

Abstract
Background and Purpose
We investigated the role of thrombin in early brain injury after subarachnoid hemorrhage (SAH).
Methods
The standard intravascular perforation model was used to produce experimental SAH in Sprague Dawley rats. Low dose (0.3mg/hr) and high dose (0.3mg/hr) of argatroban, a direct thrombin inhibitor were evaluated for effects on brain edema, blood brain barrier (BBB) disruption, apoptotic cell death, inflammatory marker and neurological outcomes after SAH.
Results
Both doses of argatroban attenuated BBB disruption, however, only high dose was effective in lowering edema in all brain regions, reducing cell death and inflammatory marker expression, and improving neurological outcomes.
Conclusions
Thrombin inhibition by argatroban improves neurological outcomes and provides neuroprotection against acute events after SAH such as BBB disruption, brain edema and cell death.
Keywords: Early brain injury, subarachnoid hemorrhage, thrombin, argatroban, brain edema, Aneurysms, Apoptosis, Blood Brain Barrier, Edema, Brain, Experimental, Inflammation, SAH, Subarachnoid Hemorrhage, Vasospasm
Early brain injury comprising of BBB disruption, brain edema and global ischemia is important in the pathophysiology of SAH; however, the mechanisms are not clearly understood.1 Thrombin, a serine protease coagulation protein, has been implicated in BBB disruption and brain edema after cerebral ischemia and intracerebral hemorrhage.2-4 We investigated the role of thrombin by using argatroban, a direct inhibitor2-4 in the standard rat intravascular perforation model for SAH.5,6
Methods
All procedures were approved by Loma Linda University animal care committee. The intravascular perforation SAH model was used as previously described in adult male Sprague Dawley rats.5,6 143 animals were divided into four groups: sham (n=24), vehicle (n=39, saline with hydrochloric acid pH 1.4-1.6), low-dose (n=35, 0.3mg/hr) and high-dose (n=45, 0.9mg/hr) argatroban delivered intraperitoneally 15 mins after SAH using osmotic minipumps (ALZET, Alza Corp, Palo Alto, CA).2 Animals having mild SAH (3 from vehicle-treated, 2 from low-dose and 2 from high-dose argatroban groups) were excluded from the study as per the SAH grading system criteria reported previously; animals obtaining grade less than 5/18 on blinded evaluation were classified as mild SAH and excluded.6 The animals were not randomly allocated however, the neurological status of the animals was evaluated by a blinded observer using an 18-point scoring scale and right forelimb placing test before euthanization.7 Brain water content (BWC) of the cerebral hemispheres, cerebellum and brain stem was examined to assess brain edema and spectrophotometric quantitation of Evan's blue dye extravasation into the cerebral hemispheres provided a measure of BBB disruption at 24hrs as described previously.5,6 Cell Death Detection ELISA kit (Roche Applied Science, Indianapolis, USA) was used to quantify cell death in left hemisphere at 24hrs and 72hrs.8 Standard western blotting protocol9 using the following antibodies: rabbit polyclonal zona occludens-1 (ZO-1) antibody from Invitrogen, mouse monoclonal IL-1β antibody, and goat polyclonal actin antibody from Santa Cruz Biotechnology, was performed on brain tissue from left hemisphere (ipsilateral to perforation) at 24hrs. All molecular studies were performed by blinded researcher. The data are expressed as mean±SEM and differences between groups were assessed with a one-way analysis of variance (ANOVA) with Holm-Sidak post-hoc analysis with P<0.05 considered statistically significant.
Results
Physiological parameters were not significantly different amongst groups. Mortality rates (calculated using Chi-square test) were not significantly different amongst the vehicle and treatment groups at 24hrs (Vehicle = 22%, Low dose Agratroban = 45% and High dose Argatroban = 40%) and 72hrs (Vehicle = 54%, High dose Argatroban = 54%). Sham animals had zero mortality. All animals subjected to experimental SAH had comparable SAH grades.6
Neurological and forelimb placement scores were significantly worse in vehicle group compared to sham over 24-72hrs. Neurological deficits were not improved by either dose of argatroban at 24hrs, however, high-dose argatroban showed significant improvement at 48 and 72 hrs after SAH (Fig 1A-D).
Figure 1. Neurological Outcomes after Argatroban Treatment.

