

Abstract
Structure-based design, synthesis and biological evaluation of a series of novel HIV-1 protease inhibitors are described. In an effort to enhance interactions with protease backbone atoms, we have incorporated stereochemically defined methyl-2-pyrrolidinone and methyl oxazolidinone as the P1′-ligands. These ligands are designed to interact with Gly-27′ carbonyl and Arg-8 side chain in the S1′-subsite of the HIV protease. We have investigated the potential of these ligands in combination with our previously developed bis-tetrahydrofuran (bis-THF) and cyclopentanyltetrahydrofuran (Cp-THF) as the P2-ligands. Inhibitor 19b with an (S)-aminomethyl-2-pyrrolidinone and a Cp-THF was shown to be the most potent compound. Inhibitor 19b maintained near full potency against multi-PI-resistant clinical HIV-1 variants. A high resolution protein-ligand X-ray crystal structure of 19b–bound HIV-1 protease revealed that the P1′-pyrrolidinone heterocycle and the P2-Cp-ligand are involved in several critical interactions with the backbone atoms in the S1’ and S2-subsites of HIV-1 protease.
Introduction
Advances in the treatment of HIV/AIDS with HIV-1 protease inhibitors in combination with reverse transcriptase inhibitors have been widely documented. The combination therapy, also known as highly active antiretroviral therapy (HAART), blocks critical viral replication at two different stages of the replication cycle.2 The HAART regimens have resulted in dramatic reduction of blood plasma viral load levels, increased CD4+ lymphocyte counts, improved life expectancy and significantly reduced HIV/AIDS-related mortality in the developed world.3 Despite these important advances, effective long-term suppression of HIV infection with HAART regimens is a complex issue in medicine for a number of reasons. These include drug side effects, poor penetration into protected HIV reservoir sites, poor oral bioavailability and interactions between drugs.4 Perhaps one of the most daunting problems in future management of HIV is the emergence of drug-resistant HIV-1 variants and the transmission of these viral strains.5,6 Thus, development of antiretroviral therapy with broad-spectrum activity and minimal drug side effects is critical for an effective management of current and future HIV/AIDS treatment. We recently reported the design and development of a number of exceedingly potent nonpeptidic HIV-1 protease inhibitors (PIs) 1–3.7–9 One of those PIs is darunavir (1, TMC-114), which was approved by the FDA in 2006 for treatment of HIV/AIDS patents who are harboring drug-resistant HIV and do not respond to other therapies.10 More recently, darunavir has received full approval for all HIV/AIDS patients.11
To combat drug resistance, our structure-based design strategies are to maximize the protease active-site interactions with the inhibitor and particularly to promote extensive hydrogen bonding with the protein backbone atoms.12 It is evident that active site backbone conformation of mutant proteases is only minimally distorted compared to that of the wild-type HIV-1 protease.13,14 Therefore, the ‘backbone binding’ strategy may be important to combat drug resistance.12 Using high resolution protein-ligand X-ray structures of 1 and 3-bound HIV-1 protease, we have shown that these PIs were engaged in extensive hydrogen bonding interactions with the backbone atoms throughout the active site cavity from the S2 to S2′ regions.9,15 To further enhance ‘backbone binding’ interactions, we became interested in designing an appropriately functionalized P1′-ligand that could interact with the backbone atoms, particularly with the Gly-27′ and Arg-8 in the S1′-subsite. This enhancement of ‘backbone binding’ interaction may lead to inhibitors with improved drug-resistance profiles. Herein, we report the design, synthesis and biological evaluation of a series of potent HIV-1 protease inhibitors that incorporated structure-based designed stereochemically defined lactam and oxazolidinone derivatives as the P1′-ligands in combination with the bis-THF or Cp-THF as the P2-ligands. Inhibitor 4 incorporating a (S)-5-aminomethyl-2-pyrrolidinone as the P1′-ligand and Cp-THF as the P2-ligand is the most potent PI in the series. Interestingly, this PI has retained full potency against a range of multi-drug-resistant HIV-1 variants. The protein-ligand X-ray structure of 4-bound HIV-1 protease revealed important molecular insight into the ligand-binding site interactions.
Chemistry
The optically active synthesis of the requisite 5-aminomethyl-2-pyrrolidinone for P1-ligands and their conversion to respective sulfonamide isostere is shown in Scheme 1. Commercially available 5-(S)-hydroxymethyl-2-pyrrolidinone 5 was reacted with tosyl chloride and triethylamine to provide the corresponding tosylate. Displacement of the resulting tosylate with sodium azide in DMF at 55 °C for 9 h provided the azide derivative in 92% yield over two steps. Catalytic hydrogenation of the azide over 10% Pd-C in ethyl acetate afforded optically active amine 6 in quantitative yield. 5-(R)-hydroxymethyl-2-pyrrolidinone (ent-5) was similarly converted to optically active amine ent-6 in comparable yield. Amine 6 was reacted with commercially available epoxide 7 in the presence of iPr2NEt (DIPEA) in 2-propanol at 70 °C for 36 h to provide epoxide opened product 8 in 85% yield.16 Amine 8 was converted to p-methoxybenezenesulfonamide derivative 9 by reaction with p-methoxybenzenesulfonyl chloride in the presence of aqueous NaHCO3 in quantitative yield. Treatment of amine 8 with p-nitrobenzenesulfonyl chloride afforded the corresponding nitrosulfonamide. Catalytic hydrogenation over 10% Pd-C gave the corresponding aniline derivative, which was reacted with benzyl chloroformate in the presence of pyridine to furnish Cbz-derivative 10 in 63% yield for 3 steps. Enantiomeric amine (ent-6) was converted to the respective methoxy and Cbz-derived 11 and 12 by analogous procedures.
Scheme 1.

Synthesis of lactam containing sulfonamide isosteres
The synthesis of various PIs incorporating methyl-pyrrolidinones as the P1′-ligand is shown in Scheme 2. Exposure of Boc-derivatives 9 and 10 to 30% CF3CO2H in CH2Cl2 at 23 °C for 40 min resulted in the respective amines 13 and 14. Alkoxycarbonylation of amine 13 with activated mixed carbonate 15,16 and 16,9 in the presence of Et3N in CH2Cl2 furnished inhibitors 17a and 17b in 98% and 87% yields, respectively.17 Alkoxycarbonylation of amine 14 with activated carbonates 15 and 16 afforded the corresponding Cbz-protected urethanes of 18a and 18b. Removal of the Cbz-group by catalytic hydrogenation over 10% Pd-C in ethyl acetate provided inhibitor 18a and 18b in 58% and 62% yields, respectively. Sulfonamide derivate 11 and 12 containing enantiomeric P1′-ligands were converted to inhibitors 19a,b and 20a,b by following analogous procedures.
Scheme 2.

Synthesis of lactam containing PIs
The synthesis of sulfonamide isosteres incorporating methyl oxazolidinone as the P1′-ligand is shown in Scheme 3. Optically active dimethyloxazolidines 21 and ent-21 were prepared by following the procedure described by Dondini and co-workers.18 Reduction of these azides by catalytic hydrogenation in methanol afforded the respective amine. Reaction of 21-derived amine with epoxide 7 in the presence of iPr2NEt in 2-propanol afforded amine 23 in 41% yield. Reaction of amine 22 with p-methoxybenzenesulfonyl chloride or p-nitrobenzenesulfonyl chloride as described previously afforded sulfonamide derivatives 23 and 24 in 80% and 92% yields, respectively. The isopropylidene functionality in 23 and 24 was converted to the corresponding oxazolidinone derivative in a three steps sequence involving: (1) treatment of 23 by a catalytic amount of p-toluenesulfonic acid (PTSA) in methanol resulted in the removal of the isopropylidene group; (2) reaction of the resulting Boc-amino alcohol with mesyl chloride in the presence of triethylamine to provide the corresponding mesylate and (3) treatment of the resulting mesylate with iPr2NEt in chloroform at reflux. This has provided oxazolidinone 25 in 45% yield over 3 steps. The nitrosulfonamide derivative 24 was similarly converted to the corresponding oxazolidinone. Catalytic hydrogenation of the resulting nitro derivative with 10% Pd-C in methanol provided aniline derivative 26 in 37% overall yield over 4 steps. Enantiomeric azide ent-21 was converted to oxazolidinone derivatives 27 and 28 by following analogous procedures.
Scheme 3.

Synthesis of sulfonamide isosteres with P1′-oxazolidinone
The synthesis of inhibitors containing oxazolidinone as P1′-ligand and bis-THF as the P2-ligand is shown in Scheme 4. Treatment of oxazoldinones 25–28 with 30% CF3CO2H in CH2Cl2 at 23 °C afforded the corresponding amines. Reaction of the resulting amines with activated mixed carbonate 15 in the presence of Et3N in CH2Cl2 afforded the target inhibitor 29–32 in excellent yields (80–90%). The structures of these inhibitors are shown in Table 1.
Scheme 4.

Synthesis of oxazolidinone-derived PIs.
Table 1.
Enzymatic inhibitory activity of lactam and oxazolidinone containing inhibitors.
| Entry | Inhibitor | Ki (nM) | IC50(µM)a |
|---|---|---|---|
| 1 | 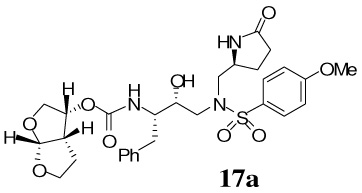 |
0.85±0.02 | 0.48±0.05 |
| 2 | 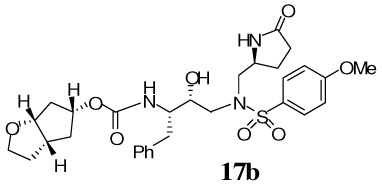 |
0.31±0.03 | 0.23±0.08 |
| 3 | 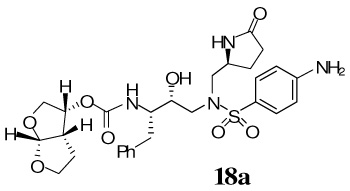 |
0.28±0.03 | >1 |
| 4 | 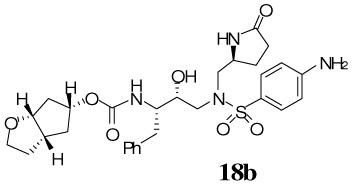 |
1.27±0.15 | >1 |
| 5 | 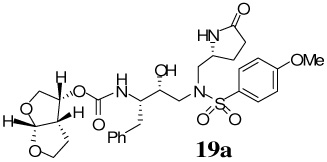 |
0.12±0.003 | 0.25±0.11 |
| 6 | 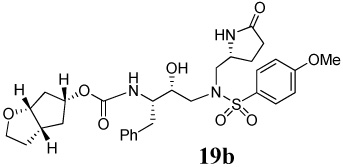 |
0.099±0.003 | 0.026±0.002 |
| 7 | 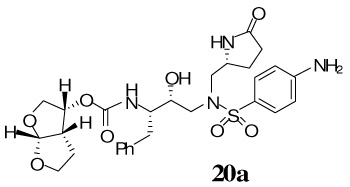 |
0.85±0.2 | >1 |
| 8 | 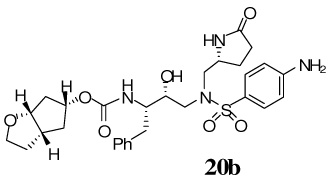 |
0.31±0.03 | 0.60±0.24 |
| 9 | 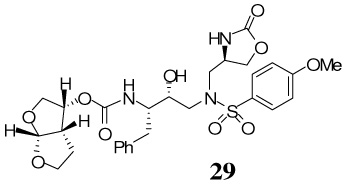 |
0.28±0.03 | 0.48±0.17 |
| 10 | 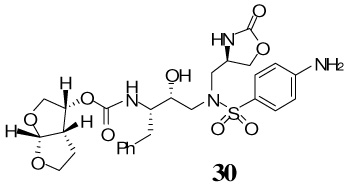 |
0.31±0.03 | >1 |
| 11 | 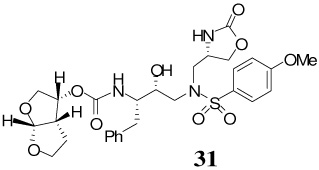 |
0.035±0.01 | 0.31±0.21 |
| 12 | 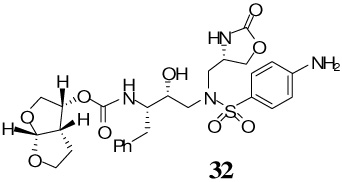 |
0.24±0.03 | >1 |
Values are means of at least two experiments.
Human T-lymphoid (MT-2) cells (2 × 103) were exposed to 100 TCID50S of HIV-Ilai and cultured in the presence of each PI, and IC50 values were determined using the MTT assay. The IC50 values of amprenavir (APV), saquinavir (SQV), and indinavir (IDV) were 0.03 µM, 0.015 µM, and 0.03 µM, respectively.
Results and Discussion
Our examination of the X-ray structure of 1-bound HIV-1 protease and its respective modeling initially suggested that a methyl-2-pyrrolidinone may interact well with residues in the S1′-site.15 As shown in Table 1, our first set of inhibitors contain a (R)-hydroxyethylamine sulfonamide isostere with either the bis-THF or Cp-THF as the P2-ligand and p-methoxysulfonamide or p-aminosulfonamide as the P2′-ligand. The enzyme inhibitory potency of these PIs was evaluated according to the procedure reported by Toth and Marshall.19 Inhibitor 17a with (S)-methyl-2-pyrrolidinone displayed an enzyme Ki of 1 nM. Inhibitor 17b with a Cp-THF showed a 3-fold improvement of potency. Antiviral activity of these inhibitors was determined in MT-2 human T-lymphoid cells exposed to HIV-1lai. Interestingly, both inhibitors have shown dramatic reduction in antiviral activity. Inhibitors 17a and 17b have shown IC50 values of 0.48 µM, and 0.23 µM, respectively. However, these inhibitors are significantly less potent compared to inhibitors with an isobutyl group as the P1′-ligand. Incorporation of p-aminosulfonamide (PIs 18a and 18b) as the P2′-ligand led to a drop in enzyme inhibitory as well as antiviral potency. Inhibitor 19a containing (R)-methyl-2-pyrrolidinone as the P1′-ligand has shown 10-fold enhancement of enzyme Ki over the (S)-isomer 17a. It showed a slight improvement in antiviral activity compared to inhibitor 17a. Inhibitor 19b with (R)-methyl-2-pyrrolidinone as the P1′-ligand and Cp-THF as the P2-ligand resulted in the most potent inhibitor in the series. It has shown an enzymatic Ki of 99 pM and a 10-fold improvement (IC50 = 0.026 µM) in antiviral activity relative to epimeric (S)-pyrrolidinone derivative 17b, suggesting an important role for the P1′-ring stereochemistry. Indeed, an X-ray structure of 19b–bound HIV-1 protease revealed that the pyrrolidinone carbonyl as well as the NH functionalities were positioned to hydrogen bond with residues in the S1′-site. Interestingly, the combination of P1′-methylpyrrolidinone and polar P2′-p-aminosulfonamide led to PIs, with subnanomolar enzyme activity. However, antiviral activity was reduced drastically. In PIs 29–32, we have incorporated both (S)- and (R)-oxazolidinone derivatives as substitutes for the respective pyrrolidinone isomers. As can be seen, oxazolidinone derivatives 29–32 have shown subnanomolar enzyme inhibitory potency. Inhibitors with p-methoxysulfonamide as the P2′-ligand displayed comparable antiviral activity relative to pyrrolidinone derivatives. Consistent with stereochemical preference, the (R)-oxazolidinone with p-methoxysulfonamide has shown better enzyme Ki values. However, the antiviral activity of these compounds is very similar. In general, both pyrrolidinone and oxazolidinone functionalities appear to be nicely accommodated in the Sl′-site.
