

Abstract
Protein arginine methyltransferases (PRMTs) are a group of eukaryotic enzymes that catalyze the methylation of Arg residues in a variety of proteins (e.g., histones H3 and H4), and their activities influence a wide range of cellular processes, including cell growth, RNA splicing, differentiation, and transcriptional regulation. Dysregulation of these enzymes has been linked to heart disease and cancer, suggesting this enzyme family as a novel therapeutic target. To aid the development of PRMT inhibitors, we characterized the substrate specificity of both the rat and human PRMT1 orthologues using histone based peptide substrates. N- and C-terminal truncations to identify a minimal peptide substrate indicate that long-range interactions between enzyme and substrate are important for high rates of substrate capture. The importance of these long-range interactions to substrate capture were confirmed by “mutagenesis” experiments on a minimal peptide substrate. Inhibition studies on S-adenosyl-homocysteine, thioadenosine, methylthioadenosine, homocysteine, and sinefungin suggest that potent and selective bisubstrate analogue inhibitor(s) for PRMT1 can be developed by linking a histone based peptide substrate to homocysteine or sinefungin. Additionally, we present evidence that PRMT1 utilizes a partially processive mechanism to dimethylate its substrates.
The protein arginine methyltransferases (PRMTs1) are a group of evolutionarily conserved S-adenosyl-l-methionine (SAM)-dependent enzymes that catalyze the direct transfer of a methyl group from SAM to one or more of the η-nitrogens of an Arg residue. This is an SN2 type reaction, and at least three products are possible, i.e., monomethyl Arg (MMA), asymmetric dimethyl Arg (ADMA), and symmetric dimethyl Arg (SDMA) (Figure 1). The PRMTs are generally classified as either type I or type II enzymes; type I PRMTs catalyze the formation of both MMA and ADMA whereas type II PRMTs catalyze the formation of MMA and SDMA (an enzyme that catalyzes the exclusive monomethyl form (MMA) has not been identified). In higher eukaryotes, there are 11 putative isozymes, with five (PRMT1, 3, 4, 6, and 8) being type I enzymes, three (PRMT5, 7, and 9) being type II, and three (PRMT2, 10, and 11) being unclassified because they have not been demonstrated to be enzymatically active (1).
Figure 1.

Reactions catalyzed by PRMT isozymes. The enzymes are classified according to the type of reaction in which they catalyze. Those which generate asymmetric dimethyl-Arg residues (ADMA) are classified as type I PRMTs, while those generating symmetric dimethyl-Arg (SDMA) residues are classified as type II PRMTs.
Numerous PRMT substrates have been identified (for reviews see (1, 2)), and while the specific effects of Arg methylation are not known in all cases, PRMT activity does influence a variety of important cellular processes including cellular growth (3, 4), nuclear/cytoplasmic protein shuttling (5–7), differentiation and embryogenesis (8–11), RNA splicing and transport (12–15), and post-transcriptional gene regulation (16). In addition, several PRMTs act as transcriptional coregulators for a number of nuclear receptors, including the estrogen and androgen receptors. PRMT1 and PRMT4/CARM1 (coactivator associated arginine methyltransferase 1) are the best characterized PRMTs, and these enzymes methylate histones H3 at Arg2, 17, and 26 (by PRMT4/CARM1) and H4 at Arg3 (by PRMT1); and their ability to methylate these substrates (and other components of the transcriptional apparatus, e.g., p300 (17, 18)) plays a critical role in coactivating nuclear receptor controlled gene transcription (17, 19–25). PRMT1 and PRMT4/CARM1 also act as transcriptional coactivators for a variety of other transcription factors, including p53 (19).
In addition to their important roles in normal cellular function, the methyltransferase activities of several PRMTs are dysregulated in human disease (26–33). For example, PRMT4/CARM1 is aberrantly expressed in prostate cancer (26, 27) and likely contributes to the proliferative capacity of prostate cancer cells through its ability to act as a transcriptional coactivator for the androgen receptor (23, 27). Knockdown of PRMT4/CARM1 expression inhibits proliferation of prostate cancer cells and induces apoptosis (27); suggesting PRMT4/CARM1 inhibitors as a novel prostate cancer therapeutic. Additionally, the fact that PRMT1 and PRMT4/CARM1 are nuclear receptor coactivators and dys-regulation of nuclear receptor signaling is a common feature of several cancers, e.g., breast cancer (34–37), suggests that inhibitors targeting these isozymes may have broad use as cancer therapeutics.
PRMT activity also appears to be dysregulated in cardiovascular disease. For example, PRMT1 and PRMT3 (both type I enzymes) are overexpressed in myocardial tissue taken from patients with coronary heart disease (38). Additionally, the levels of free ADMA, an indirect byproduct of the type I PRMT catalyzed reaction and second only to age as a predictor of mortality and cardiovascular events in patients with chronic renal failure (39), are elevated in patients suffering from atherosclerosis, hypercholesterolemia, and heart failure ((29–32) and references cited therein). Note that the levels of free ADMA appear to be dictated by the synthesis and degradation of methylated proteins and that this compound acts as an endogenous inhibitor of multiple NOS isoforms (9). It is also interesting to note that deletion of the gene encoding dimethylarginine dimethylaminohydrolase 1 (DDAH1), one of the enzymes responsible for degrading ADMA to citrulline (Cit), raises the levels of free ADMA, reduces NO signaling, and causes a variety of pathological effects on the mouse vasculature (e.g., elevated systemic and pulmonary blood pressure) (33). Thus, one would expect that inhibitors targeting one or more of PRMT isozymes would represent an effective therapy for cardiovascular disease because they would decrease the ADMA pool.
Because of the relationship between dysregulated PRMT function and human disease, we and others (40–42) are interested in developing potent and selective inhibitors targeting these enzymes. To aid these efforts, we initiated mechanistic and substrate specificity studies on PRMT1. Our initial efforts are focused on this isozyme because it is the predominant type I PRMT in mammalian cells, it is one of the best studied PRMTs at the cellular level, an X-ray crystal structure is available for the rat orthologue (43), and its methylated substrates are thought to be the major in vivo source of ADMA (44). PRMT1 is a 353 residue (40.5 kDa) enzyme that was independently identified as an interacting partner for the interferon receptor and the immediate early gene TIS21 (3, 45). This enzyme is ubiquitously expressed in adult and embryonic tissues, and although embryonic stem cells lacking this enzyme are viable, it is essential for early development because mouse PRMT1−/− knockouts die in utero (9, 44, 46). While numerous PRMT1 substrates have been identified (e.g., SAM68 (47)), its best characterized substrate, at the cellular level, is histone H4 (19, 22).
Herein, we describe the results of our efforts to characterize the substrate specificity of both the rat and human recombinant PRMT1 orthologues (rPRMT1 and hPRMT1, respectively). Our results indicate that long-range interactions are important for high affinity interactions between enzyme and substrate. These results, when combined with inhibition studies on S-adenosyl-l-homocysteine (SAH), thioadenosine (5TA), methylthioadenosine (MTA; an alkylated analogue of 5TA), and homocysteine, point the way to the development of potent and selective bisubstrate analogue inhibitors of PRMT1. Additionally, we present evidence that PRMT1 utilizes a partially processive mechanism to dimethylate its substrates.
EXPERIMENTAL PROCEDURES
General
All chemicals and N-α-Fmoc-amino acids were purchased from Sigma-Aldrich, VWR, and Novabiochem and used without further purification. 14C-labeled SAM and 14C-labeled bovine serum albumin (BSA) were purchased from Perkin-Elmer Life Sciences. Oligonucleotide primers were from Integrated DNA Technologies. Recombinant histone H4 was purified as described (48, 49). Mass spectra were acquired on a Bruker Ultraflex II MALDI-TOF mass spectrometer.
Purification of PRMTs
rPRMT1 and hPRMT1 were purified analogously to methods described in (43). Briefly, a PRMT1 expression construct, encoding a previously described His-tagged protein (43), was transformed into Escherichia coli BL21(DE3). Single colonies were used to inoculate starter cultures that were grown overnight at 37 °C. The next day these cultures were used to inoculate 4 × L of 2YT media (10 mL of culture per L of media). These large scale cultures were then grown at 37 °C until an OD600 of 0.4 was achieved. The incubation temperature was then reduced to 22 °C and isopropyl-β-d-thiogalactopyranoside added to a final concentration 0.4 mM. Cells were incubated at 22 °C overnight, at which point the cells were harvested by centrifugation for 10 min at 5000g. The resulting pellet was resuspended in 30 mL of lysis buffer (20 mM HEPES at pH 8.0, 100 mM NaCl, 5 mM imidazole, 10% glycerol, 0.5 mM phenylmethylsulfonyl fluoride). The cells were then lysed using a French pressure cell at 20,000 psi and the lysates clarified by centrifugation for 30 min at 20000g. The supernatant was applied to a nickel(II) chelating Sepharose column, and bound proteins were eluted using a stepwise imidazole gradient (5−500 mM imidazole in 20 mM HEPES at pH 8.0 plus 100 mM NaCl). The presence of PRMT1 in specific fractions was then assessed by SDS–PAGE analysis, and those fractions containing protein were dialyzed into 20 mM HEPES at pH 8.0 plus 50 mM NaCl for 2 h. The protein present in these dialyzed fractions was incubated with 100 μM SAH and further purified by FPLC using a Mono Q anion exchange column (GE Healthcare). Fractions were again analyzed by SDS–PAGE, and those containing protein were dialyzed overnight in a dialysis buffer containing 100 mM HEPES at pH 8.0, 200 mM NaCl, 1 mM DTT, 2 mM EDTA, and 10% glycerol. The dialyzed protein was then concentrated using an Amicon Centriplus centrifugal filter device with a molecular weight cutoff of 10 kDa. The concentration of the protein was determined by Bradford analysis. Enzyme aliquots were flash frozen in liquid nitrogen and stored at −80 °C. Enzyme prepared under these conditions is stable for several months. After purification on the nickel(II) chelating Sepharose and Mono Q anion exchange columns, we are consistently able to obtain highly pure enzyme in modest yield (1 mg/L of E. coli cell culture).
