

Abstract
Monocytes play a central role in defense against infection, but the mechanisms promoting monocyte recruitment and activation remain incompletely defined. Defense against Listeria monocytogenes, an intracellular bacterial pathogen, requires in vivo MCP-1 induction and CCR2-dependent recruitment of Ly6Chigh monocytes from bone marrow to sites of infection. Herein, we demonstrate that infection of bone marrow-derived macrophages with virulent L. monocytogenes induces MCP-1 expression in two phases. The first phase is rapid, induces low-level production of MCP-1, and is dependent on TLR/MyD88 signaling. The second phase promotes prolonged, higher level MCP-1 secretion and is dependent on signaling via the type I IFN receptor (IFNAR). Although attenuated L. monocytogenes strains that remain confined to the phagosome trigger TLR/MyD88-mediated signals and induce low-level MCP-1 expression, only cytosol-invasive bacteria promote IFNAR-dependent MCP-1 expression. In vivo, deficiency of either MyD88 or IFNAR signaling does not impair early monocyte emigration from bone marrow and recruitment to infected spleen. Loss of both MyD88 and IFNAR-mediated MCP-1 induction, however, results in deficient Ly6Chigh monocyte recruitment and increased susceptibility to L. monocytogenes infection. Our studies demonstrate that distinct but partially overlapping signal transduction pathways provide redundancy that ensures optimal monocyte recruitment to sites of microbial infection.
Mammalian defense against invading microbes depends on swift and coordinated recruitment of inflammatory cells to sites of infection. Among recruited inflammatory cells, monocytes expressing high levels of the Ly6C surface protein and the CCR2 chemokine receptor play a particularly important role in defense against intracellular pathogens (1). CCR2 deficiency results in diminished emigration of Ly6Chigh monocytes from the bone marrow and decreased recruitment of Ly6Chigh monocyte-derived, TNF- and iNOS-producing dendritic cells (Tip-DCs3) to Listeria monocytogenes-infected spleens (2, 3). MCP-1 is a major chemokine ligand for CCR2 (4–9), and mice deficient for MCP-1 are more susceptible to a range of microbial infections including Toxoplasma gondii and L. monocytogenes (5, 10, 11). Although MCP-1 expression is essential for optimal innate immune clearance of infection, in vivo induction of MCP-1 during infection is incompletely characterized.
L. monocytogenes is a Gram-positive bacterium that causes infections in immunocompromised humans (12, 13). In mice, following experimental inoculation, L. monocytogenes causes systemic infection that requires intact innate immune responses to control in vivo bacterial replication during the first 3 days of infection, and CD8 T cell responses to enable complete bacterial clearance from the host (13–15). MyD88-mediated TLR signaling is essential for effective innate immune control of L. monocytogenes infection, and MyD88-deficient mice are dramatically more susceptible to infection than wild-type (WT) mice (16, 17). The cellular pathogenesis of L. monocytogenes infection is well defined and involves uptake by phagocytic cells followed by bacterial escape from the phagosome into the host cell cytosol (13). Bacterial destruction of the phagosomal membrane is essential for in vivo bacterial virulence and is mediated by Listeriolysin-O (LLO), a secreted bacterial virulence protein (18). Bacterial invasion of the cytoplasm induces IRF-3-dependent expression of type I IFN (19, 20), a response that, during in vivo infection, paradoxically enhances bacterial survival (21–23) in part by nonspecifically promoting LLO-induced lymphocyte apoptosis (21, 22, 24).
During systemic L. monocytogenes infection, MCP-1/CCR2-dependent recruitment of Tip-DCs is occurring in the context of MyD88-promoted and Type I IFN-inhibited innate immune responses. How MyD88 and Type I IFN-mediated signals affect Tip-DC recruitment during in vivo infection remains incompletely defined. MCP-1 induction has been studied in great detail, and both TLR and type I IFN receptor (IFNAR) signaling can induce MCP-1 expression (25–30). In mice infected with MCMV, for example, MCP-1 induction in liver is greatly enhanced by intact IFNAR signaling (30), while our laboratory has demonstrated that MCP-1 production is modestly reduced in L. monocytogenes infected MyD88 deficient mice (11). At a cellular level, TLR-mediated signals have been demonstrated to induce MCP-1 production (25–29). This result is not surprising because characterization of the MCP-1 promoter has uncovered several NF-κB sites (31, 32). Nevertheless, the relative contributions of IFNAR and MyD88 signaling to MCP-1 induction and monocyte recruitment during L. monocytogenes infection are unknown.
We find that MyD88/TLR signals and IFNAR-mediated signals both contribute to MCP-1 induction following murine infection with virulent L. monocytogenes. However, monocyte emigration from bone marrow and recruitment to infected spleen is maintained in the absence of either MyD88 or IFNAR signaling. Deficiency of both signaling pathways, however, impairs monocyte recruitment from the bone marrow and results in diminished accumulation of Tip-DCs in the infected spleen. In addition, in contrast to IFNAR deficiency in WT mice, the absence of type I IFN signaling in mice lacking MyD88 results in increased susceptibility to L. monocytogenes infection. Our studies demonstrate a level of redundancy in the mechanisms driving monocyte recruitment and reveal, for the first time, a beneficial effect of type I IFN signaling in defense against intracellular bacterial infection.
Materials and Methods
Mice and infections
All mice used in this study were bred and maintained at the Memorial Sloan-Kettering Research Animal Resources Center. IFNAR−/− mice were purchased from B&K Universal, MyD88−/− mice were obtained from Dr. Shizuo Akira (Osaka University, Osaka, Japan), and MCP-1−/− mice were obtained from Dr. Barrett Rollins (Harvard Medical School, Boston, MA). IFNAR−/− and MyD88−/− mice strains were crossed to obtain MyD88−/− IFNAR−/− mice. MyD88−/− and MCP-1−/− mice were back-crossed over 10 generations onto the C57BL/6 background while IFNAR−/− mice crossed for seven generations to the C57BL/6 background. Mice were infected i.v. with 3000 L. monocytogenes strain 10403S. At indicated times following infection, spleens were harvested and dissociated in PBS containing 0.05% Triton X-100 and bacterial CFUs were determined by plating on brain-heart infusion agar plates.
