

Abstract
To be effective for the treatment of cancer and infectious diseases, T cell adoptive immunotherapy requires large numbers of cells with abundant proliferative reserves and intact effector functions. We are achieving these goals using a gene therapy strategy wherein the desired characteristics are introduced into a starting cell population, primarily by high efficiency lentiviral vector-mediated transduction. Modified cells are then expanded using ex vivo expansion protocols designed to minimally alter the desired cellular phenotype. In this article, we focus on strategies to (1) dissect the signals controlling T cell proliferation; (2) render CD4 T cells resistant to HIV-1 infection; and (3) redirect CD8 T cell antigen specificity.
Keywords: Keywords Lentiviral vector, CD28, PD-1, TCR, Chimeric immunoreceptor, Zinc-finger nuclease, NOG mice, Immunotherapy, Adoptive T cell therapy
Introduction
Adoptive T cell therapy is a form of transfusion therapy involving the infusion of large numbers of T cells with the aim of eliminating, or at least controlling, malignancies or infectious diseases. Successful applications of this technique include the infusion of CMV-or EBV-specific CTLs to protect immunosuppressed patients from these transplantation-associated diseases [1, 2]. Furthermore, donor lymphocyte infusions of ex vivo-expanded allogeneic T cells have been used to successfully treat hematological malignancies in patients with relapsed disease following allogeneic hematopoietic stem cell transplant [3]. However, in many other malignancies and chronic viral infections such as HIV-1, adoptive T cell therapy has achieved inconsistent and/or marginal successes. Nevertheless, there are compelling reasons for optimism on this strategy. For example, the existence of HIV-positive elite non-progressors [4], as well as the correlation between the presence of intratumoral T cells and a favorable prognosis in malignancies such as ovarian [5, 6] and colon carcinoma [7, 8], provides in vivo evidence for the critical role of the immune system in controlling both HIV and cancer.
The rapid pace of advances in the fundamental understanding of T cell biology is providing a wealth of insights into the mechanisms contributing to the low success rate of past adoptive transfer trials, and simultaneously is helping us to elucidate the “rules” governing successful adoptive immunotherapy. In retrospect, the shortcomings of previous clinical trials can be viewed from both quantitative and qualitative perspectives. First, large numbers of effector T cells, from 109 to greater than 1010, are required for a single T cell infusion. Typically, a relatively small input population is expanded in either an antigen-specific or a polyclonal fashion. Thus, the logistics of generating therapeutic cell numbers in an FDA-compliant manner are formidable, particularly for antigen-specific expansion protocols. Second, in recent years, we have witnessed an explosion of information concerning paradigm of the complexity of both CD8 and CD4 T cell subsets. The long-held TH1–TH2 CD4 T cells differentiation has been rendered far more complex by the recent discovery of both regulatory T cells [9] and Th17 cells [10]. CD8 T cell subsets are similarly complex. CD8 T cells comprise naïve, effector, effector memory, and central memory subsets [11, 12]. The ex vivo expansion systems employed in previous clinical trials, combined with the need for extensive cell replication to obtain the requisite numbers of cells, likely resulted in the generation of terminally differentiated T effector cells. These cells are characterized by the loss of CD28 expression, upregulation of CD57 expression, and increased susceptibility to activation-induced cell death [11]. In contrast, preclinical experiments have revealed that less differentiated central memory T cells (CD62L+CD28+CD27+CD57−) exerted greater antitumor effects than fully differentiated effector T cells [13].
Thus, the key to successful adoptive immunotherapy strategies appears to consist of (1) using the “right” T cell type(s) and (2) obtaining therapeutically effective numbers of these cells without compromising their effector functions or their ability to engraft within the host. This article is focused on strategies employed in our laboratory to generate the “right” cell through genetic engineering approaches, with an emphasis on redirecting the antigen specificity of CD8 T cells, and rendering CD4 T cells resistant to HIV-1 infection. The article by Paulos et al. describes the evolving process of how to best obtain therapeutically effective numbers of the “right” cells by optimizing ex vivo cell expansion strategies.
