

Abstract
AIDS has changed from a mostly male-specific health problem to one that predominantly affects females. Although sex differences in HIV-1 susceptibility are beyond doubt, the extent to which sex affects the onset and progression of AIDS has remained elusive. Here, we provide evidence for an influence of X chromosomal variation on the course of retroviral infection, both in HIV-1-infected patients and in the rhesus macaque model of AIDS. A two-stage, microsatellite-based GWAS of SIV-infected monkeys revealed MHC class I markers and a hitherto-unknown X chromosomal locus as being associated with a nominal score measuring progression to AIDS (Fisher's exact p < 10−6). The X chromosomal association was subsequently confirmed in HIV-1-infected patients with published SNP genotype data. SNP rs5968255, located at human Xq21.1 in a conserved sequence element near the RPS6KA6 and CYLC1 genes, was identified as a significant genetic determinant of disease progression in females (ANOVA p = 8.8 × 10−5), but not in males (p = 0.19). Heterozygous female carriers of the C allele showed significantly slower CD4 cell decline and a lower viral load at set point than TT homozygous females and than males. Inspection of HapMap revealed that the CT genotype is significantly more frequent among Asians than among Europeans or Africans. Our results suggest that, in addition to the individual innate and adaptive immunity status, sex-linked genetic variation impacts upon the rate of progression to AIDS. Elucidating the mechanisms underlying this sex-specific effect will promote the development of antiretroviral therapies with high efficacy in both sexes.
Introduction
Susceptibility to an infection with human immunodeficiency virus type 1 (HIV-1 [MIM 609423]) and the progression to the acquired immunodeficiency syndrome (AIDS [MIM 609423]) are influenced by (1) the pathogenicity of the virus, (2) the functionality of various innate and adaptive immune defense proteins, and (3) additional host factors with a bearing upon, for example, viral entry and replication.1 Consequently, the first genome-wide association study (GWAS) of HIV-1-infected patients unraveled single-nucleotide polymorphisms (SNPs) in the major histocompatibility complex class I region (MHC [MIM 142800]) as major determinants of both the individual viral load (VL) at set point and the rate of disease progression.2 Subsequent GWAS confirmed the prominent role of the MHC region in modulating the clinical course of HIV-1 infection.3–5 In addition, however, functional genomic studies identified hundreds of other genes that may influence the HIV-1 life cycle.6–8 This implies that a large number of host factors are likely to contribute to the variability of the HIV-related phenotypes, including genetic variants the nature and frequency of which supposedly depend upon the ethnic background of the respective patients.
One way to promote the discovery of genetic and nongenetic determinants of human disease phenotypes is their study in relevant animal models. Despite a couple of pathophysiological differences that exist between HIV-1 and simian immunodeficiency virus (SIV) infection, SIV-infected rhesus macaques still represent the most important animal model for preclinical proof-of-concept studies of HIV vaccines and microbicides and for pathogenicity studies.9–11 Therefore, rhesus macaques may potentially also provide an excellent means to identify genetic variants that are involved in a differential response to retroviral infections.
Because genome-wide SNP arrays and HapMap-like resources are not yet available for nonhuman primates, we chose to perform a step-wise microsatellite-based GWAS in two independent samples of SIV-infected rhesus macaques of Indian descent (Macaca mulatta), both of which displayed interindividual differences in terms of their progression to AIDS. Our study not only confirmed the disease relevance of genetic variation in the MHC but also identified an X chromosomal locus as being associated with the rate at which SIV infection leads to the onset of AIDS-related disease in rhesus macaques. We then successfully translated this finding into the human system by identifying an X chromosomal SNP, rs5968255, as being significantly associated with both individual VL at set point and the rate of disease progression in HIV-1-infected women, defined either by the time until treatment initiation or the time until CD4 cells have dropped below 350 μl−1.
Material and Methods
Monkey Samples
For the present study, retrospective data were compiled from different SIV infection studies on rhesus macaques of Indian descent, performed at the German Primate Center (“Deutsches Primatenzentrum,” DPZ, see Table 1 and Tables S1–S3 available online). The housing and treatment protocols of the DPZ follow the German Animal Protection Act, which in turn complies with European Union guidelines on the use of non-human primates for biomedical research and have been approved by the responsible public authority. All animals were housed, surveyed, and SIV-infected with standardized or highly similar protocols. The time point and route of SIV infection, the date of death, previous treatments, the dose of infecting virus, and the identity of the infecting virus strain itself were known for all monkeys. Necropsy results, hemograms and antibody titers were also generally available. Cell-associated viral-load data and viral RNA copy number were known for the majority of animals. Most monkeys had been infected with SIVmac251 or SIVmac239 (Tables S1–S3). Given that both strains are of similar pathogenicity,12,13 confounding of our GWAS by strain identity appears to be unlikely. Furthermore, no significant association was observed between the infecting strain and the progression score that constituted the core phenotypic measure of our study (see below), neither in stage 1 (Fisher's exact p = 0.139) nor in stage 2 (p = 0.325) of the GWAS.
Table 1.
Screening and Verification Samples of SIV-Infected Rhesus
| Sex | Stage 1 (n = 136) | Stage 2 (n = 128) | p Valuea | |
|---|---|---|---|---|
| 101 males (74%) | 110 males (86%) | 0.021 | ||
| 35 females (26%) | 18 females (14%) | |||
| Average Progression Score (± Standard Deviation) | ||||
| all | 3.15 ± 1.23 | 3.18 ± 1.07 | 0.152 | |
| males | 3.16 ± 1.22 | 3.19 ± 1.05 | 0.064 | |
| females | 3.11 ± 1.26 | 3.11 ± 1.23 | 0.323 | |
| Average Survival Time (Weeks after Infection ± Standard Error) | ||||
| all | 67.30 ± 5.09 | 83.25 ± 11.15 | 0.899 | |
| males | 70.46 ± 6.49 | 88.23 ± 12.90 | 0.841 | |
| females | 59.55 ± 7.15 | 53.90 ± 6.71 | 0.347 | |
All p values relate to the differences between stage 1 and stage 2 samples. They refer to Fisher's exact test, except for survival time for which the p value refers to a log-rank test.
Definition of Progression Score
Some 46% of the monkeys in GWAS stage 1, and 32% of animals in stage 2, were euthanized before they had developed AIDS-related symptoms or were still alive by the end of the study. In addition, ∼40% of the monkeys were immunized with a prospective AIDS vaccine before infection. None of the vaccines tested reduced VL at set point substantially but may have prolonged survival. This implies that time of survival after SIV infection alone would have been an insufficient basis for the envisaged GWAS of disease progression. To overcome this caveat, we scored disease progression on a scale of 1 to 5 points, with adjustments made for immunization effects (Figure 1). Individual scores were based upon survival time, necropsy results, antibody titer, available viral-load data, CD4+ T cell counts, and neopterin values. This scoring system reflects our long-term experience in evaluating genetic, immunological, and virological data on the course of SIV infection in rhesus macaques. The scoring system not only divides the bulk of monkeys into fast (score 2), medium (score 3), and slow progressors (score 4) but also allows a more nuanced consideration of the extreme ends of the disease spectrum (ultrafast progression: score 1, long-term nonprogression: score 5).
Figure 1.
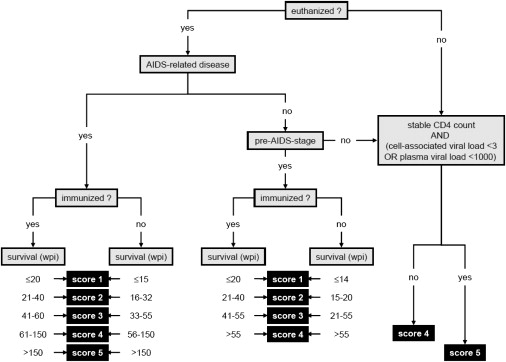
Decision-Tree-Based Definition of a Progression Score for SIV-Infected Rhesus Macaques
The diagnosis of AIDS-related disease was based upon clinical, necropsy, or histopathology findings. Animals that were euthanized with AIDS-related disease or as being at a pre-AIDS stage were scored according to their survival time after SIV infection. Monkeys not at a pre-AIDS stage after euthanasia, or not euthanized at all, were scored according to their CD4 cell decline and viral load (plasma viral load < 1000 RNA copies mL−1 or cell-associated viral load < 3 infected cells per 107 cells at set point).