Cell culture
Macrophages were grown from bone marrow precursors in antibiotic-free DMEM medium supplemented with 30% supernatant from l-cell fibroblasts and 20% FBS. On day 5, cells were harvested in ice-cold PBS, and plated at 3 × 105/well in 96-well flat-bottom plates. Bacteria were grown to log phase (A600 of 0.1), pelleted at 10,000 rpm for 10 min, and resuspended in PBS before addition to cells. WT 10403S and LLOko DP-L2161 bacteria were added to the cells at a 5:1 bacteria:cell ratio. Total volume in the wells during infection was 200 μl in 96-well plates. Infection was allowed to proceed for 1 h at which time extracellular bacteria were washed away, and gentamicin-containing medium was added to each well to prevent extracellular bacterial growth (T = 0). Supernatants were collected 1, 2, 4, and 6 h postinfection to assay for chemokines. TRIzol reagent (In-vitrogen) was added at 15 min, 30 min, 1, 2, 4, and 6 h postinfection to extract RNA from macrophages.
Flow cytometry
At various times after infection, spleens were collected, dissociated, and digested with 0.3% collagenase type 4 (Worthington Biochemical). Bone marrow cells were collected from mouse femurs as described previously (3). The following Abs were purchased from BD Pharmingen: anti-CD11b-PerCP(M1/70), anti-Ly6C-FITC(AL-21). For FACS analysis, a large FSC/SSC gate was drawn to include lymphocyte/monocyte populations.
ELISA
Murine MCP-1 was quantified using an ELISA kit from BD Biosciences. To obtain lysates for chemokine assays, organs were harvested at indicated times following infection, macerated in ice-cold PBS containing 0.01% Triton X-100, and centrifuged at 10,000 × g.
Real-time PCR analysis
Total RNA was isolated from macrophages using the TRIzol reagent (In-vitrogen). DNase-treated RNA underwent randomly primed cDNA synthesis and real-time PCR analysis. SYBR Green-based real-time PCR was performed using the DyNAmo SYBR Green qPCR Kit (Finnzymes). MCP-1-specific primers were obtained from Qiagen. Signals were normalized to GAPDH RNA (forward: 5′-ACCACAGTCCATGCCATCAC-3′; reverse: 5′-TCCACCACCCTGTTGCTGTA-3′). Normalized data were used to quantify relative levels of MCP-1 using ΔΔCt analysis.
Results
Rip2 and Trif are not required for MCP-1 expression following L. monocytogenes infection
L. monocytogenes infection induces inflammatory responses that are mediated by TLRs and MyD88. TLR-mediated recognition of microbial molecules is restricted to the cell surface or to membrane bound intracellular compartments and thus, because L. monocytogenes invades the host cell cytosol, additional innate immune sensing receptors are likely to contribute to the aggregate inflammatory response. Indeed, TLRs and Nod-like receptors (NLRs) mediate distinct transcriptional responses to L. monocytogenes (33). Nod1 and Nod2 are two cytosolic molecules that bind to peptidoglycan-derived molecules and contribute to in vivo defense against intestinal L. monocytogenes infection (34, 35) and lung Chlamydia pneumoniae infection (36). It is unclear, however, whether NLR signaling pathways promote MCP-1 production following L. monocytogenes infection. Rip2 is an intracellular kinase that mediates NLR-signaling and Rip2 deficiency abrogates Nod1 and Nod2 signaling (37, 38). Thus, we investigated in vivo induction of MCP-1 following L. monocytogenes infection in Rip2−/− mice and in mice lacking MyD88 or Trif, two intracellular adaptor molecules that mediate TLR-signaling. Spleens of Trif−/− and Rip2−/− mice that had been infected 24 h earlier with WT L. monocytogenes expressed the same level of MCP-1 as infected WT C57BL/6 mice (Fig. 1A). In contrast, and as demonstrated in previous studies from our laboratory (11), MCP-1 levels were reduced in the spleens of L. monocytogenes infected MyD88−/− mice (Fig. 1A). We next examined Tip-DC recruitment to the spleens of Rip2, Trif, and MyD88-deficient mice following L. monocytogenes infection. Following infection, the total cell numbers in the spleens of Trif−/−, Rip2−/−, and MyD88−/− mice were similar to WT mice (Fig. 1B). Tip-DCs were identified by high-level expression of Ly6C, and intermediate to high-level expression of CD11b. As demonstrated in Fig. 1, C and D, recruitment of Tip-DCs 24 h following infection was similar in WT, Trif−/−, Rip2−/−, and MyD88−/− mice. In Trif−/− and Rip2−/− mice, normal recruitment of Tip-DCs was correlated with normal splenic expression of MCP-1. However, in MyD88−/− mice, Tip-DC recruitment was normal despite a roughly 70% reduction in splenic MCP-1 levels, suggesting that the level of MCP-1 detected in the spleens of infected WT mice exceeds the amount required for Tip-DC recruitment.
Figure 1.