Site Directed Mutagenesis
An expression construct encoding the His161Tyr mutant of rPRMT1 was generated using the Quik Change Mutagenesis system (Stratagene). The sequence of the forward and reverse mutagenic primers, respectively, are 5′-CTC AAC ACT GTG CTG TAC GCT CGT GAC AAG TGG-3′ and 5′-CCA CTT GTC ACG AGC GTA CAG CAC AGT GTT GAG-3′.
Synthesis of Histone-Based Peptide Substrates
Peptides utilized in kinetic assays were synthesized using a Rainin PS3 automated peptide synthesizer. Standard Fmoc chemistry and commercially available amino acids and Wang-based resins were utilized. After synthesis, peptides were cleaved from the solid support by incubating the resin for 2 h with 20 mL of Reagent K (19 mL trifluoroacetic acid (TFA), 510 mg of phenol, 500 μL of ddH2O, 500 μL of thioanisole, and 250 μL of 1,2-ethanedithiol). The filtrate was collected and concentrated by flowing N2(g). Peptides were precipitated from this mixture with ice-cold diethyl ether, and the resulting product was centrifuged at 3000g for 10 min. The supernatant was discarded, and the diethyl ether and centrifugation process was repeated twice more. The peptide product was then dissolved in a minimal volume of water, flash frozen, and lyophilized overnight. The peptide was then purified by preparative reverse phase HPLC using a water/acetonitrile/0.05% TFA gradient. The structures of the peptides were confirmed by MALDI mass spectrometry. The expected and observed masses for the peptides used in these studies, and their respective sequences, are listed in Table 1.
Table 1.
Histone Peptide Derivatives
| Peptide | Sequence | Expected Mass (Da) | Observed Mass (Da) |
|---|---|---|---|
| RGG3 | GGRGGFGGRGGFGGRGGFG | 1669 | 1670 |
| AcH4−21 | 1-Ac-SGRGKGGKGLGKGGAKRHRKV | 2132 | 2133 |
| AcH4−21 R3K | 1-Ac-SGKGKGGKGLGKGGAKRHRKV | 2104 | 2105 |
| AcH4−18 | 1-Ac-SGRGKGGKGLGKGGAKRH | 1749 | 1750 |
| AcH4−16 | 1-Ac-SGRGKGGKGLGKGGAK | 1456 | 1457 |
| AcH4−15 | 1-Ac-SGRGKGGKGLGKGGA | 1327 | 1329 |
| AcH4−13 | 1-Ac-SGRGKGGKGLGKG | 1200 | 1201 |
| AcH4−21Δ(11−13) | 1-Ac-SGRGKGGKGL - - - GAKRHRKV | 1890 | 1891 |
| AcH4−21Δ(9−15) | 1-Ac-SGRGKGGK - - - - - - AKRHRKV | 1663 | 1664 |
| AcH4−16Δ(l) | 2-Ac-GRGKGGKGLGKGGAK | 1369 | 1370 |
| AcH4−16Δ(l-2) | 3-Ac-RGKGGKGLGKGGAK | 1312 | 1313 |
| H4−16Δ(l-2) | RGKGGKGLGKGGAK | 1157 | 1158 |
| AcH4−21 S1A | 1-Ac-AGRGKGGKGLGKGGAKRHRKV | 2116 | 2117 |
| AcH4−21 K16A | 1-Ac-SGRGKGGKGLGKGGAARHRKV | 2075 | 2076 |
| AcH4−21 R17A | 1-Ac-SGRGKGGKGLGKGGAKAHRKV | 2047 | 2048 |
| AcH4−21 H18A | 1-Ac-SGRGKGGKGLGKGGAKRARKV | 2066 | 2067 |
| AcH4−21 R19A | 1-Ac-SGRGKGGKGLGKGGAKRHAKV | 2047 | 2048 |
| AcH4−21 K20A | 1-Ac-SGRGKGGKGLGKGGAKRHRAV | 2075 | 2077 |
| AcH4−21 R17A/R19A | 1-Ac-SGRGKGGKGLGKGGAKAHAKV | 1962 | 1963 |
| AcH4−21 R17,19Cit | 1-Ac-SGRGKGGKGLGKGGAKCitHCitKV | 1995 | 1996 |
| AcH4−21 K5,8,12,16AcK | 1-Ac-SGRGKAcGGKAcGLGKAcGGAKAcRHRKV | 2300 | 2301 |
| AcH4−21 all Ac-K | l-Ac-SGRGKAcGGKAcGLGKAcGGAKAcRHRKAcV | 2342 | 2343 |
| AcH4−21 K20K(Me)3 | 1-Ac-SGRGKGGKGLGKGGAKRHRK(Me)3V | 2174 | 2175 |
| AcH4−21 R3MMA | 1-Ac-SGR(Me)GKGGKGLGKGGAKRHRKV | 2146 | 2147 |
| AcH4−21 R3ADMA | l-Ac-SGR(Me)2GKGGKGLGKGGAKRHRKV | 2160 | 2161 |
| AcH4−15R3MMA | 1-Ac-SGR(Me)GKGGKGLGKGGA | 1341 | 1343 |
Gel-Based Activity Assay
A gel based assay, previously developed for kinetic studies on histone acetyltransferases (49, 50), was adapted to quantitatively characterize PRMT1 activity. Briefly, this is a discontinuous methyltransferase assay that measures 14C-methyl-peptide (or 14C-methylprotein) production by separating the products of the reaction on 16.5% Tris-Tricine polyacrylamide gels. 14C-labeled SAM is used as the methyl donor, and the amount of radioactivity incorporated into peptide or protein substrates is quantified by phosphorimage analysis (Molecular Dynamics), using 14C-labeled BSA as an internal reference standard. Generally, peptide and 14C-methyl-SAM (15 μM (>5Km) unless otherwise noted) were incubated in assay buffer (50 mM HEPES at pH 8.0, 50 mM NaCl, 1 mM EDTA, and 0.5 mM dithiothreitol) for 10 min at 37 °C. The reaction was then initiated by the addition of hPRMT1 (212 nM final) or rPRMT1 (200 nM final). Assays were performed in duplicate, and activity generally agreed within 20% standard deviation. Both hPRMT1 and rPRMT1 activity are linear with respect to time and enzyme concentration in the range employed in the assay. The initial rates obtained from these assays were fit by nonlinear least fit squares to eq 1, using the GraFit version 5.0.11 software package (51).
| (1) |
IC50 Assays
The IC50 values were determined for sine-fungin, SAH, MTA, 5TA, and homocysteine using the enzyme assay described above. For these studies, the AcH4−21 (N-terminally acetylated) or RGG3 peptides (Table 1), inhibitor, and 14C-SAM were preincubated in assay buffer for 10 min at 37 °C prior to the initiation of the reaction by the addition of rPRMT1 (200 nM final). After incubating for an additional 20 min, the reaction samples were quenched by the addition of denaturing tris-tricine gel loading dye. Samples were processed as described above, and IC50 values were then determined by fitting the activity data to eq 2, using the GraFit version 5.0.11 software package (51), where [I] is the concentration of inhibitor.
| (2) |
Mass Spectrometry Based Methylation Assay
MS based methylation assays were performed and analyzed via matrix assisted laser desorption/ionization (MALDI) MS. For these assays, a peptide substrate and SAM were preincubated at 37 °C for 10 min in assay buffer. Subsequently, rPRMT1 (500 nM) was added to the reaction mixture and reactions were allowed to proceed for an additional 20 min. The reactions were then quenched by the addition of 3 μL of 50% TFA in ddH2O and mass spectra acquired. Samples were processed for MALDI-TOF MS analysis after purification on a C-18 zip tip. Peptides eluted from the C-18 zip tip were combined with a saturated solution of α-cyano-4-hydroxy cinnamic acid in 50% acetonitrile, 50% water, and 0.1% TFA. An aliquot of the mixture (1.5 μL) was then evaporated on a MALDI plate and analyzed in positive ion mode with the laser set at the lowest setting necessary to generate an adequate signal. The laser setting and the number of shots per scan remained constant along each set of reaction samples. After MALDI-TOF spectra were acquired and analyzed using the Flex Analysis software, the mass and intensity values of relevant peaks were analyzed to collect quantitative information for each sample. The percentage turnover was determined by dividing the intensity of the modified substrate by the sum of the intensities of the substrate and modified substrates times 100%. MALDI-MS experiments on equimolar amounts of the AcH4−21, AcH4−21 R3MMA, and AcH4−21 R3ADMA peptides yielded similar intensities; thereby indicating that Arg methylation does not cause ion suppression.