While inhibitor 31 is very potent in enzyme inhibitory assay, the significant reduction of antiviral potency is possibly due to poor cellular permeability of this polar functionality. Inhibitor 19b appeared to be most potent among the series of inhibitors examined. It exhibited comparable antiviral activity with the FDA approved PIs amprenavir, saquinavir, and indinavir in the same assay.
Inhibitor 19b was subsequently examined for its activity against a clinical wild-type X4-HIV-1 isolate (HIV-lERS104pre) along with various multidrug-resistant clinical X4- and R5- HIV-1 isolates using PBMCs as target cells.8b As can be seen in Table 2, the potency of 19b against HIV-lERio4pre (IC50 = 28 nM) was comparable to FDA approved PIs, indinavir, amprenavir and lopinavir with IC50 values 28, 25, and 30 nM, respectively. Darunavir on the other hand, is nearly 10-fold more potent (IC50 = 3.6 nM) than 19b and the above mentioned PIs. Interestingly, of all the PIs tested, indinavir was least able to suppress the replication of the multi-drug-resistant clinical isolate examined (HIV-1mdr/mm, HIV-1mdr/tm, HIV-1mdr/cand HIV-1mdr/g) with IC50 values greater than 1 µM. Both amprenavir and lopinavir displayed 10-fold or greater reduction in potency except against HIV-1MDR/G, where lopinavir showed a 5-fold reduction in potency. A more detailed virologic study using inhibitor 19b will be published elsewhere.20 Darunavir has maintained impressive activity against all the multi-drug-resistant variant. Inhibitor 19b, while less potent than darunavir, maintained near full potency against multi-drug-resistant clinical isolates examined. This impressive drug-resistance property of 19b is possibly due to its extensive interactions, particularly its ability to make extensive hydrogen-bonds throughout the active site of the protease’s backbone. Furthermore, inhibitor 19b blocked the infection and replication of each of the HIV-1nl4-3 variants exposed to and selected by up to 5 µM of saquinavir, amprenavir, indinavir, nelfinavir, or ritonavir and a 1 µM of lopinavir or atazanavir with EC50 values ranging 0.036 – 0.14 µM.20
Table 1.
Anti-HIV activity of 19b against selected clinical isolates highly resistant to multiple protease inhibitors
| Virus | Phenotype | EC50 (µM) | ||||
|---|---|---|---|---|---|---|
| IDV | APV | LPV | DRV | 19b | ||
| HIV-lERsi04Pre (wild-type) | X4 | 0.028 ± 0.005 | 0.025 ± 0.006 | 0.03 ± 0.001 | 0.0036 ± 0.0002 | 0.028 ± 0.004 |
| HIV-Itm(MDR) | X4 | >1 (>36) | 0.25 ± 0.02 (10) | 0.73 ± 0.53 (24) | 0.0036 ± 0.0002 (1) | 0.029 ± 0.004 (1) |
| HIV-ITm(MDR) | R5 | >1 (>36) | 0.32 ±0.03 (13) | 0.72 ±0.31 (24) | 0.019 ± 0.009 (5) | 0.042 ± 0.002 (2) |
| HIV-1C (MDR) | X4 | >1 (>36) | 0.35 ± 0.03 (14) | 0.32 ± 0.01 (11) | 0.015 ± 0.001 (4) | 0.023 ± 0.007 (1) |
| HIV-1G (MDR) | X4 | 0.29 ± 0.07 (10) | 0.33 ± 0.16 (13) | 0.14 ± 0.01 (5) | 0.014 ± 0.006 (4) | 0.027 ± 0.001 (1) |
Amino acid subrstitutions identified in the protease-encoding region of HIV-1ERS104preHIV-1TM, HIV-1MM, HIV-1C and HIV-1G as compared to the consensus B sequence cited from the Los Alamos data base include L63P, L10I/K14R/R41K/M46L/I54V/L63P/A71V/V82A/L90M/I93L, L10I/K43T/M46L/I54V/L63P/A71V/V82A/L90M/Q92K, L10I/I15V/K20R/L24I/M36I/M46L/I54V/I62V/L63P/K70Q/V82A/L89M, and L10I/V11I/T12E/I15V/L19I/R41K/M46L/-L63P/A71T/V82A/L90M, respectively. The EC50 values were determined by employing PHA-PBM as target cells and the inhibition of p24 Gag protein production as an endpoint. All values were determined in duplicate or triplicate, and those shown derived from the results of three independent experiments. Numbers in parentheses represent fold changes of EC50 values against each isolate compared to EC50 values against HIV-1ERS104pre. MDR, multi-drug-resistant.
X-Ray Crystallography
The binding mode of inhibitor 19b was determined from the X-ray crystal structure of its complex with wild-type HIV-1 protease. The crystal structure was solved and refined at 1.29 Å resolution with an R factor of 14.1 %. In this high resolution structure, the inhibitor was bound to the HIV-1 protease active site in two orientations with the relative occupancy of 0.8/0.2. The protease dimer comprises residues 1–99 and 1’-99’ of the two subunits, and the inhibitor binding site is formed by both subunits. The P1′-pyrrolidine ring also showed two alternate conformations with equal occupancy and related by about 18° rotation around the C12-C13 bond. A stereoview of the major conformation is shown in Figure 2 [only one conformation is shown for P1’]. As shown, extensive interactions from P2 to P2′ were observed between the inhibitor and protease active site, most notably favorable polar interactions including hydrogen bonds, weaker C-H… O and C-H… pi interactions. The isostere hydroxyl group forms asymmetric hydrogen bonds to the carboxylate oxygen atoms of the catalytic Asp25 and Asp25′ with distances of 2.4 to 3.3 Å. Also, four direct hydrogen bonds are formed between oxygens or nitrogens of the inhibitor atoms and the protease backbone atoms. These include: cyclic ether oxygen of the P2-Cp-THF and the Asp-29 NH; the urethane NH with the carbonyl oxygen of Gly-27; P2′-methoxy oxygen and Asp-30′. One conformation of the P1′-pyrrolidinone formed a hydrogen bond between the NH and the carbonyl oxygen of Gly-27′ and a water-mediated hydrogen bond between the P1′- pyrrolidinone carbonyl and the side chain of Arg-8. The other conformation of the P1’ group formed hydrophobic and C-H…O interactions with Pro-81’ and Val-82’, as shown in Figure 3. Also, there exists a tetracoordinated water-mediated interaction where the amides of Ile50 and 50′ donate hydrogen bonds and the inhibitor’s urethane carbonyl and one of the sulfonamide oxygen accept hydrogen bonds from the water molecule. These interactions are conserved in a majority of other HIV-1 protease complexes with inhibitors,21 or substrate analogs.22
Figure 2.

A stereoview of the major conformation of the X-ray structure of inhibitor 19b–bound HIV-1 protease.
Figure 3.

Protease interactions with the two alternate conformations of the inhibitor pyrrolidine ring. The inhibitor is in green bonds with thick bonds for the major and thin bonds for the minor conformations of the pyrrolidine ring. Hydrogen bonds are shown in dotted lines. Distances between donor and acceptor atoms are shown in Å.
The weaker polar interactions such as C-H…O and water–pi interactions can be analyzed accurately in this high resolution structure.23,24 These interactions are important for inhibitor-protease binding and must be considered in the design of inhibitors. The C-H…O interactions of the inhibitor with the carbonyl oxygens of Gly-48, Gly-48′ and Gly-27′ mimic the conserved hydrogen bonds obrserved peptide analog structures21,22. A conserved water-pi interaction is observed between the P2′ aromatic ring of the inhibitor and the amide of Asp29′, which is similar to the interaction with darunavir and other structure-based designed PIs from our laboratories.25,26 The bigger polar P1′ group of the 2-pyrrolidinone ring in inhibitor 19b instead of the isobutyl group in PI’s 1 and 2 introduces a new direct hydrogen bond with the backbone of HIV-1 protease and one new water-mediated hydrogen bond between the inhibitor and the side chain residue of the protease. The two alternate conformations of the P1’ group with occupancy of 0.5/0.5 provide more flexible binding within the S1’ subsite, which is likely to enhance the inhibition of resistant proteases.
As mentioned earlier, inhibitor 19b maintained near full potency against multi-drug-resistant clinical isolates examined. On the other hand, 19b is less potent than darunavir possibly due to the bigger and less optimum size of the P1′-ligand. The design strategy of incorporating new polar interactions with conserved backbone regions of the protease warrants further investigation in light of the current molecular insight into these ligand-binding site interactions.
Conclusion
We have designed a number of HIV-1 protease inhibitors with methyl-2-pyrrolidinone and methyl oxazolidinone as the P1′-ligand to enhance hydrogen bonding with the protein backbone atoms in the S1′-subsite. The ligands were synthesized in enantiomerically pure forms and a series of inhibitors were prepared and evaluated in combination with P2-bis-THF and P2-Cp-THF ligands. In general, these inhibitors exhibited enzyme inhibitory activity lower than the corresponding inhibitors with a P1′-isobutyl group. Our SAR studies indicated the importance of ligand stereochemistry and also preference for the P2-Cp-THF ligand. Interestingly, the polar P1′-ligand influenced their cellular properties. The inhibitors exhibited reduction in antiviral activity possibly due to changes in the molecule’s overall lipophilicity. Our investigation resulted in the identification of inhibitor 19b which has displayed similar antiviral potency as the other FDA approved inhibitors such as indinavir, lopinavir and amprenavir. Inhibitor 19b, however, is nearly 10-fold less potent than darunavir. Of particular importance, 19b has maintained full potency against the examined panel of multidrug-resistant HIV-1 variants. A high resolution X-ray structure of 19b–bound HIV-1 protease revealed a new hydrogen bonding of the P1′-pyrrolidinone NH with the backbone carboxyl of Gly27′. Also, there is a water mediated hydrogen bond with the pyrrolidinone carboxyl and Arg8′ side chain. Furthermore, the P1’-pyrrolidinone showed two alternate conformations that filled the S1’ subsite. These new interactions and the conformational flexibility most likely contributed to its impressive properties against multidrug-resistant clinical variants. Further investigations including optimization of ligand-binding properties are in progress.
Experimental Section
General
All moisture sensitive reactions were carried out under nitrogen or argon atmosphere. Anhydrous solvents were obtained as follows: THF, diethyl ether and benzene, distilled from sodium and benzophenone; dichloromethane, pyridine, triethylamine, and diisopropylethylamine, distilled from CaH2. All other solvents were HPLC grade. Silica gel column chromatography was performed with Whatman 240–400 mesh silica gel under low pressure. TLC was carried out with E. Merck silica gel 60-F-254 plates. 1H and 13C NMR spectra were recorded on Varian Mercury 300 and Bruker Avance 400 and 500 spectrometers. Optical rotations were measured using a Perkin-Elmer 341 polarimeter.
(S)-5-(Aminomethyl)-2-pyrrolidinone 6
To a stirred solution of (S)-5-(hydroxymethyl)-2-pyrrolidinone 5 (300 mg, 2.61 mmol) and p-toluenesulfonyl chloride (646 mg, 3.34 mmol) in CH2Cl2 (6 mL) at 0 ºC was added DMAP (64 mg, 0.52 mmol) and Et3N (472 µL, 3.34 mmol). The resulting mixture was allowed to warm to 23 ºC and stirred for 12 h. The reaction was then quenched with 7 mL of water and the aqueous layer was extracted with CH2Cl2. The combined organic extracts were washed with 1N HCl and dried over anhydrous Na2SO4. Removal of solvent under reduced pressure, followed by flash chromatography purification (100% EtOAc as the eluent) yielded (S)-toluenesulfonate (0.7 g, 93%) as a yellowish solid. Rf = 0.50 (5% MeOH in CHCl3); 1H-NMR (400 MHz, CDCl3) δ 1.75–1.80 (m, 1H), 2.19–2.35 (m, 3H), 2.44 (s, 3H), 3.85–3.92 (m, 2H), 4.00–4.03 (m, 1H), 6.49 (s, 1H), 7.35 (d, 2H, J = 8.08 Hz), 7.77 (d, 2H, J = 8.17 Hz ); 13C-NMR (100 MHz, CDCl3) δ21.68, 22.77, 29.27, 52.59, 71.97, 121.92, 130.08, 132.32, 145.37, 178.20.
To a stirred solution of the above tosylate (638 mg, 2.37 mmol) in DMF (20 mL) was added NaN3 (462 mg, 2.37 mmol). The resulting solution was stirred at 55 ºC for 9 h. Removal of solvent under reduced pressure, followed by flash chromatography purification (6% MeOH in CHCl3 as the eluent) provided the (S)-azidopyrrolidinone (330 mg, 99%) as a yellow oil. Rf = 0.50 (10% MeOH in CHCl3); 1H-NMR (400 MHz, CDCl3) δ 1.76–1.84 (m, 1H), 2.18–2.44 (m, 3H), 3.28 (dd, 1H, J = 6.5, 12.25 Hz), 3.43 (dd, 1H, J = 4.6, 12.3 Hz), 3.77–3.83 (m, 1H), 7.38 (s, 1H); 13C-NMR (100 MHz, CDCl3) δ 23.97, 29.79, 53.66, 55.89, 178.73.
