

Abstract
Apoptosis is a potent immune barrier against viral infection, and many viruses, including poxviruses, encode proteins to overcome this defense. Interestingly, the avipoxviruses, which include fowlpox and canarypox virus, are the only poxviruses known to encode proteins with obvious Bcl-2 sequence homology. We previously characterized the fowlpox virus protein FPV039 as a Bcl-2-like antiapoptotic protein that inhibits apoptosis by interacting with and inactivating the proapoptotic cellular protein Bak. However, both Bak and Bax can independently trigger cell death. Thus, to effectively inhibit apoptosis, a number of viruses also inhibit Bax. Here we show that FPV039 inhibited apoptosis induced by Bax overexpression and prevented both the conformational activation of Bax and the subsequent formation of Bax oligomers at the mitochondria, two critical steps in the induction of apoptosis. Additionally, FPV039 interacted with activated Bax in the context of Bax overexpression and virus infection. Importantly, the ability of FPV039 to interact with active Bax and inhibit Bax activity was dependent on the structurally conserved BH3 domain of FPV039, even though this domain possesses little sequence homology to other BH3 domains. FPV039 also inhibited apoptosis induced by the BH3-only proteins, upstream activators of Bak and Bax, despite interacting detectably with only two: BimL and Bik. Collectively, our data suggest that FPV039 inhibits apoptosis by sequestering and inactivating multiple proapoptotic Bcl-2 proteins, including certain BH3-only proteins and both of the critical “gatekeepers” of apoptosis, Bak and Bax.
Apoptosis is a highly conserved form of programmed cell death that plays an important role in the immune defense against pathogens. The controlled and deliberate destruction of virally infected cells comprises a potent innate immune barrier against rampant viral replication and infection. As such, many viruses, including poxviruses, encode numerous proteins that inhibit a variety of steps in the biochemical pathways that lead to cell death (29, 69).
The mitochondria, and the Bcl-2 family of proteins that preside over them, serve as an important control point in the regulation of apoptosis (87). United by the presence of one to four highly conserved Bcl-2 homology (BH) domains, the Bcl-2 family regulates the integrity of the outer mitochondrial membrane (OMM) and controls the release of apoptogenic molecules from the mitochondrial intermembrane space. Bak and Bax, the two proapoptotic Bcl-2 proteins, possess BH domains 1 to 3 and, upon activation, commit the cell to death (53, 77). Whereas Bak resides constitutively at the OMM, Bax exists in an inactive form in the cytoplasm and, upon apoptotic insult, undergoes a conformational change that exposes its C-terminal transmembrane domain and results in its relocalization to the OMM (10, 34, 41, 56). The attendant exposure of the N termini of both Bak and Bax precedes Bak and Bax homooligomerization, which facilitates mitochondrial damage and, ultimately, the release of cytochrome c (3, 4, 36, 37, 76). Cytochrome c, in turn, triggers the activation of caspases, a group of cysteine proteases responsible for dismantling the apoptotic cell (59). Bak and Bax are therefore crucial for the induction of apoptosis and, because either Bak or Bax alone is sufficient to facilitate the release of cytochrome c, both must be inactivated to effectively inhibit apoptosis (53, 77, 90). The activation of Bak and Bax is counteracted by the antiapoptotic members of the Bcl-2 family, including Bcl-2, Bcl-XL, and Mcl-1. These three proteins, which possess all four BH domains, reside at the mitochondria and prevent apoptosis by directly interacting with and inhibiting Bak and Bax or the BH3-only proteins (87). The BH3-only proteins, which possess only the BH3 domain, act as sentinels responsive to a variety of cellular stresses, including virus infection (79). Upon receipt of an apoptotic stimulus, BH3-only proteins become activated and subsequently activate Bak and Bax or inhibit the antiapoptotic function of Bcl-2, Bcl-XL, and Mcl-1 (15). Of the eight BH3-only proteins that are directly involved in the induction of apoptosis—namely, Bim, Bid, Puma, Bik, Bmf, Bad, Noxa, and Hrk—each displays a specific and characteristic ability to bind and inhibit Bcl-2 proteins (79).
Like cellular antiapoptotic Bcl-2 proteins, viral inhibitors of apoptosis have evolved especially to interfere with the activation of Bak and Bax (18, 40). For example, E1B 19K, encoded by adenovirus, and M11L, encoded by myxoma virus, bind and inactivate both Bak and Bax to inhibit apoptosis (26, 49, 65, 67, 72). Similarly, ORF125, the antiapoptotic protein encoded by the poxvirus Orf virus, also inactivates Bak and Bax, but exactly how ORF125 mediates this inactivation remains unknown (78). Although interacting with Bak and Bax is ostensibly the most direct way to prevent apoptosis, several viral antiapoptotic proteins appear to inhibit apoptosis by functioning upstream of Bak and Bax at the level of the BH3-only proteins. The vaccinia virus protein F1L, for example, interacts with Bak but not Bax, yet F1L is nonetheless capable of inactivating Bax, likely a result of F1L interacting with the BH3-only protein and Bax activator, Bim (61, 70, 74). Moreover, the Bcl-2 homolog encoded by Kaposi's sarcoma-associated herpesvirus, and BHRF-1, encoded by Epstein-Barr virus, each interact with a specific and distinct array of BH3-only proteins, yet neither protein interacts detectably with Bak or Bax (14, 27, 44). Thus, to effectively inhibit apoptosis, it may not be necessary for viral proteins to directly target Bak and Bax but, instead, to prevent the activation of Bak and Bax by interfering with the upstream BH3-only proteins (15).
Recently, our lab has shown that FPV039, encoded by fowlpox virus, localizes to the mitochondria, where it inhibits apoptosis induced by a variety of stimuli (6). Interestingly, FPV039 is the only characterized poxvirus protein that shares obvious, albeit limited, sequence homology with cellular Bcl-2 proteins (1, 6). FPV039 possesses a highly conserved BH1 and BH2 domain but lacks an obvious BH3 and BH4 domain. Importantly, however, we predicted structural homology between the Bcl-2 BH3 domain and a corresponding region in FPV039, and we validated the prediction by showing that this cryptic FPV039 BH3 domain is functionally important (6). Indeed, the ability of FPV039 to interact with the proapoptotic protein Bak is dependent on this cryptic BH3 domain (6). Thus, despite lacking sequence conservation of a highly conserved BH3 domain, FPV039 is able to interact with, and inactivate, the proapoptotic protein Bak. Nevertheless, to completely inhibit apoptosis, both Bak and Bax must be inactivated.
Accordingly, we wanted to determine whether FPV039, in addition to inactivating Bak, could inactivate Bax. We report here that FPV039 inhibited Bax activity and prevented critical steps in Bax activation. FPV039 did not appear to interact with endogenous inactive Bax; however, FPV039 was able to interact with active Bax. Moreover, FPV039 inhibited apoptosis induced by the BH3-only proteins despite interacting with only BimL and Bik. Together, these data strongly suggest FPV039 inhibits apoptosis by inactivating multiple proapoptotic Bcl-2 proteins, including the critical Bak and Bax, as well as a discrete subset of BH3-only proteins.
MATERIALS AND METHODS
Cells and viruses.
HeLa, HEK 293T, HuTK−-143B, baby green monkey kidney (BGMK), and wild-type Jurkat cells were obtained from the ATCC and maintained as previously described (6, 74, 82). Jurkat cells overexpressing Bcl-2 (Jurkat Bcl-2) were generated and maintained as previously described (7). Bak- and Bax-deficient Jurkat cells (Jurkat Bak−/−/Bax−/−) were generously provided by H. Rabinowich (University of Pittsburgh School of Medicine, Pittsburgh, PA) (73). Bak−/− BMK cells were provided by E. White (Rutgers University, Piscataway, NJ). Vaccinia virus (VV) strain Copenhagen expressing the enhanced green fluorescent protein, VV-EGFP, was provided by G. McFadden (University of Florida, Gainesville). VV strain Copenhagen lacking the F1L open reading frame, VVΔF1L (74, 75), and VV strain Copenhagen lacking the F1L open reading frame but expressing Flag-tagged FPV039(1-176), VVΔF1L-Flag-FPV039(1-176) (6), were generated as described previously. Viruses were propagated in BGMK cells and isolated as previously described (64).
Plasmids.
N1L was amplified by PCR from vaccinia virus DNA using the forward primer 5′-GGTACCATGAGGACTCTACTTATT-3′ containing a KpnI restriction site and the reverse primer 5′-GGATCCTTATTTTTCACCATATAGATC-3′ containing a BamHI restriction site. N1L PCR product was cloned into pGemT vector (Promega) and subcloned into pEGFP-C1 to generate an N-terminally tagged EGFP-N1L. To generate pcDNA3-HA-Puma, Puma was excised from pCEP4-HA-Puma (provided by B. Vogelstein, Johns Hopkins University, Baltimore, MD) using the restriction enzymes KpnI and BamHI and ligated into pcDNA3. pEGFP-Bcl-XL was generated from p(Bluescript)SK-Bcl-XL (provided by C. Bleackley, University of Alberta, Edmonton, AB, Canada) using the forward primer 5′-GAATTCATGTCTCAGAGCAACCGG-3′ containing an EcoRI restriction site and the reverse primer 5′-GGATCCTCATTTCCGACTGAAGAG-3′ containing a BamHI restriction site. pEGFP-Mcl-1 was generated by excising Mcl-1 from pCR3.1-Mcl-1 (provided by H. Rabinowich, University of Pittsburgh, Pittsburgh, PA) using the restriction enzyme EcoRI, followed by ligation into pEGFP-C3. pcDNA3-Bid-Flag was generated from pCMV5-Bid using the forward primer 5′-AAGCTTATGGACTGTGAGGTCAAC-3′ containing a HindIII restriction site and the reverse primer 5′-GGATCCTTACTTGTCGTCATCGTCTTTGTAGTCGTCCATCCCATTTCTGGC-3′ containing a BamHI restriction site. pcDNA3-tBid-Flag, comprising the caspase-8-cleaved fragment of Bid, was generated from pCMV5-Bid using the forward primer 5′-AAGCTTATGGGCAACCGCAGCAGCCAC-3′ containing a HindIII restriction site and the reverse primer 5′-GGATCCTTACTTGTCGTCATCGTCTTTGTAGTCGTCCATCCCATTTC TGGC-3′ containing a BamHI restriction site. pEGFP-FPV039(1-176), pEGFP-FPV039(1-94), pEGFP-FPV039(142-176), pEGFP-FPV039(Δ41-54), and pEGFP-F1L were generated as described elsewhere (6, 75). pcDNA3-HA-Bax and pEGFP-M11L were provided by G. McFadden (University of Florida, Gainesville); pcDNA3-Flag-BimL and pcDNA3-Bmf-T7 were provided by R. Davis (University of Massachusetts Medical School, Boston); pEGFP-Bcl-2 was provided by C. Bleackley (University of Alberta, Edmonton, AB, Canada); pcDNA3.1-Myc-Noxa was provided by D. Leaman (University of Toledo, Toledo, OH); pcDNA3-Myc-Bik was provided by E. White (Rutgers University, Piscataway, NJ); pXJ40-HA-Bad was provided by S. Baksh (University of Alberta, Edmonton, AB, Canada).
Generation of VV-Flag-F1L and VVΔF1L-FPV039(Δ41-54).