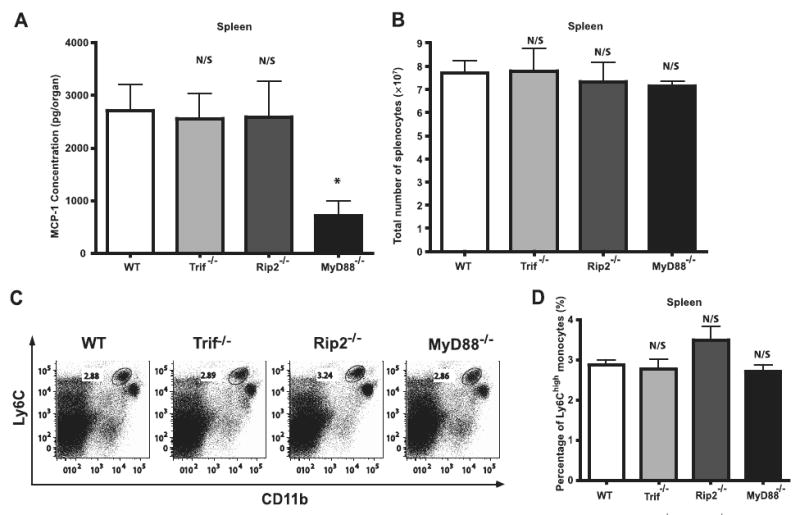
Signaling pathways involved in MCP-1 induction following L. monocytogenes infection. A, C57BL/6, Trif−/−, Rip2−/−, and MyD88−/− mice were infected i.v. with 3,000 live L. monocytogenes. Spleens were removed 24 h postinfection and MCP-1 levels were determined by ELISA. Each bar represents three mice and the experiment was repeated three times. Error bars show SD. B–D, Spleens were harvested from infected C57BL/6, Trif−/−, Rip2−/−, and MyD88−/− mice. Average total numbers of splenocytes were shown in B. Cells were stained for CD11b and Ly6C. Live lymphocyte/monocyte populations were gated and expression of CD11b and Ly6C was analyzed. Representative dot plots for three mice per group (C) and bar graphs for average monocyte percentages (D) are shown. N/S, nonsignificant as compared with WT; *, p < 0.05 as compared with WT.
MyD88 and type I IFN signals optimize MCP-1 production following infection
In vivo induction of MCP-1 is correlated with the ability of L. monocytogenes to escape the phagosome and enter the host cell cytosol. In contrast, LLO-deficient L. monocytogenes, which remains confined to the phagosome, does not induce MCP-1 expression in vivo (11). Studies from the Portnoy laboratory have demonstrated that cytosol invasion by L. monocytogenes is accompanied by the induction of IFN-β (39), a type I IFN involved in antiviral, but generally not antibacterial defense. We hypothesized that type I IFN induction provides an important in vivo stimulus for MCP-1 production. To investigate this, we measured the relative contribution of MyD88-mediated and type I IFN-mediated signaling for MCP-1 expression during L. monocytogenes infection. As a first step, bone marrow-derived macrophages (BMMØs) from C57BL/6, IFNAR−/−, MyD88−/−, and MyD88−/−IF-NAR−/− mice were infected with WT L. monocytogenes and MCP-1 protein levels were determined by ELISA (Fig. 2A). Relative to WT BMMØs, MCP-1 production at 6 h following infection was reduced by 60% in MyD88−/− BMMØs, 70% in IFNAR−/− BMMØs, and 85% in MyD88−/−IFNAR−/− BMMØs. These results indicate IFNAR- and MyD88-mediated signals both contribute to the production of MCP-1. Next, we infected C57BL/6, IFNAR−/−, MyD88−/−, and MyD88−/−IFNAR−/− mice and measured MCP-1 levels in spleens and serum by ELISA 24 h following infection. MCP-1 production was markedly reduced in MyD88−/−IFNAR−/− mice following L. monocytogenes infection, while MCP-1 protein levels were modestly reduced in both IFNAR−/− and MyD88−/− mice (Fig. 2, B and C). Loss of MyD88-mediated signals resulted in a greater decrease in MCP-1 production in vivo compared with loss of IFNAR-mediated signaling. MCP-1 production in the spleen and serum of IFNAR−/− mice was reduced by 35 and 50%, respectively (Fig. 2, B and C), while the corresponding decrease in MyD88−/− mice was 75 and 95%, respectively. Consistent with the in vitro findings using BMMØ, MCP-1 protein levels were reduced by 80 and 100% in the spleens and serum of infected MyD88−/−IFNAR−/− mice (Fig. 2, B and C). Of note, in spleen and in BMMØ, low levels of MCP-1 were detectable in the absence of both MyD88 and IFNAR-mediated signals, indicating that additional, undefined signaling pathways induce chemokine production, albeit the contribution is relatively minor.
Figure 2.

Role of MyD88 and IFNAR in MCP-1 expression. A, BMMØ from C57BL/6, IFNAR−/−, MyD88−/−, and MyD88−/−IFNAR−/− mice were infected with WT L. monocytogenes strains at a 5:1 ratio for 1 h, as described in Materials and Methods. The zero time point reflects the time 50 μg/ml gentamicin was added to medium. Supernatants were collected at indicated time and MCP-1 protein levels were quantified by ELISA. Each bar represents the average of three wells and this experiment was repeated twice. B and C, C57BL/6, IFNAR−/−, MyD88−/−, and MyD88−/−IFNAR−/− mice were infected i.v. with 3,000 live L. monocytogenes. Spleens were removed and serum was harvested 24 h postinfection. MCP-1 levels were determined by ELISA. Each bar represents three mice and the experiment was repeated three times. Error bars show SD. *, p < 0.05.