To a solution of the above azide (125 mg, 0.89 mmol) in EtOAc (10 mL) was added Pd/C (15 mg). The mixture was stirred at 23 ºC under a hydrogen filled balloon for 4 h, then filtered over Celite and the filter cake was washed with EtOAc and MeOH. Removal of solvent under reduced pressure, followed by flash chromatography purification (5% MeOH in CHCl3 as the eluent) afforded the corresponding (S)-amine 6 (105 mg, quantitive) as a yellow oil. Rf= 0.05 (20% MeOH in CHCl3); 1H-NMR (400 MHz, CDCl3) δ 1.41 (brs 2H), 1.56–1.65 (m, 1H), 2.01–2.12 (m, 1H), 2.19–2.24 (m, 2H), 2.52 (dd, 1H, J = 7.5, 12.8 Hz), 2.69 (dd, 1H, J = 4.3, 12.9 Hz), 3.50–3.57 (m, 1H), 7.90 (brs, 1H); 13C-NMR (100 MHz, CDCl3) δ 24.07, 30.29, 47.34, 57.10, 179.03.
tert-Butyl(2R,3R)-3-hydroxy-4-[((S)-5-oxopyrrolidin-2-yl)methylamino]-1-phenylbutan-2-yl-carbamate 8
To a solution of amine 6 (107 mg, 0.94 mmol) in iPrOH (5 mL) were added tert-butyl-[S-(R,R)]-(-)-(1-oxiranyl-2-phenylethyl)carbamate 7 (62 mg, 0.23 mmol) and iPr2EtN (204 µL, 1.2 mmol). The resultant mixture was stirred at 65 ºC for 18 h and then concentrated under reduced pressure. Flash chromatography purification (15% MeOH in CHCl3 as the eluent) yielded title compound 8 (76 mg, 85%). Rf= 0.47 (25% MeOH in CHCl3); 1H-NMR (400 MHz, CDCl3) δ 1.30 (s, 9H), 1.62–1.71 (m, 1H), 2.13–2.18 (m, 1H), 2.30–2.32 (m, 2 H), 2.52 (d, 1H, J = 8.86 Hz), 2.64–2.73 (m, 4H), 2.96 (d, 1H, J = 9.8 Hz), 3.54 (s, 1H), 3.72–3.75 (m, 4H) , 4.99 (brs, 1H), 7.15–7.26 (m, 5H ), 8.02 (s, 1H); 13C-NMR (100 MHz, CDCl3) δ 24.65, 28.32, 30.29, 36.37, 51.74, 54.09, 54.41, 55.30, 71.53, 79.26, 126.20, 128.29, 129.47, 138.17, 155.96, 178.98; LRMS-ESI (m/z) [M + Na]+ 400.
tert-Butyl(2R,3R)-3-hydroxy-4-(4-methoxy-N -(((S)-5-oxopyrrolidin-2- yl)methyl)phenylsulfonamido)-1-phenylbutan-2-ylcarbamate 9
To a stirred solution of amine 8 (22 mg, 0.06 mmol) in CH2Cl2 (3 mL) and aqueous sat. NaHCO3 (3 mL) was added 4-methoxybenzenesulfonyl chloride (35.6 mg, 0.17 mmol). This reaction mixture was stirred for 12 h followed by extraction of the aqueous layer with CH2Cl2; the combined organic extracts were dried over anhydrous Na2SO4. Removal of solvent under reduced pressure, followed by flash chromatography purification (6% MeOH in CHCl3 as the eluent) provided compound 9 (31 mg, quantitative). Rf= 0.40 (10% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.28 (s, 9H), 1.54–1.62 (m, 1H), 2.14–2.21 (m, 1H), 2.32–2.35 (m, 2H), 2.68–2.75 (m, 2H), 2.72 (s, 3H), 2.81–2.88 (m, 2H), 2.97–3.03 (m, 3H), 3.64–3.72 (m, 1H), 4.01–4.05 (m, 1H), 5.06 (d, 1H, J = 8.9 Hz), 6.93 (d, 2H, J = 8.6 Hz), 7.16–7.19 (m, 3H), 7.27–7.28 (m, 3H), 7.61 (d, 2H, J = 8.5 Hz); 13C-NMR (125 MHz, CDCl3) δ23.94, 28.16, 29.62, 29.93, 36.10, 53.40, 53.99, 54.57, 55.56, 56.01, 71.96, 79.51, 114.38, 126.31, 126.37, 129.37, 129.52, 137.79, 155.93, 163.15, 178.42; LRMS-ESI (m/z) [M + Na]+ 570.
tert-Butyl(2R,3R)-4-(4-Cbz-amino-N-(((S)-5-oxopyrrolidin-2-yl)methyl)phenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate 10
To a stirred solution of amine 8 (93.6 mg, 0.25 mmol) in CH2Cl2 (10 mL) and aqueous sat. NaHCO3 (10 mL) was added 4-nitrobenzenesulfonyl chloride (60 mg, 0.27 mmol). This reaction mixture was stirred for 7 h followed by extraction of the aqueous layer with CH2Cl2; the combined organic extracts were dried over anhydrous Na2SO4. Removal of solvent under reduced pressure, followed by flash chromatography purification (dry transfer, 8% MeOH in CHCl3 as the eluent) provided (S)-nitrosulfone (112 mg, 80%) as a yellowish solid. Rf= 0.56 (10% MeOH in CHCl3).
The above nitrosulfone (103 mg, 0.18 mmol) was disolved in EtOAc (20 mL) and Pd/C (11 mg) was added. The mixture was stirred under a hydrogen filled balloon for 8 h at 23 ºC. It was then filtered over Celite and the filter cake was washed with EtOAc and MeOH. Removal of solvent under reduced pressure, followed by flash chromatography purification (2.5% MeOH in CHCl3 as the eluent) afforded the corresponding (S)-amine (77 mg, 79%) as a white solid. Rf= 0.26 (5% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) 1.35 (s, 9H), 1.59–1.57 (m, 1H), 2.15–2.23 (m, 1H), 2.29–2.42 (m, 2H), 2.82–2.87 (m, 3H), 3.02 (dd, 1H, J = 4.75, 14.0), 3.13 (dd, 1H, J = 10, 13.2 Hz), 3.30 (dd, 2H, J = 1.8, 14.4 Hz), 3.73–3.82 (m, 1H), 3.90–3.95 (m, 1H), 3.99 (d, 1H, J = 6.2 Hz), 4.72 (d, 1H, J = 8.2 Hz), 6.72 (d, 2H, J = 7.9 Hz), 7.20–7.32 (m, 5H), 7.37 (s, 1H), 7.57 (d, 2H, J = 8.2 Hz); 13C-NMR (125 MHz, CDCl3) δ24.35, 28.69, 30.09, 36.58, 53.67, 54.42, 54.92, 56.66, 72.63, 79.85, 114.48, 125.07, 126.71, 128.78, 129.92, 130.09, 138.38, 151.58.
To a stirred solution of the above amine (33.1 mg, 0.0621 mmol) in CH2Cl2 (3 mL) was added pyridine (30 µL, 0.372 mmol) and benzyl chloroformate (20 µL, 0.137 mmol). This reaction mixture was stirred for 3 h, then quenched with 5 drops of benzyl amine, followed by removal of solvent under reduced pressure. Column chromatography over silica gel (2.5% MeOH in CHCl3 as the eluent) provided (S)-Cbz-amine 10 (41 mg, 99%) as a white solid. Rf= 0.37 (5% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.56 (brs, 1H), 2.12–2.18 (m, 1H), 2.26–2.41 (m, 2H), 2.71–2.78 (m, 2H), 2.79–89 (m, 1H), 3.02 (dd, 1H, J = 8.2, 18.0 Hz), 3.19–3.26 (m, 1H), 3.38 (d, 1H, J = 14.4 Hz), 3.77 (brs, 1H), 3.94–3.99 (m, 2H), 4.73 (d, 1H, J = 8.5 Hz), 5.21 (s, 2H), 7.19–7.22 (m, 3H), 7.25–7.29 (m, 3H), 7.31–7.39 (m, 4H), 7.57 (d, 2H, J = 7.6 Hz), 7.69 (d, 2H, J = 7.7 Hz); 13C-NMR (125 MHz, CDCl3) δ 24.16, 28.74, 30.12, 36.65, 53.38, 54.34, 54.89, 56.89, 67.79, 72.64, 80.01, 118.63, 126.83, 128.77, 128.85, 128.93, 129.08, 129.43, 129.99, 131.66, 136.03, 138.13, 143.07, 153.37, 156.33, 178.99; LRMS-ESI (m/z) [M + Na]+ 689.
tert-Butyl(2R,3R)-3-hydroxy-4-(4-methoxy-N-(((R)-5-oxopyrrolidin-2- yl)methyl)phenylsulfonamido)-1-phenylbutan-2-ylcarbamate 11
To a stirred solution of (R)-5-(hydroxymethyl)-2-pyrrolidinone ent-5 (500 mg, 4.34 mmol) and p-toluenesulfonyl chloride (1.08 g, 5.6 mmol) CH2Cl2 (10 mL) at 0 ºC was added DMAP (106 mg, 0.87 mmol) and Et3N (780 µL, 5.6 mmol). The resulting mixture was allowed to warm to 23 ºC and stirred for 12 h. The reaction was then quenched with 10 mL of water and the aqueous layer was extracted with CH2Cl2. The combined organic extracts were washed with 1N HCl and dried over anhydrous Na2SO4. Removal of solvent under reduced pressure, followed by flash chromatography (2.5% MeOH in CHCl3 as the eluent) yielded the (R)-toluenesulfonate (1.8 g, 93%) as a yellowish solid. Rf= 0.50 (5% MeOH in CHCl3); 1H-NMR (400 MHz, CDCl3) δ 1.70–1.79 (m, 1H), 2.13–2.35 (m, 3H), 2.42 (s, 3H), 3.84–3.89 (m, 2H), 3.96–3.04 (m, 1H), 6.76 (s, 1H), 7.33 (d, 2H, J = 8.04 Hz), 7.76 (d, 2H, J = 8.2 Hz ); 13C-NMR (100 MHz, CDCl3) δ 21.67, 22.76, 29.33, 52.61, 71.95, 127.91, 130.06, 132.30, 145.33, 178.11.
To a stirred solution of the above toluenesulfonate (1.08 g, 4.03 mmol) in DMF (30 mL) was added NaN3 (1.31 g, 20.2 mmol). The resulting solution was stirred at 55 ºC for 12 h. Solvent was then removed under reduced pressure, followed by flash chromatography purification (6% MeOH in CHCl3 as the eluent) provided the (R)-azidopyrrolidinone (558 mg, 99%) as a yellow oil. Rf = 0.44 (5% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.72–1.79 (m, 1H), 2.13–2.37 (m, 3H), 3.22 (dd, 1H, J = 6.3, 12.3 Hz), 3.37 (dd, 1H, J = 4.7, 12.3 Hz), 3.72–3.78 (m, 1H), 7.69 (s, 1H); 13C-NMR (125 MHz, CDCl3) δ 24.32, 30.26, 54.13, 56.19, 179.32.
To a solution of the above azide (528 mg, 3.77 mmol) in EtOAc (35 mL) was added Pd/C (40 mg). The mixture was stirred at 23 ºC under a hydrogenfilled balloon for 4 h, then filtered over Celite and the filter cake was washed with EtOAc and MeOH. Removal of solvent under reduced pressure, followed by flash chromatography purification (5% MeOH in CHCl3 as the eluent) afforded the (R)-amine ent-6 (257 mg, 95%) as a yellow oil. Rf= 0.05 (20% MeOH in CHCl3); 1H-NMR (400 MHz, CDCl3) δ 1.59–1.69 (m, 1H), 2.06–2.15 (m, 1H), 2.23–2.30 (m, 4H), 2.57 (dd, 1H, J = 7.6, 12.8 Hz), 2.74 (dd, 1H, J = 4.1, 12.9 Hz), 3.56–3.65 (m, 1H), 7.80 (brs, 1H); 13C-NMR (100 MHz, CDCl3) δ24.12, 30.23, 47.11, 56.84, 179.04.
To a solution of amine ent-6 (430 mg, 3.76 mmol) in iPrOH (20 mL) were added tert-butyl-[S-(R,R)]-(-)-(1-oxiranyl-2-phenylethyl)carbamate 7 (250 mg, 0.94 mmol) and iPr2EtN (1.5 mL, 8.6 mmol). The resultant mixture was stirred at 65 ºC for 36 h and then concentrated under reduced pressure. Flash chromatography purification (10% MeOH in CHCl3 as the eluent) yielded the (R)-hydroxyamine (8R) (300 mg, 84%). Rf= 0.33 (20% MeOH in CHCl3); 1H-NMR (400 MHz, CDCl3) δ1.33 (s, 9H), 1.64–1.73 (m, 1H), 2.13–2.22 (m, 1H), 2.28–2.34 (m, 2 H), 2.54 (dd, 1H, J = 9.0, 11.8 Hz), 2.65 (dd, 1H, J = 7.2, 13.2 Hz), 2.73–2.85 (m, 4H), 2.94 (dd, 1H, J = 4.4, 14.0 Hz), 3.43 (s, 1H), 3.51–3.60 (m, 1H), 3.70–3.76 (m, 1H), 3.79–3.84 (m, 1H), 5.02 (d, 1H, J = 8.9 Hz), 7.16–7.27 (m, 5H ), 7.94 (s, 1H); 13C-NMR (100 MHz, CDCl3) δ 24.64, 28.32, 30.28, 36.37, 52.14, 54.29, 54.69, 55.46, 71.62, 79.26, 126.21, 128.30, 129.47, 138.13, 155.93, 178.99; LRMS-ESI (m/z) [M + Na]+ 400.
To a stirred solution of above (R)-hydroxyamine (8R) (40 mg, 0.105 mmol) in CH2Cl2 (4 mL) and aqueous sat. NaHCO3 (4 mL) was added 4-methoxybenzenesulfonyl chloride (66 mg, 0.318 mmol). This reaction mixture was stirred for 12 h followed by extraction of the aqueous layer with CH2Cl2; the combined organic extracts were dried over anhydrous Na2SO4. Removal of solvent under reduced pressure, followed by flash chromatography purification (4% MeOH in CHCl3 as the eluent) provided compound 11 (54 mg, 93%). Rf= 0.40 (10% MeOH in CHCl3); 1H-NMR (400 MHz, CDCl3) δ 1.32 (s, 9H), 1.64–1.68 (m, 1H), 2.17–2.21 (m, 2H), 2.34–2.40 (m, 2H), 2.76–2.84 (m, 1H), 2.91–3.06 (m, 3H), 3.16–3.29 (m, 2H), 3.75–3.80 (m, 1H), 3.84 (s, 3H), 3.96–4.02 (m, 2H), 4.99 (d, 1H, J = 8.7 Hz), 6.95 (d, 2H, J = 8.8 Hz), 7.16–7.28 (m, 5H), 7.68 (d, 2H, J = 8.8 Hz), 7.93 (s, 1H); 13C-NMR (100 MHz, CDCl3) δ 24.41, 28.29, 29.99, 35.78, 35.46, 54.71, 55.21, 55.63, 55.88, 57.89, 73.94, 79.50, 114.43, 126.29,128.37, 129.31, 129.51, 138.05, 155.91, 163.12, 178.64; LRMS-ESI (m/z) [M + Na]+ 670.
tert-Butyl(2R,3R)-4-(4-Cbz-amino-N-(((R)-5-oxopyrrolidin-2-yl)methyl)phenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate 12
To a stirred solution of (R)-hydroxyamine (8R) (116 mg, 0.3 mmol) in CH2Cl2 (10 mL) and aqueous sat. NaHCO3 (10 mL) was added 4-nitrobenzenesulfonyl chloride (74 mg, 0.33 mmol). This reaction mixture was stirred for 12 h followed by extraction of the aqueous layer with CH2Cl2; the combined organic extracts were dried over anhydrous Na2SO4. Removal of solvent under reduced pressure, followed by flash chromatography purification (dry transfer, 5% MeOH in CHCl3 as the eluent) provided the (R)-nitrosulfone (164 mg, 96%) as a yellowish solid. Rf= 0.56 (10% MeOH in CHCl3).
