

Abstract
During Adeno-Associated Virus and Adenovirus (AAV/Ad) coinfection, accumulation of viral genomes and proteins can alter cellular stress responses. To determine how AAV/Ad coinfection affects the host we screened over 60 cellular proteins for their responses. AAV/Ad coinfections induce a robust DNA damage response (DDR) that is distinct from that induced by Ad infection alone. Using chemical inhibitors, deficient cell lines and siRNA knockdowns of the DDR kinases, ATM, ATR and DNA-PK, we determined that DNA-PK and ATM kinases are the initial transducers of this response. AAV/Ad coinfection induces ATM-and DNA-PK mediated phosphorylation of RPA2, NBS1, H2AX and the checkpoint kinases CHK1/2. Inhibition of one or more of the DDR kinases reduces the level of phosphorylation of downstream targets but does not dramatically reduce Ad or AAV protein expression. However, AAV DNA levels are moderately affected by kinase inhibition. These experiments provide new insights into the cellular responses to AAV/Ad coinfections.
Keywords: AAV infection, DNA damage responses
INTRODUCTION
Adeno-associated virus (AAV) belongs to the Parvoviridae family and the Dependovirus genus (Muzyczka and Berns, 2001). AAV is classified as a Dependovirus because it depends upon coinfection of another virus to efficiently propagate itself. AAV replicates most efficiently with an adenovirus (Ad) coinfection, but varying levels of helper functions are provided by other DNA viruses. The helper virus dependence is not absolute and AAV replicates at low levels when cultured cells are treated with genotoxic agents (Yakobson et al., 1989; Yakobson, Koch, and Winocour, 1987; Yalkinoglu et al., 1988). The 4,680 nucleotide, single-stranded AAV2 genome contains 145 bp inverted terminal repeat (ITR) elements that form unique T-shaped hairpins at each end. The 3′ ITR serves as the primer for DNA replication and the single strand genome is converted to a double strand conformation by cellular DNA polymerases. The AAV genome contains two translation open reading frames encoding four replication (Rep) proteins and three capsid (Cap) proteins. Expression of the largest Rep protein, Rep78, is detectable approximately four hours after infection and the remaining Rep proteins appear 8–12 h post-infection (Redemann, Mendelson, and Carter, 1989). Replicative form (RF) AAV DNA is detectable approximately 10 h post-infection and Cap proteins accumulate as AAV DNA is replicated. AAV RF DNA accumulates up to 105 copies per infected cell nucleus (Rose and Koczot, 1972). Recombinant AAV (rAAV) vectors are under intense study for treating a variety of acquired and hereditary diseases (Carter, Burstein, and Peluso, 2004).
Ad helper functions are provided by several early gene proteins that enhance AAV replication (Muzyczka and Berns, 2001). E1a proteins activate transcription of Ad early genes as well as AAV rep and cap genes (Chang, Shi, and Shenk, 1989). E1a proteins induce entry of the host cell into S-phase, which enables Ad and AAV replication. E1b-55k and E4orf6 proteins form a complex involved in the conversion of single- to double-stranded DNA and the transport of AAV mRNA from the nucleus to the cytoplasm (Samulski and Shenk, 1988). E4orf6 is also involved in cell cycle arrest by inhibiting CDC2 kinase and enhancing Cyclin A degradation (Grifman et al., 1999). This function may explain how rAAV vectors are converted to a double-stranded conformation by co-expression of E4orf6 (Ferrari et al., 1996; Fisher et al., 1996). E2a single-stranded DNA binding protein (ssDBP) enhances AAV elongation and is the only Ad protein proposed to play a direct role in AAV replication (Ward et al., 1998). E2a ssDBP also stimulates AAV gene expression (Chang and Shenk, 1990).
Host cells respond to virus infection in multiple ways to defend themselves from the deleterious effects of viral proteins and replicating viral nucleic acids. Several reviews have nicely summarized these studies (Weitzman et al., 2004; Weitzman and Ornelles, 2005). A series of cellular sensors and signal transduction pathways are activated by different types of DNA damage as well as by the accumulation of replicating viral DNA. Ataxia telangiectasia mutated (ATM), ATM-Rad3 related (ATR) or double strand DNA-activated protein kinase (DNA-PK) are considered the initial responders to cellular DNA damage (Carson et al., 2003; Collis et al., 2005; Lavin et al., 2005; Zhou et al., 2006). They belong to the phosphoinositide 3-kinase-related kinase (PI3K) family of kinases. These kinases act as signal transducers phosphorylating a variety of substrates required for DNA damage and repair (DDR). ATM and ATR are activated upon recruitment to the DNA lesion by the MRN complex of proteins. The MRN complex, comprised of MRE11, RAD50 and NBS1, is one of the primary sensors that detect double strand breaks. While the exact mechanism of DSB recognition has not been defined, it is proposed that MRN interacts with the DSBs and recruits one or more of the PI3-like protein kinases. Activation of ATM and or ATR sets off a cascade of phosphorylation events that direct the cessation of cell cycle progression, DNA repair or apoptosis. DNA-PK is another major player in the nonhomologous end joining pathway (Collis et al., 2005). The components of this pathway, DNA-PK, Ku70/80, Artemis, XRCC4 and DNA ligase IV are active in V(D)J immunoglobulin gene rearrangement. The ability of DNA-PK to function as a DDR signal transducer is evident in its multiple substrates and roles in DNA repair, apoptosis, telomere maintenance and innate immunity (Burma and Chen, 2004; Collis et al., 2005).
In an Ad infection, accumulation of replicating viral DNA results in activation of the non-homologous end joining (NHEJ) DNA repair pathway leading to concatemerization of Ad DNA and inhibition of viral replication. The (DDR) response is overcome when Ad inactivates NHEJ by targeting the Mre11-Rad50-NBS1 (MRN) complex for degradation. Using a series of Ad mutants and cell lines deficient for different DNA repair proteins, the Weitzman group showed that Ad E4 proteins inactivate the MRN complex thus preventing the NHEJ repair pathway from concatemerizing replicating Ad genomes (Carson et al., 2003; Stracker, Carson, and Weitzman, 2002). The MRN complex allowed for concatemer formation of E4 mutant viruses and colocalized to Ad replication centers. Unlike Ad E4 mutants, wild type Ad genomes did not concatemerize and mediated limited ATM activation. Thus the virus encodes factors that block concatemer formation and the DNA damage response. Two redundant functions encoded by the Ad E4 orf3 and orf6 proteins mediate these effects (Araujo et al., 2005; Evans and Hearing, 2005; Liu et al., 2005; Stracker et al., 2005). A unique characteristic of the E4orf3 protein is the reorganization of promyelocytic leukemia (PML) organizing domains (PODs) which have been implicated in a variety of cellular processes (Maul, 1998). E4 orf3 redirects the MRN complex to cytoplasmic aggresomes thus preventing their participation in NHEJ (Evans and Hearing, 2005). The E4 orf6 protein forms a complex with the E1b 55K protein that interacts with Elongins B and C, Cullin 5 and additional proteins to form an E3 ubiquitin-protein ligase that ubiquitinates one or more members of the MRN thus targeting them for degradation (Carson et al., 2003; Liu et al., 2005; Stracker, Carson, and Weitzman, 2002). The E1b 55K-E4orf6 complex also directs the degradation of p53, which is a well characterized player in the DNA repair response (Harada et al., 2002; Querido et al., 2001). Ad infection also leads to degradation of DNA ligase IV (Baker et al. 2007). Thus the inactivation of cellular DDR responses is essential for a productive Ad infection
Here we show that AAV/Ad coinfections induce a robust DDR response that is distinct from that induced by Ad infection alone. Using cell lines deficient for PI3 family kinases, drug-inhibition studies and siRNA knockdown experiments we show that DNA-PK and ATM are the principal signal transducers of this response resulting in phosphorylation of multiple downstream targets including the CHK1/2 kinases, RPA2, NBS1 and histone H2AX. Inhibition of ATM or ATR results in decreased AAV replication whereas inhibition of DNA-PK results in an increase in virus replication. These studies show that AAV and Ad coinfections trigger cellular DNA damage responses that affect the ability of the virus to replicate itself.
RESULTS
Induction of a DDR response in AAV/Ad coinfected cultures
To determine how the combined effects of AAV and Ad coinfection alter the host cell we initiated a survey of cellular protein and phosphoprotein levels. Hela cells were infected with Ad at a multiplicity of infection (m.o.i.) of 5 and AAV at an m.o.i. of 100. Cultures were harvested at 6, 12, 18, 24 and 36 h. At harvest, cellular extracts were prepared, proteins were separated by SDS-PAGE, transferred to nylon membrane and probed with antibodies specific to over 60 cellular proteins. Subtle differences were observed in the levels of several cell cycle regulation proteins. Cyclin B levels decreased slightly in AAV/Ad coinfected cells compared to Ad-infected cells whereas p21 increased slightly (data not shown). These changes have been noted previously by others (Saudan, Vlach, and Beard, 2000). One consistent change we have observed is that Cyclin A levels are consistently reduced in AAV/Ad coinfected cells compared to Ad-infected cells (Fig. 1).