Our laboratory, a part of the Translational Research Program of the Abramson Family Cancer Research Institute at the University of Pennsylvania, is charged with conducting bench to beside (and back) experiments. This has become a reality through our close and long-term relationship with the University of Pennsylvania Cell and Vaccine Production Facility (CVPF), a GMP cell manufacturing facility, which generates cell products used in multiple Phase I T cell adoptive transfer trials in both cancer and HIV [14]. Accordingly, our research focuses on defining the growth and functional properties of T cells while simultaneously focusing on generating products for evaluation in the clinic.
Infrastructure and strategy
Our laboratory’s overall strategy and flow plan for development and evaluation of engineered T cells is depicted in Fig. 1. We work almost exclusively with primary human T cells; little or no work is performed with conventional established cell lines. Thus, we benefit substantially from our close association with the UPenn Human Immunology Core. The Core performs leukaphereses on healthy donors 2–3 times a week, and provides purified peripheral blood mononuclear cell subsets, ensuring a constant influx of fresh human T cells into our laboratory. We have extensive experience in developing both bead- and cell-based artificial antigen presenting cells (aAPCs), as described in detail in the article by Paulos et al. The ability to genetically modify T cells at high efficiency is critical for virtually every project within the laboratory. We have adapted the lentiviral vector system described by Dull [15] for most, but not all, of the engineering applications in our laboratory. The versatility of this system was improved significantly by incorporating a picornavirus T2A motif within the vector backbone. Through a postulated “ribosome skip” mechanism, this sequence permits nearly equivalent expression of both cistrons in a bicistronic mRNA [16, 17]. In vivo evaluation of T cell products is carried out by the Animal Model/Biotoxicity Group within the lab, using a NOD/scid/IL-2Rγcnull (NOG) mouse model [18]. These mice, in addition to lacking mature B and T cells, have reduced levels of NK cells compared to NOD/scid mice. This strain is highly permissive to human leukocyte engraftment, and when injected with human hematopoietic stem cells, the complete human immune system can be reconstituted [18]. The Animal Model/Biotoxicity Group is also charged with carrying out FDA-mandated preclinical safety studies prior to initiation of Phase I clinical trials by the CVPF.
Fig. 1.
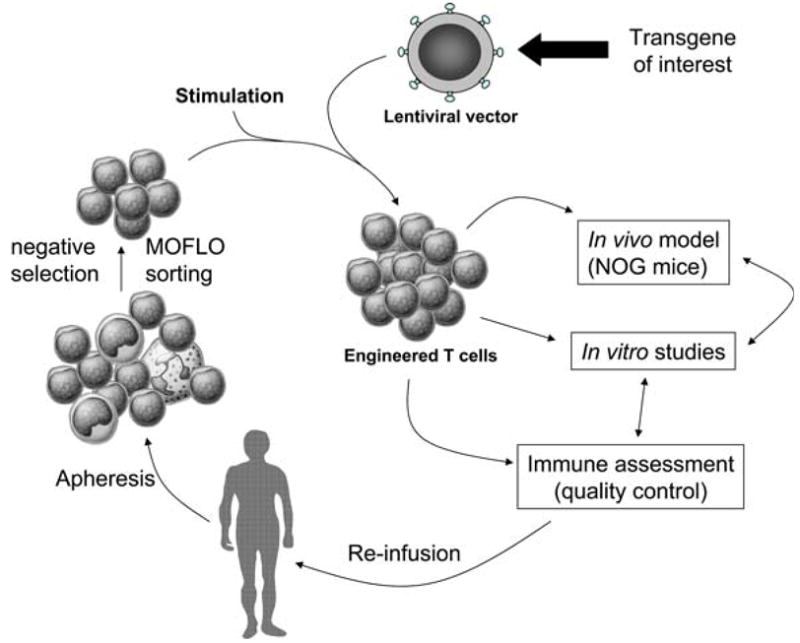
Overview of our laboratory strategy. Human mononuclear cell subsets are obtained from the apheresis product from a patient. We use magnetic bead or high-speed MOFLO sorting to obtain the required T cell subsets. After stimulation and transduction with viral vectors, the engineered cells are utilized for in vitro and in vivo studies. Finally, the technologies developed are “scaled-up” in a GMP-compliant manner to reinfuse in patients participating in clinical trials
T cell signaling in primary human T cells
Our studies elucidating the signaling pathways that control T cell activation have served to guide our translational studies, by providing insight on how to modulate the T cell response based on costimulatory receptor engagement. There are several compelling reasons to study T cell activation and signaling in primary human T cells. First, there is an emerging body of evidence suggesting that murine and human T cells are “wired” differently. Isolated murine T cells die within 48 h unless activated, whereas unstimulated human T cells can live for more than one week, suggesting that murine T cells are more sensitive to apoptosis [19, 20]. Furthermore, CD3 stimulation of murine T cells results in significant, albeit not optimal, levels of T cell expansion and IL-2 production, whereas highly purified human T cells do not expand upon CD3 stimulation and only trace levels of IL-2 are detected. This suggests that human T cells are much more dependent upon costimulatory signals [21, 22]. The hazards of using transformed cell lines to study costimulatory pathways have been previously described [23], but use of transformed cell lines to study negative regulators of T cell activation (CTLA-4, PD-1, and BTLA, for example) is even more perilous. For example, signaling molecules, such as SHIP and PTEN, which normally limit T cell proliferation, are inactivated during the transformation process [24, 25]. Since these factors are often employed by coinhibitory molecules to block T cell activation, key aspects of regulation can be overlooked.