RESULTS AND DISCUSSION
rPRMT1 Methylation of Histone H4 and Peptide-Based Substrates
Because rPRMT1 has been used for structural and molecular biology studies, we initially focused our efforts on this orthologue. Recombinant His-tagged rPRMT1 was expressed and purified using a previously described construct (43) and the steady-state kinetic parameters determined for a number of baseline substrates, including SAM (the methyl donor), recombinant histone H4 (a known PRMT1 substrate), and the RGG3 peptide (a traditionally used PRMT1 substrate whose sequence is based on an Arg rich region in fibrillarin; Table 1). A gel based radioactive assay (49) was used to monitor the transfer of the methyl group from [Me-14C]-SAM to an Arg residue in a protein or peptide substrate. The results from these initial kinetic studies indicated that both SAM and histone H4 are very good PRMT1 substrates with low micromolar Km values and kcat's in the 0.5 min−1 range (Table 2). It is also noteworthy that the data for the RGG3 peptide are consistent with those obtained using a continuous spectrophotometric assay (52), thereby validating our radioactive assay.
Table 2.
Initial Kinetic Characterization
|
Km (μM) |
kcat (min−1) |
kcat/Km (M−1·min−1) |
||||
|---|---|---|---|---|---|---|
| substrate | hPRMT1 | rPRMT1 | hPRMT1 | rPRMT1 | hPRMT1 | rPRMT1 |
| histone H4a | 3.06 ± 0.11 | 1.45 ± 0.37 | 0.44 ± 0.04 | 0.53 ± 0.04 | 1.45 × 105 | 3.68 × 105 |
| SAMb | ndd | 2.52 ± 0.54 | ndd | 0.84 ± 0.03 | ndd | 3.36 × 105 |
| RGG3a,c | 31.4 ± 5.7 | 36.1 ± 6.6 | 0.32 ± 0.02 | 0.68 ± 0.03 | 1.01 × 104 | 1.89 × 104 |
| AcH4−21a,c | 1.14 ± 0.54 | 3.55 ± 0.68 | 0.46 ± 0.02 | 0.39 ± 0.01 | 4.08 × 105 | 1.10 × 105 |
| AcH4−21 R3Ka,c | nde | nde | nde | nde | <1.58 | <0.24 |
[SAM] = 15 μM.
[AcH4−21] = 100 μM.
Sequence and mass available in Table 1.
Did not perform.
The amount of product formation was too low to accurately measure the kinetic parameters for this peptide substrate. Therefore an estimation of kcat/Km was made by dividing the maximal observed rate by the concentration of substrate.
To determine whether peptides are a good model system for identifying the substrate specificity determinants of PRMT1, the steady-state kinetic parameters were determined for the AcH4−21 peptide. The sequence of this peptide is based on the N-terminal 21 amino acids of histone H4 and contains the major in vivo site of PRMT1 catalyzed Arg methylation, i.e., Arg3 (R3) (53). Kinetic studies with this peptide revealed that the specificity constants (kcat/Km) for the AcH4−21 peptide and histone H4, which is N-terminally acetylated in vivo, are quite similar (1.10 × 105 versus 3.68 × 105, respectively), helping to validate our use of simpler peptide substrates to identify the substrate specificity determinants of PRMT1. Note that kcat/Km is used here as a measure of substrate affinity because for many enzymes Km is not a true dissociation constant.
To further establish the use of histone H4 peptides as a model system, the AcH4−21R3K peptide–the target Arg3 mutated to Lys–was synthesized and the kinetics of methylation determined. The results of these studies (Table 2) indicate that the AcH4−21 R3K peptide is a very poor substrate for rPRMT1, as evidenced by the >105-fold decrease in kcat/Km relative to the AcH4−21 peptide. The fact that these results are consistent with this site being the major site of modification in histone H4 (53) adds further support for the use of histone H4-based peptides as a model system for substrate specificity studies.
Kinetic Studies on hPRMT1
With respect to human disease and the development of a PRMT1-targeted therapeutic, the more relevant form of the enzyme is hPRMT1. The only difference between the amino acid sequences of the rat and human PRMT1 orthologues occurs at position 161, where this residue is a His in rPRMT1 and a Tyr in hPRMT1. While the importance of His161 to substrate binding is unknown, this residue is within contact distance of the RGG3 peptide in the structure of the rPRMT1·RGG3 complex (43). Thus, the determinants of substrate specificity could be dramatically different for these two orthologues. Therefore, to gain insights into the substrate specificity requirements of hPRMT1, we used site directed mutagenesis to generate a construct encoding the rPRMT1 H161Y mutant, i.e., a humanized version of rPRMT1 that we refer to hereafter as hPRMT1. This enzyme was expressed and purified using the methodology described above for rPRMT1. Interestingly, the kcat/Km values observed for all substrates tested (Table 2) were generally similar. Thus the His and Tyr are in most cases functionally equivalent, although we do note that significant differences (≤5-fold) were observed for several shorter peptides (see below).
Identification of a Minimal Peptide Substrate
To broadly identify those residues that are important for substrate recognition, a series of C-terminal truncation peptides (denoted AcH4−18, AcH4−16, AcH4−15, and AcH4−13) were synthesized (Table 1) and their ability to act as PRMT1 substrates was evaluated. Kinetic studies on these peptides (Table 3 and Figure 2) clearly indicate that long range interactions are critical for high rates of substrate capture. For example, relative to the AcH4−21 peptide, the kcat/Km values obtained for the AcH4−18 peptide are decreased 25-and 150-fold for rPRMT1 and hPRMT1, respectively. Furthermore, the AcH4−15 peptide, which lacks 3 additional residues, was an even poorer substrate as evidenced by the 62.5- and 860-fold decreases in kcat/Km for rPRMT1 and hPRMT1. To further probe the role of long range interactions in substrate recognition, we generated two mid-sequence truncation peptides, denoted AcH4−21Δ(11−13) and AcH4−21Δ(9−15), which lack glycine-rich residues 11 to 13 (GKG) and 9 to 15 (GLGKGGA), respectively, of the AcH4−21 peptide. Remarkably, the kcat/Km values obtained for these peptides were decreased by only an order of magnitude, relative to the AcH4−21 peptide. These results suggest that there is some flexibility in the positioning of the C-terminal basic residues (i.e., K16-R-H-R-K20) on the enzyme with respect to the site of methylation.
Table 3.
Effect of C-Terminal and Mid-Sequence Truncations
|
Km (μM) |
kcat (min−1) |
kcat/Km (M−1·min−1) |
||||
|---|---|---|---|---|---|---|
| substrate | hPRMT1 | rPRMT1 | hPRMT1 | rPRMT1 | hPRMT1 | rPRMT1 |
| histone H4a | 3.06 ± 0.11 | 1.45 ± 0.37 | 0.44 ± 0.04 | 0.53 ± 0.04 | 1.45 × 105 | 3.68 × 105 |
| AcH4−21a,b | 1.14 ± 0.54 | 3.55 ± 0.68 | 0.46 ± 0.02 | 0.39 ± 0.01 | 4.08 × 105 | 1.10 × 105 |
| AcH4−18a,b | 162 ± 29 | 223 ± 11 | 0.39 ± 0.02 | 0.86 ± 0.01 | 2.41 × 103 | 3.83 × 103 |
| AcH4−16a,b | 106 ± 14 | 152 ± 14 | 0.23 ± 0.01 | 0.28 ± 0.01 | 2.16 × 103 | 1.82 × 103 |
| AcH4−15a,b | 355 ± 166 | 120 ± 16 | 0.17 ± 0.05 | 0.21 ± 0.01 | 4.76 × 102 | 1.76 × 103 |
| AcH4−13a,b | 466 ± 101 | 365 ± 174 | 0.17 ± 0.02 | 0.18 ± 0.05 | 3.75 × 102 | 4.94 × 102 |
| AcH4−21Δ(11−13)a,b | 20.8 ± 2.1 | 66.6 ± 8.8 | 0.57 ± 0.01 | 0.82 ± 0.03 | 2.71 × 104 | 1.23 × 104 |
| AcH4−21Δ(9−15)a,b | 10.6 ± 1.9 | 46.7 ± 4.5 | 0.33 ± 0.01 | 0.92 ± 0.02 | 3.15 × 104 | 1.96 × 104 |
[SAM] = 15 μM.
Sequence and mass available in Table 1.
Figure 2.

Substrate specificity of human PRMT1 versus rat PRMT1. The kcat/Km values determined for histone H4 and histone H4 based peptides of varying length are depicted graphically.
