

Abstract
Cerebral palsy due to perinatal injury to cerebral white matter is usually not caused by genetic mutations, but by ischemia and/or inflammation. Here, we describe an autosomal-recessive type of tetraplegic cerebral palsy with mental retardation, reduction of cerebral white matter, and atrophy of the cerebellum in an inbred sibship. The phenotype was recorded and evolution followed for over 20 years. Brain lesions were studied by diffusion tensor MR tractography.
Homozygosity mapping with SNPs was performed for identification of the chromosomal locus for the disease. In the 14 Mb candidate region on chromosome 7q22, RNA expression profiling was used for selecting among the 203 genes in the area. In postmortem brain tissue available from one patient, histology and immunohistochemistry were performed. Disease course and imaging were mostly reminiscent of hypoxic-ischemic tetraplegic cerebral palsy, with neuroaxonal degeneration and white matter loss. In all five patients, a donor splice site pathogenic mutation in intron 14 of the AP4M1 gene (c.1137+1G→T), was identified. AP4M1, encoding for the μ subunit of the adaptor protein complex-4, is involved in intracellular trafficking of glutamate receptors. Aberrant GluRδ2 glutamate receptor localization and dendritic spine morphology were observed in the postmortem brain specimen. This disease entity, which we refer to as congenital spastic tetraplegia (CST), is therefore a genetic model for congenital cerebral palsy with evidence for neuroaxonal damage and glutamate receptor abnormality, mimicking perinatally acquired hypoxic-ischemic white matter injury.
Introduction
Tetraplegic cerebral palsy (CP) can be caused by global hypoxia-ischemia of the brain in (near) term newborns and is often associated with neonatal encephalopathy.1,2 In large collaborative studies, this type of CP represents 18% of all cases.3 Primary white matter injury is uncommon in this context. The classical symptoms are four-limb spasticity (quadriplegia) with abnormal muscular tone, spasms, involuntary movements, and muscle wasting. This type of CP is usually associated with moderate to severe learning disabilities, abnormalities of speech and vision, and epilepsy in 50% of the cases.1–3 Periventricular white matter injury (PVWMI) is often seen in preterm newborns affected by CP.2,4 PVWMI is diffuse and the culprit is the immature oligodendrocyte at ∼30 weeks of gestation. Histopathological consequences are periventricular leukomalacia (focal or widespread gliotic and/or cystic lesions) and hypomyelination.4,5 Associated neuroaxonal degeneration and loss of subplate neurons correlate with cortical and long trait degeneration on brain imaging and contribute to visual, motor, and cognitive problems.6–8 It is under debate whether the neuronal loss precedes or follows myelin loss, but the two events could be concomitant.
PVWMI is thought to result from an excessive release of glutamate, free radicals, thrombotic factors, and inflammatory cytokines. Genetic predisposition may play a role.2,9 Different glutamate receptors have been implicated in increased calcium influx, which mediates glutamate excitotoxicity, including mainly α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate type receptors.10–13 AMPA receptors are highly expressed in migrating interneurons and in immature oligodendrocytes at the time when they initiate myelination, and this might make them vulnerable to ischemic glutamate release and intracellular Ca2+overload.14 Increased recycling and switch in receptor subunit composition is observed in hypoxia-ischemia models.12,13
Glutamate receptors are selectively transported to the synaptic membrane by vesicular trafficking, under control of proteins such as TARPs and the adaptor protein (AP) complexes.12,15,16 AP complexes form a major component of the vesicle coat machinery and such as a component mediates trafficking between the trans-Golgi network (TGN), endosomes, and the plasma membrane.17,18 Four different protein complexes, AP-1 to AP-4, have been described, each with a typical heterotetrameric structure consisting of four subunits including a medium-size μ subunit. Disruption of AP-4 complex integrity in knockout mice lacking the AP-4 beta subunit (MIM 607245) leads to mislocalization of AMPA glutamate receptors in the cerebellum.16 Until now, no human disorder has been associated with mutations of the AP-4 complex subunits.19 Here, we identify a mutation in the AP4M1 gene (MIM 602296), encoding the μ subunit of the adaptor protein complex-4, as the cause of an autosomal-recessive disease presenting as tetraplegic cerebral palsy with mental retardation and evidence for glutamate-related neuronal dysfunction.
Material and Methods
Written informed consent was obtained from patients and relatives.
Fibroblast Cell Culture and RNA Isolation
Skin fibroblasts from patients and controls were cultured in 175 cm2 culture flasks in Dulbecco's modified Eagle medium (DMEM) until 80% confluence. Total RNA was extracted with Trizol reagent (GIBCO BRL Life Technologies, Gaithersburg, MD, USA) and purified with QIAGEN RNeasy mini columns (QIAGEN, Valencia, CA, USA) according to manufacturers' protocols.
DNA Isolation
DNA was isolated from blood samples with the PUREGENE Genomic DNA Isolation Kit (Gentra).
Genotyping
SNP Array Analysis
SNP genotypes were obtained in accordance with the Affymetrix (Santa Clara, CA, USA) standard protocol for the GeneChip mapping 100K Xba and Hind arrays.
In brief, 250 ng of total genomic DNA was digested with either HindIII or XbaI. Appropriate adaptors were ligated to the four base-pair overhang of the DNA fragments. A single primer was used for amplification and PCR products were purified. After fragmentation with DNase I, products were labeled with biotin and hybridized to the array. The arrays were washed and stained with streptavidin-phycoerythrin in the Affymetrix fluidics station 450 and scanned in the GeneChip Scanner 3000 G7. The images of the scans were collected and intensities were measured in the Affymetrix GeneChip operating Software (GCOS). Genotype data analysis was performed in Affymetrix GeneChip Genotyping analysis software (GTYPE) with the Dynamic Model algorithm (DM).
Microsatellite Markers
Homozygosity of the region on chromosome 7q22 was confirmed by use of two microsatellite markers (D7S657and D7S515) selected from the ABI prism set MD 10 (version 2.5) and a number of markers selected from the Marshfield map, with primers newly selected. Primers in the NRCAM gene were newly developed. Primers used are indicated in Table S1 available online.
Markers were amplified with 50 ng genomic DNA in 11 μl PCR reactions containing 1× PCR rxn buffer (Invitrogen), 1.4 mM MgCl2, 0.18 mM of each dNTPs, 0.01 μM FW primer, 0.5 μM RV primer, 0.5 μM M13 tail (either FAM, VIC or NED), and 0.04 U platinum Taq (Invitrogen). Amplification conditions were 5′ at 94°C followed by 35 cycles of 30″ at 94°C; 30″ at 55°C; and 30″ at 72°C; with a final extension for 10′ at 72°C. PCR products were pooled in panels and loaded on an ABI 3100 automated sequencer. Data were analyzed with Gene Mapper Version 2.1 software (Applied Biosystems).
Linkage Analysis
CSV files containing SNP call data and pedigree data were converted by affy2mega to files that were compatible for calculating LOD scores with Allegro.20 Mendelian inheritance check was performed for all family members, with the program PedCheck.21 SNPs showing Mendelian inconsistencies were excluded from the calculations. SNP allele frequencies were used from the Affymetrix 100K annotation file. SNPs with a minor allele frequency smaller than 0.01 as well as SNPs with a call rate lower than 95% were excluded from the calculations. Multipoint linkage analysis was performed with Allegro with a SNP spacing of 0.3 cM. LOD scores were calculated with the assumption that the disease was an autosomal-recessive disorder with 99% penetrance. The linkage analysis was performed including the consanguinity loop; individual III-3 was not included in the linkage study because of insufficient computer memory.
Haplotypes with CA repeats were constructed manually, on the basis of the minimal number of recombinations. Gene data were obtained from the NCBI Genome Browser build 36.3.
Expression Array Analysis
GeneChips (HG U133 Plus 2.0) were purchased from Affymetrix. Target preparation and microarray processing were carried out according to the manufacturer's recommendations.
In brief, 5 μg of total RNA was used for double-stranded cDNA preparation. Biotinylated cRNA was synthesized with an RNA transcript labeling kit and 20 μg of the cRNA product was chemically fragmented to ∼50–200 nucleotides. The fragmented cRNA was hybridized to an Affymetrix HG U133 Plus 2.0 chip. Chips were washed and stained with the EukGE-WS2v5 protocol on an Affymetrix fluidics 450 station. The stain included streptavidin-phycoerythrin (10 μg/mL) and biotinylated goat anti-streptavidin (3 μg/mL). Fluorescence intensities were captured with an argonion laser confocal scanner.
Expression Data Analysis
Scanned output files were analyzed with GeneChip Operating Software 1.4 (GCOS; Affymetrix). For examining the quality of the arrays, the R function AffyQC was run on the CEL files.
Data was normalized in R program with RMA (Robust Multichip Average) normalization. Data analysis was done with Omniviz software version 5.0.1 (Omniviz, Biowisdom, Maynard, MA, USA). Only the genes (=357 Probe Set Ids, according to Affymetrix annotation file: HG-U133_Plus_2.na25.annot) from the linkage region were selected for further (supervised) analysis. Differentially expressed genes between patients and controls were identified with statistical analysis of microarrays (SAM analysis). For obtaining the level of differences in expression, log2 ratios between patients and controls were calculated as well.
