

Abstract
New high-throughput sequencing technologies are generating large amounts of sequence data, allowing the development of targeted large-scale resequencing studies. For these studies, accurate identification of polymorphic sites is crucial. Heterozygous sites are particularly difficult to identify, especially in regions of low coverage. We present a new strategy for identifying heterozygous sites in a single individual by using a machine learning approach that generates a heterozygosity score for each chromosomal position. Our approach also facilitates the identification of regions with unequal representation of two alleles and other poorly sequenced regions. The availability of confidence scores allows for a principled combination of sequencing results from multiple samples. We evaluate our method on a gold standard data genotype set from HapMap. We are able to classify sites in this data set as heterozygous or homozygous with 98.5% accuracy. In de novo data our probabilistic heterozygote detection (“ProbHD”) is able to identify 93% of heterozygous sites at a <5% false call rate (FCR) as estimated based on independent genotyping results. In direct comparison of ProbHD with high-coverage 1000 Genomes sequencing available for a subset of our data, we observe >99.9% overall agreement for genotype calls and close to 90% agreement for heterozygote calls. Overall, our data indicate that high-throughput resequencing of human genomic regions requires careful attention to systematic biases in sample preparation as well as sequence contexts, and that their impact can be alleviated by machine learning-based sequence analyses allowing more accurate extraction of true DNA variants.
High-throughput sequencing is revolutionizing human genetic studies and offers the promise of deciphering the complete sequence of study subjects in the near future (Kuehn 2008; Wheeler et al. 2008) (http://www.1000genomes.org). The massively parallel sequencing technologies are also facilitating large-scale studies targeting genomic loci of biomedical interest. In targeted resequencing of human genomic regions, the goal is to catalog common or rare sequence variation in samples ascertained for a particular phenotype (Altshuler et al. 2008) or a subgroup of samples harboring haplotypes involved in population risk of disease (Lowe et al. 2007; Yeager et al. 2008). Targeted resequencing requires the enrichment of regions of interest by PCR (Brockman et al. 2008) or by capture-based methodologies (Albert et al. 2007; Gnirke et al. 2009). Both can lead to biases in coverage and allele representation. PCR-based enrichment methods often involve pooling of fragments, which are subject to sampling variation. In addition, preferential amplification can occur due to SNPs under primers (Quinlan and Marth 2007). If one allele is amplified preferentially, the ratio of reads derived from each allele will differ from the 1:1 ratio expected. When the preference for one allele is substantial or if there is a weak preference and coverage is low, heterozygous sites on that amplicon are likely to be missed by methods that do not factor in this source of bias.
Existing approaches to resequencing data analysis (Quinlan and Marth 2007; Brockman et al. 2008; Wheeler et al. 2008; Yeager et al. 2008; for review, see Stratton 2008) often require the user to set one or more global parameters, yielding results that are either very conservative (high specificity, but missing many heterozygous sites), or very liberal (high sensitivity, but often calling false heterozygous sites). It can be difficult for users to identify appropriate parameter values to achieve a desired tradeoff between high specificity and high sensitivity. Furthermore, a global decision threshold will rarely achieve the same sensitivity level across different regions, since sequence quality, coverage, and amplification bias vary locally. Regions or sites that cannot be accurately called due to low coverage, poor sequence quality, or preferential amplification are typically not distinguished from regions for which there is strong evidence that no polymorphism is present.
In this paper, we present an approach for targeted resequencing and analysis of nucleotide substitutions in data generated by massively parallel sequencing. We focus on heterozygous substitutions because they tend to be more difficult to identify than variants in the homozygous state, particularly in regions of low coverage or preferential amplification. We present a probabilistic approach to heterozygous site detection (ProbHD), which provides a heterozygosity probability for each chromosomal position. Providing probabilistic confidence scores allows the user to select an appropriate decision threshold that takes into account the goals of their application and the relative costs of false-positives and false-negatives. ProbHD also allows the identification of preferentially amplified fragments and other poorly sequenced regions. Regions that cannot be called (due to low coverage, preferential amplification, low complexity sequence, or poor alignments) are distinguished from regions that contain no heterozygous sites, thereby allowing the user to identify regions for which additional study is required. The availability of confidence scores also allows for a principled combination of sequencing results from multiple samples. We provide a Bayesian method that combines confidence scores from multiple samples of interest to estimate the probability that a site is heterozygous in all samples.
We evaluated ProbHD on genomic regions that we had previously associated with cis-regulatory variation in HapMap CEU or YRI lymphoblastoid cell lines (LCLs). These regions were discovered by allelic expression (AE) mapping as described before (Verlaan et al. 2009). Samples showing an AE phenotype and mapped to a common haplotype must be heterozygous for the variant(s) causing this differential expression. Therefore, in the targeted regions we attempted to identify all heterozygous sites, which represent potential cis-regulatory variants. However, the method may be applied to the study of other phenotypes and also used in general genotype-calling, and therefore the approaches presented below are relevant to a large body of resequencing projects.
In our study, we used long-range PCR (LR-PCR) for target preparation and high-throughput picoliter pyrosequencing (454 Life Sciences [Roche] Genome Sequencer FLX [GS-FLX] system platform). The use of HapMap cell lines (Frazer et al. 2007), for which detailed genetic variation information is available, allows for robust estimates of our ability to accurate detect heterozygous sites. Furthermore, we performed additional genotyping assays and comparison to 1000 Genomes Pilot data (http://www.1000genomes.org) to derive accurate performance statistics. The primary performance metrics we use are sensitivity and false call rate (FCR) (the fraction of heterozygous calls that are in error). Finally, we discuss the sources of errors in sample preparation, sequencing, and sequence analysis.
Results
We have developed an automated method for detecting heterozygous sites from high-throughput sequencing reads from one or more targeted regions of a single diploid sample. We developed a machine learning approach to classify each chromosomal position (a site) in the target region as heterozygous (het) or homozygous. ProbHD reports the most likely genotype, as well as the probability assigned to each genotype. Our prediction pipeline, shown in Figure 1, has four steps: sequencing, alignment, feature generation, and classification, which are briefly described below (see also Methods).
Figure 1.

Graphical overview of the four steps in the prediction pipeline. (1) Sequencing: Target regions are amplified by LR-PCR; amplicons are sequenced using a 454 GS-FLX sequencer. A set of sequence reads is generated by the 454 GS-FLX base-caller. (2) Alignment: Reads are aligned to the reference sequence and combined into a multiple sequence alignment (MSA). (3) Feature extraction: Numerical features are computed from the MSA for each site in the target region. (4) Training: Given a training set of sites with known genotypes from the HapMap database, we train a classifier to identify heterozygous sites from sequencing data. This classifier is then applied to novel data sets to identify novel SNPs.