To identify residues N-terminal to the site of methylation that are important for substrate recognition, a second series of peptide substrates, denoted AcH4−16Δ1 (H4 residues 2−16; N-terminally acetylated) and AcH4−16Δ(1−2) (H4 residues 3−16; N-terminally acetylated), was synthesized. The sequences of these peptides were designed such that a single residue is progressively removed from the N-terminus of the AcH4−16 peptide. The 16 residue peptide was used as a starting point for these studies because the AcH4−21 peptide contains two downstream Arg residues (Arg17 and Arg19) that could potentially act as alternative methyl acceptors upon the deletion of N-terminal residues, thereby complicating our kinetic analysis. Interestingly, these peptides were differentially processed by the two orthologues (Table 4). For example, the kcat/Km values for the AcH4−16Δ1 and AcH4−16Δ(1−2) peptides are decreased 5.6-fold and 16.4-fold for hPRMT1, whereas the effects of these deletions is less than 2-fold for rPRMT1. These results suggest that the backbone atoms of Ser1 and Gly2, as well as the side chain hydroxyl of Ser1, are more important for binding to hPRMT1 than to rPRMT1. The H4−16Δ(1−2) (H4 residues 3−16; unacetylated N-terminus) peptide was also synthesized to evaluate the requirement for an amide bond N-terminal to the site of Arg methylation. Interestingly, the H4−16Δ(1−2) peptide, which possesses a positively charged α-amino group, was an extremely poor substrate for both rat and human PRMT1 (kcat/Km decreased by 103- to 104-fold; Table 4), thereby indicating that interactions between the enzyme and the backbone amide N-terminal to the site of methylation are critical for substrate recognition. In summary, the results of our studies with N- and C-terminal deletion peptides indicate that the AcH4−21 peptide is methylated with comparable kinetics to histone H4, thereby defining it as the minimal peptide substrate for PRMT1.
Table 4.
Effect of N-Terminal Truncations
|
Km (μM) |
kcat (min−1) |
kcat/Km (M−1·min−1) |
||||
|---|---|---|---|---|---|---|
| substrate | hPRMT1 | rPRMT1 | hPRMT1 | rPRMT1 | hPRMT1 | rPRMT1 |
| histone H4a | 3.06 ± 0.11 | 1.45 ± 0.37 | 0.44 ± 0.04 | 0.53 ± 0.04 | 1.45 × 105 | 3.68 × 105 |
| AcH4−16a,b | 106 ± 14 | 152 ± 14 | 0.23 ± 0.01 | 0.28 ± 0.01 | 2.16 × 103 | 1.82 × 103 |
| AcH4−16Δ(1)a,b | 152 ± 34 | 141 ± 17 | 0.13 ± 0.11 | 0.68 ± 0.03 | 8.83 × 102 | 4.85 × 103 |
| AcH4−16Δ(1−2)a,b | 282 ± 79 | 510 ± 171 | 0.08 ± 0.01 | 0.74 ± 0.15 | 2.76 × 102 | 1.46 × 103 |
| H4−16Δ(1−2)a,b | ndc | ndc | ndc | ndc | <2.56 | <0.10 |
[SAM] = 15 μM.
Sequence and mass available in Table 1.
The amount of product formation was too low to accurately measure the kinetic parameters for this peptide substrate. Therefore, an estimation of kcat/Km was made by dividing the maximal observed rate by the concentration of substrate.
“Mutagenesis” Studies
The results described above suggested that both N- and C-terminal H4 peptide residues (up to 21) contribute to substrate binding. Therefore, to further examine the roles of these charged/polar residues in substrate capture, we synthesized a series of peptides in which Ser1, Lys16, Arg17, His18, Arg19, and Lys20 were systematically “mutated” to Ala. While kinetic studies with hPRMT1 on the AcH4−21S1A peptide revealed a relatively small 7.5-fold decrease in kcat/Km (Table 5), the results are gratifyingly consistent with the 5.6-fold decrease in kcat/Km observed for the AcH4−16Δ1 peptide, which lacks this amino acid. These results confirm the importance of the Ser1 hydroxyl to substrate binding.
Table 5.
Site Directed “Mutagenesis”
| substrate | Km (μM) | kcat (min−1) | kcat/Km (M−1·min−1) | kcat/Km(AcH4−21)/kcat/Km(mut) |
|---|---|---|---|---|
| AcH4−21a,b | 1.14 ± 0.54 | 0.46 ± 0.02 | 4.08 × 105 | n/a |
| AcH4−21 S1Aa,b | 7.11 ± 1.84 | 0.39 ± 0.02 | 5.45 × 104 | 7.49 |
| AcH4−21 K16Aa,b | 61.3 ± 3.2 | 0.35 ± 0.01 | 5.72 × 103 | 71.3 |
| AcH4−21 R17Aa,b | 32.7 ± 2.7 | 0.97 ± 0.02 | 1.61 × 104 | 25.3 |
| AcH4−21 H18Aa,b | 11.6 ± 3.5 | 0.38 ± 0.02 | 3.28 × 104 | 12.4 |
| AcH4−21 R19Aa,b | 41.5 ± 6.3 | 0.35 ± 0.01 | 8.48 × 103 | 48.1 |
| AcH4−21 K20Aa,b | 32.4 ± 4.3 | 0.43 ± 0.01 | 1.33 × 104 | 30.7 |
| AcH4−21 R17A/R19Aa,b | 140 ± 14 | 0.34 ± 0.03 | 3.59 × 103 | 114 |
| AcH4−21 R17Cit/R19Cita,b | 112 ± 17 | 0.56 ± 0.04 | 5.06 × 103 | 80.6 |
[SAM] = 15 μM.
Sequence and mass available in Table 1.
The kinetic parameters determined for the C-terminal mutant peptides (Table 5) are also consistent with the results obtained for the C-terminal deletion peptides. For example, relative to the parent peptide, i.e., AcH4−21, the kcat/Km values determined for R19A and K20A peptides are decreased ∼48-and ∼30-fold, respectively, values that when summed are comparable to the 150-fold decrease in kcat/Km observed for the AcH4−18 peptide, which lacks these two residues. Similarly large decreases in kcat/Km are observed for the other C-terminal mutant peptides tested. These decreases range from ∼12-fold for the AcH4−21H18A peptide to ∼71-fold for the AcH4−21K16A. While the results obtained for these “mutant” peptides (Table 5) do not identify a single residue responsible for the high affinity of the AcH4−21 peptide, the results do indicate that multiple positively charged residues at C-termini contribute to substrate binding. This presumably occurs in a synergistic fashion because the total effect of mutating residues 16 to 20 (∼190-fold) is less than the effect of deleting these residues, as is evidenced by the 860-fold decrease in kcat/Km recorded for the AcH4−15 peptide, which lacks these residues. To confirm a synergistic effect, we synthesized a peptide, denoted AcH4−21R17A/R19A, in which the two C-terminal Arg residues (i.e., Arg17 and 19) were replaced with Ala. Kinetic studies on this peptide revealed a 114-fold decrease in kcat/Km. This effect is significantly higher than the sum of the fold decreases in kcat/Km that was observed when each residue was mutated to Ala on its own (∼73-fold), consistent with the notion that C-terminal residues contribute to substrate capture in a synergistic fashion.
The fact that the C-terminal fourth of the AcH4−21 peptide is rich in positively charged residues (i.e., K16-R-H-R-K20) suggests that this region of the AcH4−21 peptide interacts with acidic residues on the surface of PRMT1. Consistent with this idea is the recent observation that multiple acidic residues on the surface of PRMT1 are important for substrate binding (54) (Figure 4). To test this hypothesis, we synthesized a peptide in which Arg17 and Arg19 were replaced with citrulline, the neutral isostere of Arg. Remarkably, the effect of this “mutation” on kcat/Km mirrors the effect of replacing both these residues with Ala (81-fold for the AcH4−21R17Cit/R19Cit peptide versus 114-fold for the AcH4−21R17A/R19A peptide), thereby confirming the importance of electrostatic interactions to substrate capture.
Figure 4.

Structure of rPRMT. A. Structure of the rPRMT1 dimer with two active sites located in the inner ring surface. B. The RGG3 peptide bound to the acidic surface of rPRMT1. Two portions of the RGG3 peptide are connected by a hypothetical linker (dashed green line). Additional acidic grooves could provide binding sites for longer peptide substrates with basic residues remote to the methylation site.
Effect of Histone Modifications on PRMT1 Activity
Histone H4 is known to be acetylated at Lys5, 8, 12, and 16 and methylated at Lys20 (22, 55, 56). Therefore, we evaluated the effect of histone H4 lysine acetylation and lysine methylation (22, 55, 56) on substrate capture by determining the kinetic parameters for peptides incorporating acetyl-Lys at positions 5, 8, 12, and 16 (the AcH4−21K5, 8, 12, 16Ac-K peptide); acetyl-Lys at Lys5, 8, 12, 16, and 20 (the AcH4−21 all Ac-K peptide); and trimethylated-Lys at position 20 (the AcH4−21K20(Me)3 peptide). The results of kinetic studies (Table 6) indicate that trimethylation of Lys20 decreases kcat/Km by ∼30-fold. While these effects on substrate capture are relatively modest, it is interesting to note that such an antagonistic effect is consistent with the in vivo roles of these modifications: trimethyl Lys20 accumulates in transcriptionally silent regions of chromatin whereas methylated Arg3 generally accumulates in transcriptionally active regions (22, 55, 56). The effects of Lys acetylation are more dramatic as evidenced by the greater than 104-fold decrease in kcat/Km. These results confirm previous studies demonstrating that histone acetylation antagonizes Arg3 methylation (22) and provide further support for the notion that modification of histones at one or more site(s) can affect their modification at a second site, i.e., cross talk.