The Affymetrix HG-U133_plus2 arrays contained one probe set for the AP4M1 gene, consisting of 11 probes, divided over exons 10–15. The percentage of present calls (∼45%), background, and ratio of GAPDH 3′ to 5′ all indicated a high quality of the samples.
Sequence Analysis
Primers used for sequence analysis are indicated in Table S2.
Amplification reactions were performed in a total volume of 25 μl, containing 1× PCR buffer (Invitrogen), 1.5 mM MgCl2, 200 μM of each dNTP, 1 μM forward primer, 1 μM reverse primer, 0.1 units Platinum Taq DNA polymerase (Invitrogen), and 25 ng genomic DNA. PCR conditions: 2′ 94°C, 30 cycles of 30″ 94°C, 30″ 60°C, 90″ 72°C with a final extension for 10′ 72°C.
PCR reactions were purified with ExoSAP-IT (USB). Direct sequencing of both strands was performed with Big Dye Terminator chemistry ver. 3.1 (Applied Biosystems) as recommended by the manufacturer.
DNA fragment analysis was performed with capillary electrophoresis on an ABI 3100 Genetic Analyzer (Applied Biosystems) and the software package Seqscape (Applied Biosystems, version 2.1).
The nomenclature of the mutation is c.1137+1G→T, where the A from translation start ATG from the transcript was taken as 1.
RT-PCR
Reverse transcriptase was performed on 2 μg RNA in a total volume of 40 μl with the iScript cDNA Synthesis Kit (BioRad) according to the manufacturer's instructions.
RT-primers spanning AP4M1 exon 10–15 are indicated in Table S3.
Amplification reactions were performed in a total volume of 25 μl, containing 1× PCR buffer (Invitrogen), 1.5 mM MgCl2, 200 μM of each dNTP, 1 μM forward primer, 1 μM reverse primer, 0.1 units Platinum Taq DNA polymerase (Invitrogen), and 1 μl cDNA. PCR conditions: 2′ 94°C, 30 cycles of 30″ 94°C, 30″ 60°C, 90″ 72°C with a final extension for 10′ 72°C.
Real-Time PCR Analysis
Relative quantification was performed with real-time PCR analysis. Reverse transcriptase was performed on 8 μg RNA with the iScript cDNA Synthesis Kit (BioRad) according to the manufacturer's instructions.
Of each cDNA 0.5 μl was used in a 25 μl reaction with 1× iTaq SYBR Green Supermix with ROX (Bio-Rad) and 200 nM of each primer. Real-time PCR was performed on a 7300 Real-Time PCR System (Applied Biosystems), cycle conditions were 10′ 95°C initial denaturation, 40 cycles 15″ denaturation, annealing, and extension and data collection 1′ 60°C.
Data were analyzed with the software package 7300 System SDS Software RQ Study Application v1.2.3 (Applied Biosystems).
For Q-PCR two primer sets for AP4M1, one primer set for the housekeeping gene ACTB (Entrez GeneID 60 [MIM 102630]) and one primer set for the housekeeping gene UBE2D2 (Entrez GeneID 7322 [MIM 602962]) were designed with the Primer Express software (Applied Biosystems). For primer sequences, see Table S4.
All reactions were performed in triplets. Relative quantification was calculated with the comparative Ct Method.
Transfection of AP4M1 in Human Embryonic Kidney HEK293 Cells
Primers to produce control and patient constructs are given in Table S5.
Amplification reactions were performed in a total volume of 25 μl, containing 1× PCR buffer (Invitrogen), 1.5 mM MgCl2, 200 μM of each dNTP, 1 μM forward primer, 1 μM reverse primer, 1 unit Platinum Taq DNA polymerase (Invitrogen), 0.1 U pfu DNA polymerase (Invitrogen), and 1 μl cDNA. PCR conditions are as follows: 2′ 94°C, 30 cycles of 30″ 94°C, 30″ 60°C, 90″ 72°C, and a final extension of 10′ 72°C.
Cloning of PCR products in pcDNA3.1_V5 His TOPO (Invitrogen) was performed according to the standard protocol.
The pcDNA3.1_V5 His_AP4M1 patient and control constructs were cotransfected with pcDNA3.1_V5 His_TSC2 in HEK293 cells with PEI (polyethylenimine) transfection.
A total of 1 μg DNA was diluted in 125 μl Dulbecco's modified Eagle medium (DMEM) containing 3 μg polyethyleneimine (Polysciences, Eppelheim, Germany).
After 15 min incubation at room temperature the DNA-polyethyleneimine complexes were added to the HEK293 cells in 35 mm diameter dishes (80% confluent). After 4 hr at 37°C, the transfection mixture was replaced with DMEM containing 10% fetal calf serum. Twenty-four hours after transfection, the expression of AP4M1_ctrl, AP4M1_pat and TSC2 (MIM 191092) were determined by immunoblotting. Antibodies, vector, and constructs used are V5 antibody obtained from Invitrogen, AP4M1_lupin antibody (courtesy of M.S. Robinson and J. Hirst), and TSC2 construct in pcDNA3.1 vector, courtesy of Mark Nellist, ErasmusMC, Rotterdam.22
SDS PAGE and Western Blot
Transfected HEK293 cells were homogenized in PBS with protease inhibitors (protease inhibitor cocktail mix complete, Roche), 1/6 v/v of homogenate was boiled in SDS-loading buffer and run on a 12% SDS-polyacrylamide gel and electroblotted onto nitrocellulose. The V5-tagged constructs were detected with mouse α-V5 (Invitrogen) and the AP4M1 constructs were detected with rabbit α-AP4M1 (courtesy of M. Robinson and J. Hirst) as the primary antibody. Horseradish peroxidase (HRP)-labeled anti-mouse and anti-rabbit Ig serum was used as secondary antibody, thereby allowing chemiluminescence detection with ECL (Amersham).
AP4M1 mRNA In Situ Hybridization on Mouse Sections
Wild-type mouse embryos (FVB/n) were used in preparing sections for the mRNA in situ hybridization experiments. Pregnant mice were killed by cervical dislocation and embryos were isolated in PBS on ice. Whole heads (E12.5, E14.5, and E16.5) or isolated brain (E18.5, P4) were snap frozen on dry ice and stored at −80°C until sectioning. Noon on the date of the vaginal plug was defined as E0.5. Housing and breeding were performed with standard conditions. The animal experiments were approved by the ethical commission for animals (Erasmus MC OZP 140-07-03).
Whole head or brain was sectioned sagittally and coronally at 12 μm with a Jung CM300 cryostat (Leica).
Probe Preparation
A DIG-UTP labeled mRNA probe, encompassing nt 1037 bp of the AP4M1 gene (primers used: 5′-AATGAGGGAACCATCTCACG-3′ [forward] and 5′-TTGCTGTGGCTTAGATGTCG-3′ [reverse]), was generated with T7 (antisense) and SP6 (sense) RNA polymerase (Roche), according to the suppliers' protocol. The mRNA in situ hybridization was performed according to Wilkinson.23 Stained sections were scanned by the NanoZoomer Digital Pathology System (Hamamatsu).
DTI Fiber Tracking
All imaging was performed on a 1.5-T GE EchoSpeed scanner (GE Medical Systems, Milwaukee, WI, USA). For DTI, an echoplanar sequence with diffusion gradients (b = 1000 s/mm2) applied in 25 noncollinear directions was used. Diffusion tensor images were transferred to a GE Advantage Windows workstation (General Electric Medical Systems, Milwaukee, WI, USA) for postprocessing with Functool 2000 software (General Electric). DTI measurements were taken from regions of interest (ROIs) positioned bilaterally within individual white matter tracts. For the placement, we used standard-size, round-shaped 30 pixel ROIs. The ROIs were used as seed points to perform fiber tracking (minimum anisotropy threshold 0.18).
Immunohistochemistry
Limited paraffin embedded brain autopsy material from patient IV-5 (age 17 months) and an aged matched control of 18 months was available of cerebral cortex and cerebellum.
Immunohistochemistry with antibodies directed against GluRδ2 (glutamate receptor delta 2) (MIM 602368), Calbindin (MIM 114050), and AP4M1 was performed in accordance with standard protocols. In brief, sections were deparaffinized, antigen retrieval was done with pressure-cooking in 0.1M sodium citrate buffer (pH 6) for 5 min, and endogenous peroxidase activity was inhibited by 30 min incubation in PBS-hydrogen peroxide (0.6%)-sodium azide solution (0.125%).
Subsequently, sections were immunoincubated with the primary antibody for 16 hr at 4°C. The Histostain-Plus broad-spectrum kit DAB (Zymed, San Francisco, CA, USA) was used as a detection system. Slides were counterstained with Mayer's Haematoxylin and mounted in Entellan.
Antibodies used were Go-anti-GluRδ2 (Grid2; Santa Cruz sc-26118) and Ra-anti-Calbindin (Chemicon AB1778).