Sequencing
Target regions, ranging in size from 8 kb to 240 kb (Table 1), were selected based on regions significantly associated with AE of particular genes in CEU and YRI HapMap LCLs (Verlaan et al. 2009). Differential AE is thought to result from heterozygous cis-acting regulatory genetic variants (i.e., heterozygous SNPs). Therefore, we aimed to identify all heterozygous SNPs in each of the resequenced regions. A LR-PCR-tiling path was designed to cover each target region, where each PCR product ranged in size from 3 kb to 10 kb. Amplicons of each tiling path were derived from genomic DNA of four distinct HapMap individuals (Supplemental Table 1). The target regions were grouped into three locus sets and were sequenced using the 454 GS-FLX system. An overview of the experimental pipeline is included in Figure 1. In total, 1220 independent LR-PCR amplicons were sequenced, containing 4905 known heterozygous and 11,813 known homozygous sites, according to HapMap.
Table 1.
Sequencing locus sets and related statistics

Sequence alignment
The FLX System software 1.1.02 (454 GS-FLX June 2007 release) was used to align each read to the reference human genome (build 36.1). Overlapping pairwise alignments were combined into a multiple sequence alignment (MSA). Because these MSAs are often inaccurate near homopolymers, we developed a new realignment strategy (called hAlign), designed to improve alignment of the 454 GS-FLX reads. Two example alignments are given in Supplemental Table 7.
Predictive features and learning method
Our approach identifies heterozygous sites based on a set of quantitative features that describe the characteristics of the aligned sequencing data at each site. The features were selected with the goal of maximizing the program's ability to identify heterozygous sites and assess the confidence of each call. The feature set includes both site-specific and amplicon-level features. Site-specific features are extracted from the MSA constructed from reads that cover a site, as well as the base quality scores assigned to each read. The amplicon-level features reflect the degree of preferential amplification observed for the corresponding LR-PCR amplicon. A complete description of all features used is given in the Supplemental material, together with the relative predictive power of each feature. Given this set of features, a random forest classifier (Breiman 2001) was trained to predict HapMap genotypes. Random forests are highly accurate and efficient machine learning predictors that have the key advantage of rarely overfitting the training data and of providing reliable confidence estimates.
PCR specificity, sensitivity, and bias
The LR-PCR protocol achieved excellent specificity, with 96% of all reads successfully mapping to one of the target regions. High sensitivity was also achieved, since >99% of amplicons yielded sequence data. Average sequence coverage ranged from 26× to 78× between sequence runs. More specifically, 95% of LR-PCR amplicons had at least 8× coverage, while 89% had at least 15× coverage (Fig. 2A). Improved quantification of the PCR amplicons in Locus set 3 compared with the two previous ones resulted in more uniform coverage across amplicons: The percentage of amplicons with at least 8× coverage increased from 95% to 98% while the percentage of amplicons with at least 15× coverage increased from 89% to 96%. However, even with quantitative pooling, a large degree of variability in coverage levels was observed: The top 80% of amplicons spanned a fourfold range of coverage (from 31× to 124× coverage).
Figure 2.

(A) Average depth of coverage obtained for each successfully amplified LR-PCR fragment. Regions covered by two overlapping amplicons in the tiling path were excluded. (B) Frequency and magnitude of amplification bias, for amplicons with at least four known heterozygous sites, and average read coverage of at least 5×.
The allele-specific amplification bias was estimated probabilistically for each amplicon, based on the relative frequency of alleles in reads covering known (or predicted) heterozygous sites. Although amplification of the two alleles is rarely perfectly equal, the estimated amplification bias is small in most cases (Fig. 2B). Only 2.7% of amplicons show evidence of strong amplification bias (i.e., <15% of reads derived from the underrepresented allele). For 94% of amplicons, the underrepresented allele still derives >35% of the reads. Allele-specific bias can be caused by heterozygosity at an unsuspected SNP within the primer used for the LR-PCR (Quinlan and Marth 2007).
Accuracy in calling known heterozygous sites
To train and test ProbHD, a gold standard data set was constructed from the HapMap database (The International HapMap Consortium 2005, 2007) (http://www.hapmap.org/). Each HapMap SNP in each sample was labeled as heterozygous or homozygous. The training set was supplemented with additional genotypes obtained from sequencing and genotyping conducted in our laboratory, resulting in a total of 12,309 homozygous and 6640 heterozygous sites.7 A cross-validation procedure was used to train the classifier and assess its ability to accurately identify heterozygous sites in a diploid genome.
For each site in our target regions, ProbHD estimates the probability that the site is heterozygous, given the observed data. In order to classify a site as heterozygous, a decision threshold must be selected. A high probability threshold results in highly accurate heterozygote calls, but with many false-negatives. Conversely, a lower threshold yields a higher sensitivity, but the FCR will also be high. This sensitivity/FCR tradeoff curve (Fig. 3A) is compared with the performance of the Mosaik/GigaBayes short-read SNP-calling pipeline by Smith et al. (2008) (see Methods). By analyzing base frequencies and quality scores independently at each site, GigaBayes is able to identify 90% of all known heterozygous sites with only a 3% FCR. In contrast, ProbHD considers additional information such as local alignment quality and preferential amplification of LR-PCR fragments, and thereby reduces the FCR by 33%, to just 2%. The estimated FCR of ProbHD remains low up to ∼95% sensitivity, at which point it starts increasing rapidly because of hard-to-call hets located in poorly aligned regions or preferential amplification.
Figure 3.

(A) Ability of three classifiers to identify known heterozygous sites. False call rate (FCR) (the fraction of called heterozygous SNPs that are known to be homozygous) is shown as a function of sensitivity (the fraction of known heterozygous sites called by each classifier). Three classifiers are compared: (1) GigaBayes; (2) ProbHD local-feature classifier, which considers all local features that could be extracted from the 454 GS-FLX generated MSA and quality scores file; and (3) ProbHD full classifier, which considers both local- and amplicon-level features from alignments generated by hAlign. (B) Estimated sensitivity and FCR for calling a site heterozygous, corrected for HapMap errors. (Dashed line) Assumes a HapMap error rate at the upper end of the 95% confidence interval; (dotted line) lower end; (solid line) no HapMap errors.
