doi: 10.1016/j.dnarep.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- Wu Y, Shin-ya K, Brosh RM., Jr FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Molecular and cellular biology. 2008;28:4116–4128. doi: 10.1128/MCB.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying CY, Gautier J. The ATPase activity of MCM2-7 is dispensable for pre-RC assembly but is required for DNA unwinding. The EMBO journal. 2005;24:4334–4344. doi: 10.1038/sj.emboj.7600892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Dutta A. An ATR- and BRCA1-mediated Fanconi anemia pathway is required for activating the G2/M checkpoint and DNA damage repair upon rereplication. Mol Cell Biol. 2006;26:4601–4611. doi: 10.1128/MCB.02141-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.