To study the signal transduction pathways initiated by ligation of CD28 family members in primary T cells, we designed a series of chimeric molecules that contained a murine CD28 extracellular domain coupled to the cytoplasmic tails of either CD28 or ICOS (Fig. 2a). The main advantage of this chimeric receptor system is that when expressed in primary human T cells, murine anti-CD28 Ab can be used to trigger our introduced molecule, and this result can be compared to the effect of antibody stimulation on the endogenous receptors. These experiments required that nearly 100% of the target cell population expressed the chimeric receptor, and they were the impetus for our initial development of high-titer lentiviral vectors, as described above.
Fig. 2.

Transduction and characterization of mCD28/h28 chimera. (a) Shown is a cartoon depicting the CD28 chimera with murine CD28 extracellular domain and human CD28 cytoplasmic tail. (b) High-titer lentiviral vector was generated and used to transduce human CD4 T cells, and these cells were stained for expression of mouse CD28 six days post-transduction. Once cells returned to a near resting state, they were stimulated with beads coated with antibodies against CD3/MHC-I, CD3/mCD28, or CD3/hCD28 for 24 h. (c, d) RNA was harvested and cDNA was synthesized and probed with primers to detect IL-2, CCR5, and 28 s rRNA. Real-time PCR amplification and product detection were performed on an ABI Prism 7700 (PE Biosystems) as recommended by the manufacturer. Results for IL-2 and CCR5 were normalized to 28 s rRNA and relative expression was determined by arbitrarily assigning the value obtained for unstimulated cells to 1
Even though lentiviral vectors can transduce non-dividing cells, they do not transduce resting T cells very well [26, 27]. Therefore, the target CD4 T cells are activated overnight prior to the addition of lentiviral vector. Using this protocol we routinely observe >90% transduction efficiency and often can see >99% of cells transduced (Fig. 2b). As a control experiment, we confirmed that the chimeric molecules containing the wildtype cytoplasmic tails were indeed mimicking the actions of the endogenous molecule. A lentiviral vector encoding the extracellular domain of murine CD28 (mCD28) and the intracellular domain of human CD28 was used to transduce primary CD4 T cells. Murine CD28 expression was detected four days later (Fig. 2b) and remained at a high-level for the duration of the experiment. When the cells were restimulated with αCD3/αmCD28-coated beads, they responded by producing IL-2 and downregulating CCR5 expression, indicating the functional integrity of the chimeric molecule (Fig. 2c, d).
Thus, the ability to efficiently modify primary human T cells has enabled us to elucidate the following aspects of costimulation: the role CD28 plays in glucose metabolism [28], decipher which domains of CD28 are required for optimal IL-2 production [29], uncover the amino acid within the PD-1 cytoplasmic tail that allows PD-1 to block T cell activation [30], establish that CTLA-4 and PD-1 use distinct mechanisms to block T cell activation [31], demonstrate that CD8 T cells do not form lipid rafts [32], and demonstrate that BTLA has multiple functional motifs that must be mutated to abrogate its inhibitory activity [33].