The effect of coverage level on prediction performance was analyzed by randomly down-sampling reads to the desired coverage and recomputing the features for each site from this smaller data set. For this analysis, only data from Locus set 3 were used, since experimental improvements in this data set allowed us to achieve more uniform coverage across all LR-PCR amplicons (∼45×). This allowed us to sample reads randomly from each sequencing run to simulate coverage levels ranging from 5× to 45× (Fig. 4A). While 5× coverage is clearly insufficient for making accurate predictions, even 15× coverage yields reasonable accuracy (94.5% sensitivity, 6% FCR when using a heterozygous confidence threshold of 0.5). Prediction accuracy increases steadily up to 30× (97% sensitivity, 3% FCR), but additional coverage yields little improvement. At this level of coverage, prediction errors are mostly due to hard-to-call SNPs near homopolymers or complete allele dropout, for which increased coverage makes little difference. For comparison, GigaBayes maintains a low FCR even at low coverage, but at the cost of very low sensitivity.8
Figure 4.

(A) Effect of coverage depth on prediction of known heterozygous sites. Sensitivity and positive predictive value (PPV, equal to 1 − FCR) are shown as a function of average depth of coverage. ProbHD results are shown with two different probability cutoffs for predicting heterozygous sites. A cutoff of c = 0.92 yields a conservative predictor that makes few false-positives, and a cutoff of c = 0.5 yields a very liberal predictor with higher sensitivity but higher FCR. Results are not corrected for HapMap errors. (B) Estimated de novo SNP-calling sensitivity and FCR, assuming 0.1% of sites are heterozygous. Well-sequenced sites are those sites with at least 13× coverage that are located on amplicons with minor allele deriving at least 25% of reads. The pronounced “elbow” is due to the severe imbalance between heterozygous and homozygous sites. Using a very conservative confidence threshold yields an error rate close to zero. However, as the threshold is lowered the percentage of homozygous sites miscalled as heterozygous sites eventually becomes nonzero. Even when the percentage of errors is quite small, the absolute number of errors quickly becomes large in comparison to the number of true hets, and the FCR climbs rapidly.
The number of errors we detect when using the HapMap test set is inflated by genotyping errors in the HapMap database. If ProbHD has a low prediction error rate, then even a small number of HapMap annotation errors can substantially inflate the observed error rate. To obtain an estimate of the true error rate on our test set, we genotyped a random set of sites (n = 62) for which our classifier gave a high-confidence prediction but where our predicted genotype did not match the genotype in the HapMap database. These additional validation results (Table 2) show that the majority of high-confidence calls that disagree with HapMap are actually correct. Out of the 52 high-confidence heterozygous calls conflicting with HapMap that were tested, 51 (98%) were actually heterozygous, while seven of 10 tested high-confidence homozygous calls were validated. Therefore, we used these numbers to factor out HapMap errors and obtain a corrected performance graph (Fig. 3B), showing that on the HapMap test set ProbHD is able to detect 95% of known heterozygotes with <1% FCR, and 98% of known heterozygotes can be identified with <7% FCR.
Table 2.
Number of sites predicted in each bin of heterozygosity probability and each HapMap genotype

aThe probabilities indicate our classifier's confidence that a site is heterozygous. The total number of sites in each category is shown, with areas of disagreement between HapMap and our classifier highlighted in boldface. The numbers in parentheses indicate the number of sites genotyped by an independent method: The first number is the number of sites for which the second technology agreed with our classifier (and HapMap is in error), and the second number is the total number of successfully re-genotyped sites.
bEach site is categorized as either homozygous or heterozygous in HapMap, and homozygous sites are separated into monomorphic (sites for which no variation was found in the entire HapMap population) and polymorphic sites.
De novo identification of polymorphic sites
All the results presented above are based on the subset of sites with known genotypes listed in the HapMap database. However, these results cannot be extended directly to de novo SNP-calling because the fraction of heterozygous sites in our HapMap training and test sets (41.5%) is much higher than the fraction we expect in our target regions (<0.1%). Consequently, the observed FCR on HapMap data is not a realistic estimate of the true FCR for de novo SNP- calling in our target region. Consider a predictor that would achieve 100% sensitivity and 1% FCR on a balanced test set (with equal numbers of heterozygous and homozygous sites). If the ratio of heterozygous sites in the target region is 1:1000, then the de novo FCR9 would actually be 91%! It is essential to correct for the differences in polymorphism rate when estimating the error rate for de novo heterozygous site prediction. Therefore, based on our estimate that 0.7% of HapMap genotypes in our target regions are incorrect (Table 2), and 1 in 1000 sites are heterozygous, a sensitivity of 93% with a FCR below 5% is predicted (Fig. 4B, solid line). We note that even better performance can be achieved by removing regions in the lower tail of coverage or extremes of allelic bias (Fig. 4B, dashed line).
We further evaluated the feasibility of ProbHD for de novo base-calling using the genotype calls from 245 kb of unique (repeat masked) sequence generated at high coverage by the 1000 Genomes Pilot Project (http://www.1000genomes.org, April 2009 release) and also sequenced in our experiments in the same individuals (NA12892 or NA12891). This comparison should allow more direct estimates of the accuracy of our method not limited by the biases outlined above. Setting two thresholds for calling bases, het probability <0.4 to call homozygous sites or het probability >0.92 to call heterozygous bases, allowed base calls for 99.4% of sequence (Supplemental Table 2). Genotype calls were made for 92% of homozygous non-ref sequence sites (homovar), and for 94% of heterozygous sites (het) reported in the 1000 Genomes database for these two individuals. Of the eight nonreference sequence homozygous sites not covered by our base-calling, six were located in a single poorly aligned genomic region. Overall base-calling agreed for >99.9% of homozygous sites (homoref/homovar). The agreement of ProbHD and 1000 Genomes genotyping calls for heterozygous and nonreference allele homozygous (homovar) sites were 89% and 98%, respectively. We note that for seven discordant sites there were independent genotyping data available, and in all cases our prediction was converging with the third method. These data suggest that ProbHD can be utilized as a general de novo base-caller in high-throughput sequencing data.
Identifying heterozygous sites shared between individuals
The probability scores generated by ProbHD are particularly valuable when combining predictions from different samples. This can be exploited for quickly cataloging common variants in the same haplotype for fine-mapping studies. A majority of the target regions (20/33) were sequenced in four individuals carrying a common haplotype associated with AE, and our heterozygosity predictor was applied to each sample. For each site, individual het probabilities and prior probabilities of polymorphism (based on dbSNP) were combined using a Bayesian approach to obtain the posterior probability that all four individuals are heterozygous.