Neurological Outcomes. Figs show impaired neurological scores and right forelimb placement test deficits in SAH+vehicle group compared to sham at 24hrs (Figs A,C) and over time course 24-72hrs (Figs B,D). Argatroban (low and high dose) did not ameliorate these neurological deficits at 24hrs but high-dose argatroban (0.9mg/kg) significantly ameliorated the neurological deficits at 48 and 72hrs after SAH. * p<0.05; ** p<0.01 vs Sham group, # p<0.05; ## p<0.01 vs Vehicle group. At 24 hrs, n as follows: sham = 24, vehicle = 24, low dose Argatroban = 18, high dose Argatroban = 24. At 48 and 72 hrs, each group had 6 animals.
BWC was significantly increased in all brain areas in vehicle group compared to sham. High-dose argatroban significantly lowered the BWC in hemispheres, cerebellum and brain stem; however, low-dose argatroban only reduced BWC in left hemisphere (ipsilateral to perforation) and cerebellum (Fig 2A). Evans blue dye extravasation was significantly increased in hemispheres in vehicle group as compared to sham and attenuated by both doses of argatroban (Fig 2B). Expression of ZO-1, a tight junction protein, was significantly decreased in vehicle group compared to sham; however reversed by high-dose but not low-dose argatroban (Fig 2C)
Figure 2. Evaluation of Brain Edema and Blood Brain Barrier Permeability.
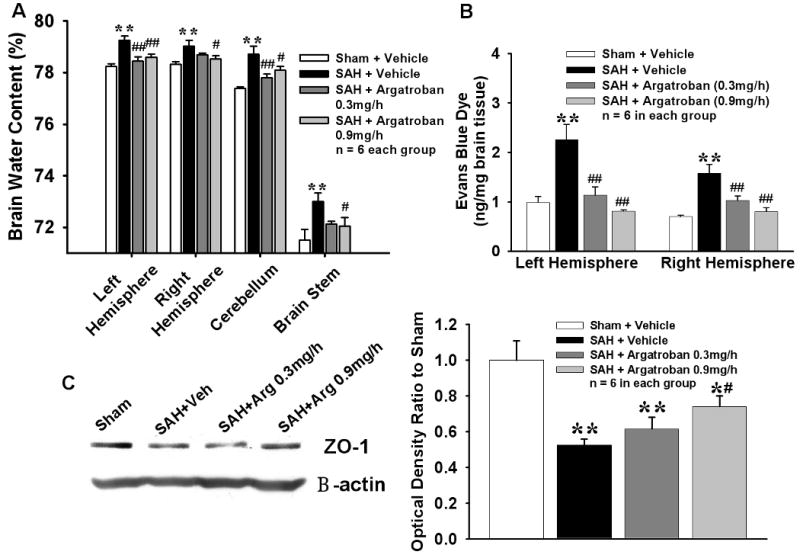
Brain Edema and Blood Brain Barrier Permeability. Figs A and B show increased water content in different areas of the brain and Evan's blue dye extravasation in hemispheres in vehicle group at 24hrs after SAH. High-dose argatroban reduced edema in all brain areas and low-dose argatroban decreased edema in left hemisphere and cerebellum; whereas both doses attenuated dye extravasation. * p<0.05; ** p<0.01 vs Sham group, # p<0.05; ## p<0.01 vs Vehicle group in Fig A. ** p<0.01 vs respective hemisphere in Sham group, ## p<0.01 vs respective hemisphere in Vehicle group. Fig C shows decreased ZO-1 protein expression in the left hemisphere (ipsilateral) in vehicle group compared to sham at 24 hrs after SAH, which was significantly reversed by high-dose argatroban treatment. * p<0.05; ** p<0.01 vs Sham group, # p<0.05; ## p<0.01 vs Vehicle group. n= 6 in each group.
Significantly higher cell death was detected at 24hrs but not at 72hrs in the vehicle group compared to sham; this was attenuated by high-dose but not low-dose argatroban (Fig 3A). Increased expression of inflammatory marker, IL1β in the vehicle group (compared to sham) at 24hrs was attenuated by high-dose but not low-dose argatroban (Fig 3B).
Figure 3. Apoptosis And Inflammatory Marker.