Figure 1. AAV/Ad-mediated DDR responses in Hela cells.
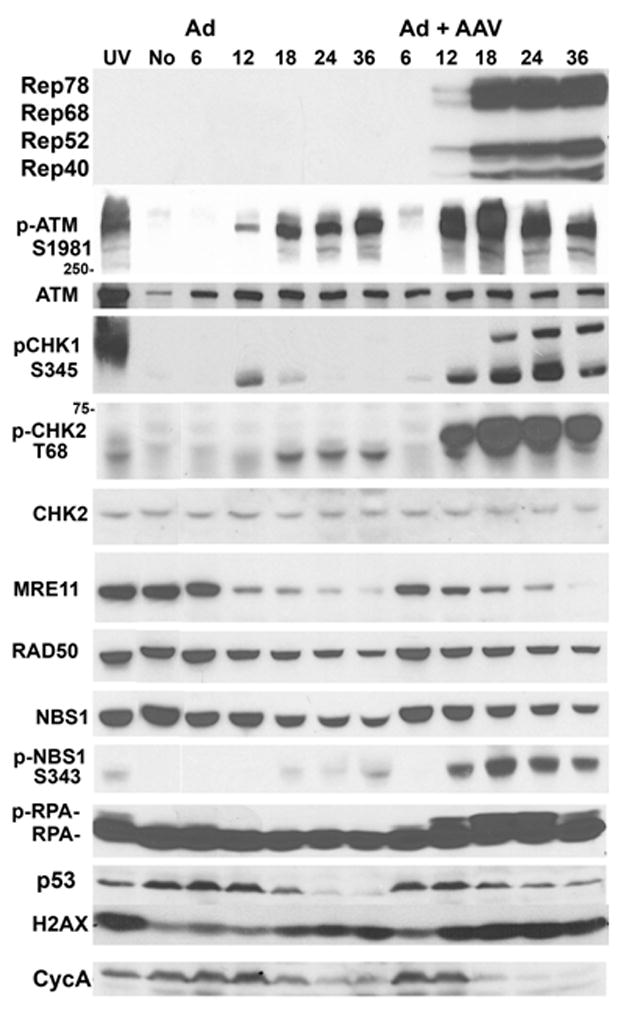
Hela cells were treated with UV light (UV), left untreated (No), infected with Ad, or coinfected with AAV and Ad. UV-treated cultures were harvested one hour after irradiation. Infected cells were harvested at 6, 12, 18, 24 and 36 hours after infection. Cells were fractionated and equal amounts of protein were separated by SDS-PAGE and immunoblotted with the antibodies indicated to the left of the figure.
The most striking changes were observed in cellular proteins involved in the DNA damage and repair response (Fig. 1). In Ad-infected cultures there was an increase in activated ATM, as determined by phosphorylation on Ser1981. However in AAV/Ad coinfected cultures there was a much stronger activation of ATM beginning at 12 h post-infection. Activation of ATM is one of the initial signaling events in response to double strand DNA breaks (Lavin et al., 2005; Shiloh, 2006). ATM activation was accompanied by a strong activation of the checkpoint kinases, CHK1 and CHK2 on Ser348 and Thr68 respectively (Fig. 1). In Ad-infected cultures there was less activation of CHK2. There was also no effect on the level of total CHK2 under any of the infection conditions. CHK1 was activated as measured by phosphorylation on S345 and occurs at the same time point as CHK2 activation. We have yet to observe a repeatable difference in the total levels of Chk2 (Fig. 1) or CHK1 (data not shown) between Ad and AAV/Ad infections (data not shown). However MRE11, RAD50 and NBS1 levels decreased as the infection progressed for both Ad and AAV/Ad coinfected cultures as has been observed by others in Ad-infected cells (Carson et al., 2003; Stracker, Carson, and Weitzman, 2002). Although NBS1 levels decreased during AAV/Ad coinfection, there was a dramatic increase in the amount of NBS1 that is phosphorylated on Ser343 beginning at the 12 h time point (pNBS1 in Fig. 1).
Replication protein A (RPA) is a single stranded (ss) DNA binding protein required for DNA replication, repair and homologous recombination. RPA is a heterotrimer composed of subunits of 70 (RPA1), 34 (RPA2) and 14 kD (RPA3), During UV- and IR-induced DNA damage responses, RPA is hyper-phosphorylated by members of the PI3-related kinase family (Wold, 1997). Hyperphosphorylation occurs primarily on the 34 kD RPA2 subunit and can be detected by altered mobility in SDS-PAGE. We have found that RPA2 mobility is altered in AAV/Ad coinfected cells and is first detected at the 12 h time point (Fig. 1). Altered RPA mobility is not observed in Ad-infected cells. Other immunoblot analyses using antibodies specific for phosphorylated Ser4 and Ser8 in RPA2, indicate that these residues are indeed phosphorylated in response to AAV/Ad coinfection (results not shown).
The cellular p53 tumor suppressor protein is a pivotal component in the cellular response to cellular stressors such as DNA damage (Meek, 2004). Both genotoxic and nongenotoxic stressors induce and stabilize p53 resulting in changes in expression of p53-responsive genes. Ad infection mediates degradation of p53 thus allowing the virus to counteract p53’s role in a variety of responses (Harada et al., 2002; Querido et al., 2001). However, in AAV/Ad coinfection there is a modest stabilization of p53 levels (Fig. 1). While the change in p53 stabilization is not as dramatic a change as that of the other DNA repair proteins, this change may contribute to the AAV-mediated cellular response to infection.
DNA damage causes phosphorylation of histone H2AX by the PI3-like kinases ATM, ATR and DNA-PK (Stucki and Jackson, 2006). Phosphorylated H2AX (γ;H2AX) contributes to DNA repair by interacting with DNA flanking the lesion. Although its role in repair is not fully understood it is thought to link the DNA damage response machinery to the lesion-containing DNA. Ad infection stimulates H2AX phosphorylation whereas the stimulation is more pronounced in an AAV/Ad coinfection (Fig. 1).
The AAV/Ad-mediated DDR response is dependent upon wild type virus coinfection
The DDR response was more pronounced in an AAV/Ad coinfection than in the absence of AAV. The response may be due to the combined effects of the AAV virion and Ad. Alternatively, a productive AAV infection may be required to cause the DDR response. To further characterize the DDR response, Hela cells were infected with AAV or a recombinant AAV vector alone or in combination with Ad infection. 18–24 hr post-infection, the cultures were harvested and analyzed by immunoblot to assess ATM, NBS1, CHK1/2, RPA2 and H2AX phosphorylation. AAV at an m.o.i. of 100 or rAAV at the same m.o.i. did not induce phosphorylation of ATM, NBS1 or CHK1/2 (Fig. 2, lanes 2 and 4 respectively). NBS1 was phosphorylated when cells are infected with Ad alone, Ad plus a rAAV vector or Ad plus AAV coinfection (Fig. 2, lanes 2, 5 and 6). However these infection conditions did not affect ATM or CHK1/2 phosphorylation. Histone H2AX was slightly phosphorylated with Ad, rAAV, wtAAV and Ad plus rAAV (lanes 2–5). The most robust DDR response was observed only with AAV and Ad coinfection in which all 5 proteins were phosphorylated (lane 6). The slight increase in H2AX induced with rAAV and wtAAV may represent the earliest stages of the DDR response and that as AAV genomes accumulate in a coinfection, the full DDR response results. Although Ad coinfection with rAAV vectors results in efficient conversion of the single strand vector DNA to a double strand conformation (Ferrari et al., 1996; Fisher et al., 1996), this was not sufficient to induce the full DDR response. To test whether ectopic AAV Rep protein expression in conjunction with Ad infection induces the DDR response, we transfected Rep-expressing plasmids into Ad-infected Hela cells. 24–48 h after transfection, the cultures were harvested and analyzed by immunoblot for phosphorylated ATM and CHK2. These experiments did not show any activation of the ATM or CHK2 kinases (data not shown). This suggests that Rep protein expression, when combined with Ad infection, does not induce the DDR response. Amplification of the AAV DNA in the presence of Ad is essential to trigger the signal transduction cascade.
Figure 2. DDR response in Hela cells infected with AAV, rAAV and Ad.
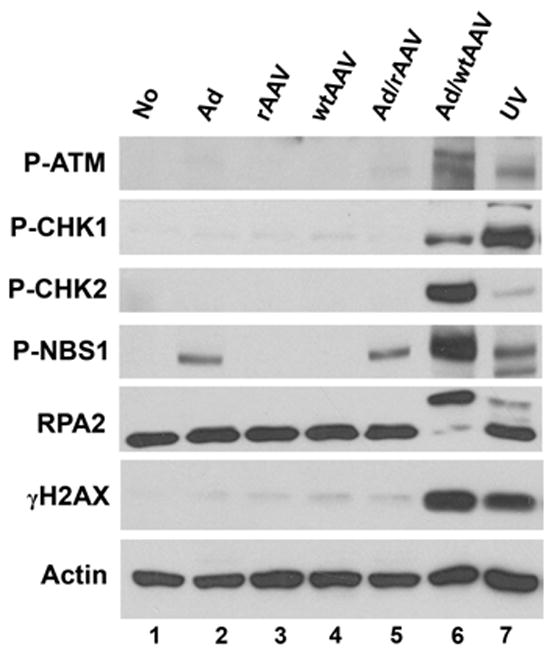
Hela cells were left uninfected (No) or were infected with Ad, a recombinant AAV vector (rAAV), wild type AAV (wtAAV) or combinations thereof. Cultures were harvested 24 hr after infection; lysates were prepared and analyzed by immunoblots using the antibodies indicated on the left side of the figure. UV-irradiated Hela cells (UV) were used as a positive control.