The diagnosis of AIDS-related disease was based upon clinical, necropsy, and histopathology findings. For monkeys to be diagnosed with AIDS-related disease, they had to suffer from anorexia, incurable diarrhea, or neurological or lung dysfunction, evidenced by ataxia or respiratory sounds. Necropsy and histology findings had to reveal one or more of the following: severe hyperplasia of secondary lymphoid tissue, severe chronic active gastroenteritis, chronic interstitial pneumonia associated with Pneumocystis jirovecii infection, giant cell disease, or encephalopathy. Lymphomas, nonbacterial thrombotic endocarditis, or artheriopathy were regarded as additional AIDS-related complications and were used for confirmation of the primary diagnosis. Notably, CD4+ T cell count was not found to be an adequate diagnostic marker of AIDS because many rapid progressors still had physiological CD4+ T cell counts by the time of euthanasia.14,15 For monkeys euthanized without a diagnosis of AIDS-related disease, necropsy and histopathology findings were used in determining whether they were at a pre-AIDS stage. The minimum prerequisite for this diagnosis was the presence of moderate to severe hyperplasia of lymphoid tissue in combination with moderate to severe chronic gastroenteritis. Animals presenting with either AIDS-related disease or at a pre-AIDS stage were scored according to their survival time after infection, taking into account whether or not they had been immunized prior to infection (Figure 1).
Monkeys that were still alive at the end of the study (all at > 100 weeks post infectio [wpi]), or that were euthanized without a diagnosis of AIDS-related disease or pre-AIDS, were scored either 4 or 5, depending upon their CD4+ T cell count and viral load (Figure 1). Score 5 (“long-term nonprogression”) was defined as the absence of any signs of immunodeficiency (i.e., a stable CD4+ T cell count), together with a cell-associated viral load < 3 infected cells per 107 cells or a plasma viral load < 1000 RNA copies mL−1 at set point. Long-term nonprogressors usually survive SIV infection substantially longer (>300 wpi) than slow progressors (score 4). Indeed, among the SIV-infected animals kept at the DPZ, this leap in survival time coincides perfectly with the differences in viral load and CD4+ T cell count defining the progression score, even at an early stage after infection.
Microsatellite Genotyping
In the screening stage (stage 1) of our microsatellite-based GWAS, 136 SIV-infected rhesus macaques were genotyped for a genome-wide set of 332 microsatellites. Human as well as Macaca mulatta-specific microsatellites that had been selected on the basis of published mapping information were used.16 All microsatellites, their chromosomal location, and the forward and reverse primers used for amplification are listed in Tables S4 and S5. For each of the 14 non-MHC markers selected for verification in 128 additional SIV-infected animals (stage 2 of our GWAS), we included two additional flanking markers located ∼250 kb up- and downstream of the original marker, respectively. These markers were identified through a systematic search for microsatellite sequences of the published Macacca mulatta genomic sequence (build 1.1). Assays were designed with the PRIMER 3 software (see Web Resources). Fragment sizes were measured by separation of PCR products on an ABI 3700 capillary sequencer (Applied Biosystems) and by subsequent data analysis with the Genemarker software (Softgenetics LLC).
Microsatellite-Based Association Analysis in Rhesus Macaques
Hemizygosity for X chromosomal microsatellites was verified in all male monkeys, and apparently heterozygous genotypes were excluded from subsequent analyses. The level of allelic association between individual markers and the progression score was assessed for statistical significance with Fisher's exact test. p values were calculated by Monte-Carlo Markov-Chain simulation. Bonferroni correction for multiple testing was carried out in stage 1 of the GWAS (Table S4 and Figure S6), although the actual selection of markers for verification in stage 2 (5% of the total markers) was based solely upon relative significance and resource availability rather than genome-wide statistical significance. In stage 2, each original marker and its two flanking neighbors were treated as one marker group (Table S5). p values were first corrected according to Holm17 within each marker group, and the most significant marker and its corrected p value were selected to represent the respective group. In a second step, the corrected p values of the group representative and the p values of three additional MHC markers were further corrected for multiple testing according to Holm.
Assessment of Population Stratification in the Monkey Verification Sample
To quantify the level of population stratification in the verification sample (stage 2), members of which originally came from four different centers, we calculated FST according to the definition of Weir and Cockerham18 by using an implementation in FSTAT (version 2.9.3; see Web Resources).
Data and Quality Control for the SNP-Based Association Analysis of the CHAVI Sample
We reanalyzed genotype data from a recent GWAS on host control determinants in HIV-1 infection, undertaken by the Centre for HIV/AIDS Vaccine Immunology (CHAVI).2 VL at set-point data and a progression phenotype, defined by the CHAVI investigators as the time until either treatment initiation or a drop in CD4 cell count below 350 cells μL−1, were available for 303 (93 females, 210 males) of the 486 original samples. For these individuals, we were provided the genotypes of all 13,821 X chromosomal SNPs generated in the CHAVI study.2 Correct sex assignment was verified with the proportion of heterozygous genotypes at all markers (average percentage expected in males: <1%, in females: ∼25%). Average pair-wise identity by state (IBS) was used for the assessment of extreme genetic similarity and dissimilarity, pointing at cognate relatives or genetic outliers, respectively. For post hoc quality control after association analysis (see below), we required nominally significant SNPs to have a call-rate ≥ 95% and to be in Hardy-Weinberg equilibrium (p > 0.05, with an exact test).19 Quality control was carried out with the R software version 2.6.2.20
Assessment of Population Stratification in the CHAVI Sample
To account for possible population stratification in the statistical analyses of the human genotypes, we used a principal component (PC)-based approach as implemented in EIGENSTRAT.21 Given that EIGENSTRAT can only handle autosomal genotypes, we assumed each male to be homozygous for each X chromosomal marker and females were doubled to ensure unchanged allele frequencies. A recent survey of genome-wide autosomal genetic variation in Europe revealed considerable variation only in the first five to six PCs.22 Therefore, we considered the first six PCs of the X chromosomal markers as sufficient to adjust our analyses of the CHAVI sample for potential population stratification as well.
Association Analysis in the CHAVI Sample
We normalized progression phenotype values by taking natural logarithms. Possible deviations from normality were tested for statistical significance with a Shapiro-Wilk test. To assess possible population stratification in the CHAVI sample, we subjected the first six PCs from the EIGENSTRAT analysis of the X chromosomal genotype data to a multiple linear regression analysis of the log-progression phenotype. AIC-based backward selection retained only the second and the sixth PC in the model. We then used ANOVA, stratified by sex and including the two significant PCs as covariates, to test for statistically significant associations between individual X chromosomal SNP genotypes and the log-progression phenotype and associations with VL at set point. All analyses were carried out with R version 2.6.2.20 The association between SNP rs5968255 and VL at set point was tested for statistical significance with linear regression modeling,2 including age at seroconversion and the two significant EIGENSTRAT PCs as covariates.
Linkage Disequilibrium Analysis
We used HaploView 4.1 separately for males and females to estimate linkage disequilibrium (LD) and to define tag SNPs.23,24 We used the CEU (Utah residents with ancestry from northern and western Europe) genotype data of the HapMap project, release 23a (phase II, March 2008) to infer which SNPs were in LD with each other at a population level.25
Kaplan-Meier Survival Analysis
In the monkey samples, we performed Kaplan-Meier survival analysis with log-rank statistics to assess the observed genotype-specific differences in survival after SIV infection for their statistical significance. In the CHAVI samples, Kaplan-Meier analyses of the progression phenotype were performed in addition to ANOVA to allow a more detailed illustration of the genotypic effects upon progression phenotype. Kaplan-Meier plots were created with R version 2.6.2 and with Sigmaplot 11 (Systat Software).
Synteny Analysis
Synteny between X chromosomal regions in humans and macaques was verified with the tuple plot program for pair-wise nucleotide sequence comparison with noise suppression.26 The human Xq21.1 region, spanning coordinates 71,657,686 to 86,891,379 in NCBI Build 36.1, was compared to the rhesus X chromosomal region spanning coordinates 71,459,512 to 86,479,549 of Mmul_051212.