Table 6.
Histone Modifications Influence H4 Arg3 Methylation
| substrate | Km (μM) | kcat (min−1) | kcat/Km (M−1·min−1) |
|---|---|---|---|
| AcH4−21a,b | 1.14 ± 0.54 | 0.46 ± 0.02 | 4.08 × 105 |
| AcH4−21 K5,8,12,16Ac-Ka,b | ndc | ndc | <10.8 |
| AcH4−21 all Ac-Ka,b | ndc | ndc | <14.4 |
| AcH4−21 K20(Me)3a,b | 29.1 ± 3.7 | 0.40 ± 0.01 | 1.38 × 104 |
[SAM] = 15 μM.
Sequence and mass available in Table 1.
The amount of product formation was too low to accurately measure the kinetic parameters for this peptide substrate. Therefore an estimation of kcat/Km was made by dividing the maximal observed rate by the concentration of substrate.
Inhibition Studies
Because SAM-dependent methyltransferases are often subject to potent product inhibition, we examined the inhibitory properties of SAH by determining the IC50 of this compound. IC50 values were also determined for thioadenosine (5TA), methylthioadenosine (MTA; an alkylated analogue of 5TA), and homocysteine–the component parts of SAH–because we are ultimately interested in using this information to aid our development of bisubstrate analogue based inhibitors of PRMT1. The IC50 value for sinefungin was also determined because this compound is a commonly used methyltransferase inhibitor. Assuming that these compounds are all competitive inhibitors of SAM binding, the IC50 data (Table 7) indicate that 5TA and MTA are rather poor PRMT1 inhibitors (Ki > 80 μM based on Dixon analysis). These results were somewhat surprising, given that the S-adenosyl moiety has been used to generate high affinity bisubstrate analogues for catechol O-methyltransferase (57, 58), and additionally calls into question the use of MTA as an in vivo inhibitor of PRMT1 activity (21). While MTA and 5TA are relatively poor PRMT1 inhibitors, the IC50 obtained with homocysteine is significantly lower. The fact that this compound binds to PRMT1 with high affinity (Ki ∼ 13.4 μM) is consistent with the observation that ADMA levels are not increased in patients with homocystinuria despite the fact that these patients are at high risk for cardiovascular disease (59). Of all the compounds tested, sinefungin was the most potent PRMT1 inhibitor with a Ki of ∼75 nM. The tight binding of this compound likely reflects the presence of a positively charged amino group that mimics the positively charged sulfonium ion in SAM. The importance of these findings to inhibitor design is discussed below.
Table 7.
Inhibition Studies on 5TA, MTA, SAH, and Sinefungin
| Inhibitor | Structure | IC50 (μM) |
|---|---|---|
| S-Adenosyl-L-Homocysteine (SAH)a,b | 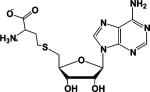 |
19.8 ± 5.57 |
| Methylthioadenosine (MTA) a,b | 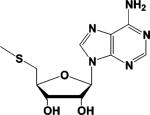 |
> 1000 |
| 5'-thioadenosine (5TA) a,b | 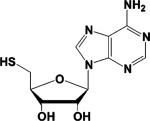 |
> 1000 |
| Homocysteine a,b | 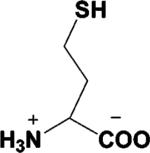 |
39.5 ± 11.1 |
| Sinefungina,c | 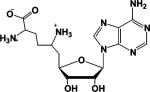 |
1.44 ± 0.41 |
[SAM] = 15 μM.
[RGG3] = 10 μM.
[AcH4−21] = 145 μM.
PRMT1 Methylates Substrates in a Partially Processive2 Fashion
Although type I PRMTs are known to catalyze the mono- and asymmetric dimethylation of Arg residues (60), it is unclear whether these two events are coupled or not, i.e., does the enzyme (i) release the monomethylated species before rebinding it to facilitate the second methylation event, i.e., a distributive mechanism, or (ii) bind to its substrates such that mono- and dimethylation occur sequentially without the release of the monomethylated species, i.e., a processive mechanism? If the first situation predominates, one would expect the monomethylated species to possess a significantly higher affinity for the enzyme; whereas if the second situation predominates, one would expect an obligate order of product release that results solely in the production of the dimethylated species.
To address this issue, we synthesized peptides in which a monomethylated Arg residue was incorporated at position 3 of the AcH4−21 and AcH4−15 peptides. The steady-state kinetic parameters were subsequently determined for these peptides, denoted AcH4−21-R3MMA and AcH4−15-R3MMA. Interestingly, the kcat/Km values obtained for the AcH4−21-R3MMA and AcH4−15-R3MMA peptides are decreased by ∼4-fold and ∼2-fold (Table 8), which is inconsistent with the idea that monomethylated substrates possess an intrinsically higher affinity for PRMT1.
Table 8.
Effect of Monomethylation on rPRMTl Kinetics
| substrate | Km (μM) | kcat (min−1) | (M−1·min−1) |
|---|---|---|---|
| AcH4−21a,b | 3.55 ± 0.68 | 0.39 ± 0.01 | 1.10 × 105 |
| AcH4−21 R3MMAa,b | 28.3 ± 4.41 | 0.79 ± 0.03 | 2.80 × 104 |
| AcH4−15a,b | 120 ± 16.0 | 0.21 ± 0.01 | 1.76 × 103 |
| AcH4−15 R3MMAa,b | 353 ± 80.2 | 0.34 ± 0.03 | 9.71 × 102 |
[SAM] = 15 μM.
Sequence and mass available in Table 1.
To further address this issue, MALDI-MS was used to monitor the methylation of peptide substrates by rPRMT1. Although obtaining quantitative results with MS methods can be challenging, under a variety of conditions (e.g., varied time, enzyme, and peptide chain length), we consistently observe the appearance of signals corresponding to both monomethyl and dimethylated Arg residues despite the presence of an excess of the unmodified peptide (Figure 3). In a separate experiment, we monitored the time course of product formation by rPRMT1. The H4 peptide (residues 1−20) was used as the substrate, and product formation was monitored by MALDI-MS (Figure 3C). Early in the reaction (∼5 min), an approximately equal amount of monomethyland dimethylated Arg residues are produced despite the presence of an excess of the unmodified substrate, consistent with the results described above. After that, the levels of dimethyl-Arg containing peptides steadily increase as the levels of the unmodified and monomethylated product decrease.
Figure 3.

rPRMT1 catalyzes H4 Arg3 methylation in a partially processive fashion. A. Methylation of the AcH4−21 peptide monitored by MALDI-MS. B. Comparison of the relative levels of monomethylated and dimethylated peptides produced as a function of peptide length. The concentrations of SAM and rPRMT1 were fixed at 250 μM and 500 nM, respectively. The concentrations of the peptide substrates were fixed at ∼2.5Km (AcH4−13 = 275 μM; AcH4−15 = 310 μM; AcH4−16 = 370 μM; AcH4−18 = 500 μM; AcH4−21 = 9 μM). C. Time course of rPRMT1 catalyzed methylation. Methods analogous to those described in (66) were used for these studies. The relative amount of each peptide species over the full time course of the reaction was expressed as a percentage of the sum of intensity of all related peaks. The methylation reaction contained 20 mM Tris pH 8.0, 200 mM NaCl, 1 mM DTT, 0.4 mM SAM, 20 μM of peptide, and 50 μg/mL of PRMT1 and was carried out at 37 °C.
Because we have already excluded the possibility that MMA containing peptides have an intrinsically higher affinity for PRMT1, these results are most consistent with a partially processive mechanism in which SAH release and SAM rebinding occurs on a time scale similar to the release of MMA containing products. While similar results have been observed for PRMT6 and interpreted as being consistent with a processive mechanism (60), i.e. dimethylation occurs without release of the monomethylated product to the bulk solution, the fact that MMA and ADMA are produced in similar quantities (60) rules out such a purely processive mechanism. While detailed kinetic analyses are required to validate the proposed mechanism, it is noteworthy that in addition to PRMT6 several Lys methyltransferases, e.g., CLR4, appear to utilize a partially processive mechanism of methyl transfer (61).