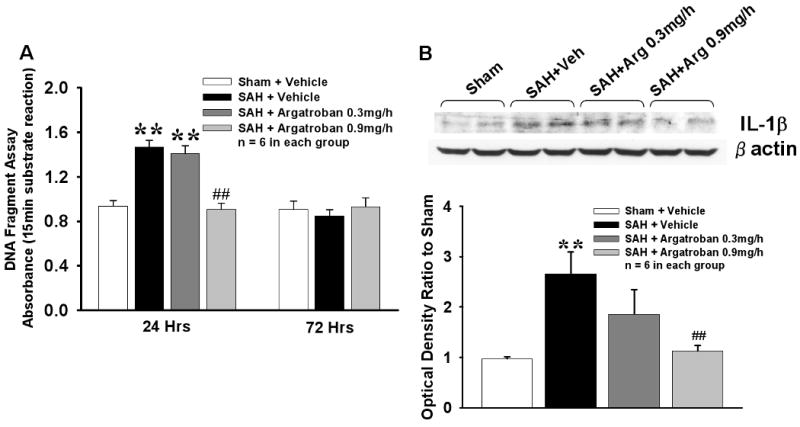
Apoptosis and Inflammatory Marker. Significantly increased apoptotic cell death and expression of IL1β were observed in vehicle group compared to sham at 24hr after SAH. High dose (0.9mg/h) but not low dose (0.3mg/h) argatroban significantly decreased apoptosis as well as IL1β expression. ** p<0.01 vs respective Sham group, ## p<0.01 vs respective Vehicle group. n = 6 in each group.
Discussion
The present study showed for the first time that thrombin plays a role in early brain injury after SAH. Brain edema following BBB disruption is a key event in early brain injury after SAH.1,4,10 High-dose of argatroban, a direct thrombin inhibitor, prevented the BBB disruption and reduced the brain edema after SAH with subsequent improvement in neurological status. High-dose argatroban also attenuated cell death and expression of inflammatory marker after SAH.
Cell death and inflammation are critical in BBB disruption and brain edema.1,4,10-12 The early brain injury peaks at 24hrs after SAH as indicated by cell death observed at 24hrs but not at 72hrs similar to previous studies; this temporal profile was also seen with brain edema and BBB disruption.11 It remains to be determined whether the anti-cell death effect of argatroban is cell specific or pan-cellular; 13 however, our BBB studies suggested that argatroban may protect the endothelial cells. Moreover, the expression of ZO-1, a tight junction protein was preserved by high-dose of argatroban after SAH indicating a protective effect on the BBB.
The lack of argatroban effect in improving neurological deficits at early time point of 24hrs could be dose related14 or due to incomplete recovery of animals from the SAH post-ictal state.11 Furthermore, the global ischemia caused by SAH may warrant further higher doses which will be examined in future studies. Argatroban has been used clinically in Japan and Korea for thromboembolic disorders and more recently for ischemic stroke.15 However, in US and Canada it has been approved only for prophylaxis and treatment of thrombosis in patients with heparin-induced thrombocytopenia.15 There are reported side-effects such as increased hemorrhagic episodes and gastrointestinal bleeding and hepatic dysfunction. In this study, we did not encounter any hemorrhagic events (brain and gastric) with both low and high dose of argatroban. This evaluation was done by gross examination post-euthanization. Other studies using the same dosage in rats have reported not encountering any intracranial or systemic rebleeding side effects.2
Macroscopically, the livers also looked normal in all treatment groups. Favorable pharmacokinetics and clinical evidence in other cerebrovascular disorders make argatroban a promising therapeutic modality to be evaluated in clinical trials for SAH.15
Conclusions
Argatroban, a direct thrombin inhibitor, ameliorated BBB disruption and brain edema with improvement in neurological outcomes and exhibited anti-cell death and anti-inflammatory effects coincident with early brain injury after SAH.
Acknowledgments
Mitsubishi Tanabe Pharma Corporation, Osaka, Japan generously provided Argatroban. Work was supported in part by grants NS45694, NS53407, and NS43338 from NIH to JHZ.