The above nitrosulfone (154 mg, 0.27 mmol) was redisolved in EtOAc (25 mL) and treated with Pd/C (16 mg) under argon. Argon was then replaced with a hydrogenfilled balloon and the reaction was allowed to stir for 12 h at 23 ºC. It was then filtered over Celite and the filter cake was washed with EtOAc and MeOH. Removal of solvent under reduced pressure, followed by flash chromatography purification (6% MeOH in CHCl3 as the eluent) afforded the corresponding (R)-aniline (123 mg, 83%) as an amorphous solid. Rf= 0.45 (10% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.31 (s, 9H), 1.58–1.64 (m, 1H), 2.15–2.21 (m, 1H), 2.29 (t, 2H, J = 8.2 Hz), 2.73–2.86 (m, 3H), 2.99 (dd, 1H, J = 4.4, 13.9 Hz), 3.23 (d, 1H, J = 13.8Hz), 3.30 (d, 1H, J = 14.8 Hz), 3.74 (brs, 1H), 3.92 (brs, 1H), 3.99 (d, 1H, J = 5.7), 4.31 (s, 1H), 5.01 (d, 1H, J = 9.1 Hz), 6.63 (d, 2H, J = 8.5 Hz), 7.16–7.21 (m, 3H), 7.24–7.27 (m, 2H), 7.48 (d, 2H, J = 8.5 Hz); 13C-NMR (125 MHz, CDCl3) δ24.35, 28.16, 29.83, 35.59, 54.66, 55.09, 55.50, 57.71, 73.83, 79.51, 113.98, 125.09, 126.23, 128.28, 129.34, 129.41, 137.83, 151.02, 155.96, 178.25; LRMS-ESI (m/z) [M + Na]+ 555.
To a stirred solution of the above (R)-aniline (101 mg, 0.19 mmol) in CH2Cl2 (15 mL) was added pyridine (34 µL, 0.41 mmol) and benzyl chloroformate (60 µL, 0.41 mmol). This reaction mixture was stirred for 1.5 h, then quenched with 3 drops of benzyl amine, followed by removal of solvent under reduced pressure. Column chromatography over silica gel (6% MeOH in CHCl3 as the eluent) provided the (R)-Cbz-amine 12 (120 mg, 95%) as a white solid. Rf= 0.35 (10% MeOH in CHCl3);1H-NMR (500 MHz, CDCl3) δ 1.29 (s, 9H), 1.55–1.65 (m, 1H), 2.15–2.23 (m, 1H), 2.26–2.31 (m, 2H), 2.60–2.75 (m, 3H), 2.97 (dd, 1H, J = 8.2, 18.1 Hz), 3.29 (d, 1H, J = 17.7 Hz), 3.36 (dd, 1H, J = 2.4, 14.9 Hz), 3.64 (s, 1H), 3.88–3.92 (m, 1H), 3.98–4.02 (m, 1H), 5.12 (d, 1H, J = 9.0 Hz), 5.17 (s, 2H), 7.14–7.19 (m, 3H), 7.21–7.28 (m, 3H), 7.30–7.37 (m, 4H), 7.54 (d, 2H, J = 8.6 Hz) 7.63 (d, 2H, J = 8.8); 13C-NMR (125 MHz, CDCl3) δ 24.82, 28.61, 30.28, 36.06, 55.14, 55.85, 55.98, 58.25, 67.65, 74.40, 80.08, 118.66, 126.76, 128.63, 128.77, 128.86, 129.03, 129.06, 129.77, 130.32, 131.28, 136.11, 138.25, 143.43, 153.68, 178.97; LRMS-ESI (m/z) [M + Na]+ 689.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl(2S,3R)-3-hydroxy-4-(4-methoxy-N-(((S)-5-oxopyrrolidin-2-yl)methyl)phenylsulfonamido)-1-phenylbutan-2-yl-carbamate 17a
A solution of compound 9 (10 mg, 0.02 mmol) in 30% trifluoroacetic acid (in CH2Cl2, 3 mL) was stirred 23 ºC for 40 min, then concentrated under reduced pressure to give the crude amine 13(S). This residue was redissolved in CH2Cl2 (3 mL), treated with Et3N (20 µL, 0.13 mmol), followed by carbonate 15 (5.5 mg, 0.02 mmol) and stirred at 23 ºC for 12 h. The reaction mixture was then concentrated under reduced pressure and the residue was purified by flash chromatography (2% MeOH in CHCl3 as the eluent) to give inhibitor 17a (11.3 mg, 98%) as a white solid, Rf= 0.48 (10% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.48 (dd, 1H, J = 5.5, 13.2 Hz), 1.58–1.68 (m, 2H), 2.17–2.26 (m, 1H), 2.34–2.49 (m, 2H), 2.76 (dd, 1H, J = 9.8, 14.0 Hz), 2.85–2.95 (m, 3H), 3.10–3.16 (m, 2H), 3.22 (dd, 1H, (dd, 1H, J = 9.9, 13.7 Hz), 3.67–3.74 (m, 2H), 3.82–3.85 (dt, 1H, J = 1.8, 8.4 Hz), 3.87 (s, 3H), 3.94 (dd, 1H, J = 6.2, 9.6 Hz), 3.96–4.01 (m, 1H), 4.04–4.08 (m, 1H), 5.0 (q, 1H, J = 6.1, 7.9 Hz), 5.64 (d, 1H, J = 5.2 Hz), 6.98 (d, 2H, J = 8.9 Hz), 7.19–7.29 (m, 3H), 7.26–7.29 (m, 2H), 7.58 (brs, 1H), 7.69 (d, 2H, J = 8.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 23.97, 25.75, 29.59, 35.94, 45.29, 53.44, 53.94, 55.16, 55.60, 56.47, 69.53, 70.88, 72.39, 73.34, 109.24, 114.42, 126.45, 128.34, 128.63, 129.26, 129.49, 137.60, 155.47, 163.24, 178.51. LRMS-ESI (m/z) [M+H]+ 604.2; HRMS-ESI (m/z) [M+H]+ calcd for C29H38N3O9S 604.2329 found 604.23
(3aS,5R,6aR)-Hexahydro-2H–cyclopenta[b]furan-5-yl-(2S,3R)-3-hydroxy-4-(4-methoxy-N-(((S)-5-oxopyrrolidin-2-yl)methyl)phenylsulfonamido)-1-phenylbutan-2-ylcarbamate 17b
A solution of compound 9 (11.8 mg, 0.02 mmol) in 30% trifluoroacetic acid (in CH2Cl2, 1.5 mL) was stirred at 23 ºC for 40 min, then concentrated under reduced pressure to give the crude amine 13S. This crude residue was redissolved in CH2Cl2 (1.5 mL), treated with Et3N (63 µL, 0.45 mmol), followed by carbonate 16 (6.4 mg, 0.02 mmol), and stirred at 23 ºC for 6 h. The reaction mixture was then concentrated under reduced pressure and the residue was purified by flash chromatography (1% MeOH in CHCl3 as the eluent) to give inhibitor 17b (11.5 mg, 87%) as a white solid, Rf= 0.35 (5% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.45 (d, 1H, J = 14.3 Hz), 1.55–1.59 (m, 1H), 1.88 (d, 1H, J = 15.1 Hz), 1.95–2.06 (m, 3H), 2.17–2.24 (m, 1H), 2.33–2.48 (m, 2H), 2.60–2.67 (m, 1H), 2.78 (dd, 1H, J = 9.1, 14.1 Hz), 2.88–2.97 (m, 2H), 3.09 (dd, 1H, J = 4.3, 14.1 Hz), 3.12–3.18 (m, 2H), 3.64–3.68 (m, 1H), 3.82–3.85 (m, 2H), 3.86 (s, 3H), 3.89–3.95 (m, 1H), 3.99–4.05 (m, 1H), 4.39–4.42 (m, 1H), 4.69 (d, 1H, J = 4.1 Hz), 4.87–4.90 (m, 1H), 4.91 (d, 1H, J = 8.9 Hz), 6.98 (d, 2H, J = 8.9 Hz), 7.20–7.23 (m, 3H), 7.27–7.30 (m, 2H), 7.42 (s, 1H), 7.70 (d, 2H, J = 8.9 Hz); 13C-NMR (125 MHz, CDCl3) δ 24.00, 29.89, 33.85, 35.99, 38.31, 39.43, 41.59, 53.26, 53.33, 53.91, 54.89, 55.56, 56.36, 67.64, 72.23, 83.71, 114.39, 126.42, 128.43, 128.78, 129.33, 129.53, 137.54, 156.18, 163.11, 178.32. LRMS-ESI (m/z) [M+Na]+ 624.0; HRMS-ESI (m/z) [M+Na]+ calcd for C30H39N3NaO8S 624.2356 found 624.2352.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl(2S,3R)-4-(4-amino-N-(((S)-5-oxopyrrolidin-2-yl)methyl)phenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate 18a
The Cbz-protected amine 10 (31 mg, 0.04 mmol) was treated with 30% trifluoroacetic acid in CH2Cl2 (6 mL) and stirred at 23 ºC for 40 min, then concentrated under reduced pressure to give the crude amine 14-(S). This residue was redissolved in CH2Cl2 (6 mL), charged with Et3N (64 µL, 0.46 mmol), followed by carbonate 15 (14 mg, 0.05 mmol), and stirred at 23 ºC for 12 h. Reaction was quenched with 3 drops of benzyl amine and concentrated under reduced pressure. Flash chromatography purification (4% MeOH in CHCl3 as the eluent) provided the Cbz-protected inhibitor (23 mg, 86%) as a white solid, Rf = 0.46 (10% MeOH in CHCl3).
To above Cbz-protected inhibitor (13.3 mg, 0.018 mmol), in EtOAc (6 mL) under argon, was added Pd/C (3 mg). The mixture was stirred at 23 ºC under a hydrogenfilled balloon for 3 h, then filtered over Celite and the filter cake was washed with EtOAc and MeOH. Removal of solvent under reduced pressure, followed by flash chromatography purification (3% MeOH in CHCl3 as the eluent) provided the title inhibitor 18a (7.4 mg, 68%) as a white solid. Rf= 0.19 (5% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.44 (d, 1H, J = 5.4 Hz), 1.59–1.69 (m, 2H), 2.16–2.25 (m, 1H), 2.36 (t, 2H, J = 7.9 Hz), 2.68 (dd, 1H, J = 9.8, 13.9 Hz), 2.85– 2.95 (m, 3H), 3.03–3.09 (m, 2H), 3.13 (dd, 1H, J = 4.4, 14.1 Hz), 3.67–3.72 (m, 2H), 3.79–3.89 (m, 3H), 3.93 (dd, 1H, J = 6.0, 9.7 Hz), 4.01–4.06 (m, 1H), 4.96 (q, 1H, J = 5.9, 7.9 Hz), 5.63 (d, 1H, J = 5.1 Hz), 6.72 (d, 2H, J = 8.2 Hz), 7.17–7.21 (m, 3H), 7.24–7.28 (m, 2H), 7.50 (d, 2H, J = 8.4 Hz); 13C-NMR (125 MHz, CDCl3) δ 24.38, 26.22, 30.07, 36.69, 45.83, 53.67, 54.28, 55.62, 56.69, 70.06, 71.51, 72.72, 73.64, 109.77, 114.63, 124.92, 126.82, 128.83, 129.72, 129.97, 130.34, 151.40, 155.98, 178.30. LRMS-ESI (m/z) [M+H]+ 589.2; HRMS-ESI (m/z) [M+H]+ calcd for C28H37N4O8S 589.2332 found 589.2336.
(3aS,5R,6aR)-Hexahydro-2H–cyclopenta[b]furan-5-yl-(2S,3R)-4-(4-amino-N-(((S)-5-oxopyrrolidin-2-yl)methyl)phenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate 18b
The cbz-protected amine 10 (29.6 mg, 0.04 mmol) was treated with 30% trifluoroacetic acid (in CH2Cl2, 6 mL) and stirred at 23 ºC for 40 min, then concentrated under reduced pressure to give the crude amine 14S. The residue was redissolved in CH2Cl2 (6 mL), charged with Et3N (31 µL, 0.22 mmol), followed by carbonate 16 (13.1 mg, 0.05 mmol), and stirred at 23 ºC for 4 h. Reaction was quenched with 2 drops of benzyl amine and concentrated under reduced pressure. Flash chromatography purification (4% MeOH in CHCl3 as the eluent) provided the cbz-protected inhibitor (26.1 mg, 82%) as a white solid, Rf = 0.49 (10% MeOH in CHCl3). To a solution of the above protected inhibitor (17 mg, 0.02 mmol), in EtOAc (5 mL) under argon, was added Pd/C (3 mg). The mixture was stirred at 23 ºC under a H2 filled balloon for 5 h, then filtered over Celite and the filter cake was washed with EtOAc and MeOH. Removal of solvent under reduced pressure, followed by flash chromatography purification (5% MeOH in CHCl3 as the eluent) provided inhibitor 18b (14 mg, 75%) as a white solid. Rf = 0.27 (10% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.41–1.45 (m, 1H), 1.56–1.67 (m, 3H), 1.85 (d, 1H, J = 13.4 Hz), 1.96–2.04 (m, 3H), 2.14–2.21 (m, 1H), 2.31-–.42 (m, 2H), 2.61 (brs, 1H), 2.74–2.85 (m, 3H), 3.09 (dd, 1H, J = 4.4, 14.4 Hz), 3.15–3.20 (m, 1H), 3.25 (d,1H, J = 14.3 Hz), 3.66–3.70 (q, 1H, J = 7.1, 7.4 Hz), 3.83–3.88 (m, 2H), 3.91–3.96 (m, 1H), 3.96–4.25 (m, 1H), 4.40 (t, 1H, J = 5.9 Hz), 4.87 (brs, 1H), 4.93 (d, 1H, J = 8.9 Hz), 6.69 (d, 2H), 7.18–7.22 (m, 3H), 7.28–7.30 (m, 2H), 7.55 (d, 2H, J = 8.4 Hz), 7.63 (s, 1H); 13C-NMR (125 MHz, CDCl3) δ 24.39, 30.12, 34.19, 36.71, 38.75, 39.85, 41.99, 53.56, 54.34, 55.31, 56.89, 68.09, 72.89, 84.17, 114.69, 125.34, 126.84, 128.87, 129.92, 130.19, 138.19, 151.43, 156.63, 178.99. LRMS-ESI (m/z) [M+H]+ 586.9; HRMS-ESI (m/z) [M+H]+ calcd for C29H39N4O7S 587.2539 found 587.2539.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl(2S,3R)-3-hydroxy-4-(4-methoxy-N-(((R)-5-oxopyrrolidin-2-yl)methyl)phenylsulfonamido)-1-phenylbutan-2ylcarbamate 19a
A solution of compound 11 (20 mg, 0.03 mmol) in 30% trifluoroacetic acid (in CH2Cl2, 3 mL) was stirred 23 ºC for 40 min, then concentrated under reduced pressure. The residue was redissolved in CH2Cl2 (3 mL), treated with Et3N (51 µL, 0.36 mmol), followed by carbonate 15 (11 mg, 0.04 mmol), and stirred at 23 ºC for 12 h. The reaction mixture was then concentrated under reduced pressure and the residue was purified by flash chromatography (2% MeOH in CHCl3 as the eluent) to give inhibitor 19a (21 mg, 92%) as a white solid, Rf = 0.41 (10% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.44 (dd, 1H, J = 5.6, 13.2 Hz), 1.57–1.67 (m, 2H), 1.81–1.91 (m, 1H), 2.19–2.27 (m, 1H), 2.33–2.41 (m, 2H), 2.73 (dd, 1H J = 10.35, 13.9 Hz), 2.83–2.91 (m, 2H), 2.95 (dd, 1H, J = 8.9, 14.9 Hz), 3.11 (dd, 1H, J = 4.2, 14.0 Hz), 3.26–3.30 (ddd, 2H, J = 2.6, 7.0, 14.3 Hz), 3.64–3.70 (m, 1H), 3.74 (dd, 1H, J = 5.5, 9.8 Hz), 3.76–3.81 (dt, 1H, J = 1.62, 8.0 Hz), 3.87 (s, 3H), 3.89–3.93 (q, 1H, J = 4.0, 5.6 Hz), 4.03–4.06 (m, 2H), 4.98–5.02 (q, 1H, J = 5.65, 7.9 Hz), 5.62 (d, 1H, J = 5.4 Hz), 6.98 (d, 2H, J = 8.9 Hz), 7.17–7.27 (m, 5H), 7.70 (d, 2H, J = 8.8 Hz), 7.89 (s, 1H); 13C-NMR (125 MHz, CDCl3) δ 24.25, 25.78, 29.89, 35.43, 45.43, 55.06, 55.37, 55.58, 55.77, 58.11, 69.56, 71.06, 73.34, 74.02, 109.29, 114.46, 126.34, 128.33, 128.88, 129.29, 129.41, 137.84, 155.52, 163.24, 178.51. LRMS-ESI (m/z) [M+Na]+ 626.3; HRMS-ESI (m/z) [M+Na]+ calcd for C29H37N3 Na O9S 626.2148 found 626.2156.