Results
Phenotype of the CST Syndrome
Family History
Clinical information of the patients with a cerebral palsy-like disease, which we define as congenital spastic tetraplegia (CST), has been collected in a clinical follow-up of 20 years and is summarized in Table 1. The patients belong to a sibship of 11 children from consanguineous Moroccan parents (Figure 1). Seven patients are affected by arterial tortuosity syndrome (ATS [MIM 208050]) (indicated with an asterisk in Figure 1), caused by a homozygous nonsense mutation in the SLC2A10 gene on chromosome 20q.24 Five siblings from this family (patients IV-1, IV-3, IV-4, IV-5, and IV-6), further referred in the paper as the “probands,” suffer from a separate neurological disorder, not segregating with ATS. Of these five patients, one was heterozygote for the SLC2A10 (MIM 606145) mutation and has no ATS symptoms (patient IV-6),24 whereas the other four have both ATS and the neurological disorder. ATS patients in general24,25 and ATS patients from this family (IV-2, IV-9, and IV-10) have no neurological symptomatology, suggesting that the neurological disorder in this family segregates as a trait distinct from ATS. Some ATS patients have been reported with enlarged head circumference.26 On the basis of a sibship of five affected probands of different sex and consanguinity of healthy parents, we assumed the neurologic disorder to be autosomal recessive.
Table 1.
Clinical Neurological Presentation
| Patient | IV-1 | IV-3 | IV-4 | IV-5a | IV-6 |
|---|---|---|---|---|---|
| Age of onset | infancy | infancy | infancy | infancy | infancy |
| Age at observation (years) | 24 | 23 | 22 | 1.5a | 21 |
| Head circumference | −1 SD | 0 SD | −2 SD | ? | −2.5 SD |
| Mental retardation | +++ | +++ | +++ | +++ | +++ |
| General tone at onset | low | low | low | low | low |
| Contractures | adducted thumbs, clubfoot | adducted thumbs | adducted thumbs | adducted thumbs, clubfeet | no |
| Pseudobulbar signs (specified) | drooling, stereotypic laughter, jaw jerk ++, gag reflex ++ | drooling, stereotypic laughter, jaw jerk ++, gag reflex ++ | drooling, stereotypic laughter, tongue thrusting, jaw jerk ++, gag reflex ++ | ?a | stereotypic laughter, jaw jerk ++, gag reflex ++ |
| Speech | never acquired | never acquired | never acquired | never acquired | dysarthric monosyllables |
| Sphincter control | absent | absent | absent | absenta | absent |
| Corticospinal involvement | limb hypertonia; >DTR; extensor plantar reflex | limb hypertonia; >DTR; extensor plantar reflex | limb hypertonia; >DTR; extensor plantar reflex | ?a | limb hypertonia; >DTR; extensor plantar reflex |
| Deambulation | never achieved | never achieved | never achieved | never achieved | never achieved |
| Purposeful hand use | no | no | No | noa | Reaches for objects |
| Loss of acquired function | standing with support at age 6 years | no | No | ?a | standing at age 7 years |
| Brain imaging | normal CT (at age 3 months) | MRI: Abn. WM, wide ventricles, cerebellar atrophy | MRI: Abn. WM, wide ventricles, normal cerebellum | n.d. | MRI: Abn. WM, wide ventricles, cerebellar atrophy |
| VEP | normal | delayed bilaterally | normal | normal | normal |
| Eye examination | n.d. | normal | n.d. | n.d. | pale optic disks |
The following symbol and abbreviations are used: ?, unknown. n.d., not determined; WM, white matter; SD, standard deviation; >DTR, high deep tendon reflexes; Abn, Abnormal; and MRI, magnetic resonance imaging.
Patient died at the age of 17 months.
Figure 1.
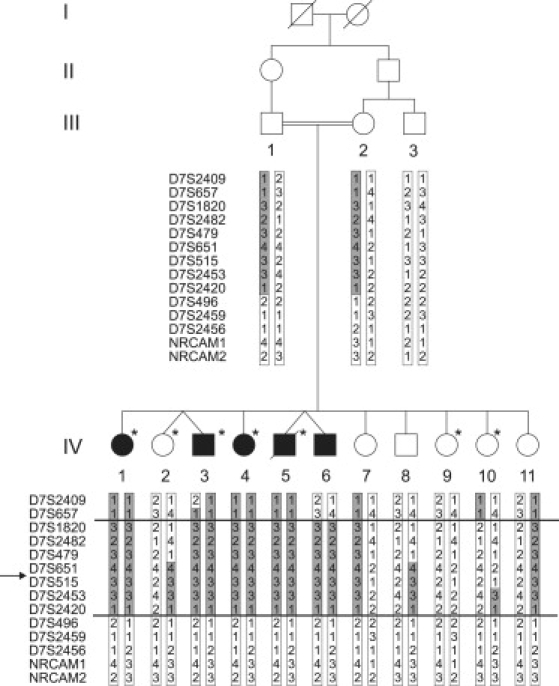
Pedigree and Haplotypes Linkage Region
Pedigree of the family with four generations (I–IV). Five siblings in generation IV (IV-1, IV-3, IV-4, IV-5, and IV-6) are affected by CST (filled symbols). Empty symbols represent those unaffected for CST; the asterisk represents those affected with arterial tortuosity syndrome.24 Haplotypes with chromosome 7 microsatellite markers are indicated. The assumed affected haplotype is indicated in gray. Recombination events delineating the region of homozygosity in the probands are indicated with a horizontal line. The proximal boundary is determined by a recombination event in individual IV-6 between D7S657 and D7S1820. A historical recombination event that was already present in both parents between markers D7S2420 and D7S496 delimits the distal border. “→” indicates the location of the AP4M1 gene.
Disease Phenotype
Prenatal ultrasound of patient IV-6 pregnancy showed ventricle dilatation in the 20th week. All five probands presented postnatally with early infantile hypotonia, delayed psychomotor development, strabismus, lack of independent walking, and severe mental retardation (total IQ 20, DSM-IV code 318.2).27 Progressive spasticity of all limbs with generalized hypertonia, high deep tendon reflexes, and Babinski signs were present by the end of the first year. Speech was absent or limited to a few meaningful but dysarthric monosyllables. Pseudobulbar signs such as drooling, stereotypic laughter, and exaggerated jaw jerk were present in all patients. They never experienced seizures. There were no clinical signs of cerebellar ataxia, although spasticity prevented adequate testing. Disease course was slowly progressive with minimal or no regression in 20 years. One patient (IV-5) died at 17 months of aspiration pneumonia (Table 1).
Patients and Clinical Investigation
Patient IV-1 has ATS. Postnatally she showed microcephaly, hypotonia, strabismus, and thumb adduction. At the age of 6 years she could stand with support, but this was lost later on; she never developed understandable speech and suffered from deep mental retardation. Neurological and metabolic investigations at that age were normal, including brain CT, muscle biopsy, skin EM, EEG, EMG, VEP, and BAER. At age 16, she was wheelchair bound with spastic tetraparesis, adducted thumbs with tenar hypoplasia, and kyphosis; she made eye contact notwithstanding the squint, was able to sit unsupported, drooled, presented with bursts of stereotypic laughter, and showed fixed contractures of large joints (Table 1). She chews and swallows normally. Acrocyanosis and skin laxity were considered features of ATS.
Patient IV-3 has ATS and was born with bilateral clubfoot. He is at the moment the most severely affected, and his psychomotor development was never higher than 6 months' level. He never achieved deambulation, unsupported sitting, or speech. From infancy, convergent bilateral strabismus was present, and this condition didn't improve after surgeries. Examination at age 22 showed normal head circumference, spastic tetraparesis with remarkable hypertonia impairing wheelchair use, tenar hypoplasia and thumb adduction, scoliosis, drooling, and strabismus; however, he chews and swallows. He makes no contact but can laugh stereotypically and has no day-night rhythm. A brain CT scan at the age of 4 months reported wide ventricles and subarachnoid spaces. A brain MRI at the age of 15 years showed diffuse cerebellar atrophy, asymmetrically enlarged lateral ventricles, thin corpus callosum, and normal signal intensity of residual white matter (Figure 2A). MR angiography detected intracerebral vessel tortuosity.
Figure 2.
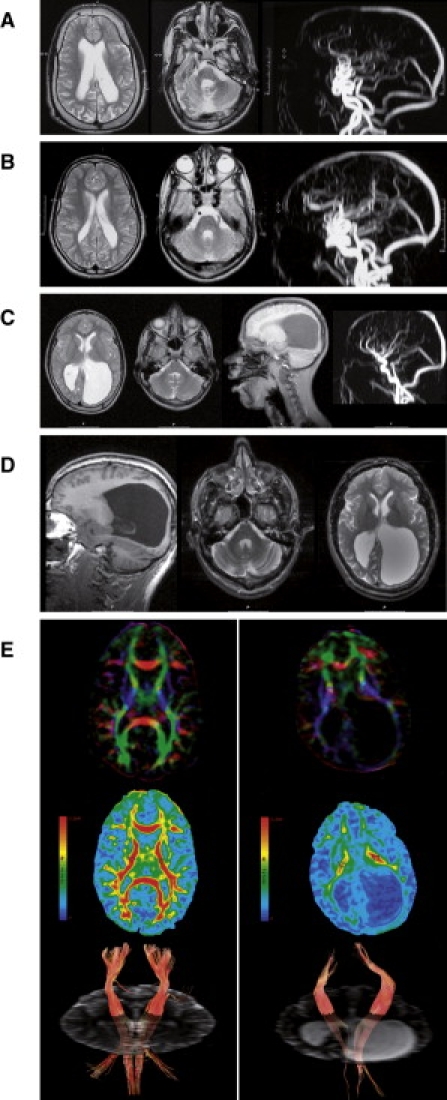
Brain Magnetic Resonance Imaging and DTI
(A) T2 weighted brain MRI of patient IV-3 at the age of 15 years shows enlarged and asymmetric lateral ventricles, thin corpus callosum, white matter loss, with normal signal intensity, thickened skull (left), diffuse cerebellar atrophy, wide IVth ventricle, and hypoplastic pons and vermis (center). Magnetic resonance angiography (MRA) shows intra- and extracranial tortuosity of the large vessels (right), in light of the fact that this proband is also affected by ATS.