Pharmacological inhibition of ATM and DNA-PK activity in AAV/Ad coinfected cells
To assess the roles of the DDR kinases in the host cell responses to AAV/Ad coinfection we used chemical inhibitors of ATM (KU55933, (Hickson et al., 2004) and DNA-PK (NU7441, (Leahy et al., 2004). We have used these compounds in Hela cells to determine how they may inhibit phosphorylation of downstream targets of ATM, ATR and DNA-PK. Hela cells were treated with the inhibitors 2 hours before infection and up until cultures were harvested 24 h later. Cellular extracts were prepared and immunoblotted to assess levels of phosphorylated CHK1, CHK2, NBS1, H2AX and RPA. Fig. 3 shows that activation of CHK1 was reduced by the ATM inhibitor, KU 55933 (KU) (lane 5), and the DNA-PK inhibitor, NU7441 (NU) (lane 7). When both NU and KU were used, activation of CHK1 was totally inhibited (lane 9). This result suggests that DNA-PK and ATM are involved in phosphorylation of CHK1. CHK2 was inhibited by the DNA-PK inhibitor (NU)(lane 7), but there was only slight inhibition from the ATM inhibitor alone (KU) (lane 5). Combining NU7441 and KU55933 resulted in complete inhibition of CHK2 activation (lane 9). This result suggests that DNA-PK may be a significant activator of CHK2 in AAV/Ad coinfection.
Figure 3. AAV/Ad-mediated DDR response in Hela cells treated with ATM and DNA-PK inhibitors.
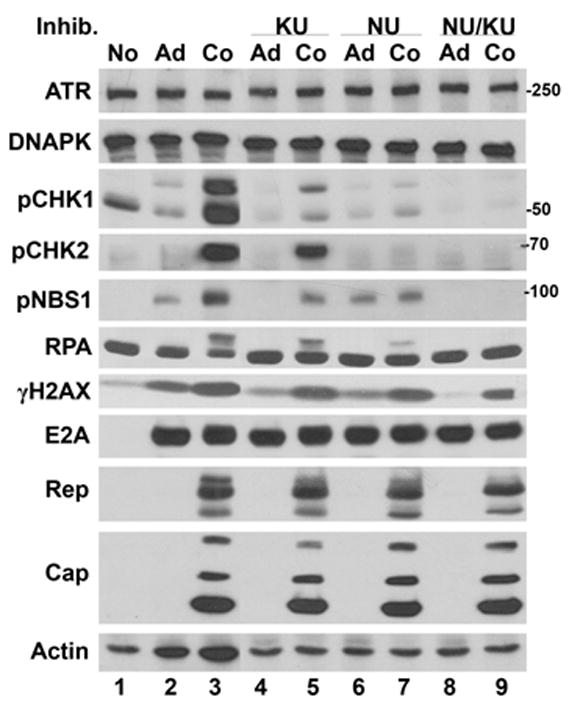
Hela cells were left uninfected (No), infected with Ad (Ad) or coinfected with AAV and Ad (Co). Parallel cultures were Ad or AAV/Ad coinfected and treated with PI3 kinase inhibitors (Inhib.), KU55933 (KU), NU7441 (NU) or both drugs (NU/KU) four hours prior to infection. Cultures were harvested 18–24 h later, cellular lysates prepared and analyzed by immunoblots using the antibodies indicated on the left side of the figure.
There was a modest level of phosphorylation of NBS1 in Ad-infected cells (Figs. 1, 2 and 3) and a more prominent phosphorylation in AAV/Ad coinfected cells. When ATM was inhibited with KU55933, phosphorylation of NBS1 was totally blocked in Ad-infected cells and reduced in AAV/Ad coinfected cultures (Fig. 3, compare lanes 4 and 5). When DNA-PK is inhibited by NU7441, NBS1 phosphorylation was not affected in Ad-infected cells (lane 6) whereas it was reduced in AAV/Ad coinfected cells (lane 7) compared to the drug-free infection (lane 3). When both ATM and DNA-PK were inhibited by NU7441 and KU55933, all NBS1 phosphorylation was inhibited (lanes 8 and 9). Although these experiments are not strictly quantitative, the results suggest that ATM mediates phosphorylation of NBS1 in Ad infections and DNA-PK mediates NBS1 phosphorylation in AAV-infected cells.
The slower migrating band of RPA2, which is the result of phosphorylation, was not affected by KU55933 (lane 5) but was partially inhibited by NU7441 (lane 7). This result suggests that DNA-PK phosphorylates RPA but that another kinase, perhaps ATR, also modifies this cellular, single strand DNA binding protein.
The effects of these small molecule inhibitors on Ad and AAV gene expression was determined by assessing the levels of AAV Rep and Cap proteins and Ad E2A. There were no apparent effects on E2A, AAV Rep and Cap proteins in all of the drug treatments. This result suggests that AAV protein expression is not dependent on DNA- PK or ATM activity.
The AAV/Ad DDR response in ATM-deficient cells
To complement the drug studies described above, we assessed DDR response signaling in cell lines lacking the ATM kinase. GM16666 and GM16667 are fibroblast cell lines from an ATM−/− patient and are transformed by the SV40 T-Ag. GM16667 contains an ATM expression plasmid whereas GM16666 contains the empty cloning vector (Ziv et al., 1997). Using the same infection and analysis procedures described above we found that AAV/Ad coinfections induced CHK1/2 and NBS1 phosphorylation in both the ATM-restored and ATM-deficient cell lines (Fig. 4, lanes 3 and 6, respectively). However when the DNA-PK inhibitor (NU7441) was used in these cell lines, CHK1 phosphorylation was reduced in the ATM-restored cells and eliminated in the ATM-deficient cells. CHK2 phosphorylation was completely inhibited in coinfected cells in the presence of the DNA-PK inhibitor (lanes 9 and 12). NBS1 phosphorylation was reduced in the ATM-restored cell line (lane 9) and abolished in the ATM-deficient cells (lane 12) in the presence of the DNA-PK inhibitor. Phosphorylated RPA was observed in both ATM cell lines but was eliminated in the presence of NU7441. There were no detectable changes in γH2AX. AAV and Ad gene expression were not affected by the absence of ATM, or ATM with inhibition of DNA-PK.
Figure 4. AAV/Ad-mediated DDR responses in ATM-deficient cell lines.
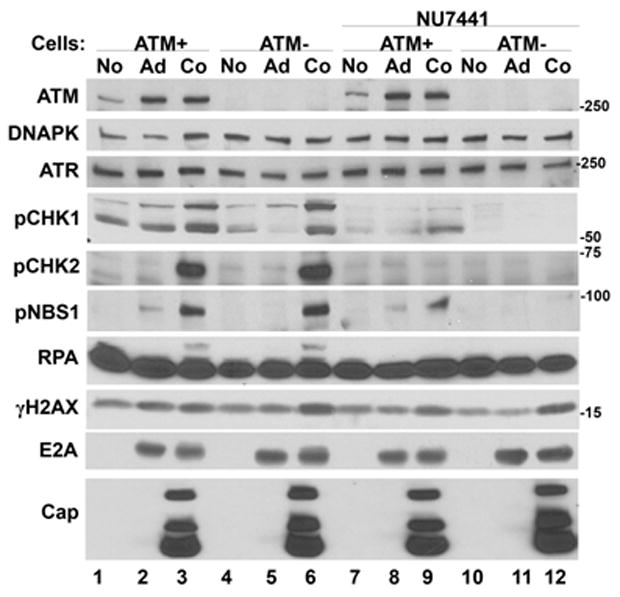
ATM-expressing (ATM+) or ATM-deficient (ATM−) cells were left uninfected (No), infected with Ad (Ad) or coinfected with AAV and Ad (Co). Parallel cultures were similarly infected and treated with NU7441 four hours prior to infection. Cultures were harvested 18–24 h later, cellular lysates prepared and analyzed by immunoblots using the antibodies indicated on the left side of the figure.
The AAV/Ad DDR response in DNA-PK-deficient cells
To complement the studies described above, we assessed the DDR response in cell lines lacking DNA-PK. MO59J cells are derived from a glioblastoma cell line that lack DNA-PK. MO59K cells were isolated from the same tumor and express normal levels of DNA-PK (Allalunis-Turner et al., 1993). AAV/Ad infections of the DNA-PK-containing cells induced phosphorylation of CHK1/2, RPA, γH2AX and NBS1 (Fig. 5, lane 3). Coinfections in cells lacking DNA-PK had reduced CHK1, NBS1 and γH2AX phosphorylation (Fig. 5, lane 6). Whereas in DNA-PK-deficient cells there was no detectable CHK2 or RPA phosphorylation and there was also a reduction in NBS1 phosphorylation (Fig. 5, lane 6). In DNA-PK-containing MO59K cells treated with the ATM inhibitor, KU55933, there was a reduction in NBS1 phosphorylation and a slight reduction in CHK1 phosphorylation whereas RPA and H2AX phosphorylation was not affected (lane 9). NBS1, RPA and H2AX phosphorylation were eliminated in AAV/Ad-infected DNA-PK-deficient cells treated with the ATM inhibitor (lane 12). CHK1 phosphorylation was substantially reduced under these conditions. AAV Cap and Ad E2A protein expression was not affected in the absence of DNA-PK or the absence of DNA-PK and ATM.