A Flag-tagged version of FPV039(Δ41-54) was generated via PCR from EGFP-FPV039(Δ41-54) using the forward primer 5′-GTCGACATGGACTACAAAGACGATGACGACAAGGCTAGTAGTAATATGAAA-3′ containing a SalI restriction site and Flag tag and the reverse primer 5′-GCGGCCGCTTACATATAAAAGGAACATAT-3′ containing a NotI restriction site. Flag-FPV039(Δ41-54) was subcloned into the pSC66 vector, which places the construct under the control of a poxviral promoter (23). To generate the recombinant VV strain Copenhagen expressing Flag-F1L, CV-1 cells were infected at a multiplicity of infection (MOI) of 0.05 with VV Copenhagen and transfected with 5 μg of pSC66-Flag-F1L using Lipofectin (Invitrogen Life Technologies). To generate the recombinant VV strain Copenhagen devoid of F1L but expressing Flag-FPV039(Δ41-54), BGMK cells were infected at an MOI of 0.05 with VVΔF1L and transfected with 5 μg linearized pSC66-Flag-FPV039(Δ41-54) using Lipofectin. Recombinant viruses were selected by growth on HuTK−-143B cells in the presence of 5′-bromodeoxyuridine (Sigma-Aldrich) and plaque purified using 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (Rose Scientific Ltd.) to visualize recombinant viruses (23). The presence of F1L or FPV039(Δ41-54) was confirmed by PCR, and protein expression was confirmed by Western blotting using the anti-Flag M2 antibody (Sigma-Aldrich).
Measurement of mitochondrial membrane potential.
Changes in the mitochondrial membrane potential were quantified by staining with tetramethylrhodamine ethyl ester (TMRE; Invitrogen Life Technologies) (24, 55). For the Bax killing assay, 1 × 106 HeLa cells were transfected with pEGFP-C3, pEGFP-Bcl-2, pEGFP-FPV039(1-176), or pEGFP-FPV039(142-176) using Lipofectamine 2000, and apoptosis was induced by transfecting cells with 0.75 μg of pcDNA3-HA-Bax for 18 h. For the BH3-only killing assays, 1 × 106 HeLa cells were transfected with pEGFP-C3, pEGFP-Bcl-2, pEGFP-Bcl-XL, pEGFP-Mcl-1, or pEGFP-FPV039(1-176) using Lipofectamine 2000, and apoptosis was induced by transfecting cells with 1 μg of pcDNA3-Flag-BimL, pcDNA3-Myc-Bik, pcDNA3-T7-Bmf, pXJ40-HA-Bad, pcDNA3-HA-Puma, pcDNA3-Bid-Flag, pcDNA3-tBid-Flag, or pcDNA3.1-Noxa. Following transfection, cells were stained with 0.2 μM TMRE (24, 55). Cells were analyzed by two-color flow cytometry (FACScan; Becton Dickinson), with TMRE fluorescence measured through the FL-2 channel equipped with a 585-nm filter (42-nm band pass) and EGFP fluorescence measured through the FL-1 channel equipped with a 489-nm filter (42-nm band pass). Data were acquired on 20,000 cells per sample with fluorescence signals at logarithmic gain, and analysis was performed using CellQuest software. The percentage of killing was calculated as the number of EGFP+ TMRE− cells divided by the total number of EGFP+ cells, and standard deviations were generated from three independent experiments.
Confocal microscopy.
HeLa cells (5 × 105) were infected with VV-EGFP, VVΔF1L, VVΔF1L-Flag-FPV039(1-176), or VVΔF1L-Flag-FPV039(Δ41-54) at an MOI of 10 for 24 h. Cells were subsequently fixed in 4% paraformaldehyde, permeabilized with 0.04% saponin, and blocked with phosphate-buffered saline plus 1% fetal bovine serum. Bax activation was detected using the anti-Bax6A7 antibody (1:500; BD Biosciences) and the anti-mouse Alexa 546 (1:400) secondary antibody (Molecular Probes). Cells were analyzed with an LSM510 laser scanning confocal microscope at 543 nm to detect Alexa 546 and at 489 nm to detect EGFP fluorescence. Confocal results were quantified as the percentage of cells displaying anti-Bax6A7 positivity over the total number of cells and given as the mean ± the standard deviation. At least 700 cells were counted per sample, and the standard deviations were calculated from at least 18 different fields of view.
Conformational analysis of Bax by flow cytometry.
Jurkat cells (1 × 106), Jurkat Bcl-2 cells, or Jurkat Bak−/−/Bax−/− cells were infected with VV-EGFP, VVΔF1L, VVΔF1L-Flag-FPV039(1-176), or VVΔF1L-Flag-FPV039(Δ41-54) for 6 h at an MOI of 5. Following infection, cells were exposed to 2 μM staurosporine (Sigma-Aldrich) for 2 h and then fixed in 0.25% paraformaldehyde. Cells were permeabilized with 500 μg/ml digitonin and stained with 2 μg/ml anti-Bax6A7 antibody (BD Biosciences) (43) or 2 μg/ml of an isotype control antibody specific for NK1.1 (PK136; provided by K. Kane, University of Alberta, Edmonton, AB, Canada) (47) and counterstained with phycoerythrin-conjugated anti-mouse antibody (Jackson ImmunoResearch Laboratories Inc.). Antibody staining was analyzed by flow cytometry (FACScan; Becton Dickinson) with fluorescence measured through the FL-2 channel equipped with a 585-nm filter (42-nm band pass). Data were acquired on 20,000 cells per sample with fluorescence signals at logarithmic gain, and analysis was performed using CellQuest software.
Bax cross-linking.
HEK293T cells (1 × 106) were transfected with pEGFP, pEGFP-FPV039(1-176), pEGFP-FPV039(Δ41-54), pEGFP-FPV039(1-94), or pEGFP-Bcl-2, and pcDNA-HA-Bax. zVAD.fmk (50 μM; Kamiya Biomedical Company) was added following transfection to prevent the downstream activation of caspases. Cells were lysed in lysis buffer containing 2% 3-(3-cholamidopropyl)-dimethylammonio-1-propanesulfonate (CHAPS; Sigma-Aldrich), 150 mM NaCl, 50 mM Tris, pH 8.0, and supplemented with EDTA-free proteinase inhibitor (Roche Diagnostics). Following lysis, nuclei and membranes were spun down, and the supernatant was incubated with 1 μM 1,6-bismaleimidohexane (BMH; Thermo Scientific) dissolved in dimethyl sulfoxide for 30 min. Supernatants were acetone precipitated, and cross-linking was quenched by the addition of sodium dodecyl sulfate (SDS) loading buffer containing 100 mM 2-mercaptoethanol. Protein samples were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed by Western blotting with an anti-hemagglutinin (HA) antibody.
Immunoprecipitations and immunoblotting.
HEK293T cells or Bak−/− BMK cells (1 × 106) were transfected using Lipofectamine 2000 and the following plasmids: pEGFP-C3, pEGFP-F1L, pEGFP-FPV039(1-176), pEGFP-FPV039(Δ41-54), pEGFP-FPV039(1-94), pEGFP-M11L, pEGFP-Bcl-2, pEGFP-Bcl-XL, pEGFP-Mcl-1, or pcDNA3-HA-Bax, and one of pcDNA3-Flag-BimL, pcDNA3-Myc-Bik, pcDNA3-Bmf-T7, pXJ40-HA-Bad, pcDNA3-HA-Puma, pcDNA3-Bid-Flag, pcDNA3-tBid-Flag, or pcDNA3.1-Noxa. zVAD.fmk (50 μM; Kamiya Biomedical Company) was added following transfection to prevent the downstream activation of caspases. Cells were lysed in 2% CHAPS lysis buffer, followed by precipitation using goat anti-EGFP antibody (provided by Luc Berthiaume, University of Alberta, Edmonton, Alberta, Canada). Similar experiments were performed in the context of viral infection. HeLa cells (7 × 106) were infected with VVΔF1L, VV-Flag-F1L, or VVΔF1L-Flag-FPV039(1-176) at an MOI of 5 for 16 h. Cells were lysed in 2% CHAPS (Sigma-Aldrich), followed by immunoprecipitation using rabbit anti-Flag M2 (Sigma-Aldrich) or mouse anti-Bax6A7 (BD Biosciences).
To assess Bax activation by immunoprecipitation, 7 × 106 HeLa cells were infected with VV-EGFP, VVΔF1L, VVΔF1L-Flag-FPV039(1-176), or VVΔF1L-Flag-FPV039(Δ41-54) at an MOI of 10 for 24 h. Cells were lysed in 2% CHAPS lysis buffer or 1% Triton X-100 lysis buffer (1% Triton X-100, 150 mM NaCl, 50 mM Tris, pH 8.0, EDTA-free proteinase inhibitor [Roche Diagnostics]) followed by precipitation using mouse anti-Bax6A7 (BD Biosciences).
Cell lysates were subjected to SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. The following antibodies were used for detection: mouse anti-EGFP antibody (1:5,000; Cedarlane Laboratories Ltd.), rabbit anti-BaxN20 (1:500; Santa Cruz), mouse anti-Bax2D2 (1:10,000; Trevigen), mouse anti-Flag HRP (1:2,000; Sigma-Aldrich), mouse anti-Myc (clone 9E10; 1:2,500), mouse anti-T7 Tag (1:5,000; Novagen), mouse anti-HA antibody (clone 12CA5; 1:4,000; Roche Diagnostics), and mouse anti-Noxa (1:300; Alexis Biochemicals). Proteins were visualized using enhanced chemiluminescence according to the manufacturer's directions (GE Healthcare).
RESULTS
FPV039 inhibits apoptosis induced by Bax.
Permeabilization of the outer mitochondrial membrane and the concomitant progression of apoptosis can be induced independently by either Bak or Bax (16, 53, 58, 77). Because we have shown previously that FPV039 inhibits apoptosis induced by overexpression of Bak (6), we wanted to determine if FPV039 could likewise inhibit apoptosis induced by Bax overexpression. HeLa cells were transfected with pEGFP, pEGFP-FPV039(1-176) (which expresses full-length FPV039), pEGFP-FPV039(142-176) (which expresses only the C-terminal transmembrane tail of FPV039), or pEGFP-Bcl-2, the prototypical cellular antiapoptotic protein, and apoptosis was triggered by expression of HA-Bax. Apoptosis was then quantified in transfected (EGFP-positive) cells by measuring the fluorescence intensity of TMRE, a fluorescent dye taken up exclusively by healthy mitochondria with an intact membrane potential (24, 55). Overexpression of Bax resulted in the artificial activation of Bax (46, 84) and subsequent loss of the mitochondrial membrane potential in 45% of cells expressing EGFP, indicating that EGFP is not able to protect cells from apoptosis (Fig. 1A). However, when EGFP-Bcl-2 was overexpressed along with Bax, apoptosis was significantly inhibited (Fig. 1A). Similarly, full-length EGFP-FPV039(1-176) also inhibited Bax-induced apoptosis, with only 10% of transfected cells exhibiting a loss in mitochondrial membrane potential (Fig. 1A). Conversely, EGFP-FPV039(142-176), the transmembrane tail of FPV039, did not protect against Bax-induced apoptosis, with more than 50% of transfected cells displaying a loss in mitochondrial membrane potential (Fig. 1A).
FIG. 1.