Because all of the structurally characterized PRMTs, i.e., rPRMT1 (43), rPRMT3 (62), and yeast RMT1/Hmt1 (63), form a ringlike dimer (Figure 4A), it is conceivable that binding of the protein substrate within this ringlike structure could facilitate the processive dimethylation of Arg residues by allowing the product of the first methylation reaction, monomethylarginine, to enter the active site of the second molecule of the dimer without releasing the substrate from the ring. In partial support of this possibility is the fact that mutants of rPRMT1, yeast RMT1/Hmt1, and PRMT4/CARM1 in which the dimerization arm is either deleted (residues 188−222), replaced with Ala residues, or mutated exist as monomers in solution and lack methyltransferase activity (43, 63, 64). The fact that the PRMT1 dimer forms oligomers in vivo and in vitro (43, 46) is also noteworthy because these higher order structures could contribute to substrate processivity. For example, the two methylation events–from zero-to-mono and from mono-to-di–could be carried out by two dimers within the same oligomer. In addition, because the majority of PRMT1 substrates contain multiple arginines in RGG or RXR contexts, a PRMT1 oligomer could methylate multiple substrate arginines simultaneously per binding event, another form of processivity. Furthermore, processivity could also be influenced, both negatively and positively, by PRMT1 interacting proteins in vivo. More study will be required to settle these issues.
CONCLUSIONS
The PRMTs, and Arg methylation in general, have garnered significant attention over the last several years because of their important roles in human cell signaling and human disease. However, the ability to fully delineate the molecular processes/pathways in which these enzymes play a role is hampered by the lack of enzyme specific inhibitors. Furthermore, the question of whether PRMTs are valid drug targets can only be fully answered by the development and testing of potent and selective PRMT inhibitors. Therefore, we initiated a program to develop PRMT-selective inhibitors. As a part of that program, we began studies on the substrate specificity and kinetics of PRMT1 catalysis to identify important features that could be exploited for the design of inhibitors targeting this isozyme.
The substrate specificity studies on the rat and human PRMT1 orthologues demonstrate that the determinants of substrate recognition are quite similar for the two enzymes (which differ only at one position), although we note that subtle differences do exist. Furthermore, our studies to identify a minimal peptide substrate indicate that regions far from the PRMT1 active site contribute to substrate recognition. These interactions are presumably mediated by acidic residues on the surface of the enzyme and positively charged residues in the C-terminal fourth of the AcH4−21 peptide because peptides incorporating neutral amino acids were poorly processed. Consistent with this hypothesis are crystal-lographic and mutational studies on PRMT1 (43, 54). For example, the RGG3 peptide appears to interact with several highly acidic meandering grooves on the surface of PRMT1 (Figure 4B) and recent mutational studies confirm the importance of acidic surface residues to substrate binding (54). However, further studies to identify the specific residues on PRMT1 that mediate these long range interactions will be required because the electron density for the RGG3 peptide was not of sufficient resolution to identify individual amino acids within this peptide; thus the N- and C-terminal portions of this peptide could not be identified. Interestingly, these results are also consistent with the fact that a number of known PRMT1 substrates contain positively charged residues far from the site of methylation (Figure 5). While the distance between these residues and the site of methylation is not fixed, this is not surprising because our results with the mid-sequence truncation peptides (Table 3) demonstrate that this need not be the case.
Figure 5.

Alignment of known PRMT1 substrates. Amino acid sequences of a subset of known PRMT1 substrates were manually aligned to show the preponderance of positively charged Arg rich sequences distal to the site of methylation. The site of methylation is highlighted in bold, and positively charged residues/regions are highlighted in yellow. The proteins presented in this alignment are hHNF4 (67), hnRNP K (68), SAM 68 (47), HMW FGF-2 (69), and HMGA1a (70).
In summary, our results demonstrate our ability to kinetically characterize PRMT1 activity, that long range interactions are important for substrate recognition, and that this enzyme catalyzes Arg methylation in a partially processive fashion. Combined with our studies on small molecule PRMT1 inhibitors, the information contained in this report will undoubtedly aid the development of a PRMT1 selective inhibitor. For example, it should be possible to generate a potent and selective bisubstrate analogue inhibitor of PRMT1 by linking the AcH4−21 peptide to a portion of homocysteine or sinefungin. Bisubstrate analogues of this type have been developed for the histone acetyltransferases (50, 65); and these compounds have shown great utility as chemical probes for studying the role of histone acetylation both in vitro and in vivo (50, 65). The fact that the AcH4−21 peptide is a much better substrate than the AcH4−15 peptide is also highly significant because it suggests the possibility of identifying inhibitors targeting regions outside the active site by screening for compounds that inhibit the methylation of the AcH4−21 peptide but not the AcH4−15 peptide.
Footnotes
This work was supported in part by the start up funds from the University of South Carolina Research Foundation (P.R.T.) and from a new investigator grant from the American Heart Association. T.C.O. is supported by an NSF GK-12 fellowship, O.O. is supported by an NIH-PREP fellowship (GM066526), and X.Z. and X.C. are supported by an NIH grant (GM068680).
Abbreviations: PRMT, protein arginine methyltransferase; hPRMT, human protein arginine methyltransferase; rPRMT, rat PRMT; SAM, S-adenosyl-l-methionine; CARM1, coactivator associated arginine methyltransferase 1; SAH, S-adenosyl-l-homocysteine; MMA, monomethylarginine; ADMA, asymmetric dimethylarginine; SDMA, symmetric dimethylarginine; 5TA, thioadenosine; MTA, methylthioadenosine; Cit, citrulline; HEPES, N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid); EDTA, ethylenediamine tetraacetic acid; MALDI, matrix assisted laser desorption/ionization.
With regard to PRMT1, a partially processive mechanism implies that the production of the asymmetrically dimethylated species does not occur in an obligate fashion but does occur in appreciable amounts, resulting in the production of both monomethylated and asymmetrically dimethylated Arg residues. In contrast, a fully processive mechanism implies that the production of asymmetrically dimethylated Arg residues occurs in an obligate fashion without the release of the monomethylated species from the enzyme; and as a result the levels of this intermediate do not rise above the concentration of the enzyme.
REFERENCES
- 1.Krause CD, Yang ZH, Kim YS, Lee JH, Cook JR, Pestka S. Protein arginine methyltransferases: evolution and assessment of their pharmacological and therapeutic potential. Pharmacol. Ther. 2007;113:50–87. doi: 10.1016/j.pharmthera.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 2.Bedford MT, Richard S. Arginine methylation an emerging regulator of protein function. Mol. Cell. 2005;18:263–272. doi: 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Lin WJ, Gary JD, Yang MC, Clarke S, Herschman HR. The mammalian immediate-early TIS21 protein and the leukemia-associated BTG1 protein interact with a protein-arginine N-methyltransferase. J. Biol. Chem. 1996;271:15034–15044. doi: 10.1074/jbc.271.25.15034. [DOI] [PubMed] [Google Scholar]
- 4.Aletta JM, Cimato TR, Ettinger MJ. Protein methylation: a signal event in post-translational modification. Trends Biochem. Sci. 1998;23:89–91. doi: 10.1016/s0968-0004(98)01185-2. [DOI] [PubMed] [Google Scholar]
- 5.McBride AE, Silver PA. State of the arg: protein methylation at arginine comes of age. Cell. 2001;106:5–8. doi: 10.1016/s0092-8674(01)00423-8. [DOI] [PubMed] [Google Scholar]