Results
Genetic Variations in the MHC Class I Region and on Chromosome X Are Associated with Disease Progression in the Rhesus Macaque Model of AIDS
We performed a microsatellite-based GWAS of host factors that influence AIDS-free survival in SIV-infected rhesus macaques of Indian origin. The markers were derived mainly from the published linkage map of the rhesus macaque.16 An initial, pedigree-based linkage analysis of the available animals was dismissed after the number of genetically verifiable relationships turned out to be too small to achieve sufficient credibility (data not shown). We therefore reverted to a two-stage association analysis, by using 136 SIV-infected monkeys for screening in stage 1 (Table 1; Tables S1 and S2). Prominent association signals obtained in stage 1 were selected for verification in stage 2, in which an independent group of 128 unrelated SIV-infected animals was analyzed (Table 1; Table S3). In order to ensure consistent phenotyping, we used a scoring system for the progression rate toward AIDS-related disease (Material and Methods; Figure 1). The samples used in stages 1 and 2 were characterized by similar average progression scores and similar average AIDS-free survival times (Table 1).
A total of 332 microsatellite markers were genotyped in stage 1 (call-rate > 90%). The average intermarker genetic distance was ∼9 centiMorgans (cM). With a Monte-Carlo Markov-Chain implementation of Fisher's exact test, 83 markers were found to exhibit a nominally significant allelic association with the progression score (p ≤ 0.05). Four of these associations remained significant after Bonferroni correction (Table 2; Table S4), namely D6S2972 (p ≤ 10−6), D6S2704 (p = 1.2 × 10−4), D2S434 (p = 2.4 × 10−5), and MML9S30 (p ≤ 10−6). Markers D2S434 and MML9S30 are located on rhesus chromosomes 12 and 9, respectively; D6S2972 and D6S2704 are located in the rhesus MHC class I region on chromosome 4 (Table 2).
Table 2.
Detection and Verification of the Top 5% Associations with Progression Score, as Obtained in Stage 1 of a Microsatellite-Based GWAS in SIV-Infected Rhesus Macaques
| Marker | Chromosome | Location (Mmul_051212) | Stage 1 p Valuea | Stage 2 p Valueb |
|---|---|---|---|---|
| D6S2972 | 4 | 29414233 | <10−6 | 5.0 × 10−3 |
| MML9S30 | 9 | 127592629 | <10−6 | 0.416 |
| D2S434 | 12 | 81515926 | 2.4 × 10−5 | 0.718 |
| D6S2704 | 4 | 29800682 | 1.2 × 10−4 | 2.9 × 10−3 |
| D8S322 | 8 | 22034079 | 8.6 × 10−4 | 0.863 |
| D7S496 | 3 | 144937483 | 1.2 × 10−3 | 0.137 |
| DXS986 | X | 78914283 | 1.2 × 10−3 | <10−6 |
| D3S1583 | 2 | 167994335 | 1.5 × 10−3 | 0.447 |
| D5S2122 | 6 | 68784187 | 1.6 × 10−3 | 0.021 |
| D7S524 | 3 | 132219500 | 1.9 × 10−3 | 0.607 |
| D1S215 | 1 | 191075285 | 2.0 × 10−3 | 0.064 |
| D2S2364 | 13 | 78819370 | 2.0 × 10−3 | 0.579 |
| MML6S8 | 6 | 91055880 | 2.0 × 10−3 | 0.556 |
| MML10S14 | 10 | 52709132 | 2.4 × 10−3 | 0.463 |
| D7S493 | 3 | 104444180 | 3.1 × 10−3 | 0.620 |
| D6S2876 | 4 | 32439777 | 3.7 × 10−3 | 0.269 |
| MML9S32 | 9 | 129952729 | 3.7 × 10−3 | 0.168 |
Fisher's exact test;
Except for the three MHC markers (D6S∗), p values represent the minimum of a set of three Holm-corrected p values, including the original marker plus two flanking markers located ∼250 kb upstream and downstream, respectively. For D6S2972, D6S2704, and D6S2876, p values are uncorrected and correspond to a single marker only. The X chromosomal marker (DXS986) that yielded a significant result in both GWAS stages is highlighted in bold.
For verification of the results of stage 1, the top 5% of markers with the smallest p values (n = 17) were genotyped in a second sample of animals. This set of markers included 3 MHC markers, 13 markers from other autosomal loci, and 1 X chromosomal marker. For each of the non-MHC markers, two additional flanking polymorphisms ∼250 kb apart from the original marker were included in the genotyping. In total, 45 microsatellites were genotyped in stage 2 (Table S5). Of these, only X chromosomal microsatellite DXS986 (p = 3.2 × 10−3) and its two neighbors (DXS986+1, p ≤ 10−6; DXS986−2, p = 6.2 × 10−5) exhibited a statistically significant association with the progression score after multiple testing correction (Table 2). The two MHC markers D6S2704 and D6S2972 retained nominal significance, whereas the phenotype association of D2S434 and MML9S30 could not be confirmed in stage 2 (Table 2; Tables S4 and S5). The verification of the MHC association was not surprising because MHC class I genes are well-established determinants for disease progression in both rhesus macaques and humans.1,13,27,28 Although some marginal population substructure became apparent in the verification sample (FST = 0.035), this was deemed too small to have inflated the significance of our results. Subgroup analyses of naive and immunized animals revealed that the association between DXS986 and the progression score achieved nominal significance in both subsets of stage 1 (p = 1.1 × 10−4 in naive animals, p = 0.027 in immunized animals). In stage 2, the two subgroup-specific p values were of borderline significance (0.05 < p < 0.10). These results argue against any notable confounding of the association between DXS986 and the progression score by the immunization state. A sex-specific subgroup analysis was not deemed meaningful because the number of females was too low in the two stages (35 and 18, respectively) to provide sufficient statistical power.
Analysis of X Chromosomal SNPs in HIV-1-Infected Patients
In order to investigate whether our results in macaques would also apply to HIV-infected humans, we obtained phenotypic and X chromosomal SNP genotype data from a recent human GWAS, carried out by the Centre for HIV/AIDS Vaccine Immunology (CHAVI).2 In the CHAVI study, the progression phenotype was defined as either the time until treatment initiation or the time until the CD4 cell count was observed or extrapolated to have dropped below 350 cells μL−1, a threshold recommended by many guidelines for the initiation of antiretroviral treatment. In the natural course of HIV-1 infection, this event often signifies the transition from a nonsymptomatic state to the phase of AIDS-defining illnesses and death, if not treated.29 We studied 303 seroconverters (93 females, 210 males) for whom the progression phenotype and information on the VL at set point were available.
After synteny between the region surrounding DXS986 in macaques and Xq21.1 in humans had been confirmed (Figure S1), we performed an association analysis (Table 3; Table S6) by using 128 SNPs from a 15 Mb region centered at DXS986 (NCBI Build 36.1 coordinates 71,657,686 to 86,891,379). The region of interest was flanked at its proximal end by MMLXS5, the nearest nonsignificant marker in stage 1 of the rhesus GWAS (Table S4). After Bonferroni correction, sex-stratified ANOVA without adjustment for population structure revealed five SNPs from the DXS986 region to be significantly associated with the log-progression phenotype in females (data not shown). When the two significant PCs were included as covariates, only the association with rs5968255 remained significant. Notably, no significant association with the log-progression phenotype was observed for males. Multiple linear regression analysis of the log-progression phenotype and the five significant SNPs around DXS986, involving AIC-based backward selection with and without inclusion of the two significant PCs, confirmed rs5968255 as the only significantly associated marker in the DXS986 region after Bonferroni correction (corrected p = 1.3 × 10−2; nominal p = 8.8 × 10−5 in females, nominal p = 0.19 in males; Table S7).
Table 3.