Engineering of CD4 T cells for immunotherapy
CD4 T cells are the primary target of HIV-1, and decreasing CD4 T cell numbers is a hallmark of advancing HIV-1 disease [34]. Thus, strategies that protect CD4 T cells from HIV-1 infection in vivo would conceivably provide sufficient immunological help to control HIV-1 infection. Our early observations that CD3/CD28 costimulation resulted in improved ex vivo expansion of CD4 T cells from both healthy and HIV-infected donors, as well as enhanced resistance to HIV-1 infection [35, 36], ultimately led to the first-in-human trial of lentiviral vector-modified CD4 T cells [37]. In this trial, CD4 T cells from HIV-positive subjects who had failed antiretroviral therapy were transduced with a lentiviral vector encoding an antisense RNA that targeted a 937 bp region in the HIV-1 envelope gene. Preclinical studies demonstrated that this antisense region, directed against the HIV-1NL4-3 envelope, provided robust protection from a broad range of both R5-and X4-tropic HIV-1 isolates [38]. One year after administration of a single dose of the gene-modified cells, four of the five enrolled patients had increased peripheral blood CD4 T cell counts, and in one subject, a 1.7 log decrease in viral load was observed. Finally, in two of the five patients, persistence of the gene-modified cells was detected one year post-infusion. These observations provided the basis for a follow-up study evaluating the effect of multiple doses of gene-modified cells. This study is being conducted in a cohort of patients who control their viremia, and it will incorporate an analytic treatment interruption to evaluate effects on viral load recrudescence and viral set point.
We are also exploring alternative gene transfer strategies by which CD4 T cells can be rendered HIV-1 resistant. For example, the Rhesus macaque TRIM5α (rhTRIM5α) gene product potently restricts HIV-1 replication at a post-entry, preintegration stage [39, 40]. However, the human ortholog of rhTRIM5α exhibits only modest HIV-1 restriction [40]. Our preclinical studies evaluated the ability of CD4 T cells transduced with lentiviral vectors encoding either rhTRIM5α or huTRIM5α323–332 to confer restriction against HIV-1 infection in adoptive transfer settings. The latter construct, a huTRIM5α derivative containing five Rhesus amino acid residues, restricts HIV-1 replication nearly as potently as rhTRIM5α [39], but its largely human composition will hopefully preclude concerns about the immunogenicity of foreign proteins introduced into human cells. We found that rhTRIM5α is effective in blocking cell-free infection in primary human T cells. However, when these rhTRIM5α-expressing cells were mixed with untransduced cells, all antiviral activity was lost, suggesting that cell-to-cell transmission could be blocked by rhTRIM5α [41]. These studies, while engendering little enthusiasm to test adoptive T cell therapy with rhTRIM5α expressing CD4 T cells, provided important information regarding how rhTRIM5a exerts its anti-viral activity. This highlights that translational research can bolster basic research in a similar manner that basic research augments translational studies.
Since its identification as the primary co-receptor involved in HIV transmission, CCR5 has attracted considerable attention as a target for HIV therapy [42, 43]. Indeed, “experiments of nature” have shown that individuals with a homozygous CCR5 Δ32 deletion are highly resistant to HIV-1 infection. Thus, we hypothesized that knocking out the CCR5 locus would generate CD4 T cells permanently resistant to infection by R5 isolates of HIV-1. To test this hypothesis we took advantage of zinc-finger nuclease (ZFN) technology [44]. ZFNs introduce sequence-specific double-strand DNA breakage, which is imperfectly repaired by non-homologous end-joining. This results in the permanent disruption of the genomic target, a process termed genome editing (Fig. 3). We generated adenoviral vectors encoding CCR5-specific ZFNs (Ad5/35-CCRZFN) and used them to transduce CD4 T cells. Adenoviral vectors were chosen for this application because ZFNs only need to be expressed transiently to perform their function. High-level (~50%) disruption of the CCR5 locus was observed following a single round of transduction with the Ad5/35-CCRZFN vectors [45]. When CCR5-ZFN-transduced cell populations were infected in vitro with R5 HIV-1 isolates, a two-fold enrichment of the gene-modified cells was observed at the end of the culture period, implying positive selection for the Δ32CCR5 phenocopy. Importantly, in mock-infected cultures, the proportion of gene-modified cells remained unchanged over the culture period, indicating that disruption of the CCR5 locus did not confer a growth disadvantage. Next, we explored the in vivo efficacy of CCR5ZFN-modified cells using the NOG mouse model. NOG mice were injected with either CCR5ZFN-modified or mock-transduced T cells, and then challenged with HIV-1-infected PBMCs. Approximately 2 months post-challenge, the percentage of CCR5 gene-modified cells was significantly increased in the peripheral blood of most animals. Furthermore, plasma viremia was significantly lower in these animals than in animals that received mock-transduced CD4 T cells. Thus, these cells provided protection against HIV-1 in vivo [45]. Currently, our laboratory is conducting FDA-mandated biotoxicity and genotoxicity studies of these cells prior to initiating a Phase I clinical trial of CCR5 gene-modified T cells.