(3aS,5R,6aR)-Hexahydro-2H–cyclopenta[b]furan-5-yl-(2S,3R)-3-hydroxy-4-(4-methoxy-N-(((R)-5-oxopyrrolidin-2-yl)methyl)phenylsulfonamido)-1-phenylbutan-2-ylcarbamate 19b
A solution of compound 11 (21 mg, 0.04 mmol) in 30% trifluoroacetic acid (in CH2Cl2, 3 mL) was stirred 23 ºC for 40 min, then concentrated under reduced pressure. The residue was redissolved in CH2Cl2 (3 mL), treated with Et3N (27 µL, 0.19 mmol), followed by carbonate 16 (12 mg, 0.04 mmol) and stirred at 23 ºC for 12 h. The reaction mixture was then concentrated under reduced pressure and the residue was purified by flash chromatography (1% MeOH in CHCl3 as the eluent) to give inhibitor 19b (21 mg, 93%) as a white solid, Rf = 0.31 (5% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.48 (d, 1H, J = 14.1 Hz), 1.58–1.62 (m, 1H), 1.83–2.04 (m, 5H), 2.19–2.25 (m, 1H), 2.31–2.41 (m, 2H), 2.59–2.67 (m, 1H), 2.73 (dd, 1H, J = 9.0, 13.9 Hz), 3.03 (dd, 1H, J = 7.0, 15.0 Hz), 3.08–3.16 (m, 2H), 3.19 (d, 1H, J = 14.9 Hz), 3.57–3.63 (m, 1H), 3.83–3.86 (m, 3H), 3.86 (s, 3H), 3.94–3.99 (m, 1H), 4.78 (d, 1H, J = 13.7 Hz), 4.90 (s, 1H), 5.36 (d, 1H, J = 8.1 Hz), 6.97 (d, 2H, J = 8.6 Hz), 7.18–7.28 (m, 5H), 7.46 (d, 1H, J = 18.4 Hz), 7.70 (d, 2H, J = 8.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 22.55, 30.00, 31.49, 33.97, 35.88, 38.25, 39.59, 41.76, 54.60, 54.91, 55.56, 56.08, 57.76, 67.72, 74.00, 83.97, 114.44, 126.21, 128.27, 128.90, 129.35, 129.47, 137.91, 156.16, 163.14, 178.52. LRMS-ESI (m/z) [M+H]+ 601.7; HRMS-ESI (m/z) [M+H]+ calcd for C30H40N3O8S 602.2536 found 602.2536.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl-(2S,3R)-4-(4-amino-N-(((R)-5-oxopyrrolidin-2-yl)methyl)phenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate 20a
A solution of the free amine 12 (15 mg, 0.03 mmol) in 30% trifluoroacetic acid (in CH2Cl2, 3 mL) was stirred at 23 ºC for 40 min, then concentrated under reduced pressure. The residue was redissolved in CH2Cl2 (3 mL), treated with Et3N (40 µL, 0.28 mmol), followed by carbonate 15 (9.2 mg, 0.03 mmol), and stirred at 23 ºC for 6 h. The reaction mixture was then concentrated under reduced pressure and the residue was purified by flash chromatography (4% MeOH in CHCl3 as the eluent) to give inhibitor 20a (12.5 mg, 94%) as a white solid, Rf = 0.26 (10% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.35 (dd, 1H, J = 5.7 Hz), 1.50–1.63 (m, 2H), 2.15–2.23 (m, 1H), 2.27–2.31 (m, 2H), 2.56–2.72 (m, 3H), 2.82–2.87 (m, 2H), 3.07 (dd, 1H, J = 4.0, 14.0 Hz), 3.36 (q, 1H, J = 1.4, 3.5 Hz), 3.36 (q, 1H, J = 2.0, 3.4 Hz), 3.60–3.65 (m, 1H), 3.70 (dd, 1H, J = 5.4, 9.8 Hz), 3.75–3.79 (dt, 1H, J = 1.96, 8.3 Hz), 3.80–3.86 (m, 1H), 3.88 (dd, 1H, J = 5.9, 9.8 Hz), 3.90–3.94 (m, 1H), 3.99–4.02 (m, 1H), 4.92–4.97 (q, 1H, J = 5.7, 8.1 Hz), 5.58 (d, 1H, J = 5.8 Hz), 5.92 (d, 1H, J = 9.5 Hz), 6.63 (d, 2H, J = 8.7 Hz), 7.13–7.22 (m, 5H), 7.46 (d, 2H, J = 8.7 Hz); 13C-NMR (125 MHz, CDCl3) δ 24.16, 25.70, 29.75, 35.39, 45.40, 55.01, 55.51, 55.61, 58.11, 69.55, 71.05, 73.19, 73.94, 109.27, 113.85, 124.27, 126.23, 128.22, 129.19, 129.35, 137.89, 151.44, 155.67, 178.63. LRMS-ESI (m/z) [M+Na]+ 611.4; HRMS-ESI (m/z) [M+Na]+ calcd for C28H36N4NaO8S 611.2152 found 611.2149.
(3aS,5R,6aR)-Hexahydro-2H–cyclopenta[b]furan-5-yl-(2S,3R)-4-(4-amino-N-(((R)-5-oxopyrrolidin-2-yl)methyl)phenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate 20b
The Cbz-amine 12 (40 mg, 0.06 mmol) was treated with 30% trifluoroacetic acid (in CH2Cl2, 6 mL) and stirred at 23 ºC for 40 min, then concentrated under reduced pressure. This residue was redissolved in CH2Cl2 (6 mL), charged with Et3N (42 µL, 0.3 mmol), followed by carbonate 16 (19 mg, 0.07 mmol), and stirred at 23 ºC for 12 h. Reaction was quenched with 3 drops of benzyl amine and concentrated under reduced pressure. Flash chromatography purification (5% MeOH in CHCl3 as the eluent) provided the Cbz-protected inhibitor (32.1 mg, 75%) as a white solid, Rf = 0.41 (10% MeOH in CHCl3).
To above Cbz-protected inhibitor (25.8 mg, 0.03 mmol), in EtOAc (5 mL) under argon, was added Pd/C (5 mg). The mixture was stirred at 23 ºC under a hydrogen filled balloon for 3 h, then filtered over Celite and the filter cake was washed with EtOAc and MeOH. Removal of solvent under reduced pressure, followed by flash chromatography purification (7.5% MeOH in CHCl3 as the eluent) provided the title inhibitor 20b (16.2 mg, 77%) as a white solid. Rf = 0.51 (15% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.46 (m, 1H), 1.58–1.64 (m, 1H), 1.77–1.84 (m, 1H), 1.86–2.04 (m, 5H), 2.15–2.23 (m, 1H), 2.32–2.36 (m, 2H), 2.59–2.65 (m, 1H), 2.68 (dd, 1H, J = 9.2, 14.0 Hz), 2.95–3.04 (m, 2H), 3.11 (d, 2H, J = 13.7 Hz), 3.18 (d, 1H, J = 14.9 Hz), 3.57–3.62 (q, 1H, J = 6.9, 7.7 Hz), 3.82–3.87 ( q, 2H, J = 6.5, 7.9 Hz), 3.92–3.97 (m, 1H), 4.30 (s, 2H), 4.37 (t, 1H, J = 5.7 Hz), 4.75 (s, 1H), 4.89 (s, 1H), 5.40 (d, 1H, J = 8.4 Hz), 6.66 (d, 2H, J = 8.6 Hz), 7.16–7.22 (m, 3H), 7.24–7.27 (m, 2H), 7.40 (s, 1H), 7.51 (d, 2H, J = 8.6 Hz) ; 13C-NMR (125 MHz, CDCl3) δ 23.72, 30.01, 33.94, 35.94, 38.18, 39.59, 41.78, 54.62, 54.85, 55.99, 57.64, 67.74, 74.04, 76.68, 84.01, 114.10, 124.98, 126.21, 128.27, 129.35, 129.48, 137.91, 151.08, 156.20, 178.40. LRMS-ESI (m/z) [M+Na]+ 609.0; HRMS-ESI (m/z) [M+Na]+ calcd for C29H38N4NaO7S 609.2359 found 609.2362.
(R)-tert-butyl-4-(((2R,3S)-3-(tert-butoxycarbonylamino)-2-hydroxy-4-phenylbutylamino)methyl)-2,2-dimethyloxazolidine-3-carboxylate 22
To a solution of the (R)-tert-butyl 4-(azidomethyl)-2,2-dimethyloxazolidine-3-carboxylate 21 (411 mg, 1.60 mmol) in MeOH (10 mL) was added Pd/C (40 mg). This mixture was stirred at 23 ºC under a hydrogen filled balloon for 2 h, then filtered over Celite, and the filter cake was washed with MeOH. Solvent was removed under reduced pressure and the resultant residue was redisolved in iPrOH, followed by addition of tert- butyl-[S-(R,R)]-(-)-(1-oxiranyl-2-phenylethyl) carbamate 7 (121 mg, 0.46 mmol). The mixture was allowed to stir at 65ºC for 24 h, solvent was then removed under reduced pressure. Flash chromatography purification (2% MeOH in CHCl3 as the eluent) yielded the (R)-amine 22 (93 mg, 41%) as a yellow oil. Rf = 0.32 (10% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.35 1(s, 18H), 1.48 (s, 24H), 1.54 (s, 3H), 1.58 (s, 3H), 2.67–2.92 (m, 10), 2.95 (d, 1H, J = 4.1 Hz), 2.98 (d, 1H, J = 4.1 Hz), 3.47 (brs, 2H), 3.78 (brs, 3H), 3.89–4.02 (m, 5H), 4.61 (brs, 1H), 4.68 (brs, 1H), 7.18–7.22 (m, 6H), 7.27–7.30 (m, 4H); 13C-NMR (125 MHz, CDCl3) δ 22.94, 24.27, 26.68, 27.46, 28.17, 28.33, 36.43, 51.45, 56.95, 65.97, 70.74, 70.90, 79.32, 79.45, 79.66, 80.35, 93.46, 93.77, 126.23, 128.30, 129.41, 137.55, 137.77, 152.62, 155.86; LRMS-ESI (m/z) [M + Na]+ 516.
(R)-tert-butyl-4-((N-((2R,3S)-3-(tert-butoxycarbonylamino)-2-hydroxy-4-phenylbutyl)-4-methoxyphenylsulfonamido)methyl)-2,2-dimethyloxazolidine-3-carboxylate 23
To a stirred solution of amine 22 (37 mg, 0.07 mmol) in CH2Cl2 (4 mL) and aqueous sat. NaHCO3 (4 mL) was added 4-methoxybenzenesulfonyl chloride (34 mg, 0.16 mmol). This reaction mixture was stirred for 12 h followed by extraction of the aqueous layer with CH2Cl2; the combined organic extracts were dried over anhydrous Na2SO4. Removal of solvent under reduced pressure, followed by flash chromatography purification (30% EtOAc in Hexane as the eluent) provided compound 23 (40 mg, 80%) as a white solid. Rf = 0.37 (40% EtOAc in Hex); 1H-NMR (500 MHz, CDCl3) δ 1.34 (s, 9H), 1.48 (s, 9H), 1.60 (s, 6H), 2.84–2.89 (m, 1H), 2.95–3.06 (m, 2H), 3.15 (dd, 1H, J = 6.5, 13.0 Hz), 3.29 (d, 1H, J = 9.7 Hz), 3.41 (d, 1H, J = 15.1 Hz), 3.77 (brs, 2H), 3.85 (s, 3H), 3.90–3.94 (m, 1H), 3.99 (d, 1H, J = 9.0 Hz), 4.2 (brs, 1H), 4.73 (brs, 0.5H), 4.98 (brs, 0.5H), 6.95 (d, 2H, J = 8.0 Hz), 7.19–7.24 (m, 3H), 7.25–7.28 (m, 2H), 7.69 (d, 2H, J = 8.9 Hz); 13C-NMR (125 MHz, CDCl3) δ 24.07, 27.42, 28.21, 28.31, 36.03, 53.30, 54.01, 55.48, 55.89, 56.38, 65.69, 72.47, 79.06, 80.97, 93.95, 114.29, 126.09, 128.15, 129.31, 129.61, 129.92, 137.80, 152.82, 155.43, 162.91; LRMS-ESI (m/z) [M + Na]+ 686.