(B) T2 weighted brain MRI of patient IV-4 at the age of 14 years shows enlarged lateral ventricles, particularly in the occipital area, with irregular contour and white matter loss with normal intensity and wide IVth ventricle (left and center). MRA shows arterial tortuosity of the neck and intracerebral vessels, related to ATS (right).
(C) Conventional MRI of patient IV-6, performed at the age of 13 years, shows on T2 weighted images (first and second from left) diffuse but asymmetric white matter loss with massive enlargement of the occipital horns. Stretched gyri and asymmetry of the thalami suggest hydrocephalus in the past (first left and second from right). Signal intensity of the residual white matter is normal. No vascular tortuosity at the angiography (right), conform to heterozygosity for ATS. Mild cerebellar atrophy is present in T1 sagittal images (second from right).
(D) Repeat MRI at age 21 years of patient IV-6 showing similar abnormalities with only minimal progression of cerebellar atrophy.
(E) DTI brain images of the patient (right panel) and age- and sex-matched control (left panel). Top images show color maps: color maps are based on major eigenvector orientation in each of the voxels with red representing right to left, green antero-posterior, and blue supero-inferior anatomical directions.28 Middle images show fractional anisotropy maps with an example of regions of interest (ROIs, round-shaped, 30 pixels) placed in the posterior limb of the internal capsule (PLIC) (circles). For isotropic diffusion, FA is zero (no anisotropy = blue). If there is a strongly preferred direction of diffusion (indicating capture of white matter tracts in the ROI), FA approaches a value of 1 (red). Bottom images show fiber track images of the corticospinal tract with ROIs as seedpoints (minimum anisotropy threshold 0.18). Mapping the directional principal eigenvectors forms the basis for tractography with the assumption that the principal eigenvector is aligned with the direction of the fiber bundle.28
Patient IV-4 has ATS. At birth her head circumference was at −2 SD and she was hypotonic. She suffered from deep mental retardation, never learned to speak or walk, but can sit on her knees. Spastic paraparesis required hip surgery. At the age of 22, she is borderline microcephalic and passive, and she sometimes articulates monosyllables and shows mouth dyskinesia, tongue thrusting, and bursts of unprovoked laughter. EEG, EMG, and VEP findings at the age of 11 years were unremarkable (Table 1). Brain MRI at the age of 15 years showed enlarged lateral ventricles, white matter loss without hyperintensity on T2 weighted images, normal cerebellum, and tortuosity of the neck and cerebral vessels at MR angiography (Figure 2B).
Patient IV-5 had ATS and was the dizygotic twin of patient IV-6. At birth in the 38th gestational week, he presented with hypotonia, normal head size, low Apgars, and clubfeet. Sliding diaphragmatic hernia and gastro esophageal reflux were later diagnosed. After gastric fundoplication surgery, he developed aspiration pneumonia and died at the age of 17 months. Autopsy revealed diffuse brain atrophy and signs of arterial tortuosity in all organs.25 His CNS material was revised (see brain pathology findings).
Patient IV-6 is the only patient without ATS (he is a carrier for this disease and heterozygous for the familial W170X SLC2A10 mutation). He is the dizygotic twin of patient IV-5. During pregnancy, prenatal ultrasound raised suspicion of hydrocephalus and porencephalic cystic enlargement of the left occipital ventricle. At birth, Apgars were normal, head circumference was at −2SD, and his weight was at 0 SD. First examination at the age of 1 year revealed psychomotor retardation. He became hypertonic, underwent Achilles tendon surgery, and learned to stand at the age of 7 years, but this was lost later on. He shows convergent strabismus (recidivating after surgery) but makes eye contact. He could speak meaningful but dysarthric words with echolalia and use a spoon at the age of 15 years, but at the age of 21 he speaks monosyllables and can accomplish simple manual tasks such as reaching for objects. He can shuffle on his knees and sit unsupported. Neurological examination shows mental retardation, spastic tetraplegia, no clear ataxia, normal pain sensation, microcephaly, drooling, and bursts of unprovoked laughter. Eye fundus shows pale optic disks. Results of neurological investigation are summarized in Table 1.
Brain MRI and Diffusion Tensor Imaging
Brain MR imaging and angiography were performed in probands IV-3, IV-4, and IV-6 (Figures 2A–2C). These showed consistent anatomical abnormalities in all patients with variable loss of white matter and cerebellar atrophy. Longitudinal follow-up of proband IV-6 at the age of 13 and 21 years showed unchanged abnormalities. The residual cerebral and cerebellar white matter showed normal signal intensity even at follow-up without signs of demyelination (Figure 2D), suggesting an early developmental defect rather than progressive neurodegeneration. Brain imaging of patient IV-2 and IV-9 with ATS only was normal, except for tortuosity of arteries (not shown).
We performed diffusion tensor imaging (DTI) with fiber tracking on MRI in proband IV-6 and an age-matched control to quantify and visualize white matter tracts. MR-DTI is based on capturing diffusion of water molecules in different orientations in lipid-rich brain tissue and can be expressed as fractional anisotropy (FA).28–31 It is the only available technique allowing in vivo qualitative and quantitative visualization of structural brain connections.32 Because DTI is a fairly new technique, extensive control data is lacking. We therefore decided to test a healthy age-matched male control under the same imaging and analysis conditions.33–35 Postprocessing analysis showed clear differences in the anisotropy indexes of the white matter tracts between patient and control, particularly in corpus callosum, internal and external capsule, indicating major white matter loss (Figure 2E). FA is believed to reflect many factors including axonal density, integrity, and degree of myelination.28 Despite the limited reference data for human age-matched FA, the low FA values in the patient and pattern of FA changes suggest that the lesions are related to a combination of axonal disarray and myelin integrity loss (Table 2).
Table 2.
FA Values of DTI for Control and Patient
| Control | PLIC | ALIC | CC Genu | EC |
|---|---|---|---|---|
| FA | 0.66 | 0.54 | 0.77 | 0.36 |
| L1 | 11.85 | 11.8 | 17 | 9.82 |
| L2 | 4.11 | 5.31 | 4.01 | 6.68 |
| L3 | 2.75 | 3.89 | 2.78 | 4.52 |
| L2+L3/2 | 3.43 | 4.60 | 3.39 | 5.6 |
| Patient | PLIC | ALIC | CC Genu | EC |
| FA | 0.46 | 0.37 | 0.42 | 0.17 |
| L1 | 10.4 | 10.04 | 16.6 | 8.09 |
| L2 | 5.05 | 5.99 | 9.02 | 6.61 |
| L3 | 4.24 | 4.79 | 7.45 | 5.71 |
| L2+L3/2 | 4.65 | 5.37 | 8.24 | 6.16 |
Mean values are shown of different DTI indices in (30 pixel size) regions of interest (ROIs) placed in the posterior limb of the internal capsule (PLIC), the anterior limb of the internal capsule (ALIC), the genu of the corpus callosum (CC), and the external capsule (EC).
“FA” stands for fractional anisotropy. FA values of 1 indicate high diffusion anisotropy, in which water diffusion is restricted to one main direction. FA values close to 0 indicate that the diffusion within the tissue is isotropic.55
L1–L3 refers to values of the eigenvectors. The eigenvalues of the diffusion tensor are the diffusion coefficients in the three principal directions of diffusivity. Lambda 1 (L1) is the principal eigenvector; it defines the main direction of diffusion of water molecules. Lambda 2 (L2) is the middle eigenvalue and lambda 3 (L3) the smallest eigenvalue. Directional diffusivities derived from DTI measurements can be separated into components parallel (λ 1) and perpendicular (λ2 and λ3) to the white matter tract. These components are referred to as the axial diffusivity (λ‖), and radial diffusivity (λ⊥ = (λ2 + λ3)/2), respectively.
The values for FA were low in the white matter tracts of the patient. A significant difference between patient and control for both axial (along the axon) and radial (perpendicular to the axon direction) diffusivity values were found, with reduced axial and increased radial diffusivity. Decrease in axial diffusivity (type 2 anisotropy loss) has been associated with axonal pathology, while increase in transverse (short axes) diffusivity (type 1 anisotropy loss) correlates with myelin damage.56,57
Molecular Analysis
Linkage and Expression Study
Multipoint linkage analysis on the neurological trait with 100 K SNP array data, including the consanguinity loop, but without individual III-3, resulted in only one significant linkage peak with a maxLOD score of 4.35. The locus is extending 16.3 cM on chromosome 7q22. (Figure S1). Haplotype analysis and fine mapping with chromosome 7 polymorphic microsatellite markers confirmed homozygosity in the patients and defined a 14 Mb candidate region between marker D7S657 to D7S496 (Figure 1), containing 203 genes (NCBI build 36.3).
In order to select putative candidate genes from the linkage region, we used mRNA expression arrays to study the transcripts in fibroblasts of probands (IV-1 and IV-3–IV-6 in Figure 1) and controls (IV-2 and IV-9 plus two independent controls). OmniViz supervised clustering and SAM analysis identified the AP4M1 gene as the strongest downregulated transcript in all neurologically compromised patients with a 2-fold downregulation of the gene (Figure S2).