Ten Most Significant Associations between X Chromosomal SNPs and the Progression Phenotype in the CHAVI Sample, with and without Stratification by Sex
| SNP | Coordinate | p Valuea | |
|---|---|---|---|
| Males | |||
| rs959060 | 86553764 | 3.1 × 10−3 | |
| rs5924105 | 86889097 | 8.3 × 10−3 | |
| rs2092545 | 83518130 | 0.016 | |
| rs1573906 | 86022244 | 0.018 | |
| rs5938679 | 75919876 | 0.022 | |
| rs2214607 | 75322143 | 0.028 | |
| rs5969113 | 86313544 | 0.034 | |
| rs4892718 | 76098975 | 0.044 | |
| rs1905182 | 82482574 | 0.051 | |
| rs5923642 | 85930864 | 0.052 | |
| Females | |||
| rs5968255 | 83141348 | 8.8 × 10−5 | |
| rs10218349 | 83276159 | 7.4 × 10−4 | |
| rs2092545 | 83518130 | 8.4 × 10−4 | |
| rs825541 | 82936504 | 8.5 × 10−4 | |
| rs1531868 | 82991939 | 8.5 × 10−4 | |
| rs10855652 | 86303530 | 1.7 × 10−3 | |
| rs1515024 | 78701951 | 1.8 × 10−3 | |
| rs527480 | 73552592 | 3.2 × 10−3 | |
| rs7879492 | 84905281 | 5.5 × 10−3 | |
| rs765076 | 83390987 | 7.5 × 10−3 | |
| Combined | |||
| rs5922075 | 83811026 | 1.1 × 10−3 | |
| rs959060 | 86553764 | 1.3 × 10−3 | |
| rs5968255 | 83141348 | 1.8 × 10−3 | |
| rs7879492 | 84905281 | 3.3 × 10−3 | |
| rs6623084 | 83850582 | 3.6 × 10−3 | |
| rs10218349 | 83276159 | 5.6 × 10−3 | |
| rs7882767 | 84775710 | 6.1 × 10−3 | |
| rs6617219 | 85444095 | 9.0 × 10−3 | |
| rs7879396 | 79513028 | 9.9 × 10−3 | |
| rs5923642 | 85930864 | 0.011 | |
Analysis of variance (ANOVA) including the two most significant EIGENSTRAT PCs as covariates.
Some of the SNPs analyzed in the CHAVI sample showed considerable allelic association with one another, thereby rendering Bonferroni correction for multiple testing overly conservative. We therefore repeated the initial association analysis of the CHAVI data with the assumption that the number of tag SNPs24 in the region or interest represented a good approximation of the number of independent hypotheses tested. Pair-wise r2 SNP tagging was carried out with HaploView 4.1.23 Employing either 0.8 or 0.5 as tagging thresholds yielded 109 and 92 tag SNPs (out of 128 SNPs) in the DXS986 region, respectively. However, even when these figures were used for relaxed multiple-testing correction, the number of significant results remained virtually unchanged (data not shown).
SNP rs5968255 Is Associated with Slower Progression in Women Only
The allele and genotype frequencies of rs5968255 in the CHAVI sample, which consists of individuals of self-reported European ancestry,2 were found to be similar to those in the European American HapMap sample (CEU). In both instances, T was the major allele (CEU frequency: 0.88) and C was the minor allele (CEU frequency: 0.12).25 Data from chimpanzee suggest that T represents the ancestral state of this SNP. When the CHAVI samples were stratified by sex, 20 out of 210 males (9.5%) were found to be hemizygous for the C allele. Of the 93 females, 77 (82.8%) were homozygous TT and the remaining 16 women (17.2%) were heterozygous CT. No homozygous CC females were observed.
The progression phenotype was indicative of a significant delay of CD4 cell loss in CT heterozygous females compared to TT homozygotes (Kaplan-Meier analysis, log rank p = 0.001; Figure 2 and Figure S2). No such effect was seen in C hemizygous males. Furthermore, heterozygous females showed a higher median progression phenotype (8.0 years, IQR: 5.1–20.4 years) than homozygotes (2.2 years, IQR: 1.3–4.4 years). No such trend was apparent among males (hemizygous T median: 2.6 years, IQR: 1.4–3.9 years; C median: 1.8 years, IQR: 1.1–2.7 years). In a linear-regression analysis, SNP rs5968255 explained between 21% (coefficient of determination R2; no adjustment for population stratification) and 25% (inclusion of the two significant EIGENSTRAT PCs) of the variance in female log-progression phenotype, indicating a comparatively good model fit. The association analysis between rs5968255 and the VL at set point also yielded sex-specific results (see Table 4, Table S7, and Figure S3). Here, CT heterozygous females showed a lower average VL than TT homozygous females (p = 0.001), and no significant difference was seen between T and C hemizygous males (p = 0.2). For both findings, an artifact due to a hidden association between rs5968255 and the two previously established genetic markers of both VL and the progression phenotype in the CHAVI sample,2 namely rs2395029 (a proxy for HCP5 [MIM 604676] and HLA-B∗5701 [MIM 142830]) and rs9261174 (ZNRD1 [MIM 607525] and RNF39 [MIM 607524]), could be systematically excluded (Tables S8 and S9). Therefore, rs5968255 represents an X-linked genetic variant that independently impacts upon the progression phenotype exclusively in HIV-1-infected females.
Figure 2.
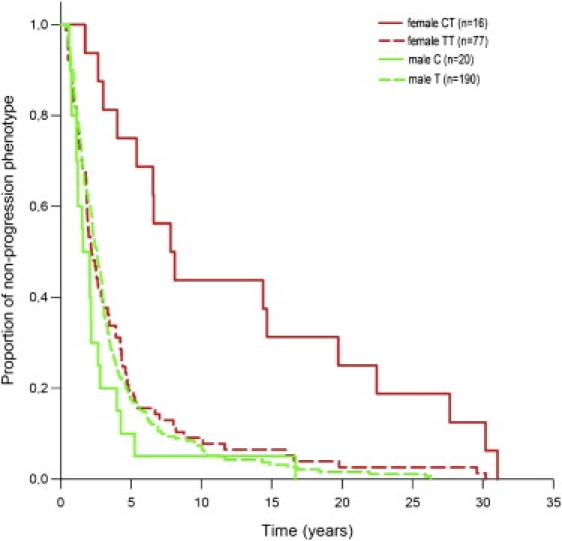
Kaplan-Meier Analysis of the Progression Phenotype, Stratified by rs5968255 Genotype and Sex
Among CT heterozygous females, the median progression time (8.0 years), i.e., the median time until treatment initiation or until a drop of the individual CD4 cell count below 350 cells μL−1 has occurred, was significantly longer (p = 0.001, log rank test) than among TT homozygotes (2.2 years), or males (T: 2.6 years, C: 1.8 years).
Table 4.
Ten Most Significant Associations between X Chromosomal SNPs and Viral Load at Set Point, with and without Stratification by Sex
| SNP | Coordinate | p Valuea | |
|---|---|---|---|
| Males | |||
| rs5981378 | 74744696 | 0.013 | |
| rs5923575 | 85772687 | 0.013 | |
| rs12014367 | 78761577 | 0.014 | |
| rs2092545 | 83518130 | 0.019 | |
| rs6617219 | 85444095 | 0.02 | |
| rs2038193 | 85174711 | 0.034 | |
| rs6647911 | 75203875 | 0.035 | |
| rs5968183 | 82634435 | 0.042 | |
| rs527480 | 73552592 | 0.043 | |
| rs2858719 | 86046788 | 0.046 | |
| Females | |||
| rs10218349 | 83276159 | 9.9 × 10−4 | |
| rs5968255 | 83141348 | 1.2 × 10−3 | |
| rs5922395 | 86357637 | 1.2 × 10−3 | |
| rs2092545 | 83518130 | 1.8 × 10−3 | |
| rs6623084 | 83850582 | 3.4 × 10−3 | |
| rs825541 | 82936504 | 4.3 × 10−3 | |
| rs1531868 | 82991939 | 4.3 × 10−3 | |
| rs4828291 | 83912613 | 5.3 × 10−3 | |
| rs5922075 | 83811026 | 6.2 × 10−3 | |
| rs5969041 | 86129916 | 0.044 | |
| Combined | |||
| rs761202 | 85819695 | 2.6 × 10−3 | |
| rs11092732 | 85073796 | 5.4 × 10−3 | |
| rs5938152 | 74799722 | 7.4 × 10−3 | |
| rs12164321 | 85766510 | 9.7 × 10−3 | |
| rs6647780 | 74740571 | 0.011 | |
| rs1938012 | 74916674 | 0.011 | |
| rs5922075 | 83811026 | 0.013 | |
| rs232957 | 76059032 | 0.013 | |
| rs5969181 | 86510305 | 0.018 | |
| rs12687256 | 75219576 | 0.018 | |
Analysis of variance (ANOVA) including the two most significant EIGENSTRAT PCs as covariates.