Fig. 3.

Gene disruption by Zinc-finger nucleases. (a) ZFNs are composed of three or four zinc-finger domains that recognize and bind to a three-base pair sequence, such that a protein including more zinc fingers targets a longer sequence and therefore has greater specificity for its target gene. The non-specific endonuclease Fok I is ligated to the zinc-finger domains and comprises the nuclease domain. (b) While one ZFN molecule binds its target sequence on one DNA strand, another ZFN molecule binds its target sequence on the opposite strand, as shown. The nuclease domains dimerize and each cleaves its own strand, producing a double-stranded break. Once the DSB is generated, it is repaired by error-prone non-homologous end-joining mechanisms, resulting in a defective gene
Genetically engineered antigen-specific CD8 T cells
Genetic modification of T cells to redirect antigen specificity is an attractive strategy compared to the lengthy process of growing T cell lines or CTL clones for adoptive transfer. Genetically modified, adoptively transferred T cells are capable of long-term persistence in humans [37, 46, 47], demonstrating the feasibility of this approach. When compared to the months it can take to generate an infusion dose of antigen-specific CTL lines or clones from a patient, a homogeneous population of redirected antigen-specific cells can be expanded to therapeutically relevant numbers in about two weeks [3]. Several strategies are being explored to bypass the need to expand antigen-specific T cells for adoptive T cell therapy. The approaches currently studied in our laboratory involve the genetic transfer of chimeric antigen receptors and supraphysiologic T cell receptors.
Chimeric antigen receptors
Chimeric antigen receptors (CARs or T-bodies) are artificial T cell receptors that combine the extracellular single-chain variable fragment (scFv) of an antibody with intracellular signaling domains, such as CD3ζ or Fc(ε)RIγ [48–50]. When expressed on T cells, the receptor bypasses the need for antigen presentation on MHC since the scFv binds directly to cell surface antigens. This is an important feature, since many tumors and virus-infected cells downregulate MHC-I, rendering them invisible to the adaptive immune system. The high-affinity nature of the scFv domain makes these engineered T cells highly sensitive to low antigen densities. In addition, new chimeric antigen receptors are relatively easy to produce from hybridomas. The key to this approach is the identification of antigens with high surface expression on tumor cells, but reduced or absent expression on normal tissues. Since one can redirect both CD4 and CD8 T cells, the T-body approach to immunotherapy represents a near universal “off the shelf” method to generate large numbers of antigen-specific helper and cytotoxic T cells.
Many T-bodies targeting diverse tumors have been developed [51], and four have been evaluated clinically [52–55]. Three of the four studies were characterized by poor transgene expression and limited T-body engraftment. However, in a study of metastatic renal cell carcinoma using a T-body directed against carbonic anhydrase IX [55], T-body-expressing cells were detectable in the peripheral blood for nearly 2 months post-administration. Furthermore, following infusion, PBMCs produced IFNγ and developed lytic functions in response to CAIX-expressing targets. However, patients in the trial also developed dose-limiting toxicity, as described below.
The major goals in the T-body field currently are to optimize their engraftment and maximize their effector functions. Our laboratory is addressing both problems simultaneously through an in-depth study of the requirements for T-body activation. We hypothesize that their limited persistence is due to incomplete cell activation due to the lack of costimulation. While naïve T cells depend on costimulation through CD28 ligation to avoid anergy and undergo full activation in response to antigen, it is recognized that effector cells also require costimulation to properly proliferate and produce cytokines [56]. Previous studies have shown that providing CD28 costimulation is crucial for the antitumoral function of adoptively transferred T cells and T-bodies [57–59]. Unlike conventional T cell activation, which requires two discrete signals, T-bodies can be engineered to provide both costimulation and CD3 signaling through one binding event.