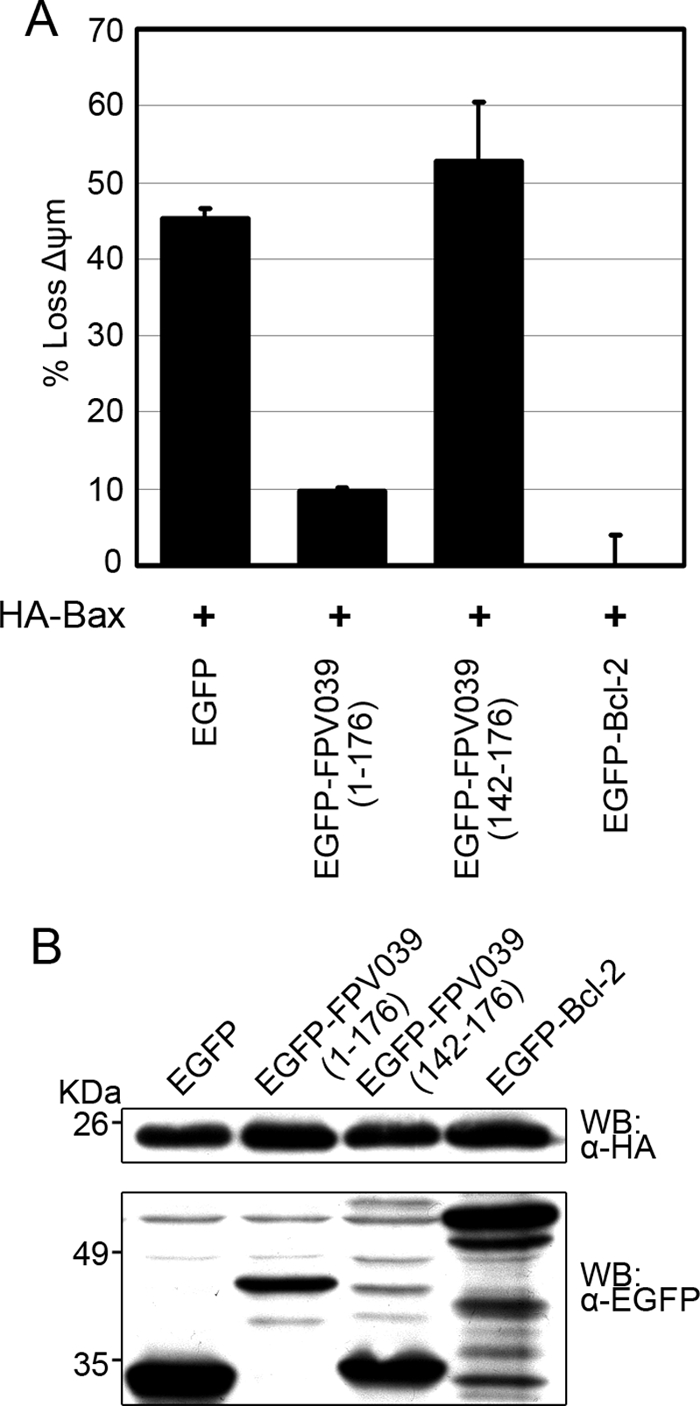
FPV039 inhibits Bax-induced apoptosis. (A) HeLa cells were cotransfected with either pEGFP, pEGFP-FPV039(1-176), pEGFP-FPV039(142-176), or pEGFP-Bcl-2 and pcDNA3-HA-Bax. Eighteen hours posttransfection, apoptosis was quantified by TMRE fluorescence in EGFP-positive cells using two-color flow cytometry. Standard deviations were calculated from three independent experiments. (B) Expression levels of the various transfected proteins. HeLa cells were transfected as above, and protein levels were determined by Western blotting (WB) of whole-cell lysates with either anti-EGFP or anti-HA antibodies.
To ensure that differences in the expression levels of either the EGFP-tagged proteins or HA-Bax were not influencing the results of this assay, we analyzed protein expression by Western blotting. EGFP-Bcl-2 was expressed at a slightly higher level than EGFP-FPV039(1-176), and this correlated with their respective abilities to inhibit apoptosis. EGFP and EGFP-FPV039(142-176) were robustly expressed; however, neither was able to inhibit apoptosis. In each case, HA-Bax was expressed to equally high levels (Fig. 1B). Together these data indicate that, in addition to inhibiting apoptosis induced by the proapoptotic protein Bak, FPV039 also inhibits apoptosis induced by the overexpression of Bax.
FPV039 inhibits the conformational activation of Bax.
Prior to the loss of mitochondrial membrane potential and the induction of apoptosis, Bax undergoes a series of conformational changes that result in its activation. In addition to the exposure of the C-terminal transmembrane domain of Bax, Bax reveals an N-terminal epitope that can be specifically detected by the antibody anti-Bax6A7 (43). Detection of this N-terminal epitope, therefore, serves as a marker for Bax activation and impending apoptosis (43). Given that FPV039 inhibited apoptosis induced by Bax overexpression, we sought to determine if FPV039 also prevented the activation of Bax. HeLa cells were infected with one of various recombinant VV at an MOI of 10, and Bax activation was visualized by confocal microscopy using the conformation-specific antibody anti-Bax6A7. Because each recombinant virus also expressed EGFP, green fluorescence served as a marker of infection. HeLa cells infected with VVΔF1L, a VV devoid of the antiapoptotic protein F1L, induced Bax activation (Fig. 2A, panels d to f), whereas both VV-EGFP, a wild-type VV, and VVΔF1L-Flag-FPV039(1-176), a VV devoid of F1L but expressing wild-type FPV039, significantly inhibited Bax activation (Fig. 2A, panels a to c and g to i). VVΔF1L-Flag-FPV039(Δ41-54), which expresses a mutated form of FPV039 lacking the putative BH3 domain, was unable to prevent Bax activation (Fig. 2A, panels j to l). These results were quantified by counting cells positive for anti-Bax 6A7 staining (Fig. 2B). To confirm the ability of FPV039 to inhibit Bax activation, we infected HeLa cells with the same panel of recombinant viruses and immunoprecipitated activated Bax from cell lysates using anti-Bax6A7. When lysed in 2% CHAPS, a detergent that does not artificially alter Bax conformation (42, 43), activated Bax was precipitated detectably from cells infected with VVΔF1L or VVΔF1L-Flag-FPV039(Δ41-54) and not from cells infected with VV-EGFP or VVΔF1L-Flag-FPV039(1-176), confirming the ability of both F1L and FPV039 to inhibit Bax activation (Fig. 2C). Conversely, when cells were lysed in 1% Triton X-100, a detergent that artificially activates Bax (42, 43), Bax was precipitated from all infected cells, validating the ability of anti-Bax6A7 to detect activated Bax (Fig. 2C). Additionally, the confocal and immunoprecipitation experiments were repeated using VVΔF1L-Flag-FPV039(1-94), a proapoptotic virus that expresses a truncated form of FPV039 lacking the BH2 and transmembrane domain (6). This virus, like VVΔF1L and VVΔF1L-Flag-FPV039(Δ41-54), was unable to inhibit Bax activation (data not shown).
FIG. 2.

FPV039 inhibits Bax activity. (A) HeLa cells were infected with VV-EGFP, VVΔF1L, VVΔF1L-Flag-FPV039(1-176), or VVΔF1L-Flag-FPV039(Δ41-54) at an MOI of 10 for 24 h. Following fixation and permeabilization, Bax activation was visualized with anti-Bax6A7. (B) Microscopy analysis was quantified as the percentage of cells displaying Bax activation (mean ± SD). (C) HeLa cells were infected as for panel A. Cells were lysed using either 2% CHAPS or 1% Triton X-100, and activated Bax was immunoprecipitated (IP) with anti-Bax6A7 and Western blotted with anti-BaxN20.
To confirm the ability of FPV039 to inhibit Bax activation we used a flow cytometry-based assay to precisely detect and quantify Bax conformational change. Jurkat cells were infected with VV-EGFP, VVΔF1L, VVΔF1L-Flag-FPV039(1-176), or VVΔF1L-Flag-FPV039(Δ41-54), and the activation of Bax was induced by treatment with staurosporine (STS), a potent apoptotic stimulus (68). Activation-associated exposure of the Bax N terminus was detected by staining with anti-Bax6A7 and quantified by flow cytometry. Jurkat cells mock infected and left untreated displayed a basal level of fluorescence attributable to nonspecific antibody binding (Fig. 3a). Upon STS treatment of mock-infected cells, however, fluorescence intensity increased, indicating that Bax became activated and underwent a conformational change exposing its N terminus to binding by the anti-Bax6A7 antibody (Fig. 3a). Conversely, cells infected with VV-EGFP completely inhibited the activation of Bax upon STS treatment due to the endogenous expression of F1L (Fig. 3b). This phenomenon was dependent on the presence of F1L, because cells infected with VVΔF1L were not protected from STS-induced Bax activation (Fig. 3c). Importantly, and in agreement with the previous experiments (Fig. 2), cells infected with VVΔF1L-Flag-FPV039(1-176) were resistant to Bax activation induced by STS (Fig. 3d), whereas cells infected with VVΔF1L-Flag-FPV039(Δ41-54) were not (Fig. 3e). Similarly, VVΔF1L-Flag-FPV039(1-94) was unable to prevent STS-induced Bax activation (data not shown). No increase in fluorescence intensity was observed in Jurkat cells stained with an isotype control antibody, confirming the specificity of anti-Bax6A7 for conformationally active Bax (Fig. 3f). Moreover, Jurkat cells deficient in both Bak and Bax did not exhibit an increase in fluorescence after infection or STS treatment, again confirming the specificity of our assay for Bax (Fig. 3g to k). Jurkat cells overexpressing antiapoptotic Bcl-2 were completely resistant to Bax activation (Fig. 3l to p). Together, these data indicate that FPV039, like both F1L and Bcl-2, is capable of inhibiting the activation-associated exposure of the Bax N terminus during virus infection and in response to potent apoptotic stimuli (Fig. 2 and 3) (42, 70).
FIG. 3.

FPV039 inhibits the conformational activation of Bax. Wild-type Jurkat cells (a to f), Jurkat cells devoid of Bak and Bax (g to k), or Jurkat cells overexpressing Bcl-2 (l to p) were infected with VV-EGFP, VVΔF1L, VVΔF1L-Flag-FPV039(1-176), or VVΔF1L-Flag-FPV039(Δ41-54) at an MOI of 5 for 6 h and then treated with 2 μM STS for 2 h to induce apoptosis. Bax activation was monitored by flow cytometry using anti-Bax6A7 or the isotype control antibody (NK1.1). Shaded histograms, untreated cells; open histograms, staurosporine-treated cells. Data are representative of at least three independent experiments.
FPV039 inhibits Bax oligomerization.
Subsequent to the conformational changes that result in Bax activation, Bax forms high-molecular-weight oligomers on the surface of the OMM that facilitate the loss of the mitochondrial membrane potential and the release of proapoptogenic cytochrome c (2-4). The formation of Bax oligomers represents the penultimate step in the induction of apoptosis by Bax. Because FPV039 inhibited the conformational activation of Bax, we wanted to determine whether FPV039 could also inhibit the consequent and climactic formation of Bax oligomers. To this end, HEK293T cells were transfected with a panel of EGFP-tagged FPV039 constructs and HA-Bax, followed by lysis with 2% CHAPS and treatment with BMH, a chemical cross-linker that irreversibly conjugates proteins at sulfhydryl groups (63). In this way, Bax oligomers could be preserved and visualized by SDS-PAGE and Western blotting (Fig. 4). Overexpression of HA-Bax alone, which is sufficient to induce apoptosis (Fig. 1), resulted in the formation of an approximately 44-kDa Bax dimer and a 66-kDa Bax trimer, both represented on Western blotting by distinct bands two and three times the size of monomeric Bax, present as a 22-kDa band. Additionally, higher-order oligomers of Bax were represented by the high-molecular-weight bands visible above 85 kDa (Fig. 4, lane 2). EGFP alone, as expected, did not prevent the formation of Bax oligomers (Fig. 4, lane 3). Importantly, EGFP-FPV039(1-176), like EGFP-Bcl-2, prevented the oligomerization of Bax (Fig. 4, lanes 4 and 7), whereas EGFP-FPV039(Δ41-54), which lacks the putative BH3 domain, and EGFP-FPV039(1-94), which lacks the BH2 and transmembrane domains, were unable to prevent Bax oligomerization (Fig. 4, lanes 5 and 6). Moreover, Western blotting of the lysates before the addition of BMH revealed that HA-Bax was expressed at similar levels in each case, as were all FPV039 constructs (Fig. 4, bottom two panels). Thus, in addition to inhibiting the conformational activation of Bax, FPV039 also inhibited Bax oligomerization, the penultimate step in the induction of apoptosis by Bax.
FIG. 4.