Sequence Analysis and Mutation Validation
Sequencing of the AP4M1 candidate locus revealed a splice donor site homozygous mutation in intron 14, c.1137+1G→T, in all five patients, whereas the parents were heterozygous (Figure 3A). Given the 100% evolutionary sequence conservation of the +1G splice donor site,36 this mutation was expected to destroy the splice donor site of intron 14. This mutation was not present in 224 ethnically matched Moroccan controls, thereby making a polymorphism unlikely.37
Figure 3.
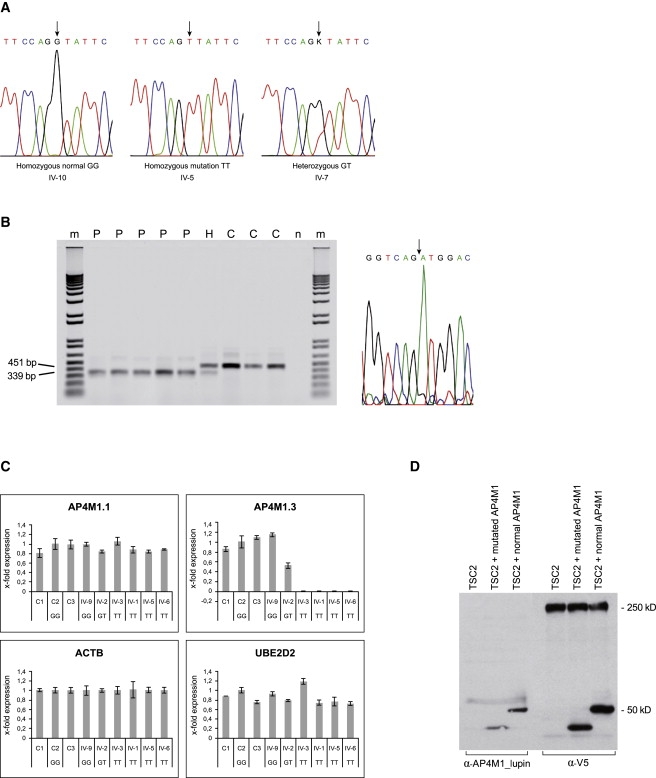
Sequencing and Functional Analysis
(A) Sequence analysis showing the AP4M1 mutation in intron 14 (c.1137+1G→T). The normal sequence from exon 14 to intron 14 is TTCCAGgtattc, with the last six bases of exon 14 indicated in capital letters and the first six bases of intron 14 in lowercase letters. Homozygous normal sequence, homozygous mutation as seen in probands IV-1, -3, -4, -5, and -6, and heterozygous mutation in carriers (parents III-1,2; sibs IV-2, -7, -8, and -11) are indicated by an arrow.
(B) The left panel shows RT-PCR on fibroblast RNA of patients IV-1, -3, -4, -5, and -6 (P), controls (C), and a heterozygous carrier (H). Normal product length of 451 bp and reduced product length of 339 bp are indicated. n = negative control without DNA, m = 1 kb plus marker (Invitrogen). In the right panel is the RT-PCR sequence showing the exon 13 to exon 15 junction fragment, GGTCAGATGGAC, present in the patients. The arrow indicates the transition from exon 13 to exon 15. Sequence data from patient IV-4 is shown. All patients showed the same sequence.
(C) qRT-PCR, expression of AP4M1 in fibroblast RNA of control samples (C1-C3, unrelated, plus family member IV-9), patients (IV-1, -3,-5, and -6), and one heterozygote (IV-2). Values are expressed as x-fold expression relative to expression of the housekeeping gene ACTB, which is arbitrarily set to 1. Expression of the housekeeping gene UBE2D2 relative to ACTB is used as positive control. Error bars indicate the standard deviation (n = 3). Expression of UBE2D2 and AP4M1.1 (primers spanning exon 2 and 3) were comparable in all control samples and patients. AP4M1.3 primers (spanning exon 14–15) showed normal mRNA expression in all control samples, 0.5-fold expression in the heterozygous family member (IV-2), and a lack of expression on all patients tested. The following abbreviations are used: GG, normal sequence; GT, heterozygous carrier; and TT, patient with homozygous mutation.
(D) Immunoblot analysis of V5-tagged wild-type and mutated AP4M1 expressed in HEK293 cells, transfected with the wild-type, and mutated AP4M1 cDNA. α-V5 as well as α-AP4M1_lupin antibodies visualized an expected protein band of 50 kDa from the wild-type cDNA construct and a band of reduced size from the mutated construct. V5-tagged cDNA constructs of TSC2 gene (250 kDa) were transfected separately and in combination with AP4M1 so that the system for transfection efficiency could be tested.
A transcript of diminished size was observed by RT-PCR on fibroblast RNA from patients compared to controls, with primers spanning AP4M1 exons 10–15, suggesting aberrant splicing in the patients (Figure 3B). Skipping of exon 14 was confirmed by sequencing the RT-PCR products. Serendipitously, all the AP4M1 probes present on the Affymetrix HG-U133_plus2 expression arrays are located at exons 10–15. A number of these probes are on exon 14, explaining why AP4M1 appeared downregulated.
Relative analysis by quantitative RT-PCR with primer sets on exons 2–3 showed the presence of equal levels of mRNA in patients and controls; however, using primer sets on exons 14–15 showed that in patients, only aberrant AP4M1 mRNA, lacking exon 14, is synthesized (Figure S3 and Figure 3C). Normal and deduced mutated AP4M1 protein is shown in Figure S4.
Because AP4M1 protein expression in control fibroblasts was too low to detect with Western blot analysis, we investigated the effect of the mutation in cell cultures by expressing V5-tagged wild-type and mutated AP4M1 (lacking exon 14) in HEK293 cells and then analyzing them via immunoblotting. α-V5 as well as α-AP4M1 antibodies showed that mutated AP4M1 induces synthesis of a protein with a lower molecular weight (44.5 kD), as predicted by abnormal splicing of exon 14, compared to the expected normal 50 kD (Figure 3D).
Developmental Expression
Given that CST is compatible with a developmental defect with onset before birth, timing and localization of the mouse ortholog Ap4m1 mRNA expression was examined by in situ hybridization during various stages of mouse brain development.
Expression of Ap4m1 in all ventricular zones was detected at embryonal stages E12.5, E14.5, E16.5, and E18.5 (Figure 4A). On E16.5, pronounced expression in the upper layer of the cortical plate is detected, broadening at E18.5 and P4. Additionally, expression was observed in the lateral and medial ganglionic eminences at E16.5. Cerebellar expression was observed at E18.5 and postnatal P4 in the external granular and Purkinje cell (PC) layer, as well as at P4 also in the internal granular layer. Early Ap4m1 expression suggests its role in cerebral and cerebellar development, and its localization corresponds to brain areas characterized by neuroglial progenitor proliferation. This suggests that AP4M1 mutation might affect neurons as well as oligodendrocytes.
Figure 4.
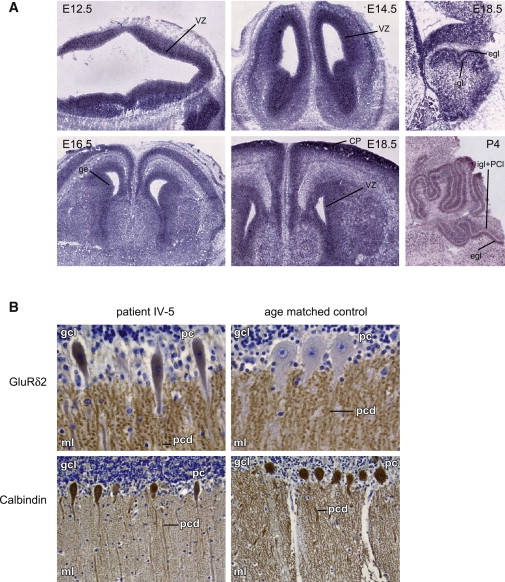
mRNA In Situ Hybridization and Immunohistochemistry
(A) mRNA ish expression patterns of Ap4m1 in mouse brain of different developmental stages (E, embryonal day; p, postnatal day). Ap4m1 expression is present in the ventricular zone of all four brain ventricles on days E12.5, E14.5, E16.5, and E18.5 (coronal sections, left and center panels), with expression in the ventricular zone of the lateral ventricles more sharply defined. On E16.5, pronounced expression in the upper layer of the cortical plate is present as well, broadening at E18.5 and P4. Also, expression is present in de lateral and medial ganglionic eminences (E16.5). Expression in the developing cerebellum was observed at E18.5 and P4 in the external granular and Purkinje cell layer and at P4 also in the internal granular layer (E18.5 and P4, right panel, both images showing sagittal sections). Control sense probe experiments were negative (data not shown). The following abbreviations are used: vz, ventricular zone; cp, cortical plate; ge, ganglionic eminence; egl, external granular layer; igl, internal granular layer; and pcl, Purkinje cell layer.
(B) Immunohistochemical staining on brain material from patient IV-5 (17 months) (left) and an age-matched control (18 months) (right). In the upper panel, a patient's cerebellum stained with GluRδ2 revealed signal in the perikarya of the PC cells and slightly abnormally ovoid shaped perikarya as compared to the age-matched control. In both patient and controls, punctuate signals are clearly seen in the molecular layer of the cerebellum, reflecting synapse-associated signaling in dendritic spines. The punctuated GluRδ2 pattern is more pronounced in the patient compared to the control. In the lower panel, staining with PC-specific calbindin antibodies shows decreased dendritic arborization in the patient as compared to the control. The following abbreviations are used: gcl, granular cell layer; ml, molecular layer; pc, Purkinje cell body; and pcd, Purkinje cell dendrites.