In our laboratory, we are evaluating the contribution of different costimulatory signaling domains to T-body function. This work was prompted by the observation that CD28 signaling is not sufficient to drive proliferation of T-bodies [60], and it is a natural outgrowth of our earlier studies showing that distinct domains within the CD28 cytoplasmic tail regulate cytokine production and control T cell survival by Bcl-xL induction [29, 61]. In contrast to CD4 T cells, human CD8 T cells do not express CD28 constitutively. Upon activation, CD8 T cells express 4–1BB, and ligation with 4–1BBL then enhances IL-2 production and Bcl-xL similar to CD28 ligation [62]. We therefore have developed a series of T-bodies that include the signaling domains from CD3ζ, CD28, and 4–1BB in all possible combinations to dissect their effects on T cell proliferation, cytokine production, and in vitro and in vivo killing (Fig. 4).
Fig. 4.

Chimeric antigen receptors. Schematic representation of chimeric antigen receptors compared to a T cell receptor. We are studying the contribution of the CD28 and 4-1BB signaling domains, alone or in combination. From the left, tripartite CAR (CD28, 4–1BB and CD3ζ), 28ζ CAR, BBζ CAR, CD3ζ CAR, and truncated-ζ CAR. T cells redirected by the T-body approach are bi-specific, since they maintain expression of their endogenous TCR
We are targeting the tumor associated antigens CD19 and mesothelin. CD19 is the earliest of the B cell-restricted antigens and is expressed on pre-B cells and most B cell malignancies, including most pro- and pre-B cell acute lymphoblastic leukemias (ALL), common ALL, non-Hodgkin-lymphomas (NHL), chronic B-lymphocytic leukemia (B-CLL), and hairy-cell leukemias (HCL) [63]. Since CD19 is continuously expressed on almost all subtypes of B lineage leukemic cells, CD19 may be a better cancer-targeting molecule than CD20. In addition, B cells that are killed by therapy targeted to CD19 should be replaced from CD19-negative precursor cells that presumably should not be killed by the treatment. Mesothelin is a GPI-linked molecule over-expressed by many lung cancers (including mesotheliomas and adenocarcinomas), in addition to pancreatic and ovarian cancers [64]. Expression on normal adult tissue is relatively low and restricted to serosal cells. Given its high expression on malignant cells and limited expression in normal tissue, mesothelin is an attractive target for redirected immunotherapy.
The specificity and efficacy of T-bodies targeting both CD19 and mesothelin have been established in vitro. Based on our data and that of others [65, 66], we hypothesize that the addition of the 4–1BB and CD28 costimulatory domains will improve the potency and persistence of the T-bodies. To address this, we have established a human xenograft model of human cancer using primary human tumors in NOG mice. We propagated human leukemia and mesothelin-positive primary tumors in the mice allowing them to grow to clinically relevant sizes before treatment to better mimic the situation on patients in the clinic. Tumor volume and peripheral blood persistence of the T-bodies over time as well as trafficking to organs and distant metastases are being measured. In addition, we are also comparing the route of administration (intraperitoneal versus intravenous versus intratumoral) and its effects on antitumor potency and persistence.
TCR-transfer immunotherapy
A different approach for redirecting specificity to T cells for adoptive immunotherapy involves the genetic transfer of full-length TCR genes. A T cell’s specificity for its cognate antigen is solely determined by its TCR. Genes encoding the α and β chains of a T cell receptor (TCR) can be isolated from a T cell specific for the antigen of interest and restricted to a defined HLA allele, inserted into a vector, and then introduced into large numbers of T cells of individual patients that share the restricting HLA allele as well as the targeted antigen. In 1999, Clay and colleagues from Rosenberg’s group at the National Cancer Institute were the first to report the transfer of TCR genes via a retroviral vector into human lymphocytes and to show that T cells gained stable reactivity to MART-1 [67]. To date, many others have shown that the same approach can be used to transfer specificity for multiple viral and tumor associated antigens in mice and human systems. These T cells gain effector functions against the transferred TCR’s cognate antigen, as defined by proliferation, cytokine production, lysis of targets presenting the antigen, trafficking to tumor sites in vivo, and clearance of tumors and viral infection.