Rapid MyD88-dependent and delayed type I IFN-dependent MCP-1 induction
To further define the relative contributions of IFNAR and MyD88-mediated signaling in MCP-1 expression, we measured induction of MCP-1 mRNA in WT, MyD88−/−, IFNAR−/−, and MyD88−/−IFNAR−/− BMMØs infected for 15 min, 30 min, 1 h, 2 h, 4 h, and 6 h with WT L. monocytogenes (Fig. 3A). In WT BMMØ, MCP-1 mRNA was induced in two phases. The first phase occurred rapidly, with measurable increases in MCP-1 mRNA 15 min following infection, and reached a plateau 30 min after infection and then declined. During this phase, MCP-1 mRNA levels in infected BMMØs were increased up to 30-fold compared with uninfected BMMØs. The second phase of MCP-1 mRNA induction began 1 h following infection and continued during the course of examination for 5 h, at which time a 50-fold increase in mRNA was measured. Analysis of MCP-1 mRNA levels in infected MyD88−/− BMMØs demonstrated that the first phase of MCP-1 mRNA induction is MyD88 dependent. The second phase of MCP-1 induction, however, is intact in MyD88−/− BMMØs and follows similar kinetics as WT BMMØs. In MyD88−/− BMMØ, MCP-1 mRNA levels remained low until 30 min following infection and then gradually increased. In contrast, in the absence of IFNAR signaling, the second phase of MCP-1 mRNA expression was abrogated while the early, MyD88-dependent phase remained intact. In MyD88−/−IFNAR−/− BMMØs, both phases are abrogated and MCP-1 mRNA remained at base level.
Figure 3.

MyD88-dependent first phase and IFNAR-dependent second phase induction of MCP-1 by L. monocytogenes. A BMMØs from C57BL/6, IFNAR−/−, MyD88−/−, and MyD88−/−IFNAR−/− mice were infected with WT L. monocytogenes strains at a 5:1 ratio for 1 h, as described in Materials and Methods. The zero time point (T = 0) reflects the time 50 μg/ml gentamicin was added to medium after 1 h infection. B and C, BMMØs from C57BL/6 mice were infected with LLOko or WT L. monocytgenes and 1 ng/ml IFN-β was added to the medium at T = 0. MCP-1 mRNA levels were measured by real-time PCR (A and B) and protein levels were quantified by ELISA (C). Each bar represents the average of three wells and this experiment was repeated three times.
To determine the role of cytosol invasion by L. monocytogenes in MCP-1 induction, we used LLO-deficient L. monocytogenes to infect BMMØ. As shown in Fig. 3B, LLOko L. monocytogenes elicited MyD88-mediated signals and induced the first phase of MCP-1 mRNA expression. However, because type I IFN production is strictly dependent on cytosol invasion, LLOko L. monocytogenes did not induce type I IFN and also did not activate the delayed phase of MCP-1 mRNA induction. To determine whether the second phase was directly induced by type I IFNs, we added IFN-β (1 ng/ml) to uninfected BMMØs or LLOko-infected BMMØs. As shown in Fig. 3B, addition of IFN-β activated the second phase of MCP-1 mRNA expression in uninfected BMMØs. When IFN-β was added to macrophages infected with LLOko bacteria, both early and delayed phases of MCP-1 mRNA induction were activated (Fig. 3B) and resulted in MCP-1 protein secretion (Fig. 3C) in quantities that approximated those detected in BMMØs infected with WT L. monocytogenes.
Normal homeostatic Ly6Chigh monocyte trafficking in MyD88−/−IFNAR−/− mice
Our previous studies demonstrated that MCP-1 is required to maintain normal levels of circulating Ly6Chigh monocytes under homeostatic conditions (5). Because MyD88- and IFNAR-mediated signals can contribute to MCP-1 expression, we decided to determine whether these signaling pathways contribute to homeostatic monocyte trafficking. Thus, we determined the frequencies of Ly6Chigh monocytes in the spleen and bone marrow of naive C57BL/6, IFNAR−/−, MyD88−/−, and MyD88−/−IFNAR−/− mice. Fig. 4 demonstrated that the frequency differences of Ly6Chigh cells in the bone marrow (Fig. 4, A and C) and spleen (Fig. 4, B and D) of naive C57BL/6, IFNAR−/−, MyD88−/−, and MyD88−/−IFNAR−/− mice were not significant. These results indicated that MyD88- and IFNAR-mediated signaling pathways are not required for maintenance of circulating monocyte frequencies in the absence of infection and suggested that MyD88- and IFNAR-independent signaling pathways stimulate homeostatic production of MCP-1.
Figure 4.
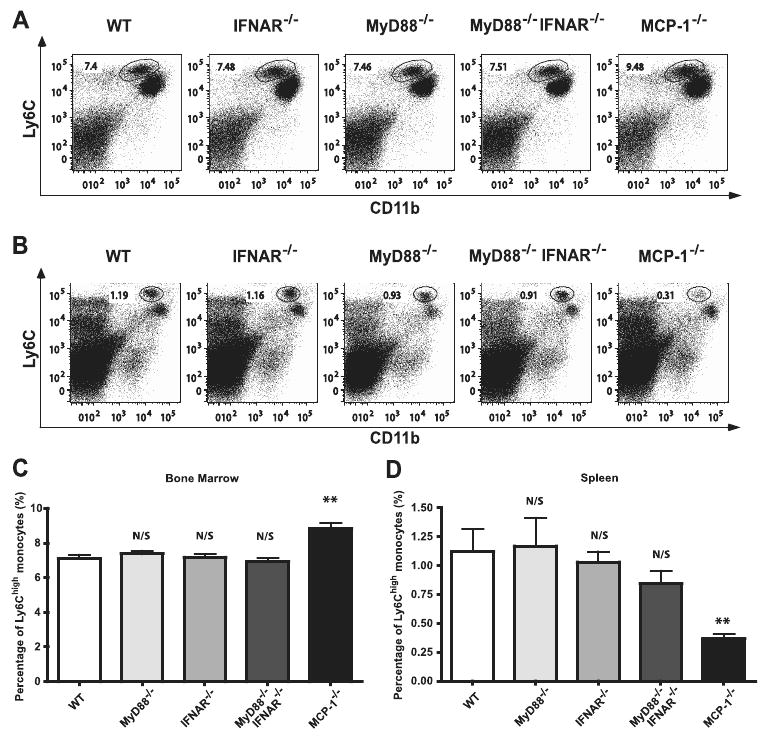
Maintenance of homeostatic monocyte trafficking is MyD88- and type I IFN-independent. Bone marrows (A) and spleens (B) were harvested from naive C57BL/6, IFNAR−/−, MyD88−/−, MyD88−/−IFNAR−/−, and MCP-1−/− mice. Cells were stained for CD11b and Ly6C. Live lymphocyte/monocyte populations were gated and expression of CD11b and Ly6C was analyzed. Representative dot plots for three mice per group are shown (A and B). Percentages of CD11bint/Ly6Chigh cells in naive C57BL/6, IFNAR−/−, MyD88−/−, MyD88−/−IFNAR−/−, and MCP-1−/− bone marrows and spleens are shown in C and D, respectively. Each bar represents the average of six to nine mice from two independent experiments. Error bars represent SD. N/S, nonsignificant as compared with WT; **, p < 0.01 as compared with WT.