Figure 5. AAV/Ad-mediated DDR responses in DNA-PK-deficient cell lines.
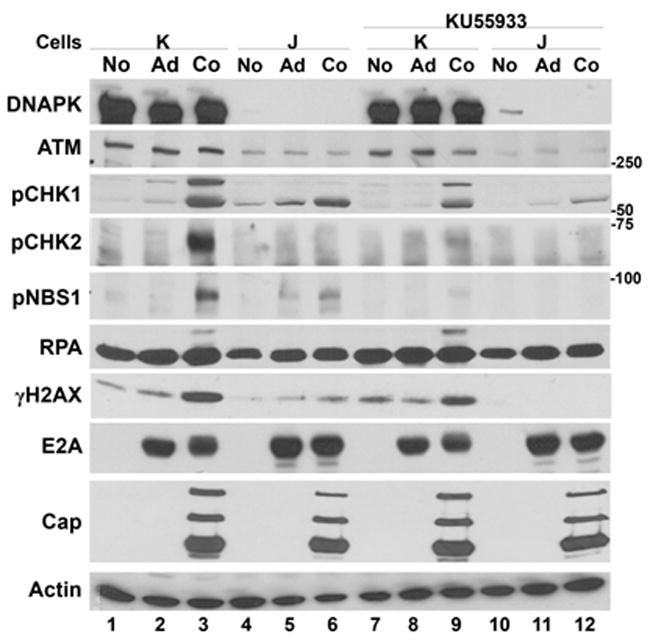
MO59K, DNA-PK-expressing cells (K) or MO59J DNA-PK-deficient (J) glioblastoma cells were left uninfected (No), infected with Ad (Ad) or coinfected with AAV and Ad (Co). Parallel cultures were similarly infected and treated with KU55933 four hours prior to infection. Cultures were harvested 18–24 h later; cellular lysates were prepared and analyzed by immunoblots using the antibodies indicated on the left side of the figure.
The AAV/Ad DDR response in ATR-FLOX cells
To determine if ATR plays a role in the AAV/Ad-mediated DDR response we performed similar experiments in ATR-FLOX cells (Cortez et al., 2001). These cells are derived from HCT116 cells in which one copy of the ATR gene has been disrupted, and the second copy of the gene contains lox sites flanking exon 2 making it susceptible to Cre-mediated deletion. Preliminary experiments showed that infection of ATR-FLOX cells with a Cre-expressing Ad vector (Ad-Cre) three days prior to AAV/Ad coinfection was the time required for maximal reduction of ATR expression resulting in up to 90% reduction [data not shown and (Cortez et al., 2001)]. Reduction of ATR expression by the Ad-Cre vector is evident in Fig. 6. (lanes 5 and 6). ATR levels were not affected when ATR-FLOX cells were infected with an Ad control vector that expresses the E. coli β-galactosidase gene instead of Cre (Fig. 6 lanes 2 and 3). There was no difference in the levels of phosphorylation of ATM, CHK2, NBS1, RPA or H2AX when comparing the Ad-Cre-treated, ATR-deficient, with the Ad-CMV-Gal-treated AAV/Ad coinfected cultures. The only noticeable difference in the AAV/Ad DDR response was a reduction of CHK1 phosphorylation in the Ad-Cre-treated cultures. As observed in the other cell lines, there were negligible effects on Ad E2A or AAV Rep and Cap protein expression.
Figure 6. AAV/Ad-mediated DDR responses in ATR-FLOX cells.

ATR-FLOX cells were infected with the Ad-CMV-Gal or Ad-Cre virus vectors at an MOI of 100. 60 hours later the cells were left uninfected (No), infected with Ad (Ad) or coinfected with AAV and Ad (Co). Cultures were harvested 18–24 h later; cellular lysates were prepared and analyzed by immunoblots using the antibodies indicated on the left side of the figure.
The AAV/Ad DDR response in Hela cells transfected with siRNAs that target ATM, ATR and DNA-PK
As an alternative means of kinase suppression Hela cells were transfected with siRNAs that target ATM, ATR and DNA-PK. Cells were transfected with siRNA on three successive days prior to virus infection and a fourth transfection after infection. 24 h after AAV/Ad infection the cultures were harvested and assessed for phosphorylation of cellular DDR proteins. Fig. 7 shows that we attained nearly 100% reduction of ATR expression (lanes 8 and 9), approximately 75% reduction of ATM (lanes 6 and 7) and 50–75% reduction of DNA-PK (lanes 10 and 11) compared to cells transfected with control siRNA (lanes 4 and 5). When ATM expression was suppressed, phosphorylation of CHK1/2, NBS1 and RPA were reduced compared to the control siRNA transfection (compare lanes 5 and 7). When DNA-PK expression was suppressed there was a reduction of CHK1/2, NBS1, RPA and H2AX phosphorylation (lane 11). When ATR expression was suppressed, phosphorylation of CHK2, NBS1 and RPA were reduced (lane 9) compared to the control siRNA. However CHK1, and perhaps RPA, phosphorylation was reduced more in the absence of ATR than when ATM and DNA-PK were suppressed (compare lane 9 to lanes 7 and 11). There were no significant effects on E2A or Rep expression in these experiments. However there was a slight reduction in Cap expression when ATM was suppressed (lane 7).
Figure 7. AAV/Ad-mediated DDR responses in siRNA-treated Hela cells.
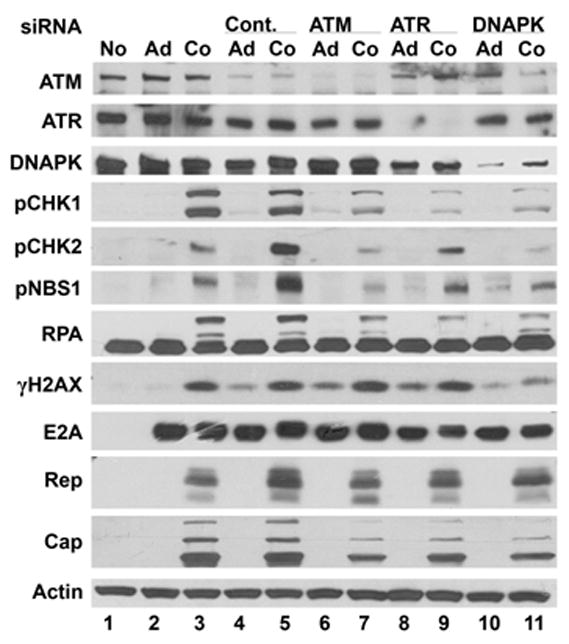
Hela cells were transfected with control siRNA (Cont.) or siRNAs designed to suppress ATM, ATR and DNA-PK expression using the protocol described in the Methods section. Cells were left uninfected (No), infected with Ad (Ad) or coinfected with AAV and Ad (Co). Cultures were harvested 18–24 h after virus infection and cellular lysates were prepared and analyzed by immunoblots using the antibodies indicated on the left side of the figure.
To assess the effects of DDR kinases on AAV DNA replication, parallel siRNA transfections and AAV/Ad coinfections were performed accompanied by treatment with the Ku55933 ATM or Nu4771 DNA-PK inhibitors. 24 h after infection, low molecular weight DNA was prepared and analyzed by Southern hybridization after agarose gel electrophoresis. Fig. 8A shows that when siRNAs targeting ATM or ATR are transfected there was a reduction in replicative form monomer (M) AAV DNA compared to control siRNA transfection. However siRNA-mediated knockdown of DNA-PK resulted in an increase in AAV RF DNA. When siATR transfection was accompanied by Ku55933-mediated inhibition of ATM, there was a further inhibition of AAV replication. However when ATR was reduced by siRNA and DNA-PK was inhibited by Nu4771, there was an increase in AAV replication suggesting that the effects on DNA-PK may be dominant compared to ATR suppression.
Figure 8. AAV DNA amplification in siRNA-treated Hela cells.