Brain Pathology
Postmortem Brain and Histology
The autopsy brain from proband IV-5, who died of pneumonia, weighted 750 g (normal at this age is 1050 g).
Widening of all ventricles and severe thinning of corpus callosum (Figures S5A–S5C), without significant thinning of the cerebral cortex were consistent with long lasting reduction of white matter. The cerebral cortex showed a normal layering without obvious loss of neurons (Figure S5D). There was marked edema of large parts of the cortex and basal ganglia, thereby causing a spongiform appearance. The neurons in these areas were shrunken as seen in agony hypoxia. The pons was significantly atrophic whereas the basal ganglia were not markedly atrophic.
The fourth ventricle was widened and although the cerebellum was not obviously atrophic, there was some thinning of the granular layer (Figure S5C). The dendate nucleus, however, showed a striking loss and degeneration of neurons, with slight gliosis (Figure S5E).
Reduced myelin with significant gliosis in the white matter of the cerebral hemispheres was noticed, with irregular thickening of axons in some long stretches (Figures S6A–S6C). No signs of active demyelination were visible (Figure S6D). The number of Purkinje cells in the cerebellum was not reduced, but sizes of the cell bodies were slightly reduced and show a somewhat ovoid appearance (Figures S6E and S6F) as shown in Figure 4B. This indicates lesions of both neurons (dentate degeneration, axonal swelling) and glia (reduced myeling, abnormal gliosis) lineage in the patient brain, but it does not clarify which event occurred first.
Immunohistochemistry
Paraffin embedded patient brain material (age 17 months) has been stored for more than 20 years. Paraffin embedded brain material of an age-matched control (age 18 months) with a storage time of 10 years was used. Despite a specific signal on Western blots, no signal could be observed with the available AP4M1 antibodies (commercially available as well as from Robinson & Hirst) in the brain of patient IV-5 and the age-matched control. The AP-4 protein complex is expressed in the central nervous system neurons and interacts with glutamate receptors 1–4 (GluR1–GluR4 [MIM 138248, 138247, 305915, and 138246]) and glutamate receptor δ2 (GluRδ2), which is exclusively expressed in cerebellar PC.38 However, except for the GluRδ2 antibodies in cerebellum, no immunoreactivity was seen in control brains with commercial glutumate receptor antibodies 1–4. We proceeded to indirectly test the effect of the AP4M1 mutation by detection and localization of its ligand GluRδ2 on cerebellum paraffin sections of patient IV-5 and two age-matched controls.
PC bodies contain low levels of immunoreactive GluRδ2 at all developmental stages.39–41 However, GluRδ2 protein is abundantly expressed on the postsynaptic sites at the dendritic spines of parallel fiber-PC synapses.42–44
In patient IV-5, GluRδ2 protein was present in the perikarya of the PC and scattered PC bodies with a somewhat abnormal ovoid shape, as compared to the control (Figure 4B, upper panel). Yap et al.38 suggested that the AP4 complex interacts with GluRδ2 and transports GluRδ2 to the dendrites. The observation of GluRδ2 staining in the PC perikarya suggests insufficient vesicle-mediated transport to the dendrite membranes in the patient and receptor retention in the perinuclear Golgi and endosomal apparatus. In both the patient and the control, punctate signals were clearly seen in the molecular layer of the cerebellum, illustrative of synapse-associated signal in dendritic spines. At higher magnification, the GluRδ2 pattern in the patient dendrites was coarse, suggesting abnormal and diminished dendritic arborization of the PC cells.
Immunohistochemical staining with PC-specific calbindin antibodies confirmed coarse spine shape and decreased dendritic arborization in the patient as compared to the control (Figure 4B, lower panel).
Both immunohistochemistry and histology are suggestive of primary neuroaxonal abnormalities, concordant with the in vivo FA data from DTI (Table 2).
Discussion
An integrative bioinformatics approach, combining homozygosity mapping of genome areas identical by descent and expression array analysis, enabled us to identify a recessive AP4M1 gene mutation in this single pedigree. The disease manifests as tetraplegic cerebral palsy; therefore, we defined it as congenital spastic tetraplegia. CST has a prenatal onset and is characterized by early hypotonia changing to spasticity within the first year, quadriplegia, and absence of sphincter control. Spastic tetraplegia is associated with lack of speech, strabismus, pseudobulbar signs, clasped thumbs, clubfeet, and severe mental retardation. Brain MRI shows diffuse loss of periventricular white matter without signs of demyelination, thereby leading to ventriculomegaly, hypoplasia of the corpus callosum, pons, dentate nucleus, vermis, and cerebellar atrophy. Quantitative analysis of DTI data indicates a combination of axonal disarray and loss of myelin integrity as expected in PVWMI. Brain pathology in one patient confirmed cerebral white matter and dentate neuronal loss, with signs of primary neuroaxonal abnormalities.
Although CST shares signs of hereditary complicated spastic paraplegia,45 disease course, neuroimaging, and lack of progressive neurodegeneration46 are reminiscent of tetraplegic cerebral palsy associated with injury to periventricular white matter after major hypoxia-ischemia.1,4,8 Age of onset in the hereditary spastic paraplegias is usually not congenital. Rare early-onset forms have been described as primary lateral sclerosis (PLS [MIM 606353]), infantile onset ascending spastic paralysis (IAHSP [MIM 607225]), and “complicated” hereditary spastic paraplegias (cSPG).45 Other types of hereditary spastic quadriplegia, like those caused by PLP1 (MIM 312920), L1CAM (MIM 307000), and MCT8 (MIM 300523) mutations, are easily differentiated by their X-linked inheritance pattern and specific signs (diffuse hypomyelination in PLP1, high-pressure hydrocephalus in L1CAM, and extrapyramidal signs in MCT8).
Glutamate excitotoxicity and abnormalities of AMPA receptors are observed in hypoxy-ischemia models of cerebral palsy.12,13 AMPA receptors are widely expressed in cerebral hemispheres. GluRδ2 and AMPA receptors colocalize to dendritic spines of rat cerebellum.42 Misrouting of GluRδ2 and AMPA receptors GluR1–GluR4 in brain of knockout mice lacking the AP-4 β-subunit has recently been shown, and this effect was reproduced by exclusive downregulation of AP-4 μ expression.16 Knockouts show swelling of PC terminal axons and accumulation of LDLR (MIM 606945), AMPA, and GluRδ2 in autophagosomes at or near the terminal ends of axons in the deep cerebellar nuclei. Lack of GluR1–GluR4 immunorectivity of commercial antibodies did not allow determination of receptor routing in our proband IV-5; however, degeneration of cerebellar dentate neurons in our patient might relate to a similar mechanism.
Brain immunohistochemistry showed abnormal staining of glutamate receptor GluRδ2 in the PC perikarya of proband IV-5, suggesting abnormal vesicle-mediated transport to the dendrites and retention at the trans-Golgi network. Also, structural changes in dendritic arborization of PC cells were observed. Abnormal dendritic arborization is seen in many developmental disorders associated with motor and cognitive abnormalities like Fragile X (MIM 300624) and Rett syndrome47,48 (MIM 312750). This indicates that neuronal dysfunction is concomitant with the apparent white matter loss on routine MRI; such a loss is possibly a secondary phenomenon.
Yap et al.38 suggested that AP-4 interacts with GluRδ2 and might be involved in glutamate trafficking in the dendritic spines of cerebellar PC. GluRδ2 plays a crucial role in motor coordination, PC synapse formation, and induction of cerebellar long-term depression (LTD), a form of synaptic plasticity underlying motor learning.
Hotfoot mutations in mice consist of various deletions in the GluRδ2 gene and result in aberrant localization of the mutant receptor, leading to profound motor disturbances. The protein is not transported beyond the endoplasmic reticulum (ER) or cis-Golgi apparatus.49 Similarly in our proband IV-5, aberrant localization of GluRδ2 has been observed in the soma of Purkinje cells.
Cerebellum and cerebral white matter of proband IV-5 showed PC with abnormal dendritic arborization, decreased amounts of synapses, and irregular thickened axons. GluRδ2 immunostaining suggested mislocalization of the GluRδ2 cargo protein to the perinuclear region of Purkinje cells as well. The effect on cerebellar function of impaired GluRδ2 transport in general and in this family in particular is unclear. Signs of cerebellar dysfunction were difficult to evaluate in our patients because of their severe quadriplegia and mental retardation.