Defective inflammatory monocyte recruitment in MyD88−/−IFNAR−/− mice but not MyD88−/− or IFNAR−/− mice
Previously, our laboratory demonstrated that MyD88-mediated signals are required for activation and differentiation of Tip-DCs, but that recruitment is MyD88 independent, despite reduced MCP-1 production following infection (11). It remains unclear, however, how much MCP-1 is required to trigger monocyte emigration from bone marrow, and whether multiple or a single signaling pathway is essential for this process. When we examined monocyte recruitment in infected IFNAR−/− mice, we observed similar results as in MyD88−/− mice, with reduced MCP-1 production but normal numbers of Ly6Chigh cells remaining in the bone marrow (Fig. 5, A and C) and trafficking to the spleen (Fig. 5, B and D). However, in the absence of both MyD88- and IFNAR-mediated signals, Tip-DC recruitment following infection was significantly decreased and similar to that detected in MCP-1−/− mice. As shown in Fig. 5C, ∼3% of Ly6Chigh monocytes emigrated from bone marrow in WT, MyD88−/−, and IFNAR−/− mice upon infection, while only 1.2% of monocytes emigrated from the bone marrow of MyD88−/−IFNAR−/− and MCP-1−/− mice. Along similar lines, frequencies of Ly6Chigh monocytes increased by nearly 2% in spleens of infected WT, MyD88−/−, and IFNAR−/− (Fig. 5D), while the increase in MyD88−/−IFNAR−/− mice and MCP-1−/− mice was only 1%. The total numbers of splenocytes in different mouse strains were similar following infection (data not shown). Thus, deficiency of both MyD88 and IFNAR signaling pathways results in a 60% reduction in monocyte egress from bone marrow and a 50% reduction of monocyte trafficking to the spleen relative to deficiency of either of these pathways alone. These results demonstrate that the MyD88-mediated and type I IFN-mediated signaling pathways play redundant roles in monocyte recruitment during infection and that either of these signaling pathways can, in the absence of the other, maintain normal inflammatory monocyte recruitment following L. monocytogenes infection, but that absence of both compromises monocyte recruitment.
Figure 5.
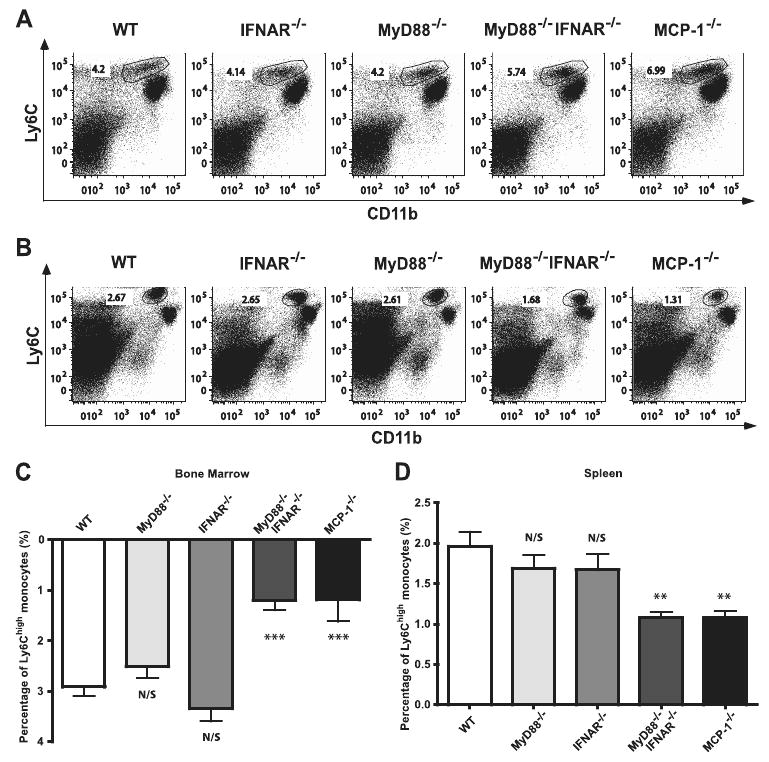
Defective inflammatory monocyte recruitment in MyD88−/−IFNAR−/− mice following infection. Bone marrows (A) and spleens (B) were harvested from C57BL/6, IFNAR−/−, MyD88−/−, MyD88−/−IFNAR−/−, and MCP-1−/− mice infected i.v. with 3,000 L. monocytogenes 24 h postinfection. Cells were stained for CD11b and Ly6C. Representative dot plots for three mice per group are shown. C and D, The differences of percentages of CD11bint/Ly6Chigh cells in bone marrows and spleens under homeostatic state (as shown in Fig. 4) vs infected state were calculated. As a result, the actual percentages of CD11bint/Ly6Chigh cells emigrated from bone marrows (C) and trafficked to spleens (D) are shown. Each bar or time point represents the average of 9–12 mice from three independent experiments. Error bars represent SD. **, p < 0.01; ***,p < 0.001 as compared with WT.