SNP rs5968255 is located in a 500 bp conserved intergenic region (Figure 3), 3.9 Mb distal to DXS986. It resides 64 kb proximal to the ribosomal protein S6 kinase 6 gene (RPS6KA6 alias RSK4 [MIM 300303]) and 138 kb distally to the cylicin 1 gene (CYLC1 [MIM 300768]). The 3′ end of an alternatively spliced CYLC1 cDNA clone (accession number BC038787) is located only 233 bp proximally to rs5968255. Because rs5968255 itself may not be causal for the observed association, we identified those SNPs in the DX6986 region that showed at least moderate LD (r2 ≥ 0.4) with rs5968255 in the HapMap CEU sample (Table S10).25 This analysis did not yield any additional association signals that would have pointed toward possible candidates for causation. Using the Genevar database,30 we also failed to obtain any evidence for an association between rs5968255 and the expression level of either CYLC1 or RPS6KA6 (data not shown).
Figure 3.
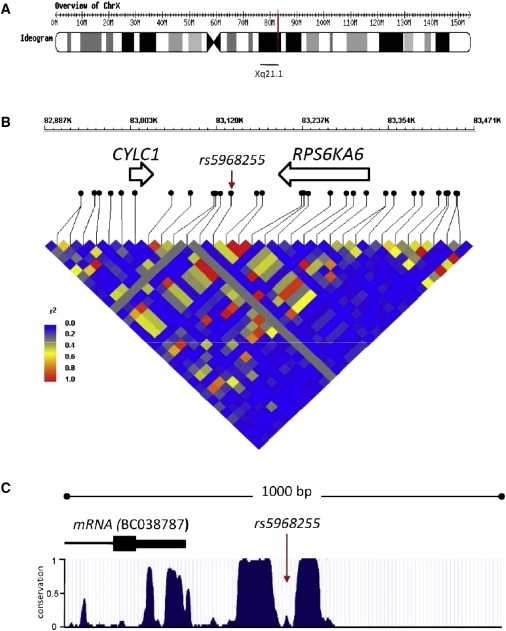
Characteristics of the Genomic Region around X-Chromosomal SNP rs5968255, Associated with the Progression Phenotype in Humans
(A) Ideogram of the human X chromosome. The position of rs5968255 at Xq21.1 (NCBI Build 36.1: 83,141,348) is indicated in red.
(B) Partial map of Xq21.1 (coordinates 82,887,000 to 83,471,000) depicting the location of genes close to rs5968255 (marked by an arrow) and the local pattern of LD in the HapMap CEU sample. This graph was created with the WGAViewer.61
(C) Evolutionary conservation within a 1 kb window centered on rs5968255 (coordinates 83,140,849 to 83,141,848); the conservation data were obtained from the UCSC Genome Browser; Vertebrate Multiz Alignment62 and PhastCons Conservation.63
HapMap Population Differences in the Allele Frequencies of SNP rs5968255
The HapMap project has been initiated to catalog common DNA sequence variation in different human population groups. The following populations have been included: YRI, Yoruba from Ibadan, Nigeria; JPT, Japanese from Tokyo; CHB, Han Chinese from Beijing, China; and CEU, CEPH (Utah residents with ancestry from northern and western Europe). Inspection of the respective genotype data revealed that C represents the minor rs5968255 allele among females in the CEU (frequency 0.15) and YRI (0.02) groups (Figure 4), but that it is a major allele in Asian females (CHB: 0.56; JPT: 0.61). Like in the CHAVI sample, no CC homozygotes were present among the CEU and YRI females. In contrast, homozygous and heterozygous female carriers of C were fairly frequent in CHB (CC: 0.35; CT: 0.43) and JPT (CC: 0.41; CT: 0.41). Similarly, C hemizygous males were a rare (0.1) group in CEU, and even rarer in YRI (0.03), but were fairly prevalent among males of Asian origin (CHB: 0.36; JPT: 0.54). Thus, on the basis of the hypothesis that the rs5968255 C allele exerts the same effect upon the progression phenotype in Asians as in Europeans (Figure 2), more HIV-1-infected females in China and Japan than in Europe would be predicted to show delayed CD4 cell decline.
Figure 4.
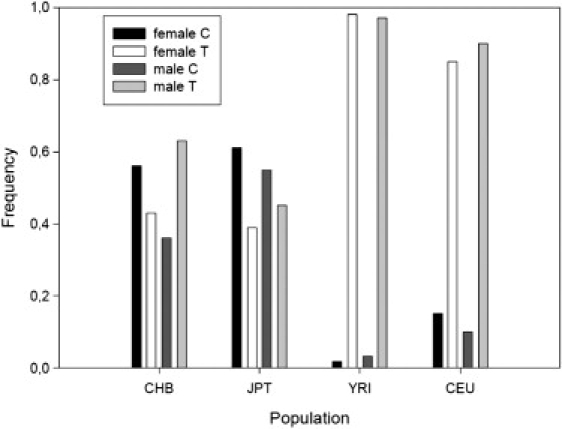
Allele Frequencies of SNP rs5968255 in HapMap
The pooled (female and male) allele frequencies differed significantly between all population groups (all Bonferroni corrected p < 0.001), except for the two Asian samples (CHB versus JPT: χ2 = 4.545, 1 d.f., corrected p = 0.198, nominal p = 0.033).
Discussion
To our knowledge, we have undertaken the first genome-wide search for host factors that impact upon disease progression in SIV-infected rhesus macaques, one of the most important animal models of human AIDS. Our data not only confirmed the importance of the MHC class I locus for this phenotype but also, to our knowledge, for the first time identified X chromosomal variation as being significantly associated with AIDS-free survival in primates. This information then allowed us to identify a variant in the orthologous Xq21.1 region as being significantly associated with a disease-progression phenotype in HIV-infected humans, an association that otherwise would have been overlooked. Over and above their scientific relevance, our results deserve specific attention because they highlight how cross-species analyses can successfully translate genetic association findings from an animal model to a human disease, even over an evolutionary distance of 25 million years.
In the screening stage (stage 1) of our rhesus GWAS, two MHC class I markers were found to be significantly associated with a score measuring differential progression to AIDS-related disease, and these polymorphisms also achieved nominal significance in the verification stage (stage 2). Such a contribution by the MHC region was not unexpected. Both markers map only 400 kb apart from each other, and flank the A region of the rhesus macaque MHC class I. Marker D6S2704 is located 150 kb distal to the ZNRD1 (zinc ribbon domain-containing protein 1 [MIM 607525]) and RNF39 (ring finger protein 39 [MIM 607524]) genes, and polymorphisms in these genes have been reported before to influence HIV-1 disease progression in humans.1,2,4,5,28 ZNRD1 was also identified in a recent search for HIV-1 dependency factors (HDFs) that may be involved in the viral life cycle.6 Inspection of the microsatellite genotypes of stage 1 revealed the presence of only four alleles of D6S2972. Two of them were perfectly associated with rhesus MHC class I allele Mamu-A∗01, which may explain the association between D6S2972 and the progression score in stage 1.12,13,28,31 In stage 2, D6S2972 was found to be less tightly associated with Mamu-A∗01, and this maybe why the phenotype association did not withstand multiple testing correction (see Figure S4). The association between D6S2704 and the progression score may also be explained in part by linkage of D6S2704 to particular MHC genes. However, because of the close physical proximity between D6S2704 and ZNRD1, the causal variation underlying the observed phenotype association must remain elusive until a more detailed analysis of the nonclassical-MHC genes within this region has been carried out.
Most importantly, an X chromosomal marker was found to be significantly associated with the progression score in both stages of our rhesus GWAS. Given that no evidence for an involvement of X chromosomal variation into the course of HIV-1 infection has been reported before, we aimed at replicating this finding in humans. In so doing, we identified SNP rs5968255 as being associated with a progression phenotype related to the rate of CD4 cell decline in HIV-1-infected patients, independent of the MHC. We found that the minor C allele of this variant is beneficial for heterozygous females, whereas the progression phenotype in women with the TT genotype was the same as that of male patients. The fact that C hemizygous males did not benefit from the protective allele suggests that its influence may depend upon the background of female physiology or genetics.32,33 No CC homozygous women could be analyzed because none were present in the CHAVI sample. Therefore, it would be too premature to speculate about a possible heterozygosity effect. Notably, our initial study of rhesus macaques revealed no significant sex difference in terms of the observed genotype-phenotype relationship (Figure S5), but this may have been due to a lack of power because females were underrepresented in both stages of the GWAS (Table 1). Such sex imbalance is a common phenomenon in potentially lethal experiments in primate research because primate centers have to retain females for breeding. Another explanation for the discrepant sex specificity in humans and monkeys could be that different genes encoded in the same X chromosomal region affect the disease differently. However, it appears more plausible that identical X-linked mechanisms influence AIDS symptom-free survival time in both species, but that the overt phenotype is modified by species-specific variants that arose during the 25 million years of evolutionary divergence between humans and macaques.