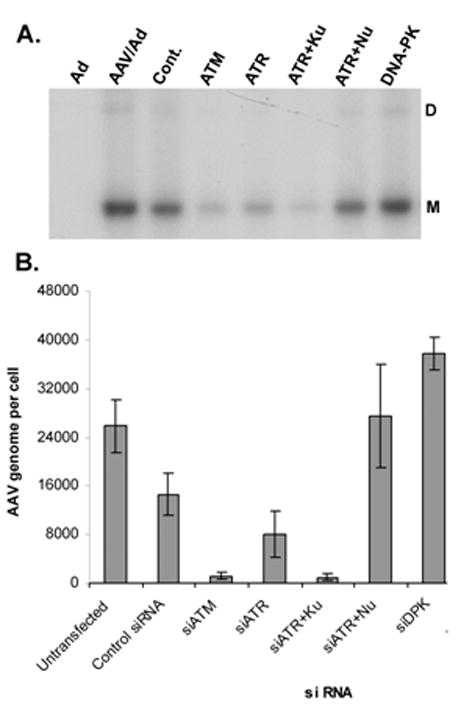
Hela cells were transfected with control siRNA (Cont.) or siRNAs designed to suppress ATM, ATR and DNA-PK expression using the protocol described in the Methods section. Cells were left uninfected (No), infected with Ad (Ad) or coinfected with AAV and Ad (Co). Cultures were harvested 18–24 h after virus infection, viral DNA was prepared and analyzed by agarose gel electrophoresis and Southern hybridization (A) or real-time PCR (B). Replicative form monomer (M) and dimer (D) DNAs are indicated to the right of the image in panel A.
To quantitatively assess the effects of kinase inhibition, the isolated AAV DNA from Fig. 8A was analyzed by real-time PCR. Knockdown of ATM by siRNA or inhibition by Ku55933 resulted in an approximate 6-fold reduction of viral DNA compared to control siRNA transfection (Fig. 8B). Knockdown of DNA-PK by siRNA transfection or inhibition by Nu4771 resulted in an approximate 2–3-fold stimulation of AAV DNA replication compared to the control siRNA transfection.
“Our observations are consistent with similar experiments performed in DNA-PK- and ATM-deficient cell lines. AAV DNA replication in DNA-PK-deficient cells was consistently higher than in the corresponding DNA-PK-containing cells. However replication was consistently lower in ATM-deficient cells compared to ATM-proficient cells (data not shown).”
Cellular localization of cellular DNA repair proteins and AAV Rep in coinfected cells
To investigate the localization of cellular DNA repair proteins in AAV/Ad coinfections, Hela cells were coinfected, fixed 24 h later and immuno-stained for Rep, and the cellular proteins. The Ad E4 orf3 protein redirects the MRN complex out of the nucleus and into cytoplasmic aggresomes thus preventing their participation in NHEJ (Evans and Hearing, 2005). Immunofluorescent staining of MRE11 and RAD50 in coinfected cells shows that AAV does not alter this relocalization. Both MRE11 and RAD50 show punctate staining that is primarily cytoplasmic whereas Rep staining is nuclear (Fig. 9). However, phosphorylated NBS1 shows more significant co-localization with the Rep proteins (Fig. 9). This co-localization suggests that phosphorylated NBS1 may be found in AAV replication centers and thus play a role in the coinfection. We also examined the cellular localization of the AAV Rep protein, ATM and DNA-PK. As described above, both of these kinases are activated by AAV/Ad coinfection. Rep and ATM showed some colocalization in AAV/Ad coinfected nuclei. Rep and DNA-PK showed abundant colocalization in coinfected cells. The significant colocalization of DNA-PK with Rep proteins suggests that the kinase plays a role in viral DNA replication.
Figure 9. Localization of AAV Rep and cellular DDR proteins.
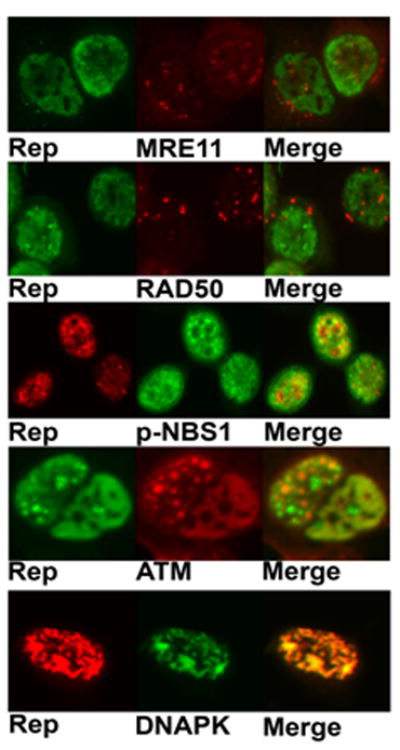
Hela cells growing in chamber slides were coinfected with AAV and Ad. 24 h post-infection the cultures were fixed and treated with primary and secondary antibodies to detect the proteins indicated below each panel. Merged images on the right are indicated.
DISCUSSION
During an AAV/Ad coinfection there is a dramatic accumulation of AAV RF genomes that have a covalently closed DNA end and a free DNA end. The accumulation of over 105 AAV genomes per infected cell (Rose and Koczot, 1972), as well as the expression of the cytotoxic Rep proteins, would likely have significant effects on the cell stress response. Our increasing understanding of how Ad affects the cellular DNA damage response is in stark contrast to what little is known about this response when AAV accompanies Ad in a coinfection; there have been no studies performed that examine if AAV alters Ad effects on the host cell. Studies with recombinant AAV (rAAV) vectors performed in the absence of Ad infection have shown that treatment of cells with DNA damaging agents, UV light, topoisomerase inhibitors and hydroxyurea; stimulate vector transduction (Alexander, Russell, and Miller, 1994; Alexander et al., 1996; Russell, Alexander, and Miller, 1995; Sanlioglu, Duan, and Engelhardt, 1999). DDR repair pathways are involved in the regulation of rAAV transduction efficiency. Using chromatin immunoprecipitation methods, Zentilin et al. showed that Ku86 and RAD52 proteins associated with rAAV DNA (Zentilin, Marcello, and Giacca, 2001). Sanlioglu et al. showed that rAAV transduction efficiency is increased in ATM-deficient fibroblasts (Sanlioglu, Benson, and Engelhardt, 2000). Recombinant AAV vectors form circular episomes upon transduction and this process depends upon functional ATM, DNA-PK, MRE11, NBS1 and the helicases BLM and WRN (Choi, McCarty, and Samulski, 2006; Sanlioglu, Benson, and Engelhardt, 2000). The DNA-PK pathway also affects AAV integration and circularization of recombinant AAV vectors (Duan, Yue, and Engelhardt, 2003; Inagaki et al., 2007; Song et al., 2004). The relevance of these vector studies to AAV/Ad coinfections remains to be determined.
Another study showed that UV-irradiated AAV or undamaged AAV, when introduced into cells in culture induces a single-strand DNA damage response with the activation of ATR signaling (Fragkos et al., 2008). Finally, one report links ectopic expression of the AAV Rep protein with ATM activation but the mechanism of this activation was not determined (Berthet et al., 2005). The relevance of these studies to our results may be limited because neither involve AAV and Ad coinfections.
The studies presented here were designed to assess host cell responses to AAV/Ad coinfection. We screened over 60 cellular proteins for how they respond to coinfection. Our initial efforts focused on cell cycle regulatory proteins and we looked for differences in the overall levels of these proteins in AAV/Ad coinfections compared to Ad infection alone. The only consistent changes we observed were modest decreases in the levels of p21, p53 and Cyclin A. The Cyclin A decrease has been observed in cells infected with AAV Rep protein-expressing retroviruses and may play a role in the Rep protein’s ability to block cell cycle proliferation (Saudan, Vlach, and Beard, 2000). Suppression of Cyclin A may contribute to cell cycle alterations during AAV/Ad coinfections.
The most dramatic changes observed were in proteins known to be altered in the cellular DNA damage responses. We observed increased phosphorylation of ATM, CHK1/2, RPA2, H2AX and NBS1 as the AAV infection progressed. ATM phosphorylation on Ser1981, CHK2 on Thr68 and CHK1 on Ser343 are all considered activating events that trigger subsequent phosphorylations of downstream targets in the DDR (Lavin et al., 2005; Shiloh, 2006). Activation of ATM is one of the initial signaling events in response to double strand DNA breaks. Activation of ATM, as determined by phosphorylation on Ser1981, has been observed in Ad-infected cultures (Carson et al., 2003). We also observed a similar activation in Ad-infected cultures however in AAV/Ad coinfected cultures there was a much stronger activation of ATM beginning at 12 h post-infection. This response corresponds with the accumulation of Rep proteins and AAV RF DNA (Redemann, Mendelson, and Carter, 1989) suggesting that the accumulation of AAV RF DNA may trigger the DDR. That active AAV replication is required for the response is further demonstrated in Fig. 2 where Ad infection alone stimulated phosphorylation of NBS1 but wild type AAV and Ad coinfection was required to elicit phosphorylation of all of the substrates that we studied.
Recently the MRN complex was shown to impede AAV replication and that MRN components accumulate at AAV replication centers in the absence of Ad infection (Schwartz et al., 2007). Our results show that MRE11, RAD50 and NBS1 levels decreased as the infection progressed for both Ad and AAV/Ad coinfected cultures as has been observed by others in Ad-infected cells (Carson et al., 2003; Stracker, Carson, and Weitzman, 2002). There was essentially no difference in the effects on the MRN complex in an AAV/Ad coinfection compared to Ad infection alone. Thus in a coinfection, the inhibition of AAV replication described by Schwartz et al. would not be observed because of the ability of Ad to suppress MRN functions
Although NBS1 levels decreased during AAV/Ad coinfection, there was a dramatic increase in the amount of NBS1 that is phosphorylated on Ser343 beginning at the 12 h time point. Phosphorylation of NBS1 by ATM is involved in the activation of the S-phase checkpoint but was not required for association with the other members of the MRN complex at DNA damage sites (Lavin, 2007). The role of phosphorylated NBS1 and whether it associates with AAV DNA remains to be determined.