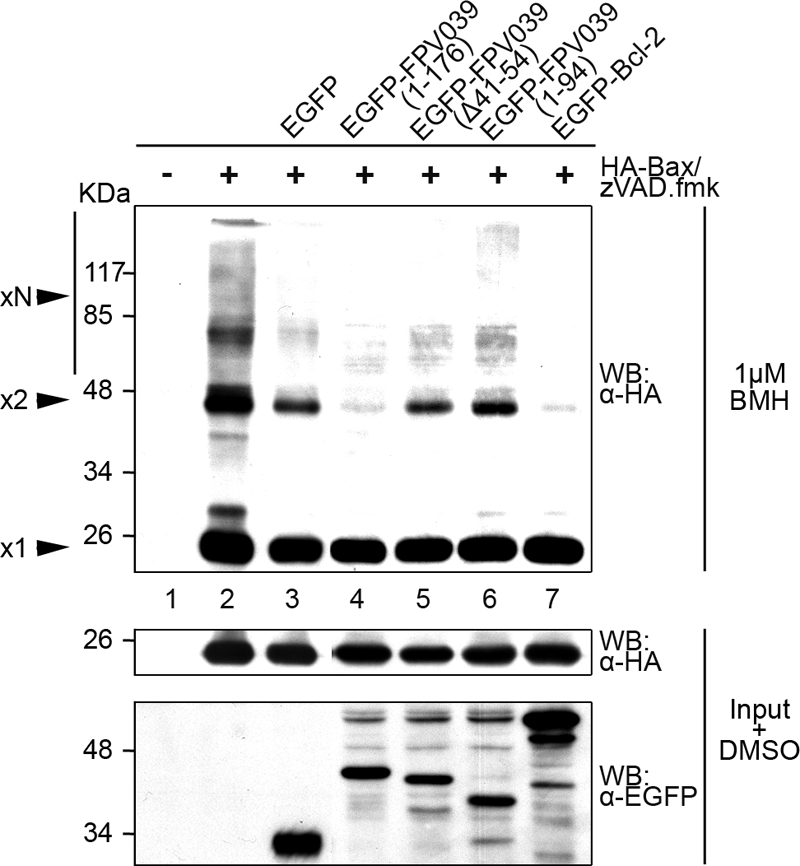
FPV039 inhibits Bax oligomerization. HeLa cells were cotransfected with pEGFP, pEGFP-FPV039(1-176), pEGFP-FPV039(Δ41-54), pEGFP-FPV039(1-94), or pEGFP-Bcl-2 and pcDNA3-HA-Bax. zVAD.fmk was added following transfection to prevent the activation of caspases. Cells were lysed in 2% CHAPS, and cell lysates were treated with 1 μM BMH and Western blotted (WB) with anti-HA to detect Bax oligomers (upper panel). A portion of the lysates (input) were treated with dimethyl sulfoxide alone and subjected to Western blotting with either anti-HA or anti-EGFP antibodies to determine expression levels of the transfected proteins (lower two panels). x1, Bax monomer; x2, Bax dimer; xN, higher-order Bax oligomers.
FPV039 interacts with Bax.
The ability of FPV039 to inhibit Bax activation and prevent Bax-induced apoptosis implied that FPV039 might interact with Bax. To determine whether FPV039 interacted with Bax, HEK293T cells were transfected with pEGFP, pEGFP-FPV039(1-176), pEGFP-FPV039(Δ41-54), pEGFP-FPV039(1-94), or pEGFP-Bcl-2 and HA-Bax, followed by lysis in 2% CHAPS. Complexes were immunoprecipitated with an anti-GFP antibody and Western blotted with an anti-Bax antibody. Using this approach, an interaction between EGFP-FPV039(1-176) and HA-Bax was detected (Fig. 5A). Importantly, FPV039 lacking the cryptic BH3 domain, EGFP-FPV039(Δ41-54), or the BH2 and transmembrane domains, EGFP-FPV039(1-94), was unable to interact with Bax, suggesting that the interaction depended on specific functional domains of FPV039 (Fig. 5A). As expected, EGFP-Bcl-2 interacted with Bax, serving as a positive control in this experiment (Fig. 5A) (86, 88). No Bax was precipitated in cells expressing HA-Bax alone or coexpressing HA-Bax along with EGFP, further confirming the specificity of the interaction between FPV039 and Bax. Although each EGFP-tagged protein was precipitated equally (Fig. 5A, second panel), lysates were also Western blotted to ensure that all proteins were expressed at comparable levels. Notably, HA-Bax was similarly expressed in each case, as were all FPV039 constructs (Fig. 5A, bottom two panels). These data provide convincing evidence that FPV039 interacts with Bax and that this interaction is dependent on Bcl-2 functional domains, including the cryptic BH3 domain.
FIG. 5.
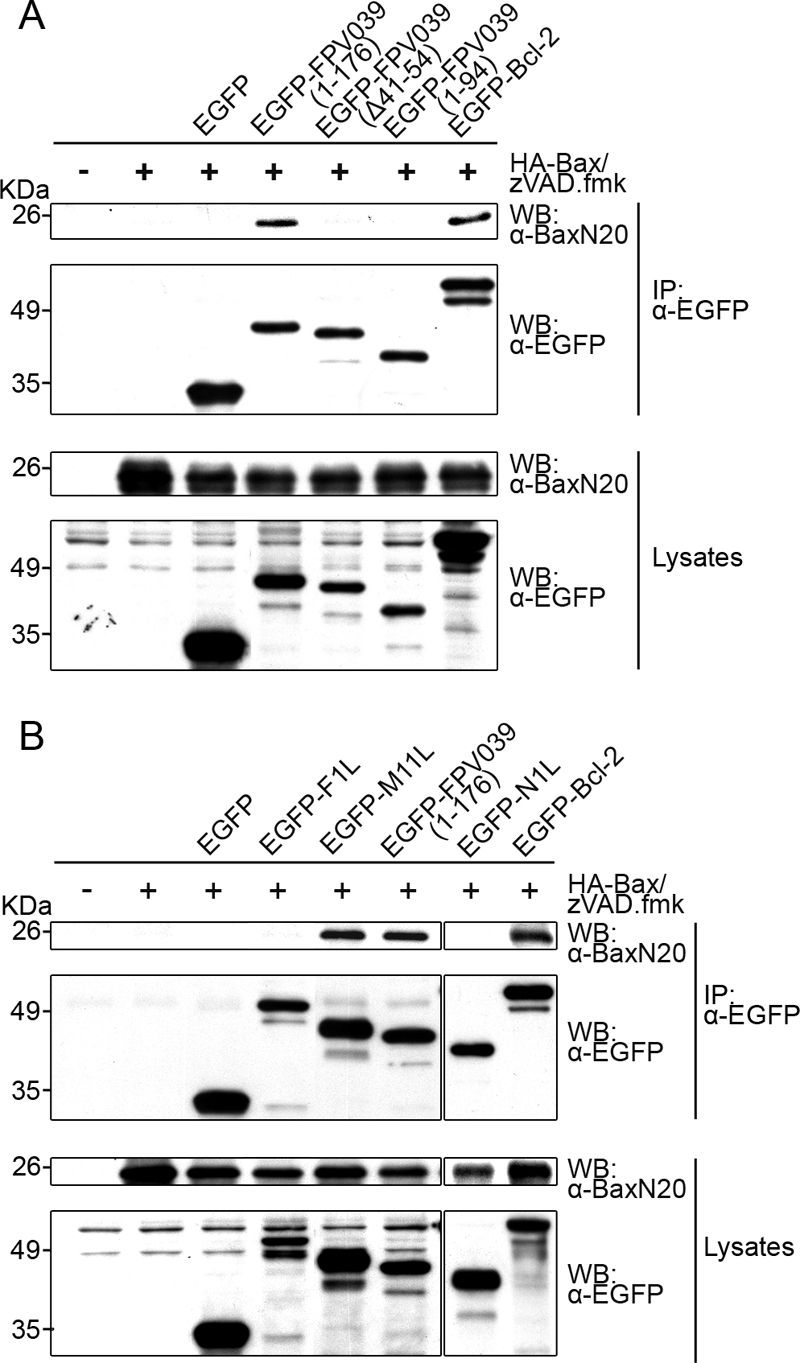
FPV039 interacts with Bax. (A) HEK293T cells were cotransfected with pEGFP, pEGFP-FPV039(1-176), pEGFP-FPV039(Δ41-54), pEGFP-FPV039(1-94), or pEGFP-Bcl-2 and pcDNA3-HA-Bax, followed by lysis with 2% CHAPS. Cell lysates were immunoprecipitated (IP) with anti-EGFP and Western blotted (WB) with either anti-BaxN20 or anti-EGFP to detect interaction. Whole-cell lysates were Western blotted with anti-BaxN20 or anti-EGFP to determine the expression levels of HA-Bax or the EGFP-tagged proteins, respectively. (B) HEK293T cells were cotransfected with pEGFP, pEGFP-F1L, pEGFP-M11L, pEGFP-FPV039(1-176), pEGFP-N1L, or pEGFP-Bcl-2 and pcDNA3-HA-Bax. Immunoprecipitations and Western blotting was performed as described for panel A. zVAD.fmk was added following transfection to prevent the activation of caspases.
In an effort to compare FPV039 with other poxviral inhibitors of apoptosis, we performed a second coimmunoprecipitation experiment and included EGFP-F1L or EGFP-N1L, both antiapoptotic proteins encoded by vaccinia virus, and EGFP-M11L, the antiapoptotic protein encoded by myxoma virus (Fig. 5B). As previously shown, M11L, like FPV039, exhibited an interaction with Bax, whereas F1L failed to interact with Bax (Fig. 5B) (49, 65, 70). Contrary to previously published results (17), N1L did not exhibit an interaction with Bax in this assay (Fig. 5B).
We excluded the possibility that FPV039 was interacting with Bax through an interaction with Bak by repeating the immunoprecipitations in Bak-deficient baby mouse kidney cells. Cells were transfected with pEGFP, pEGFP-Bcl-2, or pEGFP-FPV039(1-176) and cotransfected with pcDNA-HA-Bax. Following lysis and immunoprecipitation, Western blot analysis revealed that FPV039, like Bcl-2, was able to interact with Bax even in the absence of Bak (Fig. 6). These data collectively suggest that FPV039 interacts with Bax, a property not necessarily shared among other poxvirus inhibitors of apoptosis like F1L and N1L.
FIG. 6.
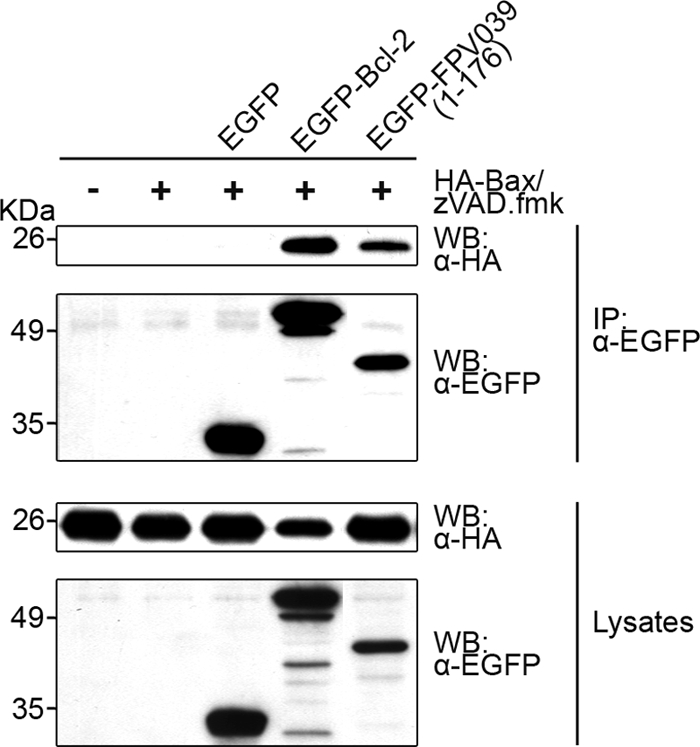
FPV039 retains an interaction with Bax in the absence of Bak. Bak−/− BMK cells were cotransfected with pEGFP, pEGFP-Bcl-2, or pEGFP-FPV039(1-176) and pcDNA3-HA-Bax, followed by lysis with 2% CHAPS. Cell lysates were immunoprecipitated (IP) with anti-EGFP and Western blotted (WB) with either anti-HA or anti-EGFP to detect interaction. Whole-cell lysates were Western blotted with anti-HA or anti-EGFP to determine the expression levels. zVAD.fmk was added following transfection to prevent the activation of caspases.