It is possible that the AP4M1 mutation affects transport of several glutamate receptors, also in cerebral hemispheres. In all four known AP complexes, the μ subunit is essential for binding of cargo proteins, like several membrane receptors.17,50
The AP4M1 gene encodes the μ subunit of the adaptor protein complex-4. The splice site mutation in CST destroys the donor splice site of intron 14, and only aberrant AP4M1 mRNA is synthesized in the patients. The mutant mRNA lacks exon 14 and has a premature stop codon in exon 15 leading to truncation of the COOH terminus, which is the site of cargo binding. The mutant mRNA in HEK293 cells leads to the synthesis of an aberrant protein. The mutation possibly causes impaired function of the AP-4 complex without disassembly or degradation of the whole complex. It is also possible that the mutated μ subunit functionally disables the whole AP-4 complex, given that a large part of the C terminus, including exons 14 and 15, belongs to the Mu homology domain profile (Prosite database, entry PS51072), which is necessary for cargo protein binding.51
Mutations in other adaptor-related complexes have been related to human disease.19 Mutations in AP1S2 (MIM 300629), the sigma 2 subunit of the AP1 complex, which is located on Xp22, are described in patients with mental retardation, delayed motor development, hydrocephalus, and basal ganglia calcifications52,53 (MIM 300630). Compound heterozygous mutations in the beta3A subunit of the AP-3 protein (MIM 603401) complex have been described in patients with Hermansky-Pudlak syndrome type 2 (MIM 608233).54 These findings reveal the physiological importance of uniquitous AP complexes in brain function, but disease pathogenesis is not understood.
We have not been able to identify CST in a second pedigree. We sequenced AP4M1 in six sporadic patients with a comparable phenotype and no history of perinatal injury, but no mutation was found. This suggests that CST is a very rare disorder.
We hypothesize that AP4M1 mutation causes a developmental defect of the brain leading to congenital spastic tetraplegia and mimicking glutamate-mediated perinatal white matter injury through early neuroaxonal degeneration and secondary white matter loss and that abnormal cycling of glutamate receptors might be the underlying pathogenic mechanism.
Supplemental Data
Supplemental Data include five tables and six figures and can be found with this article online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Allen Institute for Brain Science, http://www.brain-map.org
ExPASy, http://www.expasy.ch/prosite/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/OMIM/
Orphanet, www.orpha.net
Accession Numbers
The expression array data is deposited at the GEO database under accession number GSE16447.
Acknowledgments
We acknowledge the family for their cooperation. We kindly thank Dicky Halley and Alice Brooks for providing control Moroccan DNA samples, Edwin Mientjes for advising in qRT-PCR analysis, and Luc Nelemans, Marcel van der Weiden, Lies-Anne Severijnen, Asma Azmani, Marit de Haan, and Wasiem Sahebali for technical assistance. We thank Mark Nellist for providing the TSC2 construct. We thank Marie Claire de Wit for patient care and Frank Grosveld for critically reading the manuscript. Tom de Vries Lentsch is acknowledged for excellent graphical support. We thank Mirjam C.G.N. van Vroonhoven for computer system administration, supported by NBIC.
References
- 1.Rennie J.M., Hagmann C.F., Robertson N.J. Outcome after intrapartum hypoxic ischaemia at term. Semin. Fetal Neonatal Med. 2007;12:398–407. doi: 10.1016/j.siny.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 2.Nelson K.B. Causative factors in cerebral palsy. Clin. Obstet. Gynecol. 2008;51:749–762. doi: 10.1097/GRF.0b013e318187087c. [DOI] [PubMed] [Google Scholar]
- 3.Bax M., Tydeman C., Flodmark O. Clinical and MRI correlates of cerebral palsy: The European Cerebral Palsy Study. JAMA. 2006;296:1602–1608. doi: 10.1001/jama.296.13.1602. [DOI] [PubMed] [Google Scholar]
- 4.Volpe J.J. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr. Res. 2001;50:553–562. doi: 10.1203/00006450-200111000-00003. [DOI] [PubMed] [Google Scholar]
- 5.Folkerth R.D. Neuropathologic substrate of cerebral palsy. J. Child Neurol. 2005;20:940–949. doi: 10.1177/08830738050200120301. [DOI] [PubMed] [Google Scholar]
- 6.Deng W., Pleasure J., Pleasure D. Progress in periventricular leukomalacia. Arch. Neurol. 2008;65:1291–1295. doi: 10.1001/archneur.65.10.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McQuillen P.S., Ferreiro D.M. Perinatal subplate neuron injury: Implications for cortical development and plasticity. Brain Pathol. 2005;15:250–260. doi: 10.1111/j.1750-3639.2005.tb00528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inder T.E., Huppi P.S., Warfield S., Kikinis R., Zientara G.P., Barnes P.D., Jolesz F., Volpe J.J. Periventricular white matter injury in the premature infant is followed by reduced cerebral cortical gray matter volume at term. Ann. Neurol. 1999;46:755–760. doi: 10.1002/1531-8249(199911)46:5<755::aid-ana11>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 9.Schaefer G.B. Genetics considerations in cerebral palsy. Semin. Pediatr. Neurol. 2008;15:21–26. doi: 10.1016/j.spen.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Follett P.L., Deng W., Dai W., Talos D.M., Massillon L.J., Rosenberg P.A., Volpe J.J., Jensen F.E. Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: A protective role for topiramate. J. Neurosci. 2004;24:4412–4420. doi: 10.1523/JNEUROSCI.0477-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jensen F.E. Role of glutamate receptors in periventricular leukomalacia. J. Child Neurol. 2005;20:950–959. doi: 10.1177/08830738050200120401. [DOI] [PubMed] [Google Scholar]
- 12.Liu B., Liao M., Mielke J.G., Ning K., Chen Y., Li L., El-Hayek Y.H., Gomez E., Zukin R.S., Fehlings M.G., Wan Q. Ischemic insults direct glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. J. Neurosci. 2006;26:5309–5319. doi: 10.1523/JNEUROSCI.0567-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bell J.D., Ai J., Chen Y., Baker A.J. Mild in vitro trauma induces rapid Glur2 endocytosis, robustly augments calcium permeability and enhances susceptibility to secondary excitotoxic insult in cultured Purkinje cells. Brain. 2007;130:2528–2542. doi: 10.1093/brain/awm164. [DOI] [PubMed] [Google Scholar]
- 14.Metin C., Denizot J.P., Ropert N. Intermediate zone cells express calcium-permeable AMPA receptors and establish close contact with growing axons. J. Neurosci. 2000;20:696–708. doi: 10.1523/JNEUROSCI.20-02-00696.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Man H.Y., Ju W., Ahmadian G., Wang Y.T. Intracellular trafficking of AMPA receptors in synaptic plasticity. Cell. Mol. Life Sci. 2000;57:1526–1534. doi: 10.1007/PL00000637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsuda S., Miura E., Matsuda K., Kakegawa W., Kohda K., Watanabe M., Yuzaki M. Accumulation of AMPA receptors in autophagosomes in neuronal axons lacking adaptor protein AP-4. Neuron. 2008;57:730–745. doi: 10.1016/j.neuron.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 17.Robinson M.S. Adaptable adaptors for coated vesicles. Trends Cell Biol. 2004;14:167–174. doi: 10.1016/j.tcb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Hirst J., Bright N.A., Rous B., Robinson M.S. Characterization of a fourth adaptor-related protein complex. Mol. Biol. Cell. 1999;10:2787–2802. doi: 10.1091/mbc.10.8.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gissen P., Maher E.R. Cargos and genes: Insights into vesicular transport from inherited human disease. J. Med. Genet. 2007;44:545–555. doi: 10.1136/jmg.2007.050294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gudbjartsson D.F., Jonasson K., Frigge M.L., Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 21.O'Connell J.R., Weeks D.E. PedCheck: A program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dan H.C., Sun M., Yang L., Feldman R.I., Sui X.M., Ou C.C., Nellist M., Yeung R.S., Halley D.J., Nicosia S.V. Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J. Biol. Chem. 2002;277:35364–35370. doi: 10.1074/jbc.M205838200. [DOI] [PubMed] [Google Scholar]
- 23.Wilkinson D. Whole mount in situ hybridization of vertebrate embryos. In: Wilkinson D., editor. In Situ Hybridization: A Practical Approach. IRL Press of Oxford University Press; Oxford: 1992. pp. 75–83. [Google Scholar]
- 24.Coucke P.J., Willaert A., Wessels M.W., Callewaert B., Zoppi N., De Backer J., Fox J.E., Mancini G.M., Kambouris M., Gardella R. Mutations in the facilitative glucose transporter GLUT10 alter angiogenesis and cause arterial tortuosity syndrome. Nat. Genet. 2006;38:452–457. doi: 10.1038/ng1764. [DOI] [PubMed] [Google Scholar]