Footnotes
Conflicts of Interest Disclosures:
Takashi Sugawara: None
Vikram Jadhav: None
Robert Ayer: None
Wanqiu Chen: None
Hidenori Suzuki: None
John Zhang: Research grants awarded by NIH NS45694, NS53407, and NS43338
References
- 1.Hansen-Schwartz J, Vajkoczy P, Macdonald RL, et al. Cerebral vasospasm: looking beyond vasoconstriction. Trends Pharmacol Sci. 2007;28:252–256. doi: 10.1016/j.tips.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 2.Kitaoka T, Hua Y, Xi G, Hoff J, Keep R. Delayed argatroban treatment reduces edema in a rat model of intracerebral hemorrhage. Stroke. 2002;33:3012–3018. doi: 10.1161/01.str.0000037673.17260.1b. [DOI] [PubMed] [Google Scholar]
- 3.Nagatsuna T, Nomura S, Suehiro E, Fujisawa H, Koizumi H, Suzuki M. Systemic administration of argatroban reduces secondary brain damage in a rat model of intracerebral hemorrhage: histopathological assessment. Cerebrovasc Dis. 2005;19:192–200. doi: 10.1159/000083466. [DOI] [PubMed] [Google Scholar]
- 4.Ohyama H, Hosomi N, Takahashi T, Mizushige K, Kohno M. Thrombin inhibition attenuates neurodegeneration and cerebral edema formation following transient forebrain ischemia. Brain Res. 2001;902:264–271. doi: 10.1016/s0006-8993(01)02354-x. [DOI] [PubMed] [Google Scholar]
- 5.Park S, Yamaguchi M, Zhou C, Calvert J, Tang J, Zhang JH. Neurovascular protection reduces early brain injury after subarachnoid hemorrhage. Stroke. 2004;35:2412–2417. doi: 10.1161/01.STR.0000141162.29864.e9. [DOI] [PubMed] [Google Scholar]
- 6.Sugawara T, Ayer R, Jadhav V, Zhang JH. A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J Neurosci Methods. 2008;167:327–334. doi: 10.1016/j.jneumeth.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hua Y, Schallert T, Keep RF, Wu J, Hoff J, Xi G. Behavioral tests after intracerebral hemorrhage in the rat. Stroke. 2002;33:2478–2484. doi: 10.1161/01.str.0000032302.91894.0f. [DOI] [PubMed] [Google Scholar]
- 8.Endo H, Nito C, Kamada H, Yu F, Chan PH. Akt/GSK3beta survival signaling is involved in acute brain injury after subarachnoid hemorrhage in rats. Stroke. 2006;37:2140–2146. doi: 10.1161/01.STR.0000229888.55078.72. [DOI] [PubMed] [Google Scholar]
- 9.Ostrowski RP, Colohan AR, Zhang JH. Mechanisms of hyperbaric oxygen-induced neuroprotection in a rat model of subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2005;25:554–571. doi: 10.1038/sj.jcbfm.9600048. [DOI] [PubMed] [Google Scholar]
- 10.Sehba FA, Bederson JB. Mechanisms of acute brain injury after subarachnoid hemorrhage. Neurol Res. 2006;28:381–398. doi: 10.1179/016164106X114991. [DOI] [PubMed] [Google Scholar]
- 11.Cahill J, Calvert JW, Solaroglu I, Zhang JH. Vasospasm and p53-induced apoptosis in an experimental model of subarachnoid hemorrhage. Stroke. 2006;37:1868–1874. doi: 10.1161/01.STR.0000226995.27230.96. [DOI] [PubMed] [Google Scholar]
- 12.Sercombe R, Dinh YR, Gomis P. Cerebrovascular inflammation following subarachnoid hemorrhage. Jpn J Pharmacol. 2002;88:227–249. doi: 10.1254/jjp.88.227. [DOI] [PubMed] [Google Scholar]
- 13.Donovan F, Pike C, Cotman C, Cunningham D, et al. Thrombin induces apoptosis in cultured neurons and astrocytes via a pathway requiring tyrosine kinase and RhoA activities. J Neurosci. 1997;17:5316–5326. doi: 10.1523/JNEUROSCI.17-14-05316.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.LaMonte MP. Argatroban in thrombotic stroke. Pathophysiol Haemost Thromb. 2002;32 3:39–45. doi: 10.1159/000069108. [DOI] [PubMed] [Google Scholar]
- 15.Moledina M, Chakir M, Gandhi PJ. A synopsis of the clinical uses of argatroban. J Thromb Thrombolysis. 2001;12:141–149. doi: 10.1023/a:1012919404290. [DOI] [PubMed] [Google Scholar]