FPV039 interacts with endogenous activated Bax.
During transient transfection, Bax is overexpressed and, consequently, activated. Although we demonstrated that FPV039 interacted with Bax in the context of overexpression, we wanted to determine if FPV039 could also interact with endogenous Bax in the context of virus infection, when the majority of Bax is apparently inactive (Fig. 2 and 3). To address this question, HeLa cells were infected with VVΔF1L, VV-Flag-F1L, which expresses a Flag-tagged version of the antiapoptotic protein F1L, or VVΔF1L-Flag-FPV039(1-176), which expresses full-length Flag-FPV039. Cell lysates were first immunoprecipitated with an anti-Flag antibody and Western blotted with an anti-Bax antibody to detect an interaction. Despite robust precipitation and expression of FPV039, an interaction between FPV039 and endogenous Bax was not observed in this experiment (Fig. 7A). Similarly, F1L also failed to interact with endogenous Bax during virus infection, as previously shown (Fig. 7A) (70). Our inability to detect Bax by precipitating FPV039 suggested that the expression of FPV039 potently prevented Bax activation (Fig. 1 to 4) and therefore preempted an interaction between FPV039 and active Bax at the mitochondria. However, we wondered whether FPV039 could interact with the minute levels of endogenous, active Bax that might be present during infection. To specifically precipitate any activated Bax that might be present, we performed a reciprocal immunoprecipitation this time using anti-Bax6A7. As expected, the amount of immunoprecipitated active Bax varied among the infected samples in this experiment, with the most Bax precipitated from cells infected with the apoptotic virus VVΔF1L and dramatically less Bax precipitated from cells infected with either of the antiapoptotic viruses, VV-Flag-F1L and VVΔF1L-Flag-FPV039 (Fig. 7B, second panel). Precipitation of active Bax resulted in the coprecipitation of FPV039 (Fig. 7B), indicating, along with Fig. 5 and 6, that FPV039 interacted with endogenous active Bax. Precipitation of active Bax also coprecipitated F1L (Fig. 7B), albeit to a lesser extent, which agrees with the observed ability of F1L to interact with artificially activated Bax in the presence of the detergent Triton X-100 (70). Although we were able to detect Flag-FPV039 by precipitating active Bax (Fig. 7 B), we were unable to detect Bax by precipitating Flag-FPV039 (Fig. 7A). Since FPV039 is a potent inhibitor of Bax activation (Fig. 1 to 4), the vast majority of Bax within a cell expressing FPV039 is inactive and presumably unable to interact with FPV039. Thus, the small minority of activated Bax that interacts with FPV039 (such as might be observed in Fig. 7A) is likely below the level of detection that can be achieved by Western blotting using our antibodies. Together, these data suggest that FPV039, like Bcl-2 and Bcl-XL (9, 21, 89), is capable of interacting with activated Bax.
FIG. 7.

FPV039 interacts with active Bax in the context of virus infection. HeLa cells were mock infected or infected at an MOI of 5 with VVΔF1L, VV-Flag-F1L, or VVΔF1L-Flag-FPV039(1-176). Sixteen hours postinfection, cells were lysed in 2% CHAPS, and cell lysates were immunoprecipitated (IP) with either anti-Flag (A) or anti-Bax6A7 (B) and Western blotted (WB) with either anti-Flag or anti-Bax2D2 (A and B). Lysates were Western blotted with anti-Bax2D2 and anti-Flag (A) or anti-BaxN20 and anti-Flag (B). *, Flag-F1L (B).
FPV039 inhibits apoptosis induced by BH3-only proteins.
The BH3-only proteins are proapoptotic members of the Bcl-2 family that induce apoptosis by facilitating the activation of both Bak and Bax (15). Given that FPV039 is a potent inhibitor of apoptosis, we wondered whether FPV039 was also capable of inhibiting cell death induced by the overexpression of BH3-only proteins. To this end, HeLa cells were transfected with pEGFP, pEGFP-FPV039(1-176), or pEGFP-Bcl-2, pEGFP-Bcl-XL, or pEGFP-Mcl-1 and cotransfected with a plasmid encoding one of eight BH3-only proteins. Apoptosis was then quantified in transfected (EGFP-positive) cells by measuring the fluorescence intensity of TMRE. EGFP alone was unable to prevent apoptosis induced by the overexpression of BimL, Bik, Bmf, or Bad, each of which induced apoptosis in over 40% of the cells. Conversely, coexpression of either EGFP-Bcl-2 or EGFP-FPV039(1-176) reduced apoptosis to less than 1% (Fig. 8A to D). Similarly, full-length Bid and active tBid induced apoptosis in approximately 50% of cells coexpressing EGFP, and this was reduced to less than 10% upon expression of EGFP-Bcl-XL or EGFP-FPV039(1-176) (Fig. 8F and G). Overexpression of Puma and Noxa induced apoptosis in only 26% and 14% of cells, respectively, but this was reduced to less than 5% upon coexpression of EGFP-Bcl-2, EGFP-Mcl-1, or EGFP-FPV039(1-176) (Fig. 8E and H). Lysates of transfected cells showed that, in each case, BH3-only proteins were being expressed (Fig. 8). Together these data indicate that FPV039 is able to inhibit apoptosis induced by the overexpression of BimL, Bik, Bmf, Bad, Puma, Bid, tBid, and Noxa.
FIG. 8.

FPV039 inhibits apoptosis induced by BH3-only proteins. HeLa cells were cotransfected with pEGFP and pEGFP-FPV039(1-176) along with one of the following BH3-only proteins and the relevant cellular Bcl-2 protein: pcDNA3-Flag-BimL and pEGFP-Bcl-2 (A), pcDNA3-Myc-Bik and pEGFP-Bcl-2 (B), pcDNA3-T7-Bmf and pEGFP-Bcl-2 (C), pXJ40-HA-Bad and pEGFP-Bcl-2 (D), pcDNA3-HA-Puma and pEGFP-Bcl-2 (E), pcDNA3-Bid-Flag and pEGFP-Bcl-XL (F), pcDNA3-tBid-Flag and pEGFP-Bcl-XL (G), or pcDNA3.1-Myc-Noxa and pEGFP-Mcl-1 (H). Eighteen hours posttransfection, apoptosis was quantified by TMRE fluorescence in EGFP-positive cells using two-color flow cytometry. Standard deviations were calculated from three independent experiments. Whole-cell lysates from parallel experiments were Western blotted with the following antibodies to determine expression levels of the BH3-only proteins: anti-Flag (A, F, and G), anti-Myc (B), anti-T7 (C), anti-HA (D and E), anti-Noxa (H). *, complete protection by EGFP-Bcl-2 or EGFP-Bcl-XL.
FPV039 interacts with a subset of BH3-only proteins.
Given the ability of FPV039 to inhibit apoptosis induced by all eight BH3-only proteins tested, we next sought to determine whether FPV039 could also interact with BH3-only proteins. One BH3-only protein in particular, Bim, is thought to directly activate Bax and has been previously shown by our lab to be required for vaccinia virus-induced apoptosis (70). To determine if FPV039 was capable of interacting with Bim, HEK293T cells were transfected with pEGFP, pEGFP-F1L, pEGFP-M11L, pEGFP-FPV039(1-176), or pEGFP-Bcl-2 and cotransfected with pcDNA3-BimL. Following lysis in 2% CHAPS, EGFP was precipitated using an anti-EGFP antibody, and Western blotting was subsequently performed using both anti-Flag and anti-EGFP antibodies. As expected, EGFP-Bcl-2 exhibited an interaction with BimL, whereas EGFP alone did not (Fig. 9) (57). Interestingly, EGFP-FPV039(1-176) also precipitated BimL, suggesting that, like Bcl-2, FPV039 was capable of interacting with BimL (Fig. 9). Additionally, EGFP-M11L, which has been previously shown to interact with BimEL (49), also interacted with BimL, as did EGFP-F1L, as previously shown (70) (Fig. 9). Each EGFP-tagged protein was precipitated at equal levels (Fig. 9, second panel), and Western blotting of the lysates confirmed that the EGFP-tagged proteins, as well as BimL, were expressed at comparable levels in each case (Fig. 9, third and fourth panels).
FIG. 9.
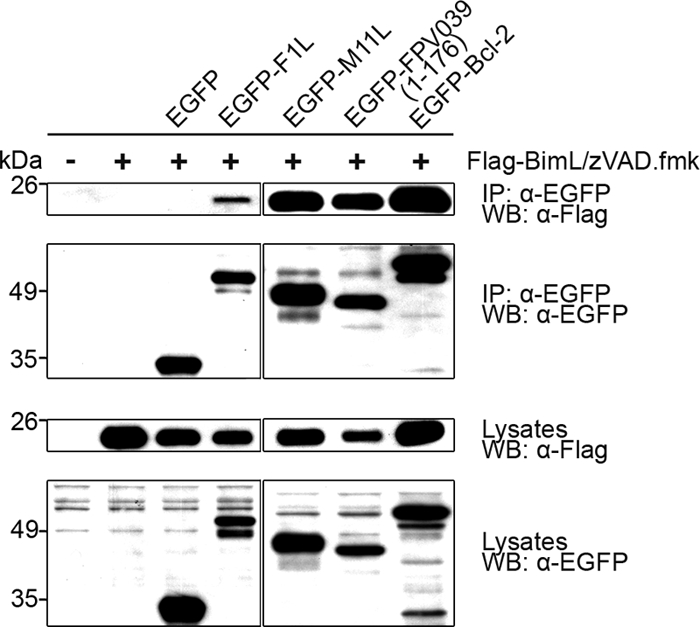
FPV039 interacts with BimL. HeLa cells were transfected with pEGFP, pEGFP-F1L, pEGFP-M11L, pEGFP-FPV039(1-176), or pEGFP-Bcl-2 and pcDNA3-Flag-BimL, followed by lysis with 2% CHAPS. Cell lysates were immunoprecipitated (IP) with anti-EGFP and Western blotted (WB) with either anti-Flag or anti-EGFP to detect interaction. Whole-cell lysates were Western blotted with anti-Flag or anti-EGFP to determine expression levels. zVAD.fmk was added following transfection to prevent the activation of caspases.
We next examined the ability of FPV039 to interact with seven other BH3-only proteins. Using a similar transfection and immunoprecipitation procedure as described for BimL, we confirmed the interaction between each BH3-only protein and the appropriate EGFP-tagged, antiapoptotic Bcl-2 family protein (Fig. 10). As expected, Bik, Bmf, Bad, and Puma all coprecipitated with EGFP-Bcl-2 (Fig. 10A to D), whereas Bid and tBid coprecipitated with EGFP-Bcl-XL (Fig. 10E and F) and Noxa coprecipitated with Mcl-1 (Fig. 10G). Interestingly, only Bik coprecipitated with EGFP-FPV039(1-176) to significant levels, suggesting that Bik and FPV039 interacted (Fig. 10A). Bmf exhibited a slight interaction with FPV039; however, the reciprocal immunoprecipitation did not detect an interaction (Fig. 10B and data not shown). Similarly, Noxa also displayed a slight interaction with FPV039, but the intensity of this interaction appeared insignificant when compared with the interaction between Noxa and Mcl-1 (Fig. 10G). In each case, EGFP was precipitated at equal levels, and the lysates showed that expression of the EGFP-tagged proteins, as well as the BH3-only proteins, was comparable (Fig. 10A to G, bottom three panels). In the case of Bmf and tBid, where the lysate levels of the two BH3-only proteins appeared lower upon cotransfection of EGFP-FPV039, we performed the reciprocal immunoprecipitation and confirmed the absence of any interaction (Fig. 10B and F and data not shown). Thus, it is clear from these data that FPV039 is capable of interacting with only two BH3-only proteins: BimL and Bik.