(R)-tert-butyl-4-((N-((2R,3S)-3-(tert-butoxycarbonylamino)-2-hydroxy-4-phenylbutyl)-4-nitrophenylsulfonamido)methyl)-2,2-dimethyloxazolidine-3-carboxylate 24
To a stirred solution of amine 22 (41 mg, 0.08 mmol) in CH2Cl2 (4 mL) and aqueous sat. NaHCO3 (4 mL) was added 4-nitrobenzenesulfonyl chloride (37 mg, 0.16 mmol). This reaction mixture was stirred for 12 h followed by extraction of the aqueous layer with CH2Cl2; the combined organic extracts were dried over anhydrous Na2SO4. Removal of solvent under reduced pressure, followed by flash chromatography purification (30% EtOAc in Hexane as the eluent) provided (R)-nitrosulfone 24 (51 mg, 92%) as a white solid. Rf = 0.52 (40% EtOAc in Hex); 1H-NMR (500 MHz, CDCl3) δ 1.35 (s, 18H), 1.49 (s, 24H), 1.62 (s, 6H), 3.43 (bd, 2H, J = 9.6 Hz), 3.55 (d, 2H, J = 15.1 Hz), 3.75 (brs, 4H), 3.95 (s, 4H), 4.28 (brs, 2H), 4.63, (d, 2H, J = 7.8 Hz), 5.16 (brs, 2H), 7.19–7.23 (m, 6H), 7.25–7.31 (m, 4H), 7.92 (d, 4H, J = 8.8 Hz), 8.30 (d, 4H, J = 8.05 Hz); 13C-NMR (125 MHz, CDCl3) δ 24.01, 27.39, 28.19, 28.29, 35.88, 53.75, 54.06, 55.55, 55.98, 65.61, 72.27, 79.47, 81.33, 94.16, 124.34, 126.31, 128.29, 128.40, 129.50, 137.46, 144.32, 149.95, 153.05, 155.53; LRMS-ESI (m/z) [M + Na]+ 701.
tert-butyl-(2S,3R)-3-hydroxy-4-(4-methoxy-N-(((R)-2-oxooxazolidin-4-yl)methyl)phenylsulfonamido)-1-phenylbutan-2-ylcarbamate 25
To (R)-toluenesulfonate 23 (32 mg, 0.05 mmol) in methanol (3 mL) was added p-toluenesulfonic acid monohydrate (2 mg), and the reaction mixture was allowed to stir at 23 ºC. After 12 h reaction reached maximum conversion and did not move any further. It was quenched with sat. aqueous NaHCO3, extracted with EtOAc, washed with water and brine, and dried over Na2SO4. Flash chromatography purification (30–50% EtOAc in Hexane) yielded 7 mg (21%) of starting material and 18 mg (58%) of the expected primary alcohol.
The above alcohol (17 mg, 0.0278 mmol), in CH2Cl2 (2 mL) and Et3N (16 µL, 0.11 mmol), was cooled to −10 ºC and treated with methanesulfonyl chloride (2.5 µL, 0.03 mmol). The reaction mixture was stirred at 0 ºC for 2 h, followed by removal of solvent via rotavap and pump. The resultant residue was redisovled in chloroform (2 mL), treated with DIPEA (19 µL, 0.11 mmol), refluxed for 8 h, and concentrated under reduced pressure. Flash chromatography purification (1% MeOH in CHCl3 as the eluent) afforded (R)-oxazolidinone 25 (12 mg, 78%) as a white solid. Rf = 0.33 (5% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.32 (s, 9H), 2.77 (dd, 1H, J = 8.4, 13.9 Hz), 2.93–3.03 (m, 3H), 3.07 (d, 1H, J = 14.5 Hz), 3.13 (dd, 1H, J = 9.3, 14.1 Hz), 3.69–3.79 (m, 2H), 3.85 (s, 3H), 3.97 (dd, 1H, J = 5.2, 8.3 Hz), 4.23–4.29 (m, 1H), 4.43 (t, 1H, J = 8.7 Hz), 6.94 (d, 2H, J = 8.8 Hz), 7.18–7.24 (m, 3H), 7.26–7.30 (m, 2H), 7.64 (d, 2H, J = 8.6 Hz); 13C-NMR (125 MHz, CDCl3) δ 28.12, 35.97, 52.02, 54.16, 54.60, 54.73, 55.55, 67.47, 71.97, 79.70, 114.42, 126.37, 128.42, 128.48, 129.30, 129.44, 137.52, 155.99, 159.44, 163.22; LRMS-ESI (m/z) [M + Na]+ 572.
tert-butyl-(2S,3R)-4-(4-amino-N-(((R)-2-oxooxazolidin-4-yl)methyl)phenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate 26
To (R)-nitrosulfone 24 (42 mg, 0.06 mmol) in methanol (4 mL) was added p-toluenesulfonic acid monohydrate (2 mg), and the reaction mixture was allowed to stir at 23 ºC. After 36 h reaction appeared to have moved only 75% to completion, and formation of free amines was evident on TLC. At this point it was quenched with sat. aqueous NaHCO3, extracted with EtOAc, washed with water and brine, and dried over Na2SO4. Flash chromatography purification (30–50% EtOAc in Hexane) yielded 7 mg (17%) of starting material and 24 mg (55%) of the expected primary alcohol.
The above alcohol (23 mg, 0.03 mmol), in CH2Cl2 (3 mL) and Et3N (20 µL, 0.14 mmol), was cooled to −10 ºC and treated with methanesulfonyl chloride (53 µL, 0.04 mmol). The reaction mixture was stirred at 0 ºC for 2 h, followed by removal of solvent via rotavap and pump. The resultant residue was redisovled in chloroform (3 mL), treated with DIPEA (25 µL, 0.14 mmol), and refluxed for 12 h; product crashed out of the chloroform and was found to be only slightly soluble in methanol. Solvent was removed under reduced pressure and the crude material was treated with methanol (3 mL) and Pd/C (3 mg). The mixture was stirred at 23 ºC under a H2 filled balloon for 2 h, then filtered over Celite and the filter cake was washed with MeOH. Removal of solvent, followed by flash chromatography purification (3% MeOH in CHCl3 as the eluent) provided (R)-oxazolidinone 26 (13 mg, 68%) as a white solid. Rf = 0.19 (5% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.31 (s, 9H), 2.73 (dd, 1H, J = 8.7, 13.6 Hz), 2.88 (dd, 1H, J = 9.6, 14.5 Hz), 2.91–2.94 (m, 1H), 2.98 (dd, 1H, J = 4.2, 13.9 Hz), 3.10 (dd, 2H, J = 9.2, 14.0 Hz), 3.70– 3.81 (m, 2H), 3.96 (dd, 1H, J = 5.2, 8.4 Hz), 4.21–4.24 (m, 1H), 4.40 (t, 1H, J = 8.7 Hz), 4.92 (d, 1H, J = 8.5 Hz), 6.63 (d, 2H, J = 8.7 Hz), 7.17–7.21 (m, 3H), 7.25–7.28 (m, 2H), 7.46 (d, 2H, J = 8.4 Hz); 13C-NMR (125 MHz, CD3OD) δ 28.13, 29.57, 35.99, 51.91, 52.00, 54.09, 54.73, 67.53, 72.12, 79.63, 113.98, 124.40, 126.31, 128.36, 129.32, 129.45, 137.62, 1551.21, 156.01, 159.52; LRMS-ESI (m/z) [M + Na]+ 557.
(tert-butyl(2S,3R)-3-hydroxy-4-(4-methoxy-N-(((S)-2-oxooxazolidin-4-yl)methyl)phenylsulfonamido)-1-phenylbutan-2-ylcarbamate 27
To a solution of the (S-)tert-butyl 4-(azidomethyl)-2,2-dimethyloxazolidine-3-carboxylate ent-21 (840 mg, 3.32 mmol) in MeOH (20 mL) was added Pd/C (80 mg). This mixture was stirred at 23 ºC under a hydrogen-filled balloon for 2 h, then filtered over Celite, and the filter cake was washed with MeOH. Solvent was removed under reduced pressure and the resultant residue was redisolved in iPrOH, followed by addition of tert-butyl-[S-(R,R)]-(-)-(1-oxiranyl-2-phenylethyl) carbamate 7 (218 mg, 0.83 mmol). The mixture was allowed to stir at 65 ºC for 24 h, solvent was then removed under reduced pressure. Flash chromatography purification (2% MeOH in CHCl3 as the eluent) yielded the corresponding (S)-amine (22S) (187 mg, 46%) as an amprphous solid. Rf = 0.28 (10% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.35 1(s, 18H), 1.48 (s, 24H), 1.54 (s, 3H), 1.58 (s, 3H), 2.64–2.91 (m, 10), 2.95–3.03 (m, 2H), 3.44 (brs, 1H), 3.50 (brs, 1H), 3.77 (brs, 3H), 3.87–4.00 (m, 5H), 4.61 (brs, 1H), 4.64 (brs, 1H), 7.18–7.22 (m, 6H), 7.27–7.30 (m, 4H); 13C-NMR (125 MHz, CDCl3) δ 22.96, 24.27, 26.69, 27.47, 28.16, 28.32, 36.48, 51.47, 56.73, 56.87, 65.95, 70.68, 79.35, 79.65, 80.34, 93.45, 93.76, 126.21, 126.30, 128.30, 129.39, 137.61, 137.79, 152.58, 155.88; LRMS-ESI (m/z) [M + Na]+ 576.
To a stirred solution of above S-amine (22S) (85 mg, 0.17 mmol) in CH2Cl2 (6 mL) and aqueous sat. NaHCO3 (6 mL) was added 4-methoxybenzenesulfonyl chloride (71 mg, 0.34 mmol). This reaction mixture was stirred for 12 h followed by extraction of the aqueous layer with CH2Cl2; the combined organic extracts were dried over anhydrous Na2SO4. Removal of solvent under reduced pressure, followed by flash chromatography purification (30% EtOAc in Hexane as the eluent) provided S-toluenesulfonate (23S) (107 mg, 93%) as a white solid. Rf = 0.40 (40% EtOAc in Hexane); 1H-NMR (500 MHz, CDCl3) δ 1.34 (s, 18H), 1.46 (s, 24H), 1.58 (s, 6H), 2.82–2.87 (m, 2H), 2.96–3.13 (m, 4H), 3.18–3.24 (m, 3H), 3.32 (brs, 1H), 3.75 (brs, 1H), 3.80 (brs, 1H), 3.84 (s, 6H), 3.89–3.93 (4H), 4.07 (d, 2H, J = 9.2 Hz), 4.16–4.19 (m, 2H), 4.65 (brs, 1H), 4.87 (d, 1H, J = 7.31 Hz), 6.93–6.97 (m, 4H), 7.18–7.23 (6H), 7.26–7.28 (m, 4H), 7.64–7.71 (4H); 13C-NMR (125 MHz, CDCl3) δ 23.98, 27.38, 28.18, 28.28, 35.68, 52.47, 54.35, 55.24, 55.47, 56.88, 65.66, 71.86, 79.19, 80.93, 93.84, 114.22, 126.13, 128.24, 129.39, 129.47, 129.64, 137.97, 152.69, 155.64, 162.85; LRMS-ESI (m/z) [M + Na]+ 686.
To above S-toluenesulfonate (23S) (96 mg, 0.14 mmol) in methanol (5 mL) was added p-toluenesulfonic acid monohydrate (4.5 mg, 0.02 mmol), and the reaction mixture was allowed to stir at 23 ºC. After 8 h reaction reached maximum conversion and did not move any further. It was quenched with sat. aqueous NaHCO3, extracted with EtOAc, washed with water and brine, and dried over Na2SO4. Flash chromatography purification (30–50% EtOAc in Hexane) yielded 13 mg (13%) of starting material and 59 mg (65%) of the expected primary alcohol.
The above alcohol (58 mg, 0.09 mmol), in CH2Cl2 (4 mL) and Et3N (33 µL, 0.23 mmol), was cooled to −10 ºC and treated with methanesulfonyl chloride (7.9 µL, 0.1 mmol). The reaction mixture was stirred at 0 ºC for 2 h, followed by removal of solvent via rotavap and pump. The resultant residue was redisovled in chloroform (4 mL), treated with DIPEA (65 µL, 0.37 mmol), refluxed for 12 h, and concentrated under reduced pressure. Flash chromatography purification (2% MeOH in CHCl3 as the eluent) afforded S-oxazolidinone 27 (44 mg, 85%) as a white solid. Rf = 0.27 (5% MeOH in CHCl3); H-NMR (500 MHz, CD3OD) δ 1.27 (s, 9H), 2.52 (dd, 1H, J = 10.95, 13.7 Hz), 2.67 (dd, 1H, J = 8.9, 14.9 Hz), 2.83 (dd, 1H, J = 6.1, 14.0 Hz), 3.10 (dd, 1H, J = 3.5, 13.8 Hz), 3.47–3.52 (m, 2H), 3.58–3.63 (m, 1H), 3.80–3.83 (m, 1H), 3.85 (s, 3H), 4.23–4.29 (m, 1H), 4.31 (dd, 1H, J = 4.8, 8.9 Hz), 4.47 (t, 1H, J = 8.6 Hz), 7.06 (d, 2H, J = 8.93 Hz), 7.12–7.16 (m, 1H), 7.20–7.24 (m, 4H), 7.75 (d, 2H, J = 8.9 Hz); 13C-NMR (125 MHz, CD3OD) δ 27.19, 35.63, 52.00, 54.38, 54.613, 54.70, 55.17, 68.04, 73.35, 78.52, 114.05, 125.57, 127.64, 128.92, 129.08, 129.31, 138.67, 156.55, 160.46, 163.36; LRMS-ESI (m/z) [M + Na]+ 572.
tert-butyl-(2S,3R)-4-(4-amino-N-(((S)-2-oxooxazolidin-4-yl)methyl)phenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate 28
To a stirred solution of S-amine (22S) (87 mg, 0.17 mmol) in CH2Cl2 (6 mL) and aqueous sat. NaHCO3 (6 mL) was added 4-nitrobenzenesulfonyl chloride (78 mg, 0.35 mmol). This reaction mixture was stirred for 12 h followed by extraction of the aqueous layer with CH2Cl2; the combined organic extracts were dried over anhydrous Na2SO4. Removal of solvent under reduced pressure, followed by flash chromatography purification (30% EtOAc in Hexane as the eluent) provided S-nitrosulfone (24S) (102 mg, 85%) as a yellow solid. Rf = 0.54 (40% EtOAc in Hex); 1H-NMR (500 MHz, CDCl3) δ 1.34 (s, 18H), 1.47 (s, 24H), 1.59 (s, 6H), 2.81 (dd, 2H, J = 8.0, 12.9 Hz), 3.03 (bd, 2H, J = 9.75 Hz), 3.16–3.36 (m, 8H), 3.82 (brs, 2H), 3.91 (dd, 4H, J = 5.15, 8.7 Hz), 4.00 (d, 2H, J = 6.2 Hz), 4.20 (q, 2H, J = 5.6, 5.7 Hz), 4.80 (brs, 1H), 4.86 (d, 1H, J = 7.2 Hz), 7.20–7.24 (m, 6H), 7.27–7.30 (m, 4H), 7.88 (d, 4H, J = 8.4 Hz), 8.29 (d, 4H, J = 8.4 Hz); 13C-NMR (125 MHz, CDCl3) δ 23.93, 27.38, 28.15, 28.27, 35.82, 52.35, 54.57, 54.97, 56.65, 65.50, 71.67, 79.57, 81.18, 94.02, 124. 21, 126.31, 128.40, 128.40, 128.51, 129.34, 137.79, 144.05, 149.93, 152.77, 155.82, 171.05; LRMS-ESI (m/z) [M + Na]+ 701.