To evaluate our ability to identify heterozygous sites occurring in multiple sequenced individuals, we constructed a test set where positive instances are sites known to be heterozygous in all four individuals (based on HapMap), and negative instances are sites where at least one individual is known to be homozygous. We find 95% of known common hets with <5% FCR (Fig. 5A). Additional genotyping experiments were conducted to validate our predictions (Table 3). Genotyping results validated our predictions for 204 out of the 206 polymorphic sites, which suggests that at a sensitivity level of 85%, the FCR is <1%.
Figure 5.

Common hets prediction. (A) Test set FCR (the percentage of predicted sites in HapMap that are not common hets) as a function of sensitivity. (B) The total number of predicted common hets as a function of desired sensitivity, based on one to four samples.
Table 3.
Sensitivity and specificity in predicting heterozygous sites shared between individuals

aThe numbers in parentheses are derived from experimental validation of de novo sites by Sequenom.
bAssumes all sites within this or a higher ranked bin are called as heterozygotes.
Sites are binned by the posterior probability that they are heterozygous in all four samples. True genotype was determined by HapMap when available. A random subset of sites with unknown genotype and predicted common-het probability >75% were genotyped in our panel of CEU and YRI HapMap samples. Of these sites, a total of 218 sites were successfully genotyped in all four samples, and 206 (95%) of these sites were found to be polymorphic in the HapMap CEU sample. Of the remaining 12 monomorphic calls, many likely represent failed genotyping assays, since in a subset of such discrepancies followed up earlier we observed concordant results with 454 GS-FLX resequencing upon redesign of the genotyping assay or by independent Sanger sequencing.
Our data also allow us to analyze the number of samples needed for cataloging common variants and the effect on follow-up genotyping in a larger sample. For example, to achieve 95% sensitivity, increasing the number of individuals sequenced from one to two almost halves the number of sites to be genotyped (Fig. 5B). Adding more individuals reduces the number of predicted sites still further, but the reduction is small in comparison.
Discussion
We show that careful consideration of alignments and systematic biases in sample preparation in targeted human resequencing experiments using second-generation sequencers allows the detection of heterozygous sites with good accuracy. ProbHD yields 85%–93% sensitivity with <5%–11% FCR in identifying heterozygous bases in human resequencing data, where about one-third of the bases have <30× coverage. The higher sensitivity and lower FCR stem from estimates based on HapMap data, whereas the more pessimistic estimates are based on direct comparison to independent (1000 Genomes Pilot Project) high-throughput sequencing data. We speculate that true sensitivity and FCR are within these limits, since independent genotyping of a subset of sites resolved most conflicts in our favor. If we exclude problem regions (low-coverage sites and preferentially amplified LR-PCR fragments), a very high sensitivity (>99%) can be obtained with a negligible FCR. In contrast to many existing approaches that report a single set of predicted hets based on a particular set of filters and parameter values (Levy et al. 2007; Wheeler et al. 2008; Yeager et al. 2008), ProbHD reports a heterozygosity probability for each site, allowing users to trade off false-positives for false-negatives as desired. We compared ProbHD with the Mosaik/GigaBayes predictor, a package designed for short-read alignment and SNP-calling (Smith et al. 2008). We showed that ProbHD has a small but significant advantage at high sequence coverage, and a larger advantage at lower coverage. Furthermore, ProbHD generates many more low-confidence scores, allowing users to achieve higher sensitivity if they are willing to tolerate a higher FCR. Finally, we show that common variants in haplotypes can be successfully cataloged with a relatively sparse sampling of haplotypes of interest, allowing investigators to fine-tune study design by balancing the cost of high-throughput sequencing and follow-up genotyping (Fig. 5B).
The FCR of ProbHD for de novo resequencing remains relatively high for three main reasons: (1) poor sequence alignment (as a result of long homopolymers or low-complexity sequences), (2) low coverage (as a result of pooling variation or difficult-to-sequence regions), and (3) preferential amplification of one allele. The impact of each source of error is detailed below.
In 454 GS-FLX reads, poor alignments typically occur in low-complexity sequence or in regions containing long homopolymers. The 454 GS-FLX sequencer, rather than sequencing a single base at a time, estimates the lengths of homopolymer runs: The longer the homopolymer run, the more uncertainty there is in the estimate of its length. To address this issue, we designed a modified alignment strategy, hAlign, for generating high-quality MSAs from 454 GS-FLX sequence reads. Using alignments generated by hAlign rather than the 454 GS-FLX-generated alignments improves predictions considerably: At 94% sensitivity, for example, the FCR dropped from 4% to 2.5%. In addition to realigning problem regions, our approach uses a more liberal filtering strategy than previously used. Rather than applying stringent filters to discard all suspect reads, we provide our classifier with features that yield quantitative measurements of local alignment quality for each site. In this way, high-coverage sites located in poorly aligned regions are never given high confidence scores, but may still be presented as candidate heterozygous sites. We note that applying other recently suggested approaches for 454 GS-FLX base-calling, quality score estimation, and read alignment (Quinlan and Marth 2007) could further reduce error associated with imprecision in alignment. In particular, the performance of the GigaBayes predictor, which relies heavily on the availability of accurate base quality estimates, may greatly benefit from improved base-calling, as previously reported (Smith et al. 2008). Homopolymers are less problematic for other high-throughput sequencing technologies (Mardis 2008), but the shorter read lengths lead to challenges in mapping and alignment of repetitive sequences (Li et al. 2008). Read mapping uncertainty, as well as any other technology-dependent quantifiable sources of uncertainty, could be easily taken into consideration by ProbHD by adding a set of additional features to the classifier.
A critical and universal challenge in targeted resequencing studies is to obtain adequate coverage for detection of heterozygotes. In shotgun sequencing of the human genome, where unequal allele representation is less of a concern than in targeted resequencing, lower coverage (<15×) should be sufficient to identify both alleles 99% of the time (Wheeler et al. 2008). In contrast, our results show that in targeted resequencing an average coverage of 15×—although sufficient to achieve reasonable predictive value—yields low sensitivity. Sensitivity continues to increase steadily even up to 30× average coverage. The improvement in sensitivity afforded by higher coverage levels is most likely due to the presence of preferentially amplified amplicons. The stronger the amplification bias, the larger the number of reads required to detect heterozygous sites.
Even when the average depth of coverage is high, and amplification is unbiased, locally low coverage is still a major cause of missed heterozygous sites. Locally low coverage is sometimes due to unusual sequence composition. For example, a 1.4-kb stretch of sequence with 74% GC content (chr5:14924400–14925800) yielded negligible coverage despite successful amplification and sequencing of flanking regions. More often, however, regions of low coverage result from uneven pooling of PCR amplicons. Our results show that efforts to derive equimolar pools result in a significant improvement in sensitivity: In the HapMap data set, the FCR at 95% sensitivity dropped from 2.2% to 0.5%. Further reductions in coverage variability would allow for higher accuracy at lower average coverage levels. However, variation in coverage is unlikely to be completely eliminated, and, therefore, to achieve high sensitivity, a classifier must be able to detect heterozygous sites even when coverage is low. ProbHD reports lower confidence in low-coverage regions, but still identifies possible heterozygous sites for further study.