Beneficial effect of type I IFN in host defense against L. monocytogenes in the absence of MyD88 signaling
IFNAR−/− mice are more resistant to L. monocytogenes infection, indicating the type I IFN production is deleterious in mice following L. monocytogenes infection (21–23). Recent studies have demonstrated that type I IFN production during infection enhances lymphocyte apoptosis and leads to IL-10 production, which hampers innate and adaptive immune mechanisms of bacterial clearance (21, 40). In our study, IFNAR signaling complements MyD88 deficiency by maintaining monocyte recruitment. This result suggested that IFNAR deficiency might render MyD88-deficient mice more susceptible to L. monocytogenes infection. To test this hypothesis we infected WT, IFNAR−/−, MyD88−/−, and IFNAR−/− MyD88−/− mice with virulent L. monocytogenes and measured in vivo bacterial growth in spleen and liver 24 h postinfection (Fig. 6). Consistent with previously published studies, IFNAR−/− mice harbored fewer viable bacteria compared with WT mice, while MyD88−/− mice had more severe infections. In contrast to IFNAR deficiency on the WT background, IFNAR deficiency in MyD88-deficient mice resulted in greater susceptibility to L. monocytogenes infection, with increased numbers of viable bacteria in spleen (Fig. 6A) and liver (Fig. 6B). These findings indicated that the detrimental effects of IFNAR-mediated signaling following L. monocytogenes infection are MyD88 dependent. In the absence of MyD88-mediated signaling, however, IFNAR-mediated signals enhance resistance to L. monocytogenes infection.
Figure 6.

Beneficial effect of type I IFN in host defense against L. monocytogenes in the absence of MyD88-mediated signaling. C57BL/6, IFNAR−/−, MyD88−/−, and MyD88−/−IFNAR−/− mice were infected i.v. with 3,000 L. monocytogenes. Spleens (A) and livers (B) were removed at 24 h postinfection, and viable bacteria were quantified. Mean numbers of colony forming units (CFU) from groups of five to ten mice from two independent experiments are shown. Error bars represent SD. *, p < 0.05 and **, p < 0.01.
Discussion
MCP-1 is a chemokine that can be produced by a remarkable range of cell types. Furthermore, a variety of exogenous and endogenous inflammatory stimuli induce MCP-1 expression. The abundance of cell populations and innate immune signaling pathways that might contribute to the production of MCP-1 following L. monocytogenes infection has made it difficult to determine their relative importance. In this report, we demonstrated that MyD88-mediated and type I IFN-mediated signals provided overlapping contributions to in vivo MCP-1 production and monocyte recruitment. In the absence of either signaling pathway, chemokine expression levels were partially reduced, but residual MCP-1 production was sufficient to mediate monocyte emigration from bone marrow. Only when both MyD88 and IFNAR-mediated signals were absent was MCP-1 expression sufficiently reduced to impair monocyte recruitment to the infected spleen.
Our previous studies demonstrated that MyD88-mediated signaling was important for maximal in vivo chemokine production and for monocyte activation at the site of infection (11). Although in vivo MCP-1 production, as measured by ELISA in spleen, liver, and serum, was diminished in the absence of MyD88-mediated signaling, monocyte recruitment from the bone marrow to the spleen occurred normally during early infection. This result indicated that residual MCP-1 production, perhaps induced by other innate immune signaling pathways, was sufficient to induce monocyte emigration from the bone marrow. To identify the signaling pathways responsible for MyD88-independent MCP-1 production and monocyte recruitment, we first examined Trif−/− mice and Rip2−/− mice. Our studies, however, indicate that neither Trif nor Rip2 contribute to MCP-1 production or monocyte recruitment following L. monocytogenes infection.
L. monocytogenes differs from many other intracellular bacterial pathogens by escaping the vacuolar compartment of infected cells, thus providing stimuli to innate immune receptors in the host cell cytosol. One result of cytosol invasion by L. monocytogenes is induction of IFN-β (39). Our initial experiments demonstrated that IFNAR-mediated signals enhanced MCP-1 production in L. monocytogenes-infected BMMØ and also in vivo following systemic infection. In the absence of MyD88 signaling, IFNAR-mediated signals induced sufficient MCP-1 production to maintain normal monocyte recruitment during the first 24 h of infection. Loss of IFNAR signaling in addition to MyD88 signaling, however, resulted in impaired monocyte recruitment. Thus, IFNAR signaling provides a backup mechanism to maintain monocyte recruitment in the absence of MyD88-mediated signals.
Type I IFNs are generally implicated in immune defense against viral infections. In the case of L. monocytogenes infection, however, production of IFN-β increases susceptibility to infection by enhancing apoptosis of lymphocytes (21–23, 40). Type I IFNs, however, have broad-ranging effects on immune responses, and thus it remained possible that enhanced lymphocyte apoptosis concealed beneficial effects of IFN-β induction. Interestingly, our results demonstrated that induction of IFN-β by L. monocytogenes can, in the right circumstances, provide protection against infection. In our studies, IFNAR deficiency rendered MyD88-deficient mice more susceptible to L. monocytogenes infection, suggesting that increased resistance of IFNAR-deficient mice is MyD88 dependent. It is possible that lymphocyte apoptosis, IL-10 secretion by macrophages ingesting apoptotic debris, or immunosuppression by IL-10 are MyD88 dependent.