Intergenic SNP rs5968255 marks a >300 kb region that must contain the causal variation underlying the sex-specific phenotype association observed in our study. Unfortunately, all SNPs in strong LD with rs5968255 were mainly intergenic or intronic and did not immediately suggest functional candidates for further investigation. Furthermore, a query of gene-expression databases indicated that rs5968255 is unlikely to influence the expression level of nearby genes (Figure 3). Therefore, the question whether RPS6KA6 or CYLC1 are functionally involved in the observed phenotype association remains open. So far, neither of the two genes has been implicated in HIV-1/SIV-induced disease. The RPS6KA6 gene encodes a member of a family of four ribosomal serine/threonine kinases (RSK1-4 [MIMs 601684, 300075, 601685, and 300303]), three of which have been studied in detail before (RSK1-3) and which were found to be involved in MAPK- (MIM 176872), ERK- (MIM 601795), and mTor-(MIM 601231) signaling important for cell proliferation and survival.34 All four proteins potentially function as mediators of the growth-factor- and stress-induced activation of transcription factors. Consequently, they have been examined in detail mainly in cancer studies but, at least indirectly, these proteins may also be involved in HIV-1 infection. For example, RSK2 can regulate cAMP regulatory element binding protein (CREB [MIM 123810]), and CREB can interact with HIV-1 protein Tat.35 RPS6KA6 may be different from the other ribosomal kinases in that it seems to be fully activated in unstimulated cells,36 and overexpression in human breast cancer cells results in suppressed proliferative activity of these cells.37 Another indirect link with HIV-induced disease may derive from the fact that overexpression of RPS6KA6 results in upregulation of claudin 2 (MIM 300520) and downregulation of the chemokine receptor CXCR4 (MIM 162643). CXCR4 is highly expressed in human malignant breast tumors and is a coreceptor of HIV-1. Furthermore, RPS6KA6 may be regulated by c-Myc (MIM 190080), which is also involved in HIV-1 expression.38 So far, it is not known whether RPS6KA6 is involved in CXCR4 receptor expression in lymphocytes, but it may be speculated that CXCR4 expression is related to progression to AIDS. HIV-1 transmission usually occurs by infection with viruses utilizing the chemokine receptor CCR5 (MIM 601373) for entry. However, after retroviral infection, HIV-1 variants arise that can use CXCR4 as a coreceptor. Although, the underlying mechanism is still enigmatic, this virus-coreceptor switch is known to be associated with the rate of progression to AIDS.39 On the other hand, even if RPS6KA6 were involved in differential CXCR4 expression in lymphocytes, thereby influencing viral coreceptor density on the cell surface, it would be unclear why this should have an effect in heterozygous women alone. It would also not explain why the RPS6KA6 gene should affect disease progression in SIV-infected monkeys because a coreceptor switch does not occur in monkeys. Claudin 2 is a tight-junction protein that is expressed, for example, in the intestine, i.e., a tissue that is severely impaired by HIV infection. Claudin proteins have been implicated in virus entry and dissemination in various infections, including HIV infection of the brain.40–43 Notably, claudin 2 is also encoded by an X chromosomal (Xq22.3) gene.
The second gene close to rs5968255 is CYLC1, proposed to encode a basic protein of the sperm head cytoskeleton. Yet, it appears to be expressed in other cell types such as brain and kidney cells and may be expressed at low levels in cells of lymphoid origin.44 Unfortunately, the function of CYLC1 has not been investigated so far so that the potential role of this gene in HIV pathogenesis must remain unclear.
Sex-specific genetic factors have been suspected for some time to influence the course of AIDS in humans,32,45,46 but these could never be established firmly by epidemiological studies. Although it seems clear that women are more vulnerable to the acquisition of HIV-1 than men,47,48 reports on sex-specific differences in HIV-1-induced disease and mortality have been controversially discussed most probably because these studies differed in terms of statistical power and prevalent socioeconomic factors, such as accessibility to the health care systems. In addition, lower viral RNA load may not always be associated with slower progression to AIDS.49 This notwithstanding, several studies suggested that women tend to progress to AIDS more slowly than men. Such a phenomenon that became even more prominent after the introduction of highly active antiretroviral therapy.50–53 Other reports repeatedly claimed that, after stratification by CD4 cell count, HIV-infected women have a lower viral load than HIV-infected men54–56 and that CD4 cell counts tend to be higher and associated with lower viral RNA levels in infected females than males.32 The latter observation is consistent with the view that the higher CD4:CD8 ratio in females is genetically determined,57 although the underlying variation is yet to be discovered. According to our results, only a genetically defined minority of women in Europe and Africa may in fact benefit from a prolonged nonsymptomatic state after HIV-1 infection. This is not surprising because, in Europe, AIDS became a medical problem only one generation ago, and its prevalence is still far too low to have induced sufficient selective pressure in favor of the C allele in the past. The reason why this allele is much more frequent in Asians is unclear, but we do not presume that it is in any way related to HIV-1 infection. Furthermore, it still remains to be investigated whether a higher frequency of Asian HIV-1-infected females indeed experience a prolonged nonsymptomatic disease state.
Sex specificity clearly adds to the complexity of the role of host factors in HIV-1 infection and, consequently, impinges upon issues such as antiretroviral treatment and vaccine efficacy. Our understanding of how different immune responses to pathogens arise in the two sexes, and specifically how HIV-1 affects the sexes differently, is still very limited.32,33 So far, such sex differences were mainly attributed to different sex steroid hormone levels or, for example, a generally higher CD4:CD8 ratio in females.32,33,57 However, the process of X chromosome inactivation (XI), and a potential skew of XI, also has to be taken into account when trying to explain the sex-specific manifestation of heritable traits.58 At least 15% of X-linked genes can escape XI and the skew of XI increases with age.59,60 Whether the observed association between rs5968255 and progression phenotype entails an interaction with XI remains to be elucidated.
To our knowledge, we have provided the first firm evidence for an association between X chromosomal genetic variation and two of the best predictors of HIV-1 disease progression, namely CD4 cell decline and VL at set point. Previous studies also suggested that several X chromosomal host genes are necessary for the HIV-1 life cycle.6 Therefore, considerably more genetic variation than uncovered by our study can still be expected to play a role in the sex specificity of HIV-1 susceptibility and the progression to AIDS in humans.
Supplemental Data
Supplemental Data include six figures and ten tables and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
FSTAT version 2.9.3, http://www2.unil.ch/popgen/softwares/
Genevar database, http://www.sanger.ac.uk/humgen/genevar/
HapMaP database, http://ftp.hapmap.org/genotypes/2008-03/forward/non-redundant/
Macacca mulatta genomic sequence, http://www.ncbi.nlm.nih.gov/projects/genome/guide/rhesus_macaque/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PRIMER 3 software, http://frodo.wi.mit.edu/
UCSC Genome Bioinformatics Site, http://genome.ucsc.edu
WGAViewer software, http://www.genome.duke.edu/centers/pg2/downloads/wgaviewer.php/
Acknowledgments
This study was supported by the German Ministry of Education and Research (BMBF) through the National Genome Research Network (NGFN), grant 0313360. We gratefully acknowledge the genotype access provided to us by the CHAVI consortium and the help of David B. Goldstein, Dongliang Ge, and Amalio Telenti. We thank Holger Thiele for retrieving microsatellites from the Macaca mulatta genome, Ivonne Heinze, Nicole Leuchte and Heidi Meyer for expert technical assistance, and Monika Franz for continuous support of the animal experiments. There are no financial conflicts of interest that might be construed to influence the results or interpretation of our manuscript.