- 25.Wessels M.W., Catsman-Berrevoets C.E., Mancini G.M., Breuning M.H., Hoogeboom J.J., Stroink H., Frohn-Mulder I., Coucke P.J., Paepe A.D., Niermeijer M.F. Three new families with arterial tortuosity syndrome. Am. J. Med. Genet. A. 2004;131:134–143. doi: 10.1002/ajmg.a.30272. [DOI] [PubMed] [Google Scholar]
- 26.Callewaert B.L., Willaert A., Kerstjens-Frederikse W.S., De Backer J., Devriendt K., Albrecht B., Ramos-Arroyo M.A., Doco-Fenzy M., Hennekam R.C., Pyeritz R.E. Arterial tortusity syndrome: Clinical and molecular findings in 12 newly identified families. Hum. Mutat. 2008;29:150–158. doi: 10.1002/humu.20623. [DOI] [PubMed] [Google Scholar]
- 27.Diagnostic and Statistical Manual of Mental Disorders DSM-IV-TR Fourth Edition . The American Psychiatry Association; Washington, DC: 2000. Text Revision. [Google Scholar]
- 28.Beaulieu C. The basis of anisotropic water diffusion in the nervous system - a technical review. NMR Biomed. 2002;15:435–455. doi: 10.1002/nbm.782. [DOI] [PubMed] [Google Scholar]
- 29.Cosottini M., Giannelli M., Siciliano G., Lazzaroti G., Michelassi M.C., Del Corona A., Bartolozzi C., Murri L. Diffusion-tensor MR imaging of corticospinal tract in amyotrophic lateral sclerosis and progressive muscular atrophy. Radiology. 2005;237:258–264. doi: 10.1148/radiol.2371041506. [DOI] [PubMed] [Google Scholar]
- 30.Mori S., Zhang J. Principles of diffusion tensor imaging and its applications to basic neuroscience research. Neuron. 2006;51:527–539. doi: 10.1016/j.neuron.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 31.Glenn O.A., Ludeman N.A., Berman J.I., Wu Y.W., Lu Y., Bartha A.I., Vigneron D.B., Chung S.W., Ferriero D.M., Barkovich A.J. Diffusion tensor MR imaging tractography of the pyramidal tracts correlates with clinical motor function in children with congenital hemiparesis. AJNR Am. J. Neuroradiol. 2007;28:1796–1802. doi: 10.3174/ajnr.A0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guye M. Imaging structural and functional connectivity: Towards a unified definition of human brain organization? Curr. Opin. Neurol. 2008;21:393–403. doi: 10.1097/WCO.0b013e3283065cfb. [DOI] [PubMed] [Google Scholar]
- 33.Qiu D., Tan L.H., Zhou K., Khong P.L. Diffusion tensor imaging of normal white matter maturation from late childhood to young adulthood: Voxel-wise evaluation of mean diffusivity, fractional anisotropy, radial and axial diffusivities, and correlation with reading development. Neuroimage. 2008;41:223–232. doi: 10.1016/j.neuroimage.2008.02.023. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y.T., Zhang C.Y., Zhang J., Li W. Age related changes of normal adult brain structure analysed with diffusion tensor imaging. Chin. Med. J. (Engl.) 2005;118:1059–1065. [PubMed] [Google Scholar]
- 35.Pfefferbaum A., Adalsteinsson E., Sullivan E.V. Replicability of diffusion tensor imaging measurements of fractional anisotropy and trace in brain. J. Magn. Reson. Imaging. 2003;18:427–433. doi: 10.1002/jmri.10377. [DOI] [PubMed] [Google Scholar]
- 36.Zhang M.Q. Statistical features of human exons and their flanking regions. Hum. Mol. Genet. 1998;7:919–932. doi: 10.1093/hmg/7.5.919. [DOI] [PubMed] [Google Scholar]
- 37.Collins J.S., Schwartz C.E. Detecting polymorphisms and mutations in candidate genes. Am. J. Hum. Genet. 2002;71:1251–1252. doi: 10.1086/344344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yap C.C., Murate M., Kishigami S., Muto Y., Kishida H., Hashikawa T., Yano R. Adaptor protein complex-4 (AP-4) is expressed in the central nervous system neurons and interacts with glutamate receptor delta2. Mol. Cell. Neurosci. 2003;24:283–295. doi: 10.1016/s1044-7431(03)00164-7. [DOI] [PubMed] [Google Scholar]
- 39.Takayama C., Nakagawa S., Watanabe M., Mishina M., Inoue Y. Developmental changes in expression and distribution of the glutamate receptor channel delta 2 subunit according to the Purkinje cell maturation. Brain Res. Dev. Brain Res. 1996;92:147–155. doi: 10.1016/0165-3806(95)00212-x. [DOI] [PubMed] [Google Scholar]
- 40.Matsuda S., Matsuda K., Yuzaki M. A new motif necessary and sufficient for stable localization of the delta2 glutamate receptors at postsynaptic spines. J. Biol. Chem. 2006;281:17501–17509. doi: 10.1074/jbc.M600240200. [DOI] [PubMed] [Google Scholar]
- 41.Kakegawa W., Miyazaki T., Emi K., Matsuda K., Kohda K., Motohashi J., Mishina M., Kawahara S., Watanabe M., Yuzaki M. Differential regulation of synaptic plasticity and cerebellar motor learning by the C-terminal PDZ-binding motif of GluRdelta2. J. Neurosci. 2008;28:1460–1468. doi: 10.1523/JNEUROSCI.2553-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Landsend A.S., Amiry-Moghaddam M., Matsubara A., Bergersen L., Usami S., Wenthold R.J., Ottersen O.P. Differential localization of delta glutamate receptors in the rat cerebellum: Coexpression with AMPA receptors in parallel fiber-spine synapses and absence from climbing fiber-spine synapses. J. Neurosci. 1997;17:834–842. doi: 10.1523/JNEUROSCI.17-02-00834.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao H.M., Wenthold R.J., Wang Y.X., Petralia R.S. Delta-glutamate receptors are differentially distributed at parallel and climbing fiber synapses on Purkinje cells. J. Neurochem. 1997;68:1041–1052. doi: 10.1046/j.1471-4159.1997.68031041.x. [DOI] [PubMed] [Google Scholar]
- 44.Yuzaki M. The delta2 glutamate receptor: 10 years later. Neurosci. Res. 2003;46:11–22. doi: 10.1016/s0168-0102(03)00036-1. [DOI] [PubMed] [Google Scholar]
- 45.Depienne C., Stevanin G., Brice A., Durr A. Hereditary spastic paraplegias: An update. Curr. Opin. Neurol. 2007;20:674–680. doi: 10.1097/WCO.0b013e3282f190ba. [DOI] [PubMed] [Google Scholar]
- 46.Crosby A.H. Disruption of cellular transport: A common cause of neurodegeneration? Lancet Neurol. 2003;2:311–316. doi: 10.1016/s1474-4422(03)00383-1. [DOI] [PubMed] [Google Scholar]
- 47.Irwin S.A., Idupulapati M., Gilbert M.E., Harris J.B., Chakravarti A.B., Rogers E.J., Crisostomo R.A., Larsen B.P., Mehta A., Alcantara C.J. Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am. J. Med. Genet. 2002;111:140–146. doi: 10.1002/ajmg.10500. [DOI] [PubMed] [Google Scholar]
- 48.Armstrong D.D., Dunn K., Antalffy B. Decreased dendritic branching in frontal, motor and limbic cortex in Rett syndrome compared with trisomy 21. J. Neuropathol. Exp. Neurol. 1998;57:1013–1017. doi: 10.1097/00005072-199811000-00003. [DOI] [PubMed] [Google Scholar]
- 49.Matsuda S., Yuzaki M. Mutation in hotfoot-4J mice results in retention of δ2 glutamate receptors in ER. Eur. J. Neurosci. 2002;16:1507–1516. doi: 10.1046/j.1460-9568.2002.02219.x. [DOI] [PubMed] [Google Scholar]
- 50.Ohno H., Stewart J., Founier M.C., Bosshart H., Rhee I., Miyatake S., Saito T., Gallusser A., Kirchhausen T., Bonifacino J.S. Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science. 1995;269:1872–1875. doi: 10.1126/science.7569928. [DOI] [PubMed] [Google Scholar]
- 51.Owen D.J., Evans P.R. A structural explanation for the recognition of tyrosine-based endocytotoc signals. Science. 1998;282:1327–1332. doi: 10.1126/science.282.5392.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tarpey P.S., Stevens C., Teague J., Edkins S., O'Meara S., Avis T., Barthorpe S., Buck G., Butler A., Cole J. Mutations in the gene encoding the sigma 2 subunit of the adaptor protein 1 complex, AP1S1, cause X-linked mental retardation. Am. J. Hum. Genet. 2006;79:1119–1124. doi: 10.1086/510137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saillour Y., Zanni G., Des Portes V., Heron D., Guibaud L., Iba-Zizen M.T., Pedespan J.L., Poirier K., Castelnau L., Julien C. Mutations in the AP1S2 gene encoding the sigma 2 subunit of the adaptor protein 1 complex AP1S2 are associated with syndromic X-linked mental retardation with hydrocephalus and calcifications in basal ganglia. J. Med. Genet. 2007;44:739–744. doi: 10.1136/jmg.2007.051334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huizing M., Scher C.D., Strove E., Fitzpatrick D.L., Hartnell L.M., Anikster Y., Gahl W.A. Nonsense mutations in ADTB3A cause complete deficiency of the beta3A subunit of adaptor complex-3 and severe Hermansky-Pudlak syndrome type 2. Pediatr. Res. 2002;51:150–158. doi: 10.1203/00006450-200202000-00006. [DOI] [PubMed] [Google Scholar]
- 55.Basser P.J., Pierpaoli C. Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J. Magn. Reson. B. 1996;111:209–219. doi: 10.1006/jmrb.1996.0086. [DOI] [PubMed] [Google Scholar]
- 56.Song S.K., Sun S.W., Ramsbottom M.J., Chang C., Russel J., Cross A.H. Dysmyelination revealed through MRI as increased radial (but unchanged axial) diffusion of water. Neuroimage. 2002;17:1429–1436. doi: 10.1006/nimg.2002.1267. [DOI] [PubMed] [Google Scholar]
- 57.Sun S.W., Liang H.F., Trinkaus K., Cross A.H., Armstrong R.C., Song S.K. Noninvasive detection of cuprizone induced axonal damage and demyelination in the mouse corpus callosum. Magn. Reson. Med. 2006;55:302–308. doi: 10.1002/mrm.20774. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.