BMMØ are commonly used in in vitro systems to examine chemokine and cytokine production. However, correlations between results of in vitro and in vivo experiments remain far from consistent. In our studies, we also detected some disparities between in vitro and in vivo studies. For example, MyD88 and type I IFN signals play equivalent roles promoting in vitro MCP-1 production, while MyD88-mediated signals make greater contributions to in vivo induction of MCP-1. Although macrophages can be an important in vivo source of MCP-1 following infection, other cell types may be equally important sources of MCP-1. Another message from our study is that chemokine expression levels do not directly correlate with the magnitude of monocyte recruitment. In mice deficient for either MyD88 or IFNAR, for example, we detected reduced levels of MCP-1 production, but monocyte recruitment remained intact. This result indicates that the amount of chemokine produced in vivo and quantified in serum and tissues greatly exceeds the concentration required for cell trafficking. It is possible that chemokines promoting monocyte trafficking function in very restricted environments, such as at the boundaries between the cellular and vascular compartments in the bone marrow. If this is the case, the magnitude of chemokine secretion in tissues and serum, while likely reflecting the intensity of infection, does not contribute to the trafficking of cells to specific sites of infection. Determining where and when chemokines provide instruction to trafficking cells remains an important challenge.
Footnotes
This work was supported by the National Institutes of Health (R37AI039031, to E.G.P.) and a Feodor-Lynen postdoc fellowship from Humboldt foundation (to K.B.).
Abbreviations used in this paper: Tip-DC, TNF- and iNOS-producing dendritic cell; IFNAR, type I IFN receptor; WT, wild type; NLR, Nod-like receptor; LLO, Listerio-lysin-O; BMMØ, bone marrow macrophage.
Disclosures: The authors have no financial conflict of interest.
References
- 1.Serbina NV, Jia T, Hohl TM, Pamer GE. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 3.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 4.Rollins BJ. Chemokines. Blood. 1997;90:909–928. [PubMed] [Google Scholar]
- 5.Jia T, Serbina NV, Brandl K, Zhong MX, Leiner IM, Charo IF, Pamer EG. Additive roles for MCP-1 and MCP-3 in CCR2-mediated recruitment of inflammatory monocytes during Listeria monocytogenes infection. J Immunol. 2008;180:6846–6853. doi: 10.4049/jimmunol.180.10.6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117:902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rollins BJ. JE/MCP-1: an early-response gene encodes a monocyte-specific cytokine. Cancer Cells. 1991;3:517–524. [PubMed] [Google Scholar]
- 8.Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, North R, Gerard C, Rollins BJ. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med. 1998;187:601–608. doi: 10.1084/jem.187.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurihara T, Warr G, Loy J, Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med. 1997;186:1757–1762. doi: 10.1084/jem.186.10.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robben PM, LaRegina M, Kuziel WA, Sibley LD. Recruitment of Gr-1+ monocytes is essential for control of acute toxoplasmosis. J Exp Med. 2005;201:1761–1769. doi: 10.1084/jem.20050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serbina NV, Kuziel W, Flavell R, Akira S, Rollins B, Pamer EG. Sequential MyD88-independent and -dependent activation of innate immune responses to intracellular bacterial infection. Immunity. 2003;19:891–901. doi: 10.1016/s1074-7613(03)00330-3. [DOI] [PubMed] [Google Scholar]
- 12.Gellin BG, Broome CV. Listeriosis. J Am Med Assoc. 1989;261:1313–1320. [PubMed] [Google Scholar]
- 13.Pamer EG. Immune responses to Listeria monocytogenes. Nat Rev Immunol. 2004;4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 14.Busch DH, Pamer EG. T lymphocyte dynamics during Listeria monocytogenes infection. Immunol Lett. 1999;65:93–98. doi: 10.1016/s0165-2478(98)00130-8. [DOI] [PubMed] [Google Scholar]
- 15.Bhardwaj V, Kanagawa O, Swanson PE, Unanue ER. Chronic Listeria infection in SCID mice: requirements for the carrier state and the dual role of T cells in transferring protection or suppression. J Immunol. 1998;160:376–384. [PubMed] [Google Scholar]
- 16.Edelson BT, Unanue ER. MyD88-dependent but Toll-like receptor 2-independent innate immunity to Listeria: no role for either in macrophage listericidal activity. J Immunol. 2002;169:3869–3875. doi: 10.4049/jimmunol.169.7.3869. [DOI] [PubMed] [Google Scholar]
- 17.Seki E, Tsutsui H, Tsuji NM, Hayashi N, Adachi K, Nakano H, Futatsugi-Yumikura S, Takeuchi O, Hoshino K, Akira S, et al. Critical roles of myeloid differentiation factor 88-dependent proinflammatory cytokine release in early phase clearance of Listeria monocytogenes in mice. J Immunol. 2002;169:3863–3868. doi: 10.4049/jimmunol.169.7.3863. [DOI] [PubMed] [Google Scholar]
- 18.Bielecki J, Youngman P, Connelly P, Portnoy DA. Bacillus subtilis expressing a haemolysin gene from Listeria monocytogenes can grow in mammalian cells. Nature. 1990;345:175–176. doi: 10.1038/345175a0. [DOI] [PubMed] [Google Scholar]
- 19.Stockinger S, Materna T, Stoiber D, Bayr L, Steinborn R, Kolbe T, Unger H, Chakraborty T, Levy DE, Muller M, Decker T. Production of type I IFN sensitizes macrophages to cell death induced by Listeria monocytogenes. J Immunol. 2002;169:6522–6529. doi: 10.4049/jimmunol.169.11.6522. [DOI] [PubMed] [Google Scholar]