To above S-nitrosulfone (24S) (92 mg, 0.13 mmol) in methanol (5 mL) was added p-toluenesulfonic acid monohydrate (4 mg, 0.02 mmol), and the reaction mixture was allowed to stir at 23 ºC. After 18 h reaction appeared to have moved only 50% to completion, and formation of free amines was evident on TLC. At this point it was quenched with sat. aqueous NaHCO3, extracted with EtOAc, washed with water and brine, and dried over Na2SO4. Flash chromatography purification (30–50% EtOAc in Hexane) yielded 32 mg (35%) of starting material and 44 mg (51%) of the expected primary alcohol.
The above alcohol (43 mg, 0.06 mmol), in CH2Cl2 (3 mL) and Et3N (24 µL, 0.17 mmol), was cooled to −10 ºC and treated with methanesulfonyl chloride (5.5 µL, 0.07 mmol). The reaction mixture was stirred at 0 ºC for 2 h, followed by removal of solvent via rotavap and pump. The resultant residue was redisovled in chloroform (2 mL), treated with DIPEA (29 µL, 0.17 mmol), and refluxed for 12 h; product crashed out of the chloroform and was found to be only slightly soluble in methanol. Solvent was removed under reduced pressure and the crude material was treated with methanol (4 mL) and Pd/C (5 mg). The mixture was stirred at 23 ºC under a hydrogen filled balloon for 3 h, then filtered over Celite and the filter cake was washed with MeOH. Removal of solvent, followed by flash chromatography purification (3% MeOH in CHCl3 as the eluent) provided S-oxazolidinone 28 (21 mg, 58%) as a white solid. Rf = 0.21 (5% MeOH in CHCl3); 1H-NMR (500 MHz, 1:1 CD3OD/CDCl3) δ1.24 (s, 9H), 2.54 (d, 1H), 2.56 (dd, 1H, J = 8.7, 14.9 Hz), 2.70 (dd, 1H, J = 7.6, 14.3 Hz), 2.97 (dd, 1H, J = 4.0, 13.9 Hz), 3.32 (dd, 1H, J = 5.0, 14.3 Hz), 3.38 (dd, 1H, J = 3.1, 14.8 Hz), 3.64–3.70 (m, 1H), 3.78–3.83 (m, 1H) 4.05 (dd, 1H, J = 5.3, 8.8 Hz), 4..16–4.20 (m, 1H), 4.39 (t, 1H, J = 8.7 Hz), 6.59 (d, 2H, J = 8.7 Hz), 7.09–7.15 (m, 3H), 7.18–7.21 (m, 2H), 7.40 (d, 2H, J = 8.7 Hz); 13C-NMR (125 MHz, 1:1 CD3OD/CDCl3) δ 31.88, 33.40, 39.42, 53.38, 56.90, 58.52, 58.84, 59.77, 71.89, 77.48, 83.34, 117.62, 127.44, 130.02, 132.05, 133.11, 133.20, 141.93, 156.09, 160.19, 164.05; LRMS-ESI (m/z) [M + Na]+ 557.
(3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl-(2S,3R)-3-hydroxy-4-(4-methoxy-N-(((R)-2-oxooxazolidin-4-yl)methyl)phenylsulfonamido)-1-phenylbutan-2-ylcarbamate 29
A solution of S-aminosulfone 25 (10.9 mg, 0.02 mmol) in 30% trifluoroacetic acid (in CH2Cl2, 2 mL) was stirred at 23 ºC for 40 min, then concentrated under reduced pressure. The residue was redisolved in CH2Cl2 (2 mL), treated with Et3N (30 µL, 0.21 mmol), followed by carbonate 15 (6.3 mg, 0.02 mmol) and stirred at 23 ºC for 12 h. The reaction mixture was then concentrated under reduced pressure and the residue was purified by flash chromatography (3% MeOH in CHCl3 as the eluent) to give inhibitor 29 (10 mg, 85%) as a white solid, Rf = 0.54 (10% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.39 (dd, 1H, J = 5.5, 13.1 Hz), 1.56–1.63 (m, 1H), 2.67 (dd, 1H, J = 10.0, 14.0 Hz), 2.85–2.90 (m, 1H), 2.95–3.01 (m, 2H), 3.10 (dd, 1H, J = 4.1, 14.1 Hz), 3.20 (d, 1H, J = 12.8 Hz), 3.28 (dd, 1H, J = 9.2, 14.1 Hz), 3.63–3.72 (m, 1H), 3.73 (dd, 1H, J = 5.2, 9.8 Hz), 3.79 (dt, 1H, J = 1.6, 8.2 Hz), 3.86 (s, 3H), 3.88–3.95 (m, 3H), 4.04 (dd, 1H, J = 5.0, 8.8 Hz), 4.25 (brs, 1H), 4.28–4.34 (m, 1H), 4.47 (t, 1H, J = 8.7 Hz), 4.99 (q, 1H, J = 5.6, 13.4 Hz), 5.38 (d, 1H, J = 9.8 Hz), 5.63 (d, 1H, J = 5.2 Hz), 6.87 (s, 1H), 6.97 (d, 2H, J = 8.9 Hz), 7.16–7.22 (m, 3H), 7.24–7.28 (m, 2H), 7.69 (d, 2H, J = 8.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 25.81, 35.92, 45.47, 51.96, 54.18, 55.09, 55.30, 55.63, 67.61, 69.62, 71.13, 72.58, 73.42, 109.29, 114.54, 126.44, 128.43, 128.47, 129.23, 129.44, 137.58, 155.64, 159.60, 163.35; LRMS-ESI (m/z) [M+Na]+ 628.2; HRMS-ESI (m/z) [M+Na]+ calcd for C28H35N3O10S 628.1941 found 628.1937.
(3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl-(2S,3R)-4-(4-amino-N-(((R)-2-oxooxazolidin-4-yl)methyl)phenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate 30
A solution of the free amine 26 (12 mg, 0.02 mmol) in 30% trifluoroacetic acid in CH2Cl2, (2 mL) was stirred at 23 ºC for 40 min, then concentrated under reduced pressure. The residue was redisolved in CH2Cl2 (2 mL), treated with carbonate 15 (7.1 mg, 0.02 mmol) and Et3N (40 µL, 0.28 mmol), and stirred at 23 ºC for 12 h at 40 ºC. The reaction mixture was then concentrated under reduced pressure and the residue was purified by flash chromatography (5% MeOH in CHCl3 as the eluent) to give inhibitor 30 (11 mg, 85%) as a white solid, Rf = 0.26 (10% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.38 (dd, 1H, J = 5.6, 13.2 Hz), 1.54–1.63 (m, 1H), 2.61 (dd, 1H, J = 9.5, 13.9 Hz), 2.83– 2.91 (m, 2H), 3.10 (d, 2H, J = 13.3 Hz), 3.18 (dd, 1H, J = 9.6, 13.9 Hz), 3.62–3.70 (m, 2H), 3.78–3.84 (m, 3H), 3.88 (dd, 1H, J = 6.0, 9.8 Hz), 3.97 (dd, 1H, J = 5.0, 8.9 Hz), 4.21–4.27 (m, 1H), 4.41 (t, 1H, J = 8.7 Hz), 4.93 (q, 1H, J = 5.7, 13.6 Hz), 5.60 (d, 1H, J = 5.25 Hz), 5.69 (d, 1H, J = 8.8 Hz), 6.64 (d, 2H, J = 8.7 Hz), 7.16–7.19 (m, 3H), 7.21–7.26 (m, 2H), 7.45 (d, 2H, J = 8.7 Hz); 13C-NMR (125 MHz, CDCl3) δ 25.73, 29.56, 36.12, 45.37, 51.74, 54.14, 54.93, 55.18, 67.50, 69.57, 70.87, 71.05, 73.21, 109.26, 113.92, 123.90, 126.33, 128.33, 129.21, 129.45, 137.73, 151.44, 155.60, 159.65; LRMS-ESI (m/z) [M+Na]+ 613.2; HRMS-ESI (m/z) [M+Na]+ calcd for C27H34N4O9S 613.1944 found 613.1939.
(3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl(2S,3R)-3-hydroxy-4-(4-methoxy-N-(((S)-2-oxooxazolidin-4-yl)methyl)phenylsulfonamido)-1-phenylbutan-2-ylcarbamate 31
A solution of S-aminosulfone 27 (16.5 mg, 0.03 mmol) in 30% trifluoroacetic acid (in CH2Cl2, 3 mL) was stirred at 23 ºC for 40 min, then concentrated under reduced pressure. The residue was redisolved in CH2Cl2 (3 mL), treated with Et3N (41 µL, 0.29 mmol), followed by carbonate 15 (10 mg, 0.03 mmol), and stirred at 23 ºC for 12 h. The reaction mixture was then concentrated under reduced pressure and the residue was purified by flash chromatography (3% MeOH in CHCl3 as the eluent) to give inhibitor 31 (16 mg, 90%) as a white solid, Rf = 0.5 (10% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.32 (dd, 1H, J = 5.4, 13.2 Hz), 1.53–1.62 (m, 1H), 2.62 (dd, 1H, J = 11.0, 13.8 Hz), 2.71 (dd, 1H, J = 7.25, 14.6 Hz), 2.84–2.89 (m, 2H), 3.10 (dd, 1H, J = 3.6, 14.0 Hz), 3.61–3.67 (m, 1H), 3.74–3.82 (m, 2H), 3.86 (s, 3H), 3.87 (dd, 2H, J = 5.8, 10.1 Hz), 3.94–3.98 (m, 1H), 4.00–4.05 (m, 2H), 4.27–4.33 (m, 1H), 4.45 (t, 1H, J = 8.8 Hz), 4.99 (q, 1H, J = 5.3, 7.7 Hz), 5.60 (d, 1H, J = 5.3 Hz), 5.68 (d, 1H, J = 9.2 Hz), 6.98 (d, 2H, J = 8.8 Hz), 7.15–7.20 (m, 1H), 7.22–7.27 (m, 4H), 7.69 (d, 2H, J = 8.8 Hz); 13C-NMR (125 MHz, CDCl3) δ 25.78, 43.46, 45.57, 53.23, 54.68, 54.96, 55.62, 56.58, 57.59, 69.60, 71.20, 73.33, 73.58, 109, 31, 114.54, 126.32, 128.32, 129.23, 129.45, 137.85, 155.94, 159.61, 163.38; LRMS-ESI (m/z) [M+Na]+ 628.2; HRMS-ESI (m/z) [M+Na]+ calcd for C28H35N3O10S 628.1941 found 628.1943.
(2R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl-(2S,3R)-4-(4-amino-N-(((S)-2-oxooxazolidin-4-yl)methyl)phenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate 32
A solution of the free amine 29 (15 mg, 0.03 mmol) in 30% trifluoroacetic acid (in CH2Cl2, 3 mL) was stirred at 23 ºC for 40 min, then concentrated under reduced pressure. The residue was redisolved in CH2Cl2 (3 mL), charged with Et3N (60 µL, 0.42 mmol), followed by carbonate 15 (8.9 mg, 0.03 mmol), and stirred at 23 ºC for 12 h at 40 ºC. The reaction mixture was then concentrated under reduced pressure and the residue was purified by flash chromatography (5% MeOH in CHCl3 as the eluent) to give inhibitor 32 (13 mg, 80%) as a white solid, Rf = 0.32 (10% MeOH in CHCl3); 1H-NMR (500 MHz, CDCl3) δ 1.33 (dd, 1H, J = 8.5 Hz), 1.53–1.62 (m, 1H), 2.59 (dd, 1H, J = 10.5, 13.9 Hz), 2.66 (dd, 1H, J = 7.4, 14.7 Hz), 2.77 (dd, 1H, J = 8.9, 14.5 Hz), 2.82– 2.89 (m, 1H), 3.10 (dd, 1H, J = 3.4, 14.0 Hz), 3.36 (dd, 1H, J = 3.5, 14.3 Hz), 3.44 (dd, 1H, J = 4.5, 14.6 Hz), 3.62–3.67 (m, 1H), 3.72 (dd, 1H, J = 5.0, 9.9 Hz), 3.77 (dt, 1H, J = 1.8, 8.2 Hz), 3.87 (dd, 1H, J = 5.8, 9.9 Hz), 3.92–3.98 (m, 2H), 3.99 (dd, 1H, J = 5.0, 9.0 Hz), 4.15–4.29 (m, 1H), 4.43 (t, 1H, J = 8.8 Hz), 4.97 (q, 1H, J = 5.4, 13.2 Hz), 5.60 (d, 1H, J = 5.3 Hz), 6.66 (d, 2H, J = 8.7 Hz), 7.15–7.18 (m, 1H), 7.20–7.25 (m, 4H), 7.49 (d, 2H, J = 8.6 Hz); 13C-NMR (125 MHz, CDCl3) δ 25.76, 29.56, 34.75, 45.51, 53.33, 54.67, 55.03, 56.62, 67.57, 69.60, 71.18, 73.47, 109.31, 114.02, 124.27, 126.30, 128.30, 129.22, 129.42, 137.86, 151.29, 155.83, 159.65; LRMS-ESI (m/z) [M+Na]+ 613.2; HRMS-ESI (m/z) [M+Na]+ calcd for C27H34N4O9S 613.1944 found 613.1938.