- 6.Yun CY, Fu XD. Conserved SR protein kinase functions in nuclear import and its action is counteracted by arginine methylation in Saccharomyces cerevisiae. J. Cell Biol. 2000;150:707–718. doi: 10.1083/jcb.150.4.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen EC, Henry MF, Weiss VH, Valentini SR, Silver PA, Lee MS. Arginine methylation facilitates the nuclear export of hnRNP proteins. Genes Dev. 1998;12:679–691. doi: 10.1101/gad.12.5.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Torres-Padilla ME, Parfitt DE, Kouzarides T, Zernicka-Goetz M. Histone arginine methylation regulates pluripotency in the early mouse embryo. Nature. 2007;445:214–218. doi: 10.1038/nature05458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pawlak MR, Scherer CA, Chen J, Roshon MJ, Ruley HE. Arginine N-methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol. Cell. Biol. 2000;20:4859–4869. doi: 10.1128/mcb.20.13.4859-4869.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yadav N, Lee J, Kim J, Shen J, Hu MC, Aldaz CM, Bedford MT. Specific protein methylation defects and gene expression perturbations in coactivator-associated arginine methyltransferase 1-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 2003;100:6464–6468. doi: 10.1073/pnas.1232272100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen SL, Loffler KA, Chen D, Stallcup MR, Muscat GE. The coactivator-associated arginine methyltransferase is necessary for muscle differentiation: CARM1 coactivates myocyte enhancer factor-2. J. Biol. Chem. 2002;277:4324–4333. doi: 10.1074/jbc.M109835200. [DOI] [PubMed] [Google Scholar]
- 12.Friesen WJ, Massenet S, Paushkin S, Wyce A, Dreyfuss G. SMN, the product of the spinal muscular atrophy gene, binds preferentially to dimethylarginine-containing protein targets. Mol. Cell. 2001;7:1111–1117. doi: 10.1016/s1097-2765(01)00244-1. [DOI] [PubMed] [Google Scholar]
- 13.Meister G, Fischer U. Assisted RNP assembly: SMN and PRMT5 complexes cooperate in the formation of spliceosomal UsnRNPs. EMBO J. 2002;21:5853–5863. doi: 10.1093/emboj/cdf585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lukong KE, Richard S. Arginine methylation signals mRNA export. Nat. Struct. Mol. Biol. 2004;11:914–915. doi: 10.1038/nsmb1004-914. [DOI] [PubMed] [Google Scholar]
- 15.Matter N, Herrlich P, Konig H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature. 2002;420:691–695. doi: 10.1038/nature01153. [DOI] [PubMed] [Google Scholar]
- 16.Li H, Park S, Kilburn B, Jelinek MA, Henschen-Edman A, Aswad DW, Stallcup MR, Laird-Offringa IA. Lipopolysaccharide-induced methylation of HuR, an mRNA-stabilizing protein, by CARM1. Coactivator-associated arginine methyltransferase. J. Biol. Chem. 2002;277:44623–44630. doi: 10.1074/jbc.M206187200. [DOI] [PubMed] [Google Scholar]
- 17.Xu W, Chen H, Du K, Asahara H, Tini M, Emerson BM, Montminy M, Evans RM. A transcriptional switch mediated by cofactor methylation. Science. 2001;294:2507–2511. doi: 10.1126/science.1065961. [DOI] [PubMed] [Google Scholar]
- 18.Chevillard-Briet M, Trouche D, Vandel L. Control of CBP co-activating activity by arginine methylation. EMBO J. 2002;21:5457–5466. doi: 10.1093/emboj/cdf548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.An W, Kim J, Roeder RG. Ordered Cooperative Functions of PRMT1, p300, and CARM1 in Transcriptional Activation by p53. Cell. 2004;117:735–748. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 20.Mowen KA, Schurter BT, Fathman JW, David M, Glimcher LH. Arginine methylation of NIP45 modulates cytokine gene expression in effector T lymphocytes. Mol. Cell. 2004;15:559–571. doi: 10.1016/j.molcel.2004.06.042. [DOI] [PubMed] [Google Scholar]
- 21.Mowen KA, Tang J, Zhu W, Schurter BT, Shuai K, Herschman HR, David M. Arginine methylation of STAT1 modulates IFNalpha/beta-induced transcription. Cell. 2001;104:731–741. doi: 10.1016/s0092-8674(01)00269-0. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, Huang Z-Q, Xia L, Feng Q, Erdjument-Bromage H, Strahl BD, Briggs SD, Allis CD, Wong J, Tempst P, Zhang Y. Methylation of Histone H4 at Arginine 3 Facilitating Transcriptional Activation by Nuclear Hormone Receptor. Science. 2001;293:853–857. doi: 10.1126/science.1060781. [DOI] [PubMed] [Google Scholar]
- 23.Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, Stallcup MR. Regulation of transcription by a protein methyltransferase. Science. 1999;284:2174–2177. doi: 10.1126/science.284.5423.2174. [DOI] [PubMed] [Google Scholar]
- 24.Bauer UM, Daujat S, Nielsen SJ, Nightingale K, Kouzarides T. Methylation at arginine 17 of histone H3 is linked to gene activation. EMBO Rep. 2002;3:39–44. doi: 10.1093/embo-reports/kvf013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma H, Baumann CT, Li H, Strahl BD, Rice R, Jelinek MA, Aswad DW, Allis CD, Hager GL, Stallcup MR. Hormone-dependent, CARM1-directed, arginine-specific methylation of histone H3 on a steroid-regulated promoter. Curr. Biol. 2001;11:1981–1985. doi: 10.1016/s0960-9822(01)00600-5. [DOI] [PubMed] [Google Scholar]
- 26.Hong H, Kao C, Jeng MH, Eble JN, Koch MO, Gardner TA, Zhang S, Li L, Pan CX, Hu Z, MacLennan GT, Cheng L. Aberrant expression of CARM1, a transcriptional coactivator of androgen receptor, in the development of prostate carcinoma and androgen-independent status. Cancer. 2004;101:83–89. doi: 10.1002/cncr.20327. [DOI] [PubMed] [Google Scholar]
- 27.Majumder S, Liu Y, Ford OH, 3rd, Mohler JL, Whang YE. Involvement of arginine methyltransferase CARM1 in androgen receptor function and prostate cancer cell viability. Prostate. 2006;66:1292–1301. doi: 10.1002/pros.20438. [DOI] [PubMed] [Google Scholar]
- 28.Boger RH, Sydow K, Borlak J, Thum T, Lenzen H, Schubert B, Tsikas D, Bode-Boger SM. LDL cholesterol upregulates synthesis of asymmetrical dimethylarginine in human endothelial cells: involvement of S-adenosylmethionine-dependent methyltransferases. Circ. Res. 2000;87:99–105. doi: 10.1161/01.res.87.2.99. [DOI] [PubMed] [Google Scholar]
- 29.Tran CT, Leiper JM, Vallance P. The DDAH/ADMA/NOS pathway. Atheroscler. Suppl. 2003;4:33–40. doi: 10.1016/s1567-5688(03)00032-1. [DOI] [PubMed] [Google Scholar]
- 30.Vallance P, Leiper J. Cardiovascular biology of the asymmetric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arterioscler. Thromb. Vasc. Biol. 2004;24:1023–1030. doi: 10.1161/01.ATV.0000128897.54893.26. [DOI] [PubMed] [Google Scholar]
- 31.Leiper J, Murray-Rust J, McDonald N, Vallance P. S-nitrosylation of dimethylarginine dimethylaminohydrolase regulates enzyme activity: further interactions between nitric oxide synthase and dimethylarginine dimethylaminohydrolase. Proc. Natl. Acad. Sci. U.S.A. 2002;99:13527–13532. doi: 10.1073/pnas.212269799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vallance P, Leone A, Calver A, Collier J, Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet. 1992;339:572–575. doi: 10.1016/0140-6736(92)90865-z. [DOI] [PubMed] [Google Scholar]
- 33.Leiper J, Nandi M, Torondel B, Murray-Rust J, Malaki M, O'Hara B, Rossiter S, Anthony S, Madhani M, Selwood D, Smith C, Wojciak-Stothard B, Rudiger A, Stidwill R, McDonald NQ, Vallance P. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat. Med. 2007;13:198–203. doi: 10.1038/nm1543. [DOI] [PubMed] [Google Scholar]
- 34.Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, Sauter G, Kallioniemi OP, Trent JM, Meltzer PS. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–968. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- 35.Torchia J, Rose DW, Inostroza J, Kamei Y, Westin S, Glass CK, Rosenfeld MG. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature. 1997;387:677–684. doi: 10.1038/42652. [DOI] [PubMed] [Google Scholar]
- 36.Lee SK, Anzick SL, Choi JE, Bubendorf L, Guan XY, Jung YK, Kallioniemi OP, Kononen J, Trent JM, Azorsa D, Jhun BH, Cheong JH, Lee YC, Meltzer PS, Lee JW. A nuclear factor, ASC-2, as a cancer-amplified transcriptional coactivator essential for ligand-dependent transactivation by nuclear receptors in vivo. J. Biol. Chem. 1999;274:34283–34293. doi: 10.1074/jbc.274.48.34283. [DOI] [PubMed] [Google Scholar]
- 37.Zhu Y, Qi C, Jain S, Le Beau MM, Espinosa R, 3rd, Atkins GB, Lazar MA, Yeldandi AV, Rao MS, Reddy JK. Amplification and overexpression of peroxisome proliferator-activated receptor binding protein (PBP/PPARBP) gene in breast cancer. Proc. Natl. Acad. Sci. U.S.A. 1999;96:10848–10853. doi: 10.1073/pnas.96.19.10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen X, Niroomand F, Liu Z, Zankl A, Katus HA, Jahn L, Tiefenbacher CP. Expression of nitric oxide related enzymes in coronary heart disease. Basic Res. Cardiol. 2006;101:346–353. doi: 10.1007/s00395-006-0592-5. [DOI] [PubMed] [Google Scholar]