The third factor that results in missed heterozygous sites is preferential amplification. Although complete allele dropout is relatively rare (only 3% of amplicons in our data set), it precludes detection of heterozygous variants, and thus has a large effect on sensitivity. It is essential to identify complete allele dropout so that these amplicons can be targeted for follow-up by alternative methods. Detection of complete allele dropout requires a set of known heterozygous sites. If no prior information is available, it could be beneficial to generate high-density genotyping data in parallel with full sequence. Alternatively, complete allele dropout can be virtually eliminated by a redundant design of LR-PCR primers. It is highly unlikely that two independent sets of primers would both contain SNPs, and so—assuming complete allele dropout is due to SNPs under primers and occurs in 3% of amplicons—fewer than 1 in 1000 amplicons would exhibit near complete allele dropout.
Even if complete allele dropout is eliminated, smaller biases will still occur. Almost undetectable allele bias can occur early in the LR-PCR reaction, and later be amplified to detectable levels. Although smaller biases are less catastrophic, they are more common: In our data, 9.5% of amplicons showed significant10 deviation from the expected 1:1 ratio. A failure to recognize these smaller biases will lead to overly conservative probability estimates, and thus, missed heterozygotes. In particular, a statistical method that assumes that the ratio of alleles is 1:1 (Li et al. 2008; Wheeler et al. 2008) will yield misleading statistics. Our method does not make any assumptions about the ratio of alleles, but uses prior information (known hets) and empirical data (predicted hets) to estimate the amplification bias for each fragment, allowing detection of amplification bias even when no heterozygotes sites are known a priori. Of all the features used by our classifier, the amplicon-level features resulted in the largest prediction improvement (Fig. 3A), revealing the importance of quantifying preferential amplification.
Coverage variation and preferential allele representation introduced in sample preparation are independent of the next-generation sequencing technology used. Similar to LR-PCR, microarray- or solution-based sequence capture methods for target enrichment (Albert et al. 2007; Gnirke et al. 2009) show wide variability in coverage per base. Furthermore, a specific bias toward calling heterozygous sites reference allele homozygotes due to polymorphisms under capture probes has been reported (Gnirke et al. 2009). Reported specificity of sequence capture-based genotype calls was high (>99%) but resulted in only ∼64% sensitivity between technical replicates (i.e., a large fraction of genotypes were not in high-coverage sequence in both samples and genotypes were not called) (Gnirke et al. 2009). In contrast, ProbHD allows a user to distinguish strong negative evidence from a lack of positive evidence, which enables a user to determine the specific regions for which additional sequencing is necessary. Training ProbHD to include features for specific biases in the newer capture technologies could allow more efficient extraction of genotypes from these data as well.
While related computational approaches can be compared on the same data, it is difficult to generalize from results on a set of sites with known genotype to results for de novo SNP-calling. First, the genotypes in these data sets were obtained by technologies that have nonnegligible error rates, which could be higher than the error rates in resequencing. We find particularly high error rates for sites assigned monomorphic status in the HapMap population. Indeed, based on our in-house genotyping and Sanger sequencing of discrepancies between ProbHD predictions and HapMap (Table 2), we derived an estimate that 0.5%–0.7% of HapMap genotypes are incorrect, closely approximating rates reported by others (0.6%) (Frazer et al. 2007; Gnirke et al. 2009).
A second challenge in estimating error rates for de novo SNP-calling arises because the proportion of homozygous and heterozygous sites in the set of known SNPs in our test set is not representative of genomic DNA. Although this bias does not affect our ability to estimate the false-positive rate (FPR)11 (the fraction of homozygous sites that are erroneously reported as heterozygous), such a metric is not appropriate when the majority of examples are negative (i.e., homozygous). In such cases, the FPR will always be low as long as few positives are predicted. Instead, we argue that for measuring performance in de novo SNP-calling, it is important to report FCR (the fraction of heterozygous calls that are erroneous) rather than specificity or FPR. However, unlike the FPR, the FCR depends on the fraction of sites that are heterozygous. Thus, it cannot be directly estimated from a data set comprised of prespecified sets of SNPs with known genotypes but unrepresentative heterozygous to homozygous ratio, such as prior genotyping data (Levy et al. 2007; Wheeler et al. 2008) or resequencing limited to short polymorphic sequences (Brockman et al. 2008). In order to determine the proportion of calls that are false, adjustments need to be made for the polymorphism rate. We evaluated ProbHD for de novo base-calling by comparing with the genotype calls derived from sequence generated at high coverage by the 1000 Genomes Pilot Project and avoided the need for such adjustments. The lower than estimated performance of ProbHD when compared with 1000 Genomes Pilot Project high-coverage sequence data could be due to limited overlap of data (∼0.1% of our data set). Furthermore, the April 2009 release of 1000 Genomes Pilot data does not provide quality scores for genotypes and, in a number of cases of apparently missed or false-positive base substitutions by ProbHD calls, we observed evidence of more complex substitutions in sequence alignments (Supplemental Table 6). The maturation of shotgun sequencing data across population samples will allow not only accurate measurements of ProbHD performance but also much more realistic training sets for development of machine learning-based sequence calls.
Overall, our analyses indicate that probabilistic approaches for calling sequence variants in high-throughput sequence data allow efficient identification of heterozygous sites, and show how confounders such as preferential representation of alleles can be integrated into the analyses. The sensitivity and FCR estimates in our data set for single sample or multiple samples will help in study designs to maximize utility of targeted sequencing of human genome in fine-mapping of loci identified in association studies as well as in next-generation, sequence-based, disease association studies.
Methods
Loci and sample selection
Target regions were selected based on allelic expression mapping in CEU and YRI LCLs as previously described (Pastinen et al. 2005; Verlaan et al. 2009). These experiments can map relative differences in expression of alleles within a sample to local variants that explain such differences in the population. In most cases, a single common haplotype was shared in a heterozygous state by all samples showing differences in relative expression in alleles. If the allelic expression trait observed in the population has a common cause (a single or group of variants) across such samples, then all “candidate” sites should be present in heterozygous state in the selected samples. While the latter assumptions are important for identification of causal variants for allelic expression, the choice of the haplotypes, samples, and loci should not affect the algorithms described in this paper. Therefore, the sample selection can be thought to represent a more general case of haplotype-based selection of samples from a population. The loci chosen for resequencing and their coordinates are shown in Table 1. Individuals sequenced for each locus (n = 4) are further delineated in Supplemental Table 1.