FIG. 10.

FPV039 interacts with a subset of BH3-only proteins. HEK293T cells were cotransfected with pEGFP and pEGFP-FPV039(1-176) along with one of the following BH3-only proteins and the relevant cellular Bcl-2 protein: pcDNA3-Myc-Bik and pEGFP-Bcl-2 (A), pcDNA3-T7-Bmf and pEGFP-Bcl-2 (B), pXJ40-HA-Bad and pEGFP-Bcl-2 (C), pcDNA3-HA-Puma and pEGFP-Bcl-2 (D), pcDNA3-Bid-Flag and pEGFP-Bcl-XL (E), pcDNA3-tBid-Flag and pEGFP-Bcl-XL (F), pcDNA3.1-Myc-Noxa and pEGFP-Mcl-1 (G). zVAD.fmk was added following transfection to prevent the activation of caspases. Cells were lysed with 2% CHAPS and immunoprecipitated (IP) with anti-EGFP. IP samples and whole-cell lysates were Western blotted (WB) with anti-EGFP and anti-Myc (A), anti-T7 (B), anti-HA (C and D), anti-Flag (E and F), or anti-Noxa (G).
DISCUSSION
To prevent the premature suicide of an infected cell, many viruses encode proteins that specifically inhibit the activity of Bak and Bax, the two critical proapoptotic Bcl-2 family proteins (18, 40). Poxviruses, in particular, encode a variety of antiapoptotic proteins, the majority of which bear little sequence similarity to each other or the Bcl-2 proteins they inactivate (25, 75, 78). In fact, only the Avipoxvirus genus, of which fowlpox virus is the prototypical member, encode proteins with obvious sequence homology to Bcl-2 family members (1, 71). We have previously shown that FPV039, encoded by fowlpox virus, interacts constitutively with Bak and inhibits Bak activity to abrogate apoptosis (6); however, because Bak and Bax often function redundantly, both Bak and Bax must be inactivated to inhibit apoptosis (53, 77, 90). Here we have shown that FPV039 also inhibits Bax activation and Bax-induced apoptosis.
The induction of apoptosis by Bax is a complex and multistep process involving numerous conformational changes and the relocalization of Bax from the cytosol to the OMM. FPV039 inhibited apoptosis induced by Bax overexpression (Fig. 1) and prevented the formation of Bax oligomers (Fig. 4), which ultimately facilitate the release of cytochrome c and the commitment to apoptosis (2-4). Moreover, in the context of virus infection, FPV039 functionally replaced F1L, the endogenous inhibitor of apoptosis in vaccinia virus, and prevented the activation-associated exposure of the Bax N terminus (Fig. 2 and 3), a step that is thought to occur concomitantly with Bax relocalization to the OMM and prior to Bax oligomerization (10, 34, 41, 51, 56, 83). We also demonstrated that FPV039 interacted with Bax during transient transfection (Fig. 5), when Bax was overexpressed and activated. Additionally, FPV039 interacted with endogenous Bax during virus infection, but only when active Bax was specifically precipitated (Fig. 7), suggesting that FPV039 interacts with active but not inactive Bax. Further, FPV039 inhibited apoptosis induced by the upstream activators of Bax and Bak, the BH3-only proteins, and interacted with at least two of them, BimL and Bik.
The ability of FPV039 to interact with active Bax and inhibit Bax activation was dependent on a structural element homologous to the BH3 domain α-helix of Bcl-2 (6). Although FPV039 possesses a highly conserved BH1 and BH2 domain, FPV039 lacks obvious BH3 and BH4 domains. Nonetheless, FPV039 is predicted to be comprised of eight α-helices, each of which closely corresponds to the α-helices in Bcl-2 and one of which corresponds precisely with the α2-helix that makes up the Bcl-2 BH3 domain (1, 6). Deleting this putative BH3 domain abrogated the ability of FPV039 to inhibit Bax activation (Fig. 2, 3, and 4) and prevented FPV039 from interacting with both Bak (6) and Bax (Fig. 5). In cellular antiapoptotic Bcl-2 proteins, the BH3, BH2, and BH1 domains form a hydrophobic pocket that binds the amphipathic BH3 α-helix of the proapoptotic Bcl-2 family members, thus forming the basis of the interactions that occur among this family (60). Despite the lack of sequence homology in the FPV039 BH3 domain, this domain was critical to the function of FPV039, which, along with the apparent structural homology, strongly suggest that this domain contributes to the formation of a hydrophobic pocket involved in interacting with proapoptotic Bcl-2 proteins. Curiously, like FPV039, most other viral Bcl-2 (vBcl-2) homologues lack a conserved BH3 domain sequence (18, 40), although the evolutionary advantage, if any, of such an adaptation is unclear.
Indeed, the structure of vBcl-2 proteins appears to play a critical and interesting role in their ability to function. It has recently been shown that the antiapoptotic proteins F1L and M11L, which lack obvious sequence homology with each other or cellular Bcl-2 proteins, both share striking structural similarity to Bcl-2 family members (22, 49, 50). Like FPV039, both F1L and M11L interact constitutively with Bak (6, 61, 72, 74), and all three proteins appear to interact with activated Bax (65, 70) (Fig. 5 and 7). Another recently characterized vaccinia virus protein, N1L, also possesses structural homology to cellular Bcl-2 proteins; however, contrary to what has been previously shown, we were unable to detect an interaction between Bax and N1L (5, 17) (Fig. 5B). It may be that N1L along with B14 and A52, two other vaccinia virus proteins that structurally resemble Bcl-2 family members, inhibit NF-κB instead of apoptosis (20, 35, 45). Regardless, the structure of these vBcl-2 proteins, including FPV039, is certainly important to their ability to interact with and inhibit the proapoptotic Bcl-2 proteins.
Direct targeting of Bax and Bak is not the only strategy employed by viruses to inhibit apoptosis. Many vBcl-2 proteins modulate the apoptotic cascade by interfering instead with the BH3-only proteins, the upstream activators of Bak and Bax. We demonstrated that FPV039 inhibited apoptosis induced by the overexpression of eight BH3-only proteins (Fig. 8) and interacted detectably with two of them, BimL and Bik (Fig. 9 and 10). Bim has been implicated in the direct activation of Bax (11, 30, 39, 48), and the importance of BimL as an activator of Bax during virus infection is underscored by the ability of F1L to inhibit Bax activation by inactivating BimL (70). Likewise, Bik has also been shown to trigger the activation of Bax (31, 32). Besides F1L and FPV039, several other vBcl-2 proteins have been shown to interact with one or more of the BH3-only proteins. Here we show that M11L interacted with BimL (Fig. 9), and it has been previously shown that M11L interacts strongly with BimEL but not at all or weakly with other BH3-only proteins (49). Similarly, A179L, encoded by African swine fever virus, and E1B19K, encoded by adenovirus, interact with Bak and Bax as well as certain BH3-only proteins to inhibit apoptosis (28, 38, 66, 85). Still other vBcl-2 proteins inhibit apoptosis apparently via interactions exclusively with BH3-only proteins and not with Bak or Bax. For example, Kaposi's sarcoma-associated herpesvirus Bcl-2 and BHRF1, both encoded by herpesviruses, do not interact with Bak or Bax but instead with various BH3-only proteins (27). It is well known that cellular antiapoptotic Bcl-2 proteins each bind to a specific subset of BH3-only proteins (12, 13, 79-81). A similar specificity appears to govern vBcl-2 proteins, including FPV039; however, the BH3-only binding profile among each vBcl-2 protein differs considerably. Indeed, the determinants of, and the functional basis for, the disparate binding profiles of vBcl-2 proteins remain to be determined. Nonetheless, the ability of viral proteins to inhibit cell death is clearly not confined to their ability to interact with the two gatekeepers of apoptosis, Bak and Bax: inactivating the upstream BH3-only proteins may be an equally effective strategy.
Taken together, our data suggest a model for FPV039-mediated inhibition of apoptosis that involves both indirect and direct inactivation of Bak and Bax, the two critical proapoptotic Bcl-2 proteins. The ability of FPV039 to interact with Bim and Bik, two BH3-only proteins known to activate Bax, suggests that FPV039 may counter apoptosis by inhibiting Bim and Bik and preventing the eventual activation of Bax. In particular, Bim appears to be a crucial activator of apoptosis in response to vaccinia virus infection (70), and several other vBcl-2 proteins, including F1L and M11L, demonstrate an interaction with Bim (Fig. 9), suggesting that the ability of FPV039 to interact robustly with BimL is physiologically relevant. Thus, it may be that by sequestering and inactivating certain BH3-only proteins, like BimL and Bik, FPV039 is able to indirectly inhibit Bax activation during virus infection. Conceivably, because inactive Bax resides in the cytoplasm (41, 83) and FPV039 localizes to the OMM (6), suppression of Bax activation by BH3-only proteins would keep Bax and FPV039 physically separated, explaining our inability to coprecipitate Bax when precipitating Flag-tagged FPV039 during virus infection (Fig. 7A). Indeed, such a phenomenon has been previously described for the cellular antiapoptotic proteins Bcl-2 and Bcl-XL. Both Bcl-2 and Bcl-XL interact with Bax only upon receipt of an apoptotic stimulus (9, 21, 89), when Bax becomes activated and translocates to the OMM, or in the context of detergents like Triton X-100 that artificially promote Bax heterodimerization (42, 43). Curiously, although FPV039 inhibited apoptosis induced by all eight BH3-only proteins, FPV039 only interacted with BimL and Bik. It is possible that sequestration of BimL and Bik is enough to inhibit Bax activation, but it is also possible that FPV039 interacted weakly or transiently with other BH3-only proteins and our system was not sensitive enough to detect those interactions. Intriguingly, however, the ability of FPV039 to interact with active Bax may be physiologically significant in conditions where (i) Bax becomes activated by BH3-only proteins not repressed by FPV039 or where (ii) Bax becomes activated by cellular factors that are independent of regulation by Bcl-2 family members (52). Indeed, Bcl-XL can sequester tBid to prevent Bax activation and, in cases where Bax becomes activated regardless, Bcl-XL can also sequester active Bax to prevent its subsequent oligomerization (9, 54). In the case of Bcl-XL, sequestration of both tBid and active Bax each contribute significantly to the inhibition of apoptosis (9). It therefore seems likely that FPV039 may inhibit the induction of apoptosis in a manner similar to Bcl-XL. Given the ability of FPV039 to interact with Bak (6), as well as active Bax and certain BH3-only proteins, it is evident that FPV039 targets multiple proapoptotic Bcl-2 family members to inhibit apoptosis.
It is interesting that FPV039, which interacts with chicken Bak (6), presumably evolved to counteract the avian members of the Bcl-2 family, yet FPV039 also inhibits apoptosis and interacts with human Bcl-2 proteins in human cells. Although this underscores the evolutionary relatedness of the apoptotic program, it is possible that FPV039 may exhibit slightly different binding profiles in avian cells. Unfortunately, only Bmf (33) and Bid (19) homologues have been characterized in chicken cells, and an incompletely annotated chicken genome coupled with a paucity of reagents have so far prevented a detailed examination of avian apoptosis.
FPV039, along with M11L and F1L, are powerful tools for dissecting the complex biochemical mechanisms that control cellular fate in all metazoans. These well-characterized viral inhibitors of apoptosis, which may share a common ancestor (6), have evolved to inhibit apoptosis in discrete yet functionally equivalent manners, and understanding precisely how they achieve this inhibition will help elucidate the role that apoptosis plays in infection and disease, including cancer. Furthermore, the recent interest in fowlpox virus as a vaccine vector and gene therapy tool (8, 62) necessitates the study of fowlpox viral immunomodulatory proteins like FPV039.