The diversity of substrates phosphorylated in response to AAV/Ad infection suggested that other members of the PI3 family kinases including DNA-PK and ATR, may also be involved in AAV/Ad-mediated DNA damage responses. Due to an absence of reliable antibodies that recognize activate ATR and DNA-PK, we used several approaches to identify the kinases that initiate the AAV/Ad-mediated DDR. Pharmacological inhibition of PI3 family kinases has been dependent on the relatively nonspecific inhibitors, caffeine, wortmannin and LY294002. Several new inhibitors have been identified that block kinase activity with more specificity. NU7441 is a selective inhibitor of DNA-PK (IC50=14 nM) (Leahy et al., 2004). KU55933 is a selective inhibitor of ATM (IC50=13 nM) (Hickson et al., 2004). Using these compounds in combination with cell lines that lack the PI3 family kinases and siRNA knockdown experiments we were able to determine which of these kinases are involved in the AAV/Ad DDR. DNA-PK is the predominant kinase that responds to AAV/Ad coinfection in that it apparently phosphorylates all five of the downstream substrates of the PI3 kinases (Fig. 10). Although ATM can phosphorylate both CHK1/2 in a coinfection, DNA-PK is the predominant modifier of the checkpoint kinases that transduce the DDR to other substrates. Activation of the ATR kinase appears to play only a minor role in response to coinfection in that it is involved only in CHK1 phosphorylation.
Figure 10. Signal transduction induced by AAV replication.
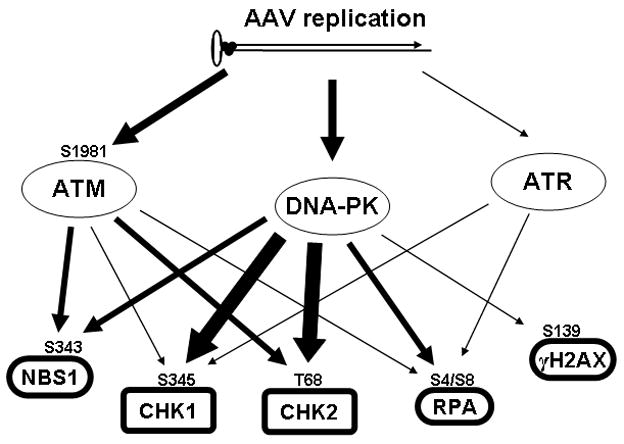
An AAV replicative form monomer with a trimer of attached Rep proteins is indicated at the top of the figure. AAV replication activates initial DDR kinases which in turn results in phosphorylation of downstream substrates.
The Rep proteins are found throughout the nucleus of AAV/Ad coinfected cells. Early in infection, regions of the nucleus contain foci of Rep proteins and AAV DNA that are sites of virus replication and assembly (Bevington et al., 2007; Hunter and Samulski, 1992; Weitzman, Fisher, and Wilson, 1996). Our observation that the AAV Rep proteins co-localize with DNA-PK, and to a lesser extent with ATM, suggests that these kinases play some role in AAV replication or assembly. “DNA-PK is known to phosphorylate numerous proteins involved in DNA repair and recombination (Burma and Chen, 2004; Collis et al., 2005). We also have preliminary evidence that DNA-PK phosphorylates the AAV Rep78 protein on Ser197 (data not shown). We have also shown that phosphorylation of the AAV Rep proteins alters their interactions with the AAV DNA terminal repeats (Collaco, Prasad, and Trempe, 1997). Thus this regulation may play an essential role in viral DNA replication.”
Our results demonstrate a robust cellular DDR response to AAV/Ad coinfection. Surprisingly, elimination of one or two of the PI3 kinases had only a modest effect on viral DNA replication. Removal of ATM alone results in a 2–10-fold decrease in AAV replication. Removal of DNA-PK results in a modest 2–3-fold increase in replication.. Removal of ATR results in a 0–2-fold decrease. The loss of ATR can be counteracted by pharmacological suppression of DNA-PK resulting in an overall increase in replication. We have not yet been successful in obtaining adequate reductions of all three kinases to assess AAV DNA replication under those conditions. The mechanism of how the reduction of ATM and DNA-PK affect virus replication is not clear. As noted earlier, rAAV vector circularization and integration depend upon functional ATM, DNA-PK, MRE11, NBS1 and the helicases BLM and WRN (Choi, McCarty, and Samulski, 2006; Duan, Yue, and Engelhardt, 2003; Inagaki et al., 2007; Sanlioglu, Benson, and Engelhardt, 2000; Song et al., 2004). Loss of DNA-PK would result in less circularization and possibly more DNA replication. How the loss of ATM results in a decrease in replication remains to be seen.
These studies present the first evidence of DNA damage and repair responses that occur in cells coinfected with AAV and Ad. The response is distinctly different from that observed in Ad infected cultures. Further characterization of these responses along with the identification of other substrates modified by the PI3 and checkpoint kinases will provide valuable insights into how AAV replicates to very high titers in coinfected cells.
MATERIALS AND METHODS
Cell lines
Hela cells (American Type Culture Collection) were grown as a monolayer at 37°C (5% CO2 atmosphere) in Eagle’s minimum essential medium (MEM) supplemented with 10%(v/v) fetal bovine serum (FBS), 2 mM L-glutamine, 25 U/mL penicillin, 25 μg/mL streptomycin, 2.5 μg/mL amphotericin B, and 100 μg/mL gentamicin. SV40-transformed AT cell lines, GM16666 and GM16667, were obtained from the Coriell Institute. GM16667 were derived from GM16666 and carry an ATM gene expression vector that corrects the AT−/− phenotype (Ziv et al. Oncogene 15:159 ’97). AT cells were grown in DMEM containing high glucose, 10 %(v/v) fetal bovine serum (FBS), 2 mM L-glutamine, 25 U/mL penicillin, 25 μg/mL streptomycin, 2.5 μg/mL amphotericin B, 100 μg/mL gentamicin and 100 μg/mL of hygromycin. MO59J (lacking DNA-PK) and MO59K (normal DNA-PK) cells were obtained from American Type Culture Collection (ATCC) and were grown in MEM (containing Earle’s Salts) with 2.5 mM L-glutamine and supplemented with 0.05 mM non-essential amino acids,15% FBS, 25 U/mL penicillin, 25 μg/mL streptomycin, 2.5 μg/mL amphotericin B and100 μg/mL gentamicin. ATR-FLOX cells (CRL-2780) were obtained from ATCC and were propagated in McCoy’s 5A medium containing 10% FBS, 25 U/mL penicillin, 25 μg/mL streptomycin, 2.5 μg/mL amphotericin B and100 μg/mL gentamicin.
Virus preparation and infections
AAV2 and Ad5 were prepared by methods previously described (Carter et al., 1979; Casper et al., 2005; Winters and Russell, 1971). Ad infections were performed at a multiplicity of infection (m.o.i.) of 1 or 5. AAV infections were performed at an m.o.i. of 50 or 100. Ad-CRE (Cat. No. 1700) and Ad-CMV-Gal (Cat. No. 1080) were obtained form Vector Biolabs.
UV treatment
For UV treatment of cells, the media was carefully removed and saved in a sterile tube. Cells were washed gently with Phosphate Buffered Saline (PBS). The PBS was aspirated and the cells were UV-treated at 100 mJ/cm2 in a UV-Stratalinker (Stratagene). In case of multi-well plates, the untreated wells (containing media) were covered with aluminum foil. After cross-linking, the media was added back to the dish and the cells were incubated for another 2–21/2 hours at 37°C before harvesting and extraction.
Drug treatment
The ATM inhibitor (KU55933) and the DNA-PK inhibitor (NU7441) were kindly provided by Dr. Graeme Smith (KuDOS Pharmaceuticals, Cambridge, UK). The inhibitors were added to cells where indicated (final concentration of 10 μM) 4 hrs prior to infection and left on for the duration of the experiment.