- 39.Zoccali C, Bode-Boger S, Mallamaci F, Benedetto F, Tripepi G, Malatino L, Cataliotti A, Bellanuova I, Fermo I, Frolich J, Boger R. Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: a prospective study. Lancet. 2001;358:2113–2117. doi: 10.1016/s0140-6736(01)07217-8. [DOI] [PubMed] [Google Scholar]
- 40.Cheng D, Yadav N, King RW, Swanson MS, Weinstein EJ, Bedford MT. Small molecule regulators of protein arginine methyltransferases. J. Biol. Chem. 2004;279:23892–23899. doi: 10.1074/jbc.M401853200. [DOI] [PubMed] [Google Scholar]
- 41.Spannhoff A, Heinke R, Bauer I, Trojer P, Metzger E, Gust R, Schule R, Brosch G, Sippl W, Jung M. Target-based approach to inhibitors of histone arginine methyltransferases. J. Med. Chem. 2007;50:2319–2325. doi: 10.1021/jm061250e. [DOI] [PubMed] [Google Scholar]
- 42.Spannhoff A, Machmur R, Heinke R, Trojer P, Bauer I, Brosch G, Schule R, Hanefeld W, Sippl W, Jung M. A novel arginine methyltransferase inhibitor with cellular activity. Bioorg. Med. Chem. Lett. 2007 doi: 10.1016/j.bmcl.2007.05.088. in press. [DOI] [PubMed] [Google Scholar]
- 43.Zhang X, Cheng X. Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure. 2003;11:509–520. doi: 10.1016/s0969-2126(03)00071-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang J, Frankel A, Cook RJ, Kim S, Paik WK, Williams KR, Clarke S, Herschman HR. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J. Biol. Chem. 2000;275:7723–7730. doi: 10.1074/jbc.275.11.7723. [DOI] [PubMed] [Google Scholar]
- 45.Abramovich C, Yakobson B, Chebath J, Revel M. A protein-arginine methyltransferase binds to the intracytoplasmic domain of the IFNAR1 chain in the type I interferon receptor. EMBO J. 1997;16:260–266. doi: 10.1093/emboj/16.2.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang J, Gary JD, Clarke S, Herschman HR. PRMT 3, a type I protein arginine N-methyltransferase that differs from PRMT1 in its oligomerization, subcellular localization, substrate specificity, and regulation. J. Biol. Chem. 1998;273:16935–16945. doi: 10.1074/jbc.273.27.16935. [DOI] [PubMed] [Google Scholar]
- 47.Cote J, Boisvert FM, Boulanger MC, Bedford MT, Richard S. Sam68 RNA binding protein is an in vivo substrate for protein arginine N-methyltransferase 1. Mol. Biol. Cell. 2003;14:274–287. doi: 10.1091/mbc.E02-08-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luger K, Rechsteiner TJ, Richmond TJ. Preparation of nucleosome core particle from recombinant histones. Methods Enzymol. 1999;304:3–19. doi: 10.1016/s0076-6879(99)04003-3. [DOI] [PubMed] [Google Scholar]
- 49.Thompson PR, Kurooka H, Nakatani Y, Cole PA. Transcriptional coactivator protein p300. Kinetic characterization of its histone acetyltransferase activity. J. Biol. Chem. 2001;276:33721–33729. doi: 10.1074/jbc.M104736200. [DOI] [PubMed] [Google Scholar]
- 50.Lau OD, Kundu TK, Soccio RE, Ait-Si-Ali S, Khalil EM, Vassilev A, Wolffe AP, Nakatani Y, Roeder RG, Cole PA. HATs off: selective synthetic inhibitors of the histone acetyltransferases p300 and PCAF. Mol. Cell. 2000;5:589–595. doi: 10.1016/s1097-2765(00)80452-9. [DOI] [PubMed] [Google Scholar]
- 51.Leatherbarrow RJ. Erathicus Software. Staines, U.K: 2004. [Google Scholar]
- 52.Dorgan KM, Wooderchak WL, Wynn DP, Karschner EL, Alfaro JF, Cui Y, Zhou ZS, Hevel JM. An enzyme-coupled continuous spectrophotometric assay for S-adenosylmethionine-dependent methyltransferases. Anal. Biochem. 2006;350:249–255. doi: 10.1016/j.ab.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 53.Strahl BD, Briggs SD, Brame CJ, Caldwell JA, Koh SS, Ma H, Cook RG, Shabanowitz J, Hunt DF, Stallcup MR, Allis CD. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr. Biol. 2001;11:996–1000. doi: 10.1016/s0960-9822(01)00294-9. [DOI] [PubMed] [Google Scholar]
- 54.Lee DY, Ianculescu I, Purcell D, Zhang X, Cheng X, Stallcup MR. Surface-scanning mutational analysis of protein arginine methyltransferase 1: roles of specific amino acids in methyltransferase substrate specificity, oligomerization, and coactivator function. Mol. Endocrinol. 2007;21:1381–1393. doi: 10.1210/me.2006-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarg B, Helliger W, Talasz H, Koutzamani E, Lindner HH. Histone H4 hyperacetylation precludes histone H4 lysine 20 trimethylation. J. Biol. Chem. 2004;279:53458–53464. doi: 10.1074/jbc.M409099200. [DOI] [PubMed] [Google Scholar]
- 56.Sarg B, Koutzamani E, Helliger W, Rundquist I, Lindner HH. Postsynthetic trimethylation of histone H4 at lysine 20 in mammalian tissues is associated with aging. J. Biol. Chem. 2002;277:39195–39201. doi: 10.1074/jbc.M205166200. [DOI] [PubMed] [Google Scholar]
- 57.Lerner C, Masjost B, Ruf A, Gramlich V, Jakob-Roetne R, Zurcher G, Borroni E, Diederich F. Bisubstrate inhibitors for the enzyme catechol-O-methyltransferase (COMT): influence of inhibitor preorganisation and linker length between the two substrate moieties on binding affinity. Org. Biomol. Chem. 2003;1:42–49. doi: 10.1039/b208690p. [DOI] [PubMed] [Google Scholar]
- 58.Masjost B, Ballmer P, Borroni E, Zurcher G, Winkler FK, Jakob-Roetne R, Diederich F. Structure-based design, synthesis, and in vitro evaluation of bisubstrate inhibitors for catechol O-methyltransferase (COMT) Chem Eur. J. 2000;6:971–982. doi: 10.1002/(sici)1521-3765(20000317)6:6<971::aid-chem971>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 59.Wilcken DE, Wang J, Sim AS, Green K, Wilcken B. Asymmetric dimethylarginine in homocystinuria due to cystathionine beta-synthase deficiency: relevance of renal function. J. Inherited Metab. Dis. 2006;29:30–37. doi: 10.1007/s10545-006-0208-0. [DOI] [PubMed] [Google Scholar]
- 60.Frankel A, Yadav N, Lee J, Branscombe TL, Clarke S, Bedford MT. The novel human protein arginine N-methyltransferase PRMT6 is a nuclear enzyme displaying unique substrate specificity. J. Biol. Chem. 2002;277:3537–3543. doi: 10.1074/jbc.M108786200. [DOI] [PubMed] [Google Scholar]
- 61.Dirk LM, Flynn EM, Dietzel K, Couture JF, Trievel RC, Houtz RL. Kinetic manifestation of processivity during multiple methylations catalyzed by SET domain protein methyltransferases. Biochemistry. 2007;46:3905–3915. doi: 10.1021/bi6023644. [DOI] [PubMed] [Google Scholar]
- 62.Zhang X, Zhou L, Cheng X. Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. EMBO J. 2000;19:3509–3519. doi: 10.1093/emboj/19.14.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weiss VH, McBride AE, Soriano MA, Filman DJ, Silver PA, Hogle JM. The structure and oligomerization of the yeast arginine methyltransferase, Hmt1. Nat. Struct. Biol. 2000;7:1165–1171. doi: 10.1038/82028. [DOI] [PubMed] [Google Scholar]
- 64.Higashimoto K, Kuhn P, Desai D, Cheng X, Xu W. Phosphorylation-mediated inactivation of coactivator-associated arginine methyltransferase 1. Proc. Natl. Acad. Sci. U.S.A. 2007;104:12318–12323. doi: 10.1073/pnas.0610792104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zheng Y, Thompson PR, Cebrat M, Wang L, Devlin MK, Alani RM, Cole PA. Selective HAT inhibitors as mechanistic tools for protein acetylation. Methods Enzymol. 2004;376:188–199. doi: 10.1016/S0076-6879(03)76012-1. [DOI] [PubMed] [Google Scholar]
- 66.Zhang X, Yang Z, Khan SI, Horton JR, Tamaru H, Selker EU, Cheng X. Structural basis for the product specificity of histone lysine methyltransferases. Mol. Cell. 2003;12:177–185. doi: 10.1016/s1097-2765(03)00224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barrero MJ, Malik S. Two functional modes of a nuclear receptor-recruited arginine methyltransferase in transcriptional activation. Mol. Cell. 2006;24:233–243. doi: 10.1016/j.molcel.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chiou YY, Lin WJ, Fu SL, Lin CH. Direct Mass-Spectrometric Identification of Arg296 and Arg299 as the Methylation Sites of hnRNP K Protein for Methyltransferase PRMT1. Protein J. 2007;26:87–93. doi: 10.1007/s10930-006-9049-9. [DOI] [PubMed] [Google Scholar]
- 69.Klein S, Carroll JA, Chen Y, Henry MF, Henry PA, Ortonowski IE, Pintucci G, Beavis RC, Burgess WH, Rifkin DB. Biochemical analysis of the arginine methylation of high molecular weight fibroblast growth factor-2. J. Biol. Chem. 2000;275:3150–3157. doi: 10.1074/jbc.275.5.3150. [DOI] [PubMed] [Google Scholar]
- 70.Zou Y, Webb K, Perna AD, Zhang Q, Clarke S, Wang Y. A Mass Spectrometric Study on the in Vitro Methylation of HMGA1a and HMGA1b Proteins by PRMTs: Methylation Specificity, the Effect of Binding to AT-Rich Duplex DNA, and the Effect of C-Terminal Phosphorylation. Biochemistry. 2007;46:7896–7906. doi: 10.1021/bi6024897. [DOI] [PubMed] [Google Scholar]