Acknowledgments
This research was supported by grants from the Canadian Institutes of Health Research, the Howard Hughes Medical Institute, and the Alberta Heritage Foundation for Medical Research (AHFMR). L.B. was supported by a Canada Graduate Doctoral Scholarship from the National Sciences and Engineering Council of Canada (NSERC-CGSD) and an AHFMR studentship. K.V. was supported by an AHFMR fellowship. S.C. was supported by an NSERC-CGSD. M.B. is a Senior Scholar of the Alberta Heritage Foundation for Medical Research, a Canadian Institutes for Medical Research New Investigator, and a Howard Hughes Medical Institute Scholar in Infection and Parasitology.
Footnotes
Published ahead of print on 13 May 2009.
REFERENCES
- 1.Afonso, C. L., E. R. Tulman, Z. Lu, L. Zsak, G. F. Kutish, and D. L. Rock. 2000. The genome of fowlpox virus. J. Virol. 743815-3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Annis, M. G., E. L. Soucie, P. J. Dlugosz, J. A. Cruz-Aguado, L. Z. Penn, B. Leber, and D. W. Andrews. 2005. Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J. 242096-2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antonsson, B., S. Montessuit, S. Lauper, R. Eskes, and J. C. Martinou. 2000. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem. J. 345271-278. [PMC free article] [PubMed] [Google Scholar]
- 4.Antonsson, B., S. Montessuit, B. Sanchez, and J. C. Martinou. 2001. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J. Biol. Chem. 27611615-11623. [DOI] [PubMed] [Google Scholar]
- 5.Aoyagi, M., D. Zhai, C. Jin, A. E. Aleshin, B. Stec, J. C. Reed, and R. C. Liddington. 2007. Vaccinia virus N1L protein resembles a B cell lymphoma-2 (Bcl-2) family protein. Protein Sci. 16118-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banadyga, L., J. Gerig, T. Stewart, and M. Barry. 2007. Fowlpox virus encodes a Bcl-2 homologue that protects cells from apoptotic death through interaction with the proapoptotic protein Bak. J. Virol. 8111032-11045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barry, M., J. A. Heibein, M. J. Pinkoski, S. F. Lee, R. W. Moyer, D. R. Green, and R. C. Bleackley. 2000. Granzyme B short-circuits the need for caspase 8 activity during granule-mediated cytotoxic T-lymphocyte killing by directly cleaving Bid. Mol. Cell. Biol. 203781-3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beukema, E. L., M. P. Brown, and J. D. Hayball. 2006. The potential role of fowlpox virus in rational vaccine design. Expert Rev. Vaccines 5565-577. [DOI] [PubMed] [Google Scholar]
- 9.Billen, L. P., C. L. Kokoski, J. F. Lovell, B. Leber, and D. W. Andrews. 2008. Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol. 6e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cartron, P. F., H. Arokium, L. Oliver, K. Meflah, S. Manon, and F. M. Vallette. 2005. Distinct domains control the addressing and the insertion of Bax into mitochondria. J. Biol. Chem. 28010587-10598. [DOI] [PubMed] [Google Scholar]
- 11.Cartron, P. F., T. Gallenne, G. Bougras, F. Gautier, F. Manero, P. Vusio, K. Meflah, F. M. Vallette, and P. Juin. 2004. The first alpha helix of Bax plays a necessary role in its ligand-induced activation by the BH3-only proteins Bid and PUMA. Mol. Cell 16807-818. [DOI] [PubMed] [Google Scholar]
- 12.Certo, M., V. Del Gaizo Moore, M. Nishino, G. Wei, S. Korsmeyer, S. A. Armstrong, and A. Letai. 2006. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9351-365. [DOI] [PubMed] [Google Scholar]
- 13.Chen, L., S. N. Willis, A. Wei, B. J. Smith, J. I. Fletcher, M. G. Hinds, P. M. Colman, C. L. Day, J. M. Adams, and D. C. Huang. 2005. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 17393-403. [DOI] [PubMed] [Google Scholar]
- 14.Cheng, E. H., J. Nicholas, D. S. Bellows, G. S. Hayward, H. G. Guo, M. S. Reitz, and J. M. Hardwick. 1997. A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc. Natl. Acad. Sci. USA 94690-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chipuk, J. E., and D. R. Green. 2008. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 18157-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chittenden, T., E. A. Harrington, R. O'Connor, C. Flemington, R. J. Lutz, G. I. Evan, and B. C. Guild. 1995. Induction of apoptosis by the Bcl-2 homologue Bak. Nature 374733-736. [DOI] [PubMed] [Google Scholar]
- 17.Cooray, S., M. W. Bahar, N. G. Abrescia, C. E. McVey, N. W. Bartlett, R. A. Chen, D. I. Stuart, J. M. Grimes, and G. L. Smith. 2007. Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J. Gen. Virol. 881656-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cuconati, A., and E. White. 2002. Viral homologs of BCL-2: role of apoptosis in the regulation of virus infection. Genes Dev. 162465-2478. [DOI] [PubMed] [Google Scholar]
- 19.Diaz-Gil, G., F. Gomez-Esquer, D. Agudo, J. Delcan, F. Martinez-Arribas, C. Rivas, J. Schneider, M. A. Palomar, and R. Linares. 2006. Characterization of a human Bid homologue protein from Gallus gallus. Gene 37226-32. [DOI] [PubMed] [Google Scholar]
- 20.DiPerna, G., J. Stack, A. G. Bowie, A. Boyd, G. Kotwal, Z. Zhang, S. Arvikar, E. Latz, K. A. Fitzgerald, and W. L. Marshall. 2004. Poxvirus protein N1L targets the I-κB kinase complex, inhibits signaling to NF-κB by the tumor necrosis factor superfamily of receptors, and inhibits NF-κB and IRF3 signaling by toll-like receptors. J. Biol. Chem. 27936570-36578. [DOI] [PubMed] [Google Scholar]
- 21.Dlugosz, P. J., L. P. Billen, M. G. Annis, W. Zhu, Z. Zhang, J. Lin, B. Leber, and D. W. Andrews. 2006. Bcl-2 changes conformation to inhibit Bax oligomerization. EMBO J. 252287-2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Douglas, A. E., K. D. Corbett, J. M. Berger, G. McFadden, and T. M. Handel. 2007. Structure of M11L: a myxoma virus structural homolog of the apoptosis inhibitor, Bcl-2. Protein Sci. 16695-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Earl, P. L., B. Moss, L. S. Wyatt, and M. W. Carroll. 1998. Generation of recombinant vaccinia viruses, p.16.17.1-16.17.19. In R. B. Frederick, M. Ausubel, Robert E. Kingston, David D. Moore, J. G. Seidman, John A. Smith, and Kevin Struhl (ed.), Current protocols in molecular biology. John Wiley and Sons, Inc., New York, NY.
- 24.Ehrenberg, B., V. Montana, M. D. Wei, J. P. Wuskell, and L. M. Loew. 1988. Membrane potential can be determined in individual cells from the Nernstian distribution of cationic dyes. Biophys. J. 53785-794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Everett, H., M. Barry, S. F. Lee, X. Sun, K. Graham, J. Stone, R. C. Bleackley, and G. McFadden. 2000. M11L: a novel mitochondria-localized protein of myxoma virus that blocks apoptosis of infected leukocytes. J. Exp. Med. 1911487-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farrow, S. N., J. H. White, I. Martinou, T. Raven, K. T. Pun, C. J. Grinham, J. C. Martinou, and R. Brown. 1995. Cloning of a bcl-2 homologue by interaction with adenovirus E1B 19K. Nature 374731-733. [DOI] [PubMed] [Google Scholar]
- 27.Flanagan, A. M., and A. Letai. 2008. BH3 domains define selective inhibitory interactions with BHRF-1 and KSHV BCL-2. Cell Death Differ. 15580-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galindo, I., B. Hernaez, G. Diaz-Gil, J. M. Escribano, and C. Alonso. 2008. A179L, a viral Bcl-2 homologue, targets the core Bcl-2 apoptotic machinery and its upstream BH3 activators with selective binding restrictions for Bid and Noxa. Virology 375561-572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galluzzi, L., C. Brenner, E. Morselli, Z. Touat, and G. Kroemer. 2008. Viral control of mitochondrial apoptosis. PLoS Pathog 4e1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gavathiotis, E., M. Suzuki, M. L. Davis, K. Pitter, G. H. Bird, S. G. Katz, H. C. Tu, H. Kim, E. H. Cheng, N. Tjandra, and L. D. Walensky. 2008. BAX activation is initiated at a novel interaction site. Nature 4551076-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gillissen, B., F. Essmann, V. Graupner, L. Starck, S. Radetzki, B. Dorken, K. Schulze-Osthoff, and P. T. Daniel. 2003. Induction of cell death by the BH3-only Bcl-2 homolog Nbk/Bik is mediated by an entirely Bax-dependent mitochondrial pathway. EMBO J. 223580-3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gillissen, B., F. Essmann, P. G. Hemmati, A. Richter, A. Richter, I. Oztop, G. Chinnadurai, B. Dorken, and P. T. Daniel. 2007. Mcl-1 determines the Bax dependency of Nbk/Bik-induced apoptosis. J. Cell Biol. 179701-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gomez-Esquer, F., M. A. Palomar, I. Rivas, J. Delcan, R. Linares, and G. Diaz-Gil. 2008. Characterization of the BH3 protein Bmf in Gallus gallus: identification of a novel chicken-specific isoform. Gene 40721-29. [DOI] [PubMed] [Google Scholar]
- 34.Goping, I. S., A. Gross, J. N. Lavoie, M. Nguyen, R. Jemmerson, K. Roth, S. J. Korsmeyer, and G. C. Shore. 1998. Regulated targeting of BAX to mitochondria. J. Cell Biol. 143207-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Graham, S. C., M. W. Bahar, S. Cooray, R. A. Chen, D. M. Whalen, N. G. Abrescia, D. Alderton, R. J. Owens, D. I. Stuart, G. L. Smith, and J. M. Grimes. 2008. Vaccinia virus proteins A52 and B14 Share a Bcl-2-like fold but have evolved to inhibit NF-κB rather than apoptosis. PLoS Pathog 4e1000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Griffiths, G. J., B. M. Corfe, P. Savory, S. Leech, M. D. Esposti, J. A. Hickman, and C. Dive. 2001. Cellular damage signals promote sequential changes at the N-terminus and BH-1 domain of the pro-apoptotic protein Bak. Oncogene 207668-7676. [DOI] [PubMed] [Google Scholar]
- 37.Griffiths, G. J., L. Dubrez, C. P. Morgan, N. A. Jones, J. Whitehouse, B. M. Corfe, C. Dive, and J. A. Hickman. 1999. Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J. Cell Biol. 144903-914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han, J., P. Sabbatini, and E. White. 1996. Induction of apoptosis by human Nbk/Bik, a BH3-containing protein that interacts with E1B 19K. Mol. Cell. Biol. 165857-5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harada, H., B. Quearry, A. Ruiz-Vela, and S. J. Korsmeyer. 2004. Survival factor-induced extracellular signal-regulated kinase phosphorylates BIM, inhibiting its association with BAX and proapoptotic activity. Proc. Natl. Acad. Sci. USA 10115313-15317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hardwick, J. M., and D. S. Bellows. 2003. Viral versus cellular BCL-2 proteins. Cell Death Differ 10(Suppl. 1)S68-S76. [DOI] [PubMed] [Google Scholar]
- 41.Hsu, Y. T., K. G. Wolter, and R. J. Youle. 1997. Cytosol-to-membrane redistribution of Bax and Bcl-XL during apoptosis. Proc. Natl. Acad. Sci. USA 943668-3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hsu, Y. T., and R. J. Youle. 1998. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J. Biol. Chem. 27310777-10783. [DOI] [PubMed] [Google Scholar]
- 43.Hsu, Y. T., and R. J. Youle. 1997. Nonionic detergents induce dimerization among members of the Bcl-2 family. J. Biol. Chem. 27213829-13834. [DOI] [PubMed] [Google Scholar]