Sample preparation and DNA sequencing
A tiling path of 3- to 10-kb LR-PCR amplicons was designed to fully cover each target region. Each LR-PCR amplicon was independently produced. To obtain more material and reduce the background level of genomic DNA starting material, we performed a second round of LR-PCR of the product obtained from the initial LR-PCR, using the same conditions and primers. The final LR-PCR amplicons from multiple target regions were then pooled together using normalized quantities. For Locus sets 1 and 2, amplicon quantities were quantified with a Nanodrop spectrophotometer, while Locus set 3 quantities were quantified with a PicoGreen spectrophotometer. The libraries were then amplified and sequenced on a 454 GS-FLX instrument.
Image and signal processing were carried out using the GS-FLX System version 1.1.02 (454 Life Sciences [Roche], June 2007 release), with default parameter settings. Flowgrams were converted to nucleotide sequences and each base is assigned a quality score. The reads are filtered using the default FLX System software quality control filters, and primer sequences are trimmed from the ends of reads.
Read mapping and alignment
Mapping and alignment of reads to the human reference sequence was conducted by the FLX System Reference Mapper software, with default parameters. All reads were mapped to the targeted regions of the human reference genome (NCBI Build 35 for Locus sets 1 and 2, and NCBI Build 36 for Locus set 3). Partially mapped reads are retained, but reads that cannot be mapped uniquely within the target region are discarded.
Multiple sequence alignments are initially constructed from the 454 GS-FLX Instrument pairwise alignments, such that all read bases aligned to the same base in the reference are aligned together, and all bases inserted i bases after a reference base are aligned together. However, these alignments often contain errors, especially near long homopolymers and SNPs. The 454 GS-FLX sequencer, rather than sequencing a single base at a time, estimates the lengths of homopolymer runs. The longer the homopolymer run, the more uncertainty there will be in the estimate of its length. Incorrect read alignment near long homopolymers results in a significant number of miscalls. Thus, we designed an alternative alignment procedure for 454 GS-FLX reads. Our modified alignment algorithm, hAlign, uses a progressive alignment strategy, and imposes different indel and mismatch penalties in homopolymer regions. In particular, reduced penalties are imposed for gaps occurring within homopolymers.
Poorly aligned portions of the target region are first selected for realignment. The read sequences overlapping these regions are collected. The reference and reads are then input to hAlign. The realignment procedure is based on a modified Needleman–Wunsch alignment algorithm (Needleman and Wunsch 1970). First, a preprocessing step identifies long near-homopolymeric regions, allowing for intermittent and nonconsecutive occurrences of other nucleotides. A specialized gap scoring scheme was designed for these regions, reducing gap opening and extension penalties to better model length variations of the regions. Also, when nucleotides other than the repeated base occur in these regions, matches of these nucleotides are weighted more heavily. Reads are progressively aligned, in an order that is based on pairwise alignment score with the reference.
Progressive alignments are sensitive to the ordering of the sequences to be aligned. “Noisy” reads often resulted in alignments where the two alleles of a heterozygous position appeared in separate columns. These positions commonly covaried (i.e., a gap is seen in one column whenever a nucleotide is seen in the other). We thus introduced a post-processing step in the alignment procedure to screen the initial realignments for such column pairs and reprioritize the order of the alignment of the read sequences, postponing the alignment of the reads causing the problem. Full details about the realignment procedure are available in the Supplemental material.
Feature selection
The multiple sequence alignments produced by hAlign are used to define site-specific and amplicon-level features from which heterozygosity predictions will be made. A column of the MSA that contains variant bases may indicate a heterozygous site. However, observed variant bases may also be a result of sequencing or alignment errors. When calling heterozygotes, the main sources of error and uncertainty include low depth of coverage, low-quality reads/bases, incorrectly mapped reads, preferential amplification, and poor alignment. Poor alignments can occur as a result of neighboring polymorphisms, low-complexity sequence, and neighboring homopolymers. Thus, the types of local features used to predict heterozygotes in 454 GS-FLX data include: (1) total number of reads covering the site (forward and reverse), (2) number of reads with a variant allele at the site (forward and reverse), (3) number of variants that occur near the end of their read, (4) base quality scores of each allele, (5) local alignment quality, and (6) length of neighboring homopolymers. The complete list of features is described in the Supplemental material.
When predicting heterozygous sites based on reads derived from amplicons, it is also necessary to consider the possibility that unknown SNPs within the PCR primer sites caused preferential amplification of one allele (Quinlan and Marth 2007). Thus, global amplicon-level features are also provided to the classifier. These features include our maximum a posteriori estimate of the fraction of reads originating from the nonpreferred allele, as well as the confidence interval on this estimate.
Training and test sets
The data set comprised all sites in each sample with known genotypes in the HapMap database (release 22 for Locus set 1 and 2 and release 23 for Locus set 3). According to the genotype, each site was labeled as heterozygous, homozygous reference (i.e., identical to the reference genome), or homozygous variant.
The training set consisted of these instances: a number of additional polymorphic12 sites genotyped at our center using Sequenom panels, and a set of genotypes obtained using Sanger-chemistry sequencing. A small fraction of HapMap genotypes that conflicted with our Sequenom results were corrected or removed from the training set, but remained unaltered in the test set.
This training set contained very few sites with low coverage. However, we wanted our classifier to generate accurate confidence values even for sites covered by very few reads. For this reason the training set was supplemented with additional training instances, generated by randomly down-sampling reads (to obtain coverage levels of 5×, 10×, 15×, 20×, and 30×) and then recomputing feature values for sites with known genotypes. Each site is thus represented several times in our training data, at different coverage levels.
Learning and cross-validation procedure
Our classifier was benchmarked against the test set using a fivefold cross-validation procedure. The set of sites with known genotypes was randomly divided into five equal-sized subsets. Each subset in turn was used for testing. Any training instance matching a test instance (i.e., with the same chromosomal position and sample ID) was removed from the training set, and the remainder of the training instances was used for training. The performances on the five test sets were averaged to obtain an overall error estimate.
The randomForest package (Breiman 2001) in the R statistical software package (http://www.r-project.org) was used to train a classifier to distinguish between heterozygous and homozygous sites. The classifier builds 250 unpruned decision trees, each from a different random subset of the training instances and features. Specifically, each tree is trained on a random subset of five features, and from a random, balanced set of examples (equal number of hets and non-hets). The proportion of trees that classify a site as heterozygous is used as an estimate of the probability that the site is a het.