- 20.Stockinger S, Reutterer B, Schaljo B, Schellack C, Brunner S, Materna T, Yamamoto M, Akira S, Taniguchi T, Murray PJ, et al. IFN regulatory factor 3-dependent induction of type I IFNs by intracellular bacteria is mediated by a TLR- and Nod2-independent mechanism. J Immunol. 2004;173:7416–7425. doi: 10.4049/jimmunol.173.12.7416. [DOI] [PubMed] [Google Scholar]
- 21.Carrero JA, Calderon B, Unanue ER. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med. 2004;200:535–540. doi: 10.1084/jem.20040769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, Perry AK, Nguyen BO, Lane TF, Taniguchi T, et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med. 2004;200:437–445. doi: 10.1084/jem.20040712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Auerbuch V, Brockstedt DG, Meyer-Morse N, O'Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med. 2004;200:527–533. doi: 10.1084/jem.20040976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carrero JA, Calderon B, Unanue ER. Listeriolysin O from Listeria monocytogenes is a lymphocyte apoptogenic molecule. J Immunol. 2004;172:4866–4874. doi: 10.4049/jimmunol.172.8.4866. [DOI] [PubMed] [Google Scholar]
- 25.Kopydlowski KM, Salkowski CA, Cody MJ, van Rooijen N, Major J, Hamilton TA, Vogel SN. Regulation of macrophage chemokine expression by lipopolysaccharide in vitro and in vivo. J Immunol. 1999;163:1537–1544. [PubMed] [Google Scholar]
- 26.Kumar MV, Nagineni CN, Chin MS, Hooks JJ, Detrick B. Innate immunity in the retina: Toll-like receptor (TLR) signaling in human retinal pigment epithelial cells. J Neuroimmunol. 2004;153:7–15. doi: 10.1016/j.jneuroim.2004.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boyd JH, Divangahi M, Yahiaoui L, Gvozdic D, Qureshi S, Petrof BJ. Toll-like receptors differentially regulate CC and CXC chemokines in skeletal muscle via NF-κB and calcineurin. Infect Immun. 2006;74:6829–6838. doi: 10.1128/IAI.00286-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colotta F, Borre A, Wang JM, Tattanelli M, Maddalena F, Polentarutti N, Peri G, Mantovani A. Expression of a monocyte chemotactic cytokine by human mononuclear phagocytes. J Immunol. 1992;148:760–765. [PubMed] [Google Scholar]
- 29.Tsuboi N, Yoshikai Y, Matsuo S, Kikuchi T, Iwami K, Nagai Y, Takeuchi O, Akira S, Matsuguchi T. Roles of toll-like receptors in C-C chemokine production by renal tubular epithelial cells. J Immunol. 2002;169:2026–2033. doi: 10.4049/jimmunol.169.4.2026. [DOI] [PubMed] [Google Scholar]
- 30.Hokeness KL, Kuziel WA, Biron CA, Salazar-Mather TP. Monocyte chemoattractant protein-1 and CCR2 interactions are required for IFN-α/β-induced inflammatory responses and antiviral defense in liver. J Immunol. 2005;174:1549–1556. doi: 10.4049/jimmunol.174.3.1549. [DOI] [PubMed] [Google Scholar]
- 31.Boekhoudt GH, Guo Z, Beresford GW, Boss JM. Communication between NF-κB and Sp1 controls histone acetylation within the proximal promoter of the monocyte chemoattractant protein 1 gene. J Immunol. 2003;170:4139–4147. doi: 10.4049/jimmunol.170.8.4139. [DOI] [PubMed] [Google Scholar]
- 32.Ping D, Jones PL, Boss JM. TNF regulates the in vivo occupancy of both distal and proximal regulatory regions of the MCP-1/JE gene. Immunity. 1996;4:455–469. doi: 10.1016/s1074-7613(00)80412-4. [DOI] [PubMed] [Google Scholar]
- 33.Leber JH, Crimmins GT, Raghavan S, Meyer-Morse NP, Cox JS, Portnoy DA. Distinct TLR- and NLR-mediated transcriptional responses to an intracellular pathogen. PLoS Pathog. 2008;4:e6. doi: 10.1371/journal.ppat.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim YG, Park JH, Shaw MH, Franchi L, Inohara N, Nunez G. The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to Toll-like receptor ligands. Immunity. 2008;28:246–257. doi: 10.1016/j.immuni.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 35.Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Flavell RA. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 36.Shimada K, Chen S, Chen PW, Sorrentino R, Alsabeh R, Slepenkin AV, Peterson E, Doherty TM, Underhill D, Crother TR, Arditi M. The NOD/RIP2 pathway is essential for host defenses against Chlamydophila pneumoniae lung infection. PLoS Pathog. 2009;5:e1000379. doi: 10.1371/journal.ppat.1000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park JH, Kim YG, McDonald C, Kanneganti TD, Hasegawa M, Body-Malapel M, Inohara N, Nunez G. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol. 2007;178:2380–2386. doi: 10.4049/jimmunol.178.4.2380. [DOI] [PubMed] [Google Scholar]
- 38.Park JH, Kim YG, Shaw M, Kanneganti TD, Fujimoto Y, Fukase K, Inohara N, Nunez G. Nod1/RICK and TLR signaling regulate chemokine and antimicrobial innate immune responses in mesothelial cells. J Immunol. 2007;179:514–521. doi: 10.4049/jimmunol.179.1.514. [DOI] [PubMed] [Google Scholar]
- 39.O'Riordan M, Yi CH, Gonzales R, Lee KD, Portnoy DA. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc Natl Acad Sci USA. 2002;99:13861–13866. doi: 10.1073/pnas.202476699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carrero JA, Calderon B, Unanue ER. Lymphocytes are detrimental during the early innate immune response against Listeria monocytogenes. J Exp Med. 2006;203:933–940. doi: 10.1084/jem.20060045. [DOI] [PMC free article] [PubMed] [Google Scholar]