Immunoblot analyses
Cells were harvested 18–20 hrs post infection and extracts were prepared depending on the protein to be detected. ATM, ATR, DNA-PK, Mre11, Rad50, pNbs1, Nbs1, Chk2, pChk2, AAV Rep/Cap and Adenovirus E2a proteins were detected using the IPP (Immunoprecipitation) buffer extraction. Chk1, pChk1, p53, p34 and γH2A.X were detected using 1X SDS Sample Buffer (Cell Signaling) extraction method as described below. For IPP extracts, cells were resuspended (150 μl per 1.2 million cells) in chilled IPP Buffer [50 mM Tris (pH=8), 150 mM NaCl, 20 mM EDTA, 0.5% NP-40, 20 mM DTT; pH=8] supplemented with protease and phosphatase inhibitors [1mm PMSF, 1 μg/ml Pepstatin A, 1 mM Benzamidine, 10 μM Leupeptin, 1 mM Sodium orthovanadate, 1 mM Sodium fluoride, 1 mM Sodium pyrophosphate, 1.75 mM B-glycerophosphate]. The suspensions were vortexed vigorously for 15 seconds and kept on ice for 60 minutes with occasional vortexing. Extracts were sonicated for 6 × 1 sec pulses at level 3 (Fisher Scientific Sonic Dismembrator). Extracts were centrifuged at 16,000×g for 10 min at 4°C. Supernatants were separated from pellets and protein assays were performed to determine protein concentration. For Denaturing extracts, cells were resuspended (150 μl per 1.2 million cells) in 1X SDS sample Buffer [62.5 mM Tris-HCl (pH=6.8), 2% SDS, 10% glycerol, 50 mM DTT, 5% B-ME, 0.01% bromophenol blue]. Lysates were vortexed briefly and sonicated (12 × 1 sec pulses: level 4). Extracts were heated to 100°C in a water bath for 10 min. and allowed to cool for 5 min. Extracts were centrifuged at 16,000×g for 10 min at RT. Supernatants were separated from pellets and protein assays were performed to determine protein concentration. For western blots of P-I-3 kinases, 8% polyacrylamide (stock = 49:1, acrylamide:bisacrylamide) SDS gels were used. For the remaining proteins, 10% polyacrylamide (stock = 29:1, acrylamide:bisacrylamide) SDS gels were used. Western blots were performed using different antibodies according to the manufacturer’s recommendations.
Antibodies
The following antibodies were used for western blot analysis: ATM (A300-135A, Bethyl Laboratories Inc.), p-ATM (200–301–400, Rockland), ATR (SC-1887, Santa Cruz Biotechnology), DNA-PK (sc-9051, Santa Cruz Biotechnology, Inc.), Rad50 (GTX70228, GeneTex, Inc.), Mre11 (GTX70212, GeneTex, Inc.), Nbs-1 (GTX70224, GeneTex, Inc.), p-Nbs1 (sc-12936-R, Santa Cruz Biotechnology, Inc.), Chk2 (KAM-CC-1113, Assay Designs), pChk2 (2661S, Cell Signaling Technology), Chk1 (KAM-CC-111, Assay Designs), p-Chk1 (2348S, Cell Signaling Technology), p53 (OP09–100ug, Calbiochem), RPA/p34 (MS-691-P1ABX, Thermo Scientific) Actin (sc-1615 Santa Cruz Biotechnology, Inc.), AAV Cap (Mouse monoclonal, Intergen), AAV Rep (Rabbit polyclonal, Rockland), Ad E2a (Dr. Tom Shenk), γH2A.X (ab18311, Abcam).
siRNA transfection
The siRNA transfections were performed using HiPerfect transfection reagent (Qiagen) according to the manufacturer’s protocol. The siRNAs were prepared according to the manufacturers (Qiagen) recommendation; ATM (Hs_ATM_8; SI00604730), ATR (Hs_ATR_12; SI02664347), DNA-PK (Hs_PRKDC_6; SI02224236), Control (1022076). The cells were sequentially transfected four times and the viral infections were performed just before the final transfection. The cells were harvested and extra-chromosomal DNA was isolated using a QIAamp DNA mini kit (Qiagen) and used for real time PCR as described below..
Real Time PCR
Replicative genome copy number was determined by quantitative real time PCR of wt AAV genomes. Primers for PCR were designed to amplify a 301 bp fragment. The forward primer was 5′-AACTGGTTCGCGGTCACAA-3′ (AAV nt 708) and the reverse primer was 5′-ACCCGACCAGCTCCATGTAC-3′ (AAV nt 1008). Primers were used at a concentration of 1.5 μM and Power Sybr Green master mix (Applied Biosystems) was used to perform PCR. Amplification was initiated at 95°C for 10min to activate the polymerase followed by 40 amplification cycles [95°C for 30 sec, 54°C for 45 sec and 72°C for 45 sec]. Genome copy number was determined by comparison to a standard curve plotted after amplification of the same fragment from plasmid pNTC244 at 10-fold serial dilutions starting from 1×1010 copies to 1×102 copies. All experimental and serial dilution templates were run in triplicates. Data was analyzed using 7500 system SDS software (Applied Biosystems).
Acknowledgments
We thank Drs. Kandace Williams and Stephen Patrick for many helpful discussions and providing us with several important antibodies. The work was supported in part by NIH grants AI51471 and GM64765.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander IE, Russell DW, Miller AD. DNA-damaging agents greatly increase the transduction of nondividing cells by adeno-associated virus vectors. J Virol. 1994;68(12):8282–7. doi: 10.1128/jvi.68.12.8282-8287.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander IE, Russell DW, Spence AM, Miller AD. Effects of gamma irradiation on the transduction of dividing and nondividing cells in brain and muscle of rats by adeno-associated virus vectors. Hum Gene Ther. 1996;7(7):841–50. doi: 10.1089/hum.1996.7.7-841. [DOI] [PubMed] [Google Scholar]
- Allalunis-Turner MJ, Barron GM, Day RS, 3rd, Dobler KD, Mirzayans R. Isolation of two cell lines from a human malignant glioma specimen differing in sensitivity to radiation and chemotherapeutic drugs. Radiat Res. 1993;134(3):349–54. [PubMed] [Google Scholar]
- Araujo FD, Stracker TH, Carson CT, Lee DV, Weitzman MD. Adenovirus type 5 E4orf3 protein targets the Mre11 complex to cytoplasmic aggresomes. J Virol. 2005;79(17):11382–91. doi: 10.1128/JVI.79.17.11382-11391.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthet C, Raj K, Saudan P, Beard P. How adeno-associated virus Rep78 protein arrests cells completely in S phase. Proc Natl Acad Sci U S A. 2005;102(38):13634–9. doi: 10.1073/pnas.0504583102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevington JM, Needham PG, Verrill KC, Collaco RF, Basrur V, Trempe JP. Adeno-associated virus interactions with B23/Nucleophosmin: identification of sub-nucleolar virion regions. Virology. 2007;357(1):102–13. doi: 10.1016/j.virol.2006.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burma S, Chen DJ. Role of DNA-PK in the cellular response to DNA double-strand breaks. DNA Repair (Amst) 2004;3(8–9):909–18. doi: 10.1016/j.dnarep.2004.03.021. [DOI] [PubMed] [Google Scholar]
- Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD. The Mre11 complex is required for ATM activation and the G2/M checkpoint. Embo J. 2003;22(24):6610–20. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter BC, Laughlin CA, delaMaza LM, Myers M. Adeno-associated virus auto-interference. Virology. 1979;92:449–462. doi: 10.1016/0042-6822(79)90149-1. [DOI] [PubMed] [Google Scholar]
- Carter BJ, Burstein H, Peluso RW. Adeno-associated virus and AAV vectors for gene delivery. In: Templeton-Smith N, editor. Gene and Cell Therapy: Therapeutic Mechanisms and Strategies. 2 . Dekker; New York: 2004. pp. 53–101. [Google Scholar]
- Casper JM, Timpe JM, Dignam JD, Trempe JP. Identification of an adeno-associated virus Rep protein binding site in the adenovirus E2a promoter. J Virol. 2005;79(1):28–38. doi: 10.1128/JVI.79.1.28-38.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LS, Shenk T. The adenovirus DNA-binding protein stimulates the rate of transcription directed by adenovirus and adeno-associated virus promoters. J Virol. 1990;64(5):2103–9. doi: 10.1128/jvi.64.5.2103-2109.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LS, Shi Y, Shenk T. Adeno-associated virus P5 promoter contains an adenovirus E1A-inducible element and a binding site for the major late transcription factor. J Virol. 1989;63(8):3479–88. doi: 10.1128/jvi.63.8.3479-3488.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi VW, McCarty DM, Samulski RJ. Host cell DNA repair pathways in adeno-associated viral genome processing. J Virol. 2006;80(21):10346–56. doi: 10.1128/JVI.00841-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaco R, Prasad KMR, Trempe JP. Phosphorylation of the adeno-associated virus replication proteins. Virology. 1997;(23):332–336. doi: 10.1006/viro.1997.8563. [DOI] [PubMed] [Google Scholar]
- Collaco RF. Ph.D. Medical College of Ohio. Toledo; Ohio: 1999. [Google Scholar]