References
- 1.O'Brien S.J., Nelson G.W. Human genes that limit AIDS. Nat. Genet. 2004;36:565–574. doi: 10.1038/ng1369. [DOI] [PubMed] [Google Scholar]
- 2.Fellay J., Shianna K.V., Ge D., Colombo S., Ledergerber B., Weale M., Zhang K., Gumbs C., Castagna A., Cossarizza A. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Limou S., Le Clerc S., Coulonges C., Carpentier W., Dina C., Delaneau O., Labib T., Taing L., Sladek R., Deveau C. Genomewide association study of an AIDS-nonprogression cohort emphasizes the role played by HLA genes (ANRS Genomewide Association Study 02) J. Infect. Dis. 2009;199:419–426. doi: 10.1086/596067. [DOI] [PubMed] [Google Scholar]
- 4.van Manen D., Kootstra N.A., Boeser-Nunnink B., Handulle M.A., van't Wout A.B., Schuitemaker H. Association of HLA-C and HCP5 gene regions with the clinical course of HIV-1 infection. AIDS. 2009;23:19–28. doi: 10.1097/QAD.0b013e32831db247. [DOI] [PubMed] [Google Scholar]
- 5.Catano G., Kulkarni H., He W., Marconi V.C., Agan B.K., Landrum M., Anderson S., Delmar J., Telles V., Song L. HIV-1 disease-influencing effects associated with ZNRD1, HCP5 and HLA-C alleles are attributable mainly to either HLA-A10 or HLA-B∗57 alleles. PLoS ONE. 2008;3:e3636. doi: 10.1371/journal.pone.0003636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brass A.L., Dykxhoorn D.M., Benita Y., Yan N., Engelman A., Xavier R.J., Lieberman J., Elledge S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 7.Konig R., Zhou Y., Elleder D., Diamond T.L., Bonamy G.M., Irelan J.T., Chiang C.Y., Tu B.P., De Jesus P.D., Lilley C.E. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135:49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou H., Xu M., Huang Q., Gates A.T., Zhang X.D., Castle J.C., Stec E., Ferrer M., Strulovici B., Hazuda D.J. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 2008;4:495–504. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 9.Ahmed R.K., Biberfeld G., Thorstensson R. Innate immunity in experimental SIV infection and vaccination. Mol. Immunol. 2005;42:251–258. doi: 10.1016/j.molimm.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 10.Morgan C., Marthas M., Miller C., Duerr A., Cheng-Mayer C., Desrosiers R., Flores J., Haigwood N., Hu S.L., Johnson R.P. The use of nonhuman primate models in HIV vaccine development. PLoS Med. 2008;5:e173. doi: 10.1371/journal.pmed.0050173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watkins D.I., Burton D.R., Kallas E.G., Moore J.P., Koff W.C. Nonhuman primate models and the failure of the Merck HIV-1 vaccine in humans. Nat. Med. 2008;14:617–621. doi: 10.1038/nm.f.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muhl T., Krawczak M., Ten Haaft P., Hunsmann G., Sauermann U. MHC class I alleles influence set-point viral load and survival time in simian immunodeficiency virus-infected rhesus monkeys. J. Immunol. 2002;169:3438–3446. doi: 10.4049/jimmunol.169.6.3438. [DOI] [PubMed] [Google Scholar]
- 13.Sauermann U., Siddiqui R., Suh Y.S., Platzer M., Leuchte N., Meyer H., Matz-Rensing K., Stoiber H., Nurnberg P., Hunsmann G. Mhc class I haplotypes associated with survival time in simian immunodeficiency virus (SIV)-infected rhesus macaques. Genes Immun. 2008;9:69–80. doi: 10.1038/sj.gene.6364448. [DOI] [PubMed] [Google Scholar]
- 14.Hirsch V.M., Santra S., Goldstein S., Plishka R., Buckler-White A., Seth A., Ourmanov I., Brown C.R., Engle R., Montefiori D. Immune failure in the absence of profound CD4+ T-lymphocyte depletion in simian immunodeficiency virus-infected rapid progressor macaques. J. Virol. 2004;78:275–284. doi: 10.1128/JVI.78.1.275-284.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sauermann U., Stahl-Hennig C., Stolte N., Muhl T., Krawczak M., Spring M., Fuchs D., Kaup F.J., Hunsmann G., Sopper S. Homozygosity for a conserved Mhc class II DQ-DRB haplotype is associated with rapid disease progression in simian immunodeficiency virus-infected macaques: Results from a prospective study. J. Infect. Dis. 2000;182:716–724. doi: 10.1086/315800. [DOI] [PubMed] [Google Scholar]
- 16.Rogers J., Garcia R., Shelledy W., Kaplan J., Arya A., Johnson Z., Bergstrom M., Novakowski L., Nair P., Vinson A. An initial genetic linkage map of the rhesus macaque (Macaca mulatta) genome using human microsatellite loci. Genomics. 2006;87:30–38. doi: 10.1016/j.ygeno.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 17.Holm S. A simple sequentially rejective multiple test procedure. Scandinavian Journal of Statistics. 1979;6:65–70. [Google Scholar]
- 18.Weir B.S., Cockerham C.C. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 19.Wigginton J.E., Cutler D.J., Abecasis G.R. A note on exact tests of Hardy-Weinberg equilibrium. Am. J. Hum. Genet. 2005;76:887–893. doi: 10.1086/429864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.R Development Core Team . R Foundation for Statistical Computing; Vienna, Austria: 2008. R: A language and environment for statistical computing. [Google Scholar]
- 21.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 22.Lao O., Lu T.T., Nothnagel M., Junge O., Freitag-Wolf S., Caliebe A., Balascakova M., Bertranpetit J., Bindoff L.A., Comas D. Correlation between genetic and geographic structure in Europe. Curr. Biol. 2008;18:1241–1248. doi: 10.1016/j.cub.2008.07.049. [DOI] [PubMed] [Google Scholar]
- 23.Barrett J.C., Fry B., Maller J., Daly M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 24.Carlson C.S., Eberle M.A., Rieder M.J., Yi Q., Kruglyak L., Nickerson D.A. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am. J. Hum. Genet. 2004;74:106–120. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szafranski K., Jahn N., Platzer M. tuple_plot: Fast pairwise nucleotide sequence comparison with noise suppression. Bioinformatics. 2006;22:1917–1918. doi: 10.1093/bioinformatics/btl277. [DOI] [PubMed] [Google Scholar]
- 27.Martin M.P., Carrington M. Immunogenetics of viral infections. Curr. Opin. Immunol. 2005;17:510–516. doi: 10.1016/j.coi.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 28.O'Connor D.H., Mothe B.R., Weinfurter J.T., Fuenger S., Rehrauer W.M., Jing P., Rudersdorf R.R., Liebl M.E., Krebs K., Vasquez J. Major histocompatibility complex class I alleles associated with slow simian immunodeficiency virus disease progression bind epitopes recognized by dominant acute-phase cytotoxic-T-lymphocyte responses. J. Virol. 2003;77:9029–9040. doi: 10.1128/JVI.77.16.9029-9040.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korenromp E.L., Williams B.G., Schmid G.P., Dye C. Clinical prognostic value of RNA viral load and CD4 cell counts during untreated HIV-1 infection - a quantitative review. PLoS ONE. 2009;4:e5950. doi: 10.1371/journal.pone.0005950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stranger B.E., Nica A.C., Forrest M.S., Dimas A., Bird C.P., Beazley C., Ingle C.E., Dunning M., Flicek P., Koller D. Population genomics of human gene expression. Nat. Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pal R., Venzon D., Letvin N.L., Santra S., Montefiori D.C., Miller N.R., Tryniszewska E., Lewis M.G., VanCott T.C., Hirsch V. ALVAC-SIV-gag-pol-env-based vaccination and macaque major histocompatibility complex class I (A∗01) delay simian immunodeficiency virus SIVmac-induced immunodeficiency. J. Virol. 2002;76:292–302. doi: 10.1128/JVI.76.1.292-302.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fish E.N. The X-files in immunity: Sex-based differences predispose immune responses. Nat. Rev. Immunol. 2008;8:737–744. doi: 10.1038/nri2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ober C., Loisel D.A., Gilad Y. Sex-specific genetic architecture of human disease. Nat. Rev. Genet. 2008;9:911–922. doi: 10.1038/nrg2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carriere A., Ray H., Blenis J., Roux P.P. The RSK factors of activating the Ras/MAPK signaling cascade. Front. Biosci. 2008;13:4258–4275. doi: 10.2741/3003. [DOI] [PubMed] [Google Scholar]