Mosaik/GigaBayes predictions
454 GS-FLX reads and base quality scores were aligned against the selected regions of the reference human genome using MosaikAligner 0.9.0891 with arguments recommended for 454 GS-FLX read alignment: “-hs 15 -mmp 0.05 -a all -m unique -mhp 100 -act 26 -mmal -p 2.” Roughly 95% of reads uniquely align to the desired target region. Alignments were sorted and assembled using MosaikSorter and MosaikAssembler. Heterozygous sites were predicted using GigaBayes 0.4.1 with arguments “–sample single–PSL 0.001–ploidy diploid–QRL 1–QAL 1.” We note that the values of QRL and QAL (minimum read base quality and minimum aggregate allele quality, respectively) were set much lower than the defaults (10 and 40, respectively). This allowed increased sensitivity and did not affect FCR.
Validation by Sequenom and Sanger sequencing
Validation of candidate heterozygous sites was done using the Sequenom MassARRAY iPLEX Gold (Sequenom Inc.). Allele detection was performed using matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry.
A subset of sites that could not be validated either on HapMap or by Sequenom genotyping was further tested by Sanger sequencing of PCR amplicons using Big Dye Terminator protocols (Applied Biosystems) and capillary sequencing in Applied Biosystems 3730 XL DNA instrument (detailed protocols available upon request).
Estimating corrected, de novo prediction performance
To estimate de novo prediction performance, we corrected for errors in the HapMap database as well as the low rate of heterozygosity in genomic DNA compared with our test set. Based on the validation results shown in Table 2, we estimated the number of genotypes for which HapMap, rather than our classifier, is in error. We assumed that HapMap is always correct in cases where our classifier has low confidence (<85% for heterozygous calls, <95% for homozygous calls). For confident calls, we estimate the number of genotypes for which HapMap is in error based on the 95% confidence interval computed from the number and proportion of validated calls in each probability bin. This confidence interval yields a lower and upper bound on the number of HapMap genotypes in error. Summing the estimated number of errors from each confidence bin yields an estimate of the total proportion of HapMap genotypes in error: 0.98% –1.2% for “monomorphic” sites, and 0.33% –0.5% for polymorphic sites, yielding an overall HapMap error rate of 0.5%–0.7% for our target regions.
Given an estimate of the total number of erroneous HapMap genotypes, we estimated our predictor's error rates for a range of probability thresholds. Predicted probabilities were divided into 100 discrete bins, and the number of hets and non-hets in each bin was tallied. The distribution of HapMap errors among the bins was calculated by assuming false-positive errors (sites incorrectly labeled heterozygous) follow the probability distribution displayed by known homozygous sites, and similarly for false-negative errors. The number of observed errors in each bin was reduced by the estimated number of HapMap errors in that bin. Finally, the number of het and non-het sites in each bin was corrected for the expected heterozygosity rate. Known heterozygous sites from HapMap comprise 0.055% of target sites. Assuming HapMap SNPs account for half of all SNPs in the region suggests an approximate heterozygosity rate of 1:1000 (compared with 294:1000 in the HapMap set).
1000 Genomes Pilot data comparison
Two samples (NA12892 and NA12891) included in the high-coverage sequencing of the 1000 Genomes Pilot Project were also included in a subset of our sequencing experiments. We compared data from 245 kb of unique sequence included in chr2:38064246–38177190 (NA12892), chr12:9713048–9790776 (NA12891), chr10:6126099–6162015 (NA12891), chr2:191605785–191739895 (NA12892), and chr7:128356196–128487322 (NA12892). Genotype calls for 1000 Genomes in these samples were downloaded from ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/ for the April 2009 release (L Brooks, G McVean, and G Abecasis, pers. comm.). Sites with no genotyping calls were considered homozygous for reference allele. A comparison of ProbHD predictions and 1000 Genomes genotypes is given in Supplemental Table 2.
Identifying preferentially amplified fragments
For each LR-PCR amplicon, we estimate a parameter r, the fraction of reads that were derived from the nonpreferentially amplified allele. When r = 0.5, both chromosomes were amplified equally, and when r = 0 only one chromosome was amplified. We would like to find the posterior distribution of r, given the data D = (D1,…,Dl), where Di represents the data at position i in the MSA, and l is the length of the amplicon. If we make the simplifying assumption that sequencing and alignment errors are independent at neighboring locations in the amplicon, then we can estimate the posterior probability density function as follows:
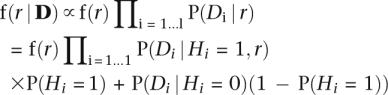 |
where Hi = 1 if site i is heterozygous and Hi = 0 otherwise. A uniform distribution on (0,0.5) was used as the prior f(r). The probability that site i is heterozygous, P(Hi = 1), is computed from the probability estimated by the random forest classifier as described above, assuming every amplicon was amplified in an unbiased fashion (i.e., r = 0.5). These raw probabilities are adjusted to reflect estimated HapMap error rates and estimated heterozygosity rates, as described above. For any site with a known genotype, we set P(r) = 0.99 if the site is heterozygous and P(r) = 0.01 if the site is homozygous. However, when testing our classifier, we took care to avoid using known genotypes when computing amplicon-level features. For each site in the training and test set, amplicon-level features were computed without knowledge of that site's genotype.
The data Di are summarized with two statistics: ni, the total number of reads aligned at position i in the MSA, and ki, the frequency of the second most common base observed in position i. We assume the probability of observing the data Di given that site i is heterozygous is P(Di | Hi = 1,r) = P(X = ki) + P(X = ni − ki), where X follows a binomial distribution X ∼ Bin(ni,r). The probability of Di = (ni,mi,ki) given that the site is homozygous is modeled as a mixture of this binomial distribution and a uniform distribution: P(Di | Hi = 0) = λ (P(X = ki) + P(X = ni − ki)) + (1 − λ) / (⌊ni / 2− + 1). Parameters were selected to maximize the likelihood of the observed data for known homozygous sites in the training data (p = 0.015 and λ = 0.85). For training a classifier, the posterior distribution was summarized with five statistics: the maximum a posteriori value of r, the probability that r ≥ 0.35, the probability that r < 0.15, and the upper and lower bounds of the 90% confidence interval for the parameter r.