- Collis SJ, DeWeese TL, Jeggo PA, Parker AR. The life and death of DNA-PK. Oncogene. 2005;24(6):949–61. doi: 10.1038/sj.onc.1208332. [DOI] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294(5547):1713–6. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- Duan D, Yue Y, Engelhardt JF. Consequences of DNA-dependent protein kinase catalytic subunit deficiency on recombinant adeno-associated virus genome circularization and heterodimerization in muscle tissue. J Virol. 2003;77(8):4751–9. doi: 10.1128/JVI.77.8.4751-4759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JD, Hearing P. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J Virol. 2005;79 (10):6207–15. doi: 10.1128/JVI.79.10.6207-6215.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari FK, Samulski T, Shenk T, Samulski RJ. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J Virology. 1996;70:3227–3234. doi: 10.1128/jvi.70.5.3227-3234.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher KJ, Gao GP, Weitzman MD, DeMatteo R, Burda JF, Wilson JM. Transduction with recombinant adeno-associated virus for gene therapy is limited by leading -strand synthesis. J Virology. 1996;70:520–532. doi: 10.1128/jvi.70.1.520-532.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragkos M, Breuleux M, Clement N, Beard P. Recombinant adeno-associated viral vectors are deficient in provoking a DNA damage response. J Virol. 2008;82(15):7379–87. doi: 10.1128/JVI.00358-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifman M, Chen NN, Gao GP, Cathomen T, Wilson JM, Weitzman MD. Overexpression of cyclin A inhibits augmentation of recombinant adeno-associated virus transduction by the adenovirus E4orf6 protein. J Virol. 1999;73 (12):10010–9. doi: 10.1128/jvi.73.12.10010-10019.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada JN, Shevchenko A, Shevchenko A, Pallas DC, Berk AJ. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J Virol. 2002;76(18):9194–206. doi: 10.1128/JVI.76.18.9194-9206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64(24):9152–9. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- Hunter LA, Samulski RJ. Colocalization of adeno-associated virus Rep and capsid proteins in the nuclei of infected cells. J Virol. 1992;66(1):317–24. doi: 10.1128/jvi.66.1.317-324.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki K, Ma C, Storm TA, Kay MA, Nakai H. The role of DNA-PKcs and artemis in opening viral DNA hairpin termini in various tissues in mice. J Virol. 2007;81(20):11304–21. doi: 10.1128/JVI.01225-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin MF. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene. 2007;26(56):7749–58. doi: 10.1038/sj.onc.1210880. [DOI] [PubMed] [Google Scholar]
- Lavin MF, Birrell G, Chen P, Kozlov S, Scott S, Gueven N. ATM signaling and genomic stability in response to DNA damage. Mutat Res. 2005;569(1–2):123–32. doi: 10.1016/j.mrfmmm.2004.04.020. [DOI] [PubMed] [Google Scholar]
- Leahy JJ, Golding BT, Griffin RJ, Hardcastle IR, Richardson C, Rigoreau L, Smith GC. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg Med Chem Lett. 2004;14(24):6083–7. doi: 10.1016/j.bmcl.2004.09.060. [DOI] [PubMed] [Google Scholar]
- Liu Y, Shevchenko A, Shevchenko A, Berk AJ. Adenovirus exploits the cellular aggresome response to accelerate inactivation of the MRN complex. J Virol. 2005;79(22):14004–16. doi: 10.1128/JVI.79.22.14004-14016.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul GG. Nuclear domain 10, the site of DNA virus transcription and replication. Bioessays. 1998;20(8):660–7. doi: 10.1002/(SICI)1521-1878(199808)20:8<660::AID-BIES9>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Meek DW. The p53 response to DNA damage. DNA Repair (Amst) 2004;3(8–9):1049–56. doi: 10.1016/j.dnarep.2004.03.027. [DOI] [PubMed] [Google Scholar]
- Muzyczka N, Berns KI. Parvoviridae: The viruses and their replication. In: Knipe DM, Howley PM, editors. Fields Virology. 4 . Vol. 2. Lippincott Williams and Wilkins; Philadelphia: 2001. pp. 2327–2359. [Google Scholar]
- Querido E, Blanchette P, Yan Q, Kamura T, Morrison M, Boivin D, Kaelin WG, Conaway RC, Conaway JW, Branton PE. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev. 2001;15(23):3104–17. doi: 10.1101/gad.926401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redemann BE, Mendelson E, Carter BJ. Adeno-associated virus rep protein synthesis during productive infection. J Virology. 1989;63:873–882. doi: 10.1128/jvi.63.2.873-882.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JA, Koczot F. Adenovirus-associated virus multiplication. VII. Helper requirement for viral deoxyribonucleic acid and ribonucleic acid synthesis. J Virology. 1972;10:1–8. doi: 10.1128/jvi.10.1.1-8.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell DW, Alexander IE, Miller AD. DNA synthesis and topoisomerase inhibitors increase transduction by adeno-associated virus vectors. Proc Natl Acad Sci U S A. 1995;92(12):5719–23. doi: 10.1073/pnas.92.12.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samulski RJ, Shenk T. Adenovirus E1B 55-Mr polypeptide facilitates timely cytoplasmic accumulation of adeno-associated virus mRNAs. J Virol. 1988;62:206–210. doi: 10.1128/jvi.62.1.206-210.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanlioglu S, Benson P, Engelhardt JF. Loss of ATM function enhances recombinant adeno-associated virus transduction and integration through pathways similar to UV irradiation. Virology. 2000;268(1):68–78. doi: 10.1006/viro.1999.0137. [DOI] [PubMed] [Google Scholar]
- Sanlioglu S, Duan D, Engelhardt JF. Two independent molecular pathways for recombinant adeno-associated virus genome conversion occur after UV-C and E4orf6 augmentation of transduction. Hum Gene Ther. 1999;10(4):591–602. doi: 10.1089/10430349950018661. [DOI] [PubMed] [Google Scholar]
- Saudan P, Vlach J, Beard P. Inhibition of S-phase progression by adeno-associated virus Rep78 protein is mediated by hypophosphorylated pRb. EMBO J. 2000;19:4351–4361. doi: 10.1093/emboj/19.16.4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz RA, Palacios JA, Cassell GD, Adam S, Giacca M, Weitzman MD. The Mre11/Rad50/Nbs1 complex limits adeno-associated virus transduction and replication. J Virol. 2007;81(23):12936–45. doi: 10.1128/JVI.01523-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci. 2006;31(7):402–10. doi: 10.1016/j.tibs.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Song S, Lu Y, Choi YK, Han Y, Tang Q, Zhao G, Berns KI, Flotte TR. DNA-dependent PK inhibits adeno-associated virus DNA integration. Proc Natl Acad Sci U S A. 2004;101(7):2112–6. doi: 10.1073/pnas.0307833100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418(6895):348–52. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- Stracker TH, Lee DV, Carson CT, Araujo FD, Ornelles DA, Weitzman MD. Serotype-specific reorganization of the Mre11 complex by adenoviral E4orf3 proteins. J Virol. 2005;79(11):6664–73. doi: 10.1128/JVI.79.11.6664-6673.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucki M, Jackson SP. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 2006;5(5):534–43. doi: 10.1016/j.dnarep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Ward PFBD, O’Donnell ME, Berns KI. Role of the adenovirus DNA-binding protein in in vitro adeno-associated virus DNA replication. J Virology. 1998;72:420–427. doi: 10.1128/jvi.72.1.420-427.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzman MD, Carson CT, Schwartz RA, Lilley CE. Interactions of viruses with the cellular DNA repair machinery. DNA Repair (Amst) 2004;3(8–9):1165–73. doi: 10.1016/j.dnarep.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Weitzman MD, Fisher KJ, Wilson JM. Recruitment of wild-type and recombinant adeno-associated virus into adenovirus replication centers. J Virol. 1996;70:1845–1854. doi: 10.1128/jvi.70.3.1845-1854.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzman MD, Ornelles DA. Inactivating intracellular antiviral responses during adenovirus infection. Oncogene. 2005;24(52):7686–96. doi: 10.1038/sj.onc.1209063. [DOI] [PubMed] [Google Scholar]
- Winters WD, Russell WC. Studies on the assembly of adenovirus in vitro. J Gen Virol. 1971;10:181–194. doi: 10.1099/0022-1317-10-2-181. [DOI] [PubMed] [Google Scholar]
- Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- Yakobson B, Hrynko TA, Peak MJ, Winocour E. Replication of adeno-associated virus in cells irradiated with UV light at 254 nm. J Virol. 1989;63(3):1023–30. doi: 10.1128/jvi.63.3.1023-1030.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakobson B, Koch T, Winocour E. Replication of adeno-associated virus in synchronized cells without the addition of a helper virus. J Virol. 1987;61(4):972–81. doi: 10.1128/jvi.61.4.972-981.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalkinoglu AO, Heilbronn R, Burkle A, Schlehofer JR, zur Hausen H. DNA amplification of adeno-associated virus as a response to cellular genotoxic stress. Cancer Res. 1988;48(11):3123–9. [PubMed] [Google Scholar]
- Zentilin L, Marcello A, Giacca M. Involvement of cellular double-stranded DNA break binding proteins in processing of the recombinant adeno-associated virus genome. J Virol. 2001;75(24):12279–87. doi: 10.1128/JVI.75.24.12279-12287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Lim CU, Li JJ, Cai L, Zhang Y. The role of NBS1 in the modulation of PIKK family proteins ATM and ATR in the cellular response to DNA damage. Cancer Lett. 2006;243(1):9–15. doi: 10.1016/j.canlet.2006.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv Y, Bar-Shira A, Pecker I, Russell P, Jorgensen TJ, Tsarfati I, Shiloh Y. Recombinant ATM protein complements the cellular A-T phenotype. Oncogene. 1997;15(2):159–67. doi: 10.1038/sj.onc.1201319. [DOI] [PubMed] [Google Scholar]