- 44.Huang, Q., A. M. Petros, H. W. Virgin, S. W. Fesik, and E. T. Olejniczak. 2003. Solution structure of the BHRF1 protein from Epstein-Barr virus, a homolog of human Bcl-2. J. Mol. Biol. 3321123-1130. [DOI] [PubMed] [Google Scholar]
- 45.Jacobs, N., N. W. Bartlett, R. H. Clark, and G. L. Smith. 2008. Vaccinia virus lacking the Bcl-2-like protein N1 induces a stronger natural killer cell response to infection. J. Gen. Virol. 892877-2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kitanaka, C., T. Namiki, K. Noguchi, T. Mochizuki, S. Kagaya, S. Chi, A. Hayashi, A. Asai, Y. Tsujimoto, and Y. Kuchino. 1997. Caspase-dependent apoptosis of COS-7 cells induced by Bax overexpression: differential effects of Bcl-2 and Bcl-xL on Bax-induced caspase activation and apoptosis. Oncogene 151763-1772. [DOI] [PubMed] [Google Scholar]
- 47.Koo, G. C., and J. R. Peppard. 1984. Establishment of monoclonal anti-Nk-1.1 antibody. Hybridoma 3301-303. [DOI] [PubMed] [Google Scholar]
- 48.Kuwana, T., L. Bouchier-Hayes, J. E. Chipuk, C. Bonzon, B. A. Sullivan, D. R. Green, and D. D. Newmeyer. 2005. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 17525-535. [DOI] [PubMed] [Google Scholar]
- 49.Kvansakul, M., M. F. van Delft, E. F. Lee, J. M. Gulbis, W. D. Fairlie, D. C. Huang, and P. M. Colman. 2007. A structural viral mimic of prosurvival bcl-2: a pivotal role for sequestering proapoptotic bax and bak. Mol. Cell 25933-942. [DOI] [PubMed] [Google Scholar]
- 50.Kvansakul, M., H. Yang, W. D. Fairlie, P. E. Czabotar, S. F. Fischer, M. A. Perugini, D. C. Huang, and P. M. Colman. 2008. Vaccinia virus anti-apoptotic F1L is a novel Bcl-2-like domain-swapped dimer that binds a highly selective subset of BH3-containing death ligands. Cell Death Differ. 151564-1571. [DOI] [PubMed] [Google Scholar]
- 51.Lalier, L., P. F. Cartron, P. Juin, S. Nedelkina, S. Manon, B. Bechinger, and F. M. Vallette. 2007. Bax activation and mitochondrial insertion during apoptosis. Apoptosis 12887-896. [DOI] [PubMed] [Google Scholar]
- 52.Leber, B., J. Lin, and D. W. Andrews. 2007. Embedded together: the life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis 12897-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lindsten, T., A. J. Ross, A. King, W. X. Zong, J. C. Rathmell, H. A. Shiels, E. Ulrich, K. G. Waymire, P. Mahar, K. Frauwirth, Y. Chen, M. Wei, V. M. Eng, D. M. Adelman, M. C. Simon, A. Ma, J. A. Golden, G. Evan, S. J. Korsmeyer, G. R. MacGregor, and C. B. Thompson. 2000. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell 61389-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lovell, J. F., L. P. Billen, S. Bindner, A. Shamas-Din, C. Fradin, B. Leber, and D. W. Andrews. 2008. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 1351074-1084. [DOI] [PubMed] [Google Scholar]
- 55.Metivier, D., B. Dallaporta, N. Zamzami, N. Larochette, S. A. Susin, I. Marzo, and G. Kroemer. 1998. Cytofluorometric detection of mitochondrial alterations in early CD95/Fas/APO-1-triggered apoptosis of Jurkat T lymphoma cells. Comparison of seven mitochondrion-specific fluorochromes. Immunol. Lett. 61157-163. [DOI] [PubMed] [Google Scholar]
- 56.Nechushtan, A., C. L. Smith, Y. T. Hsu, and R. J. Youle. 1999. Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J. 182330-2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.O'Connor, L., A. Strasser, L. A. O'Reilly, G. Hausmann, J. M. Adams, S. Cory, and D. C. Huang. 1998. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 17384-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oltvai, Z. N., C. L. Milliman, and S. J. Korsmeyer. 1993. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74609-619. [DOI] [PubMed] [Google Scholar]
- 59.Ow, Y. P., D. R. Green, Z. Hao, and T. W. Mak. 2008. Cytochrome c: functions beyond respiration. Nat. Rev. Mol. Cell Biol. 9532-542. [DOI] [PubMed] [Google Scholar]
- 60.Petros, A. M., E. T. Olejniczak, and S. W. Fesik. 2004. Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta 164483-94. [DOI] [PubMed] [Google Scholar]
- 61.Postigo, A., J. R. Cross, J. Downward, and M. Way. 2006. Interaction of F1L with the BH3 domain of Bak is responsible for inhibiting vaccinia-induced apoptosis. Cell Death Differ. 131651-1662. [DOI] [PubMed] [Google Scholar]
- 62.Skinner, M. A., S. M. Laidlaw, I. Eldaghayes, P. Kaiser, and M. G. Cottingham. 2005. Fowlpox virus as a recombinant vaccine vector for use in mammals and poultry. Expert Rev. Vaccines 463-76. [DOI] [PubMed] [Google Scholar]
- 63.Smyth, D. G., O. O. Blumenfeld, and W. Konigsberg. 1964. Reactions of N-ethylmaleimide with peptides and amino acids. Biochem. J. 91589-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stuart, D., K. Graham, M. Schreiber, C. Macaulay, and G. McFadden. 1991. The target DNA sequence for resolution of poxvirus replicative intermediates is an active late promoter. J. Virol. 6561-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Su, J., G. Wang, J. W. Barrett, T. S. Irvine, X. Gao, and G. McFadden. 2006. Myxoma virus M11L blocks apoptosis through inhibition of conformational activation of Bax at the mitochondria. J. Virol. 801140-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sundararajan, R., A. Cuconati, D. Nelson, and E. White. 2001. Tumor necrosis factor-alpha induces Bax-Bak interaction and apoptosis, which is inhibited by adenovirus E1B 19K. J. Biol. Chem. 27645120-45127. [DOI] [PubMed] [Google Scholar]
- 67.Sundararajan, R., and E. White. 2001. E1B 19K blocks Bax oligomerization and tumor necrosis factor alpha-mediated apoptosis. J. Virol. 757506-7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tamaoki, T., H. Nomoto, I. Takahashi, Y. Kato, M. Morimoto, and F. Tomita. 1986. Staurosporine, a potent inhibitor of phospholipid/Ca++ dependent protein kinase. Biochem. Biophys. Res. Commun. 135397-402. [DOI] [PubMed] [Google Scholar]
- 69.Taylor, J. M., and M. Barry. 2006. Near death experiences: poxvirus regulation of apoptotic death. Virology 344139-150. [DOI] [PubMed] [Google Scholar]
- 70.Taylor, J. M., D. Quilty, L. Banadyga, and M. Barry. 2006. The vaccinia virus protein F1L interacts with Bim and inhibits activation of the pro-apoptotic protein Bax. J. Biol. Chem. 28139728-39739. [DOI] [PubMed] [Google Scholar]
- 71.Tulman, E. R., C. L. Afonso, Z. Lu, L. Zsak, G. F. Kutish, and D. L. Rock. 2004. The genome of canarypox virus. J. Virol. 78353-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang, G., J. W. Barrett, S. H. Nazarian, H. Everett, X. Gao, C. Bleackley, K. Colwill, M. F. Moran, and G. McFadden. 2004. Myxoma virus M11L prevents apoptosis through constitutive interaction with Bak. J. Virol. 787097-7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang, G. Q., E. Wieckowski, L. A. Goldstein, B. R. Gastman, A. Rabinovitz, A. Gambotto, S. Li, B. Fang, X. M. Yin, and H. Rabinowich. 2001. Resistance to granzyme B-mediated cytochrome c release in Bak-deficient cells. J. Exp. Med. 1941325-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wasilenko, S. T., L. Banadyga, D. Bond, and M. Barry. 2005. The vaccinia virus F1L protein interacts with the proapoptotic protein Bak and inhibits Bak activation. J. Virol. 7914031-14043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wasilenko, S. T., T. L. Stewart, A. F. Meyers, and M. Barry. 2003. Vaccinia virus encodes a previously uncharacterized mitochondrial-associated inhibitor of apoptosis. Proc. Natl. Acad. Sci. USA 10014345-14350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wei, M. C., T. Lindsten, V. K. Mootha, S. Weiler, A. Gross, M. Ashiya, C. B. Thompson, and S. J. Korsmeyer. 2000. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 142060-2071. [PMC free article] [PubMed] [Google Scholar]
- 77.Wei, M. C., W. X. Zong, E. H. Cheng, T. Lindsten, V. Panoutsakopoulou, A. J. Ross, K. A. Roth, G. R. MacGregor, C. B. Thompson, and S. J. Korsmeyer. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292727-730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Westphal, D., E. C. Ledgerwood, M. H. Hibma, S. B. Fleming, E. M. Whelan, and A. A. Mercer. 2007. A novel Bcl-2-like inhibitor of apoptosis is encoded by the parapoxvirus ORF virus. J. Virol. 817178-7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Willis, S. N., and J. M. Adams. 2005. Life in the balance: how BH3-only proteins induce apoptosis. Curr. Opin. Cell Biol. 17617-625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Willis, S. N., L. Chen, G. Dewson, A. Wei, E. Naik, J. I. Fletcher, J. M. Adams, and D. C. Huang. 2005. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 191294-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Willis, S. N., J. I. Fletcher, T. Kaufmann, M. F. van Delft, L. Chen, P. E. Czabotar, H. Ierino, E. F. Lee, W. D. Fairlie, P. Bouillet, A. Strasser, R. M. Kluck, J. M. Adams, and D. C. Huang. 2007. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315856-859. [DOI] [PubMed] [Google Scholar]
- 82.Wilton, B. A., S. Campbell, N. Van Buuren, R. Garneau, M. Furukawa, Y. Xiong, and M. Barry. 2008. Ectromelia virus BTB/kelch proteins, EVM150 and EVM167, interact with cullin-3-based ubiquitin ligases. Virology 37482-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wolter, K. G., Y. T. Hsu, C. L. Smith, A. Nechushtan, X. G. Xi, and R. J. Youle. 1997. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 1391281-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xiang, J., D. T. Chao, and S. J. Korsmeyer. 1996. BAX-induced cell death may not require interleukin 1 beta-converting enzyme-like proteases. Proc. Natl. Acad. Sci. USA 9314559-14563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yasuda, M., P. Theodorakis, T. Subramanian, and G. Chinnadurai. 1998. Adenovirus E1B-19K/BCL-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J. Biol. Chem. 27312415-12421. [DOI] [PubMed] [Google Scholar]
- 86.Yin, X. M., Z. N. Oltvai, and S. J. Korsmeyer. 1994. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature 369321-323. [DOI] [PubMed] [Google Scholar]
- 87.Youle, R. J., and A. Strasser. 2008. The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 947-59. [DOI] [PubMed] [Google Scholar]
- 88.Zha, H., C. Aime-Sempe, T. Sato, and J. C. Reed. 1996. Proapoptotic protein Bax heterodimerizes with Bcl-2 and homodimerizes with Bax via a novel domain (BH3) distinct from BH1 and BH2. J. Biol. Chem. 2717440-7444. [DOI] [PubMed] [Google Scholar]
- 89.Zhang, Z., S. M. Lapolla, M. G. Annis, M. Truscott, G. J. Roberts, Y. Miao, Y. Shao, C. Tan, J. Peng, A. E. Johnson, X. C. Zhang, D. W. Andrews, and J. Lin. 2004. Bcl-2 homodimerization involves two distinct binding surfaces, a topographic arrangement that provides an effective mechanism for Bcl-2 to capture activated Bax. J. Biol. Chem. 27943920-43928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zong, W. X., T. Lindsten, A. J. Ross, G. R. MacGregor, and C. B. Thompson. 2001. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 151481-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]