Combining individual scores to identify common hets
Given 454 sequence data for k individuals of interest, for each site i in the reference sequence, we want to estimate the probability that site i is a common het, i.e., that it is heterozygous in all k individuals, given (Di1,…,Dik), the 454 data for site i. The probability that the site is heterozygous in all k sequenced individuals is:
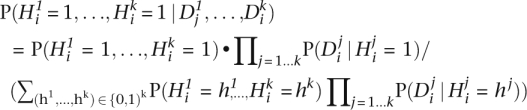 |
In order to compute this probability we must determine the prior probability of observing each combination of heterozygotes and homozygotes at a random site, in these k individuals. This probability will depend on whether the site is polymorphic. If P(Yi = 1) is the prior probability that site i is polymorphic in the population, then P(Hi1 = h1,…,Hik = hk) = P(Hi1 = h,…,Hik = hk | Yi = 1)P(Yi = 1) + P(Hi1 = h1,…,Hik = hk|Yi = 0)P(Yi = 0). Note that P(Hi1 = h1,…,Hik = hk | Yi = 0) is nonzero only if h1 = … = hk = 0. We estimate P(Hi1 = h1,…,Hik = hk | Yi = 1) empirically from the set of polymorphic sites in our training set.
To estimate the probability P(Di | Hi = h), we summarize the sequencing data Di with a single statistic, ci, the probability that site i is heterozygous as estimated by the single-individual het predictor described above: P(Di | Hi = h) ≈ P(Ci = ci | Hi = h), which is estimated empirically on held-out HapMap training data. The prior that a site is polymorphic in these samples, P(Yi = 1), is set heuristically to 0.25 for SNPs previously reported in dbSNP, and 0.0015 otherwise.
Correlation between primer design and preferential amplification
We sequenced every target region in four individuals. In most cases a single tiling path was used in all four individuals, but in some cases different primers were selected. The 1220 amplicons generated in our experiments can be grouped into 418 distinct sets, according to primer design, where each primer design was used with one to four samples. Of the 1220 amplicons, we detected 36 amplicons (2.95%) for which one allele was severely underrepresented. However, the 36 failed amplicons represent only 21 of the 418 primer sets, which is substantially fewer sets than would be expected if amplicons failed independently in each sample. In other words, amplicons generated from the same primers, but in different individuals, have preferential amplification ratios that are more correlated than would be expected by chance. This suggests that preferential amplification can be reduced by creating two independent tiling paths for each target region.
Acknowledgments
This work was funded by Genome Quebec and Genome Canada. T.P. holds a Canada Research Chair in Human Genomics (CRC, tier 2). We thank the 1000 Genomes Project for allowing us to use the pilot project data for validation.
Footnotes
Notice that the numbers of homozygous and heterozygous sites are much more balanced than in actual genomic data—this is addressed in the section on de novo het-calling.
At low coverage (<15×), the distribution of GigaBayes scores does not allow much of a trade-off between sensitivity and FCR, as the majority of hets are assigned score 0.
If f is the FCR on a balanced test set, and s is the sensitivity obtained on that set, then the expected FCR on a data set with a polymorphism rate of p is (1 − p) • f / ((1 − p) • f + p • s).
The upper bound of the 90% Bayesian credible interval on the fraction of reads derived from the underrepresented allele is <0.45.
Adding to the confusion, the term false-positive rate (FP/(FP+TN)) is sometimes used to denote what we call false call rate (FP/(FP+TP)), and specificity is sometimes used to refer to 1 − false call rate. Most often, terms are used without a clear explanation of the mathematical quantity represented.
For training the classifier, only sites classified as polymorphic by Sequenom were included.
[Supplemental material is available online at http://www.genome.org. Alignment and SNP-calling software is available at http://www.mcb.mcgill.ca/∼blanchem/reseq.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.092072.109.
References
- Albert TJ, Molla MN, Muzny DM, Nazareth L, Wheeler D, Song X, Richmond TA, Middle CM, Rodesch MJ, Packard CJ, et al. Direct selection of human genomic loci by microarray hybridization. Nat Methods. 2007;4:903–905. doi: 10.1038/nmeth1111. [DOI] [PubMed] [Google Scholar]
- Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008;322:881–888. doi: 10.1126/science.1156409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. Random forests. Mach Learn. 2001;45:5–32. [Google Scholar]
- Brockman W, Alvarez P, Young S, Garber M, Giannoukos G, Lee WL, Russ C, Lander ES, Nusbaum C, Jaffe DB. Quality scores and SNP detection in sequencing-by-synthesis systems. Genome Res. 2008;18:763–770. doi: 10.1101/gr.070227.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnirke A, Melnikof A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennel T, Giannoukos G, Fisher S, Russ C, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1229–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–862. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn BM. 1000 Genomes Project promises closer look at variation in human genome. JAMA. 2008;300:2715. doi: 10.1001/jama.2008.823. [DOI] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe CE, Cooper JD, Brusko T, Walker NM, Smyth DJ, Bailey R, Bourget K, Plagnol V, Field S, Atkinson M, et al. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet. 2007;39:1074–1082. doi: 10.1038/ng2102. [DOI] [PubMed] [Google Scholar]
- Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- Pastinen T, Ge B, Gurd S, Gaudin T, Dore C, Lemire M, Lepage P, Harmsen E, Hudson TJ. Mapping common regulatory variants to human haplotypes. Hum Mol Genet. 2005;14:3963–3971. doi: 10.1093/hmg/ddi420. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Marth GT. Primer-site SNPs mask mutations. Nat Methods. 2007;4:192. doi: 10.1038/nmeth0307-192. [DOI] [PubMed] [Google Scholar]
- Smith R, Quinlan AR, Peckham HE, Makowsky K, Tao W, Woolf B, Shen L, Donahue WF, Tusneem N, Stromberg MP, et al. Rapid whole-genome mutational profiling using next-generation sequencing technologies. Genome Res. 2008;18:1638–1642. doi: 10.1101/gr.077776.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton M. Genome resequencing and genetic variation. Nat Biotechnol. 2008;26:65–66. doi: 10.1038/nbt0108-65. [DOI] [PubMed] [Google Scholar]
- Verlaan DJ, Ge B, Grundberg E, Hoberman R, Lam KC, Koka V, Dias J, Gurd S, Martin NW, Mallmin H, et al. Targeted screening of cis-regulatory variation in human haplotypes. Genome Res. 2009;19:118–127. doi: 10.1101/gr.084798.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen YJ, Makhijani V, Roth GT, et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- Yeager M, Xiao N, Hayes RB, Bouffard P, Desany B, Burdett L, Orr N, Matthews C, Qi L, Crenshaw A, et al. Comprehensive resequence analysis of a 136 kb region of human chromosome 8q24 associated with prostate and colon cancers. Hum Genet. 2008;124:161–170. doi: 10.1007/s00439-008-0535-3. [DOI] [PMC free article] [PubMed] [Google Scholar]