- 35.Xing J., Ginty D.D., Greenberg M.E. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science. 1996;273:959–963. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- 36.Dummler B.A., Hauge C., Silber J., Yntema H.G., Kruse L.S., Kofoed B., Hemmings B.A., Alessi D.R., Frodin M. Functional characterization of human RSK4, a new 90-kDa ribosomal S6 kinase, reveals constitutive activation in most cell types. J. Biol. Chem. 2005;280:13304–13314. doi: 10.1074/jbc.M408194200. [DOI] [PubMed] [Google Scholar]
- 37.Thakur A., Sun Y., Bollig A., Wu J., Biliran H., Banerjee S., Sarkar F.H., Liao D.J. Anti-invasive and antimetastatic activities of ribosomal protein S6 kinase 4 in breast cancer cells. Clin. Cancer Res. 2008;14:4427–4436. doi: 10.1158/1078-0432.CCR-08-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang G., Espeseth A., Hazuda D.J., Margolis D.M. c-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. J. Virol. 2007;81:10914–10923. doi: 10.1128/JVI.01208-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Connor R.I., Sheridan K.E., Ceradini D., Choe S., Landau N.R. Change in coreceptor use correlates with disease progression in HIV-1-infected individuals. J. Exp. Med. 1997;185:621–628. doi: 10.1084/jem.185.4.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evans M.J., von Hahn T., Tscherne D.M., Syder A.J., Panis M., Wolk B., Hatziioannou T., McKeating J.A., Bieniasz P.D., Rice C.M. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 41.Lu T.S., Avraham H.K., Seng S., Tachado S.D., Koziel H., Makriyannis A., Avraham S. Cannabinoids inhibit HIV-1 Gp120-mediated insults in brain microvascular endothelial cells. J. Immunol. 2008;181:6406–6416. doi: 10.4049/jimmunol.181.9.6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verma S., Lo Y., Chapagain M., Lum S., Kumar M., Gurjav U., Luo H., Nakatsuka A., Nerurkar V.R. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: Transmigration across the in vitro blood-brain barrier. Virology. 2009;385:425–433. doi: 10.1016/j.virol.2008.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng J., Xie Y., Campbell R., Song J., Massachi S., Razi M., Chiu R., Berenson J., Yang O.O., Chen I.S. Involvement of claudin-7 in HIV infection of CD4(-) cells. Retrovirology. 2005;2:79. doi: 10.1186/1742-4690-2-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su A.I., Wiltshire T., Batalov S., Lapp H., Ching K.A., Block D., Zhang J., Soden R., Hayakawa M., Kreiman G. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Squires K.E. Treating HIV infection and AIDS in women. AIDS Read. 2003;13 228–234, 239–240. [PubMed] [Google Scholar]
- 46.Squires K.E. Gender differences in the diagnosis and treatment of HIV. Gend. Med. 2007;4:294–307. doi: 10.1016/s1550-8579(07)80060-x. [DOI] [PubMed] [Google Scholar]
- 47.Madkan V.K., Giancola A.A., Sra K.K., Tyring S.K. Sex differences in the transmission, prevention, and disease manifestations of sexually transmitted diseases. Arch. Dermatol. 2006;142:365–370. doi: 10.1001/archderm.142.3.365. [DOI] [PubMed] [Google Scholar]
- 48.Quinn T.C., Overbaugh J. HIV/AIDS in women: An expanding epidemic. Science. 2005;308:1582–1583. doi: 10.1126/science.1112489. [DOI] [PubMed] [Google Scholar]
- 49.Mofenson L.M., Harris D.R., Rich K., Meyer W.A., Read J.S., Moye J., Nugent R.P., Korelitz J., Bethel J., Pahwa S. Serum HIV-1 p24 antibody, HIV-1 RNA copy number and CD4 lymphocyte percentage are independently associated with risk of mortality in HIV-1-infected children. National Institute of Child Health and Human Development Intravenous Immunoglobulin Clinical Trial Study Group. AIDS. 1999;13:31–39. doi: 10.1097/00002030-199901140-00005. [DOI] [PubMed] [Google Scholar]
- 50.Collazos J., Asensi V., Carton J.A. Sex differences in the clinical, immunological and virological parameters of HIV-infected patients treated with HAART. AIDS. 2007;21:835–843. doi: 10.1097/QAD.0b013e3280b0774a. [DOI] [PubMed] [Google Scholar]
- 51.Jarrin I., Geskus R., Bhaskaran K., Prins M., Perez-Hoyos S., Muga R., Hernandez-Aguado I., Meyer L., Porter K., del Amo J. Gender differences in HIV progression to AIDS and death in industrialized countries: Slower disease progression following HIV seroconversion in women. Am. J. Epidemiol. 2008;168:532–540. doi: 10.1093/aje/kwn179. [DOI] [PubMed] [Google Scholar]
- 52.Nicastri E., Angeletti C., Palmisano L., Sarmati L., Chiesi A., Geraci A., Andreoni M., Vella S. Gender differences in clinical progression of HIV-1-infected individuals during long-term highly active antiretroviral therapy. AIDS. 2005;19:577–583. doi: 10.1097/01.aids.0000163934.22273.06. [DOI] [PubMed] [Google Scholar]
- 53.Nicastri E., Leone S., Angeletti C., Palmisano L., Sarmati L., Chiesi A., Geraci A., Vella S., Narciso P., Corpolongo A. Sex issues in HIV-1-infected persons during highly active antiretroviral therapy: A systematic review. J. Antimicrob. Chemother. 2007;60:724–732. doi: 10.1093/jac/dkm302. [DOI] [PubMed] [Google Scholar]
- 54.Napravnik S., Poole C., Thomas J.C., Eron J.J. Gender difference in HIV RNA levels: A meta-analysis of published studies. J. Acquir. Immune Defic. Syndr. 2002;31:11–19. doi: 10.1097/00126334-200209010-00002. [DOI] [PubMed] [Google Scholar]
- 55.Sterling T.R., Vlahov D., Astemborski J., Hoover D.R., Margolick J.B., Quinn T.C. Initial plasma HIV-1 RNA levels and progression to AIDS in women and men. N. Engl. J. Med. 2001;344:720–725. doi: 10.1056/NEJM200103083441003. [DOI] [PubMed] [Google Scholar]
- 56.Foca M., Moye J., Chu C., Matthews Y., Rich K., Handelsman E., Luzuriaga K., Paul M., Diaz C. Gender differences in lymphocyte populations, plasma HIV RNA levels, and disease progression in a cohort of children born to women infected with HIV. Pediatrics. 2006;118:146–155. doi: 10.1542/peds.2005-0294. [DOI] [PubMed] [Google Scholar]
- 57.Amadori A., Zamarchi R., De Silvestro G., Forza G., Cavatton G., Danieli G.A., Clementi M., Chieco-Bianchi L. Genetic control of the CD4/CD8 T-cell ratio in humans. Nat. Med. 1995;1:1279–1283. doi: 10.1038/nm1295-1279. [DOI] [PubMed] [Google Scholar]
- 58.Lyon M.F. Evolution of X-chromosome inactivation in mammals. Nature. 1974;250:651–653. doi: 10.1038/250651a0. [DOI] [PubMed] [Google Scholar]
- 59.Carrel L., Willard H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- 60.Knudsen G.P., Pedersen J., Klingenberg O., Lygren I., Orstavik K.H. Increased skewing of X chromosome inactivation with age in both blood and buccal cells. Cytogenet. Genome Res. 2007;116:24–28. doi: 10.1159/000097414. [DOI] [PubMed] [Google Scholar]
- 61.Ge D., Zhang K., Need A.C., Martin O., Fellay J., Urban T.J., Telenti A., Goldstein D.B. WGAViewer: Software for genomic annotation of whole genome association studies. Genome Res. 2008;18:640–643. doi: 10.1101/gr.071571.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blanchette M., Kent W.J., Riemer C., Elnitski L., Smit A.F., Roskin K.M., Baertsch R., Rosenbloom K., Clawson H., Green E.D. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.