Determination of X-ray structure of 19b–bound HIV protease (WT)
The stabilized HIV-1 protease with the substitutions of Q7K, L33I, L63I, C67A and C95A that reduce autoproteolysis and aggregation27 was expressed and purified as described28. These mutations do not alter the inhibitor binding site and the stabilized protease has kinetic parameters and stability indistinguishable from those of the unsubstituted enzyme27,28. Inhibitor 19b was dissolved in dimethylsulfoxide (DMSO). Crystals were grown by the hanging drop vapor diffusion method using 1:5 molar ratio of protease (at 3.9 mg/mL) to inhibitor. The reservoir contained 0.1 M citrate phosphate buffer, pH 5.0, 0.35 M NaCl and 4% DMSO. Crystals were mounted on a nylon loop and flash-frozen in liquid nitrogen with a cryoprotectant of 30% (v/v) glycerol. X-ray diffraction data were collected on the SER-CAT beamLine of the Advanced Photon Source, Argonne National Laboratory. Diffraction data were processed using HKL2000 to 1.29 Å resolution.28 Data were reduced in space group P21212 with unit cell dimensions of a = 58.11 Å, b = 86.42 Å, c = 45.97 Å with one dimer in the asymmetric unit. The structure was solved by molecular replacement using the CCP4i29,30, using WT/GRL0255 complex (PDB 3DJK) as a starting model. 26 The structure was refined using SHELX9731 and refitted manually using the molecular graphics program COOT33. Alternate conformations were modeled for the protease residues where obrserved in the electron density maps. Anisotropic atomic displacement parameters (B-factors) were refined for all atoms including solvent molecules. Hydrogen atoms were automatically added by SHELXL in the last round of the refinement. The identity of ions and other solvent molecules from the crystallization conditions was deduced from the shape and peak height of the 2Fo-Fc and Fo-Fc electron density, the hydrogen bond interactions and interatomic distances. The solvent molecules were one sodium ion, two chloride ions, one glycerol molecule and 207 water (including partial occupancy sites). The final R was 14.1 % for working set and Rfree was 18.2 % for all data between 10 and 1.29 Å resolution. The rmsd values from ideal bonds and angle distances were 0.013 Å and 0.031 Å, respectively. The average B-factor was 15.9 and 22.7 Å2 for protease main chain and side chain atoms, respectively, 14.1 Å2 for inhibitor atoms and 32.1 Å2 for water atoms. The crystallographic coordinates and structure factors have been deposited in the Protein Databank (PDB) with access code 3H5B.34
Supplementary Material
Figure 1.

Structures of inhibitors 1–4.
Acknowledgements
The research was supported by grants from the National Institutes of Health (GM53386, AKG and GM62920, IW). This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health and in part by a Grant-in-aid for Scientific Research (Priority Areas) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Monbu Kagakusho), a Grant for Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan (Kosei Rohdosho: H15-AIDS-001), and the Grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Re emerging Infectious Diseases (Renkei Jigyo: No. 78, Kumamoto University) of Monbu-Kagakusho. We thank the staff at the SER-CAT beamline at the Advanced Photon Source, Argonne National Laboratory, for assistance during X-ray data collection. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Abbreviations
- HIV
human immunodeficiency virus
- bis-THF
bis-tetrahydrofuran
- Cp-THF
cyclopentanyltetrahydrofuran
- PI
protease inhibitor
- HAART
highly active antiretroviral therapy
- APV
amprenavir
- DRV
darunavir
- SQV
saquinavir
- IDV
indinavir
- LPV
lopinavir
- RTV
ritonavir
Footnotes
The PDB accession code for 19b-bound HIV-1 protease X-ray structure is 3H5B.
Supporting Information Available. HPLC and HRMS data of inhibitors. This material is available free of charge via the Internet at http://pubrs.acs.org.
References
- 1.Flexner C. HIV-protease inhibitors. N. Engl. J. Med. 1998;338:1281–1292. doi: 10.1056/NEJM199804303381808. [DOI] [PubMed] [Google Scholar]; (b) Cihlar T, Bischofberger N. Annual Reports in Medicinal Chemistry. Vol. 35. San Diego,CA: Academic Press; 2000. Chapter 16: Recent developments in antiretroviral therapies; pp. 177–189. [Google Scholar]
- 2.Sepkowitz KA. AIDS-the first 20 years. N. Engl. J. Med. 2001;344:1764–1772. doi: 10.1056/NEJM200106073442306. [DOI] [PubMed] [Google Scholar]
- 3.UNAIDS/WHO Report on Annual AIDS Epidemic Update; United Nations Publications: December, 2008 http://www.unaids.org/epi/2008/.
- 4.(a) Waters L, Nelson M. Why do patients fail HIV therapy? Int. J. Clin. Pract. 2007;61:983–990. doi: 10.1111/j.1742-1241.2007.01383.x. [DOI] [PubMed] [Google Scholar]; (b) Grabar S, Pradier C, Le Corfec E, Lancar R, Allavena C, Bentata M, Berlureau P, Dupont C, Fabbro-Peray P, Poizot-Martin I, Costagliola D. Factors associated with clinical and virological failure in patients receiving a triple therapy including a protease inhibitor. AIDS. 2000;14:141–149. doi: 10.1097/00002030-200001280-00009. [DOI] [PubMed] [Google Scholar]
- 5.(a) Pillay D, Bhaskaran K, Jurriaans S, Prins M, Masquelier B, Dabis F, Gifford R, Nielsen C, Pedersen C, Balotta C, Rezza G, Ortiz M, de Mendoza C, Kucherer C, Poggensee G, Gill J, Porter K. The impact of transmitted drug resistance on the natural history of HIV infection and response to first-line therapy. AIDS. 2006;20:21–28. doi: 10.1097/01.aids.0000196172.35056.b7. [DOI] [PubMed] [Google Scholar]; (b) Wainberg MA, Friedland G. Public health implications of antiretroviral therapy and HIV drug resistance. J. Am. Med. Ass. 1998;279:1977–1983. doi: 10.1001/jama.279.24.1977. [DOI] [PubMed] [Google Scholar]
- 6.Yerly S, Kaiser L, Race E, Bru JP, Clavel F, Perrin L. Transmission of antiretroviral drug resistant HIV-1 variants. Lancet. 1999;354:729–733. doi: 10.1016/S0140-6736(98)12262-6. [DOI] [PubMed] [Google Scholar]
- 7.(a) Ghosh AK, Kincaid JF, Cho W, Walters DE, Krishnan K, Hussain KA, Koo Y, Cho H, Rudall C, Holland L, Buthod J. Potent HIV protease inhibitors incorporating high-affinity P2-ligands and (R)-(hydroxyethylamino)sulfonamide isostere. Bioorg. Med. Chem. Lett. 1998;8:687–690. doi: 10.1016/s0960-894x(98)00098-5. [DOI] [PubMed] [Google Scholar]; (b) Ghosh AK, Shin D, Swanson L, Krishnan K, Cho H, Hussain KA, Walters DE, Holland L, Buthod J. Structure-based design of non-peptide HIV protease inhibitors. Farmaco. 2001;56:29–32. doi: 10.1016/s0014-827x(01)01025-4. [DOI] [PubMed] [Google Scholar]; (c) Ghosh AK, Sridhar PR, Kumaragurubaran N, Koh Y, Weber IT, Mitsuya H. Bis-tetrahydrofuran: A privileged ligand for darunavir and a new generation of HIV protease inhibitors that combat drug resistance. ChemMedChem. 2006;1:939–950. doi: 10.1002/cmdc.200600103. [DOI] [PubMed] [Google Scholar]
- 8.(a) Yoshimura K, Kato R, Kavlick MF, Nguyen A, Maroun V, Maeda K, Hussain KA, Ghosh AK, Gulnik SV, Erickson JW, Mitsuya H. A potent human immunodeficiency virus type 1 protease inhibitor, UIC-94003 (TMC-126), and selection of a novel (A28S) mutation in the protease active site. J. Virol. 2002;76:1349–1358. doi: 10.1128/JVI.76.3.1349-1358.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents. Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky A, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. Structure-Based Design of Novel HIV-1 Protease Inhibitors To Combat Drug Resistance. J. Med. Chem. 2006;49:5252–5261. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- 10.FDA approved Darunavir on June 23, 2006: FDA approved new HIV treatment for patients who do not respond to existing drugs. Please see: http://www.fda.gov/bbrs/topics/NEWS/2006/NEW01395.htmL
- 11.On October 21, 2008, FDA granted traditional approval to Prezista (darunavir), co-administered with ritonavir and with other antiretroviral agents, for the treatment of HIV-1 infection in treatment-experienced adult patients. In addition to the traditional approval, a new dosing regimen for treatment-naïve patients was approved.
- 12.Ghosh AK, Chapsal BD, Weber IT, Mitsuya H. Design of HIV protease inhibitors targeting protein backbone: An effective strategy for combating drug resistance. Acc. Chem. Res. 2008;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- 13.Hong L, Zhang XC, Hartsuck JA, Tang J. Crystal structure of an in vivo HIV-1 protease mutant in complex with saquinavir: Insights into the mechanisms of drug resistance. Protein Sci. 2000;9:1898–1904. doi: 10.1110/ps.9.10.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Tie Y, Kovalevsky AY, Boross P, Wang YF, Ghosh AK, Tozser J, Harrison RW, Weber IT. Atomic resolution crystal structures of HIV-1 protease and mutants V82A and I84V with saquinavir. Proteins. 2007;67:232–242. doi: 10.1002/prot.21304. [DOI] [PubMed] [Google Scholar]; (b) Kovalevsky AY, Tie Y, Liu F, Boross PI, Wang YF, Leshchenko S, Ghosh AK, Harrison RW, Weber IT. Effectiveness of nonpeptide clinical inhibitor TMC-114 on HIV-1 protease with highly drug resistant mutations D30N, I50V, and L90M. J. Med. Chem. 2006;49:1379–1387. doi: 10.1021/jm050943c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Tie Y, Boross PI, Wang YF, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. High resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multi-drug-resistant clinical strains. J. Mol. Biol. 2004;338:341–352. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]; (b) Amano M, Koh Y, Das D, Li J, Leschenko S, Wang YF, Boross PI, Weber IT, Ghosh AK, Mitsuya H. A novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI), GRL-98065, is potent against multiple-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents Chemother. 2007;51:2143–2155. doi: 10.1128/AAC.01413-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Ghosh AK, Leshchenko S, Noetzel M. Stereoselective Photochemical 1,3-Dioxalene Addition to 5-Alkoxymethyl-2(5H)-Furanone: Synthesis of Bis-Tetrahydrofuranyl Ligand for HIV Protease Inhibitor UIC-94017 (TMC-114) J. Org. Chem. 2004;69:7822–7829. doi: 10.1021/jo049156y. [DOI] [PubMed] [Google Scholar]; (b) Ghosh AK, Li J, Ramu SP. A Stereoselective Anti-aldol Route to (3R,3aS,6aR)-Tetrahydro-2H-furo[2,3-b]furan-3-ol: A Key Ligand for a New Generation of HIV Protease Inhibitors. Synthesis. 2006:3015–3019. doi: 10.1055/s-2006-942547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Ghosh AK, Duong TT, McKee SP. Di(2-pyridyl) carbonate promoted alkoxycarbonylation of amines: A convenient synthesis of functionalized carbamates. Tetrahedron Lett. 1991;32:4251–4254. doi: 10.1016/S0040-4039(00)92141-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghosh AK, Duong TT, McKee SP, Thompson WJ. N,N‘-dissuccinimidyl carbonate: A useful reagent for alkoxycarbonylation of amines. Tetrahedron Lett. 1992;33:2781–2784. doi: 10.1016/S0040-4039(00)78856-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Dondoni A, Perrone D. Synthesis of 1,1-Dimethylethyl (S)-4-Formyl-2,2-Dimethyl-3-Oxazolidinecarboxylate by Oxidation of the Alcohol. Org. Synth. :2004. [Google Scholar]; Coll. Vol. 10:320. [Google Scholar]; Org. Synth. 2000;77:64. [Google Scholar]; (b) Dondoni A, Giovannini PP, Massi A. Assembling Heterocycle-Tethered C-Glycosyl and α-Amino Acid Residues via 1,3-Dipolar Cycloaddition Reactions. Org. Lett. 2004;6:2929–2932. doi: 10.1021/ol048963g. [DOI] [PubMed] [Google Scholar]
- 19.Toth MV, Marshall GR. A simple, continuous fluorometric assay for HIV protease. Int. J. Pep. Protein Res. 1990;36:544–550. doi: 10.1111/j.1399-3011.1990.tb00994.x. [DOI] [PubMed] [Google Scholar]
- 20.Koh Y, Das D, Leshchenko S, Nakata H, Ogata-Aoki H, Amano M, Nakayama M, Ghosh AK, Mitsuya H. GRL-02031: A Novel Nonpeptide Protease Inhibitor (PI) Containing A Stereochemistry Defined Fused Cyclopentanyltetrahydrofuran (Cp-THF) Potent Against Multi-PI-Resistant HIV-1 In Vitro. Antimicrob. Agents Chemother. 2009 doi: 10.1128/AAC.00689-08. Accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gustchina A, Sansom C, Prevost M, Richelle J, Wodak S, Wlodawer A, Weber I. Energy calculations and analysis of HIV-1 protease-inhibitor crystal structures. Protein Eng. 1994;7:309–317. doi: 10.1093/protein/7.3.309. [DOI] [PubMed] [Google Scholar]
- 22.Tie Y, Boross PI, Wang YF, Gaddis L, Liu F, Chen X, Tozser J, Harrison RW, Weber IT. Molecular basis for subrstrate recognition and drug resistance from 1.1 to 1.6 Å resolution crystal structures of HIV-1 protease mutants with subrstrate analogs. FEBS J. 2005;272:5265–5277. doi: 10.1111/j.1742-4658.2005.04923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Panigrahi S, Desiraju G. Strong and weak hydrogen bonds in the protein-ligand interface. Proteins. 2007;67:128–141. doi: 10.1002/prot.21253. [DOI] [PubMed] [Google Scholar]
- 24.Steiner T. Hydrogen bonds from water molecules to aromatic acceptors in very high-resolution protein crystal structures. Biophys. Chem. 2002;95:195–201. doi: 10.1016/s0301-4622(01)00256-3. [DOI] [PubMed] [Google Scholar]
- 25.Wang YF, Tie Y, Boross PI, Tozser J, Ghosh AK, Harrison RW, Weber IT. Potent new antiviral compound shows similar inhibition and structural interactions with drug resistant mutants and wild type HIV-1 protease. J. Med. Chem. 2007;50:4509–4515. doi: 10.1021/jm070482q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh AK, Gemma S, Baldridge A, Wang YF, Kovalevsky AY, Koh Y, Weber IT, Mitsuya H. Flexible cyclic ethers/polyethers as novel P2-ligands for HIV-1 protease inhibitors: Design, synthesis, biological evaluation, and protein-ligand X-ray studies. J. Med. Chem. 2008;51:6021–6033. doi: 10.1021/jm8004543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Louis JM, Clore GM, Gronenborn AM. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat. Struct. Biol. 1999;6:868–875. doi: 10.1038/12327. [DOI] [PubMed] [Google Scholar]
- 28.Mahalingam B, Louis JM, Hung J, Harrison RW, Weber IT. Structural implications of drug resistant mutants of HIV-1 protease: High resolution crystal structures of the mutant protease/subrstrate analog complexes. Proteins. 2001;43:455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- 29.Otwinowski Z, Minor W. Processing of X-ray diffraction data in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 30.Collaborative Computational Project, Number 4 The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 31.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta. Cryst. 2003;D59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 32.Sheldrick GM, Schneider TR. SHELXL: High resolution refinement. Methods in Enzymology. 1997;277:319–343. [PubMed] [Google Scholar]
- 33.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 34.Emsley P, Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 35.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.