In 2006, Rosenberg’s group redirected patients’ PBLs with the naturally occurring, MART-1-specific TCR reported in 1999 by Clay. In the first clinical trial to test TCR-transfer immunotherapy, these modified T cells were infused into melanoma patients [68]. While the transduced T cells persisted in vivo, only two of the 17 patients had an objective response to this therapy. One issue revealed by the study was the poor expression of the transgenic TCRs by the transferred T cells. Nonetheless, the results from this trial showed the potential of TCR-transfer immunotherapy as a safe form of therapy for cancer and highlighted the need to optimize such therapy to attain maximum potency.
One limitation of TCR immunotherapy is the very nature of the TCR-pMHC interaction. Compared to antibodies, TCRs have a relatively low affinity for their cognate antigen with KDs typically within the range of 1 to 100 μM [69–71]. One way to improve the effectiveness of TCR transfer therapy is to improve the functional avidity of the TCR-transduced T cells. Previous studies have shown that high-avidity CTLs offer superior protection against viral infections because they are able to recognize lower Ag densities and initiate lysis more rapidly than low-avidity CTLs [72]. While several methods to improve the functional avidity of TCR-transduced T cells have been suggested, including the use of optimal transfer vectors and strategies to prevent gene silencing, the most obvious way to improve functional avidity is to select a TCR with high-affinity for its cognate antigen. While the chances of finding and cloning such a TCR from a patient’s sample are highly unlikely, this setback has recently been overcome by using directed evolution and phage display affinity maturation. Phage display can be applied to any TCR and can be used to screen library sizes of 1010 variants. This approach has produced TCRs with picomolar affinities, representing almost a millionfold improvement in affinity compared to the parent TCR [73, 74]. This affinity and its related antigen binding half-life are within the range of those reported for therapeutically applied antibodies [75]. High-affinity TCRs, expressed as soluble molecules, can detect very low levels of peptide-MHC-I complexes on APCs and show no cross-reactivity to endogenous peptides [74]. In our laboratory, we are studying the feasibility of expressing these high-affinity TCRs on primary T cells for adoptive immunotherapy.
High-affinity TCR-transfer into primary T cells
To study the effect of supraphysiological TCR affinity on primary T cells, we selected an HIV-1 p17Gag77–85 (SLYNTVATL; SL9)-specific TCR and three derived mutants with increased affinity [76]. High levels of expression of the TCRs were achieved by introducing the codon-optimized, full-length TCRα and TCRβ chain genes into a lentiviral vector driven via the EF1-α promoter. The TCRα and TCRβ chains are separated by the picornavirus T2A sequence to allow coordinate expression of both TCR chains from a single transcript [16]. When transferred by lentiviral transduction into primary CD8 T cells, these TCR constructs are expressed on the T cell surface at physiological levels, as assessed by comparing TCR and CD3 expression between transduced and untransduced T cells. The transduced cells show no apparent defects in proliferation. The high-affinity TCRs are able to re-direct CD4 T cells to HLA-A*02; thus we can generate antigen-specific T cell help with this strategy.
T cells expressing the high-affinity SL9-specific TCRs are able to detect very low densities of cognate antigen on APCs and are capable of producing multiple cytokines, having a higher frequency of polyfunctional effector CTLs. When compared to cells expressing the parental TCR, the high-affinity TCR-bearing cells are better able to control spreading of HIV infection in vitro. In addition, the high-affinity TCR-transduced T cells can recognize common SL9 CTL-escape mutations [76]. These common epitope mutations affect TCR recognition rather than HLA-A*02 binding [77]. We are in the process of testing the ability of high-affinity transgenic T cells to control HIV in vivo in our NOG mouse model. A recent study measuring the anti-HIV-1 T cell response in a cohort of untreated HIV-1 infected individuals found that the magnitude and breadth of the HIVgag immune response correlated with low viral loads [78]. With the knowledge gained from these studies with HIVgag as a model antigen, we are now studying supraphysiologic TCRs specific for the HLA-A*02-restricted tumor associated antigens NY-ESO-1, hTERT I540, WT-1, and gp100.
Safety concerns for engineered T cells
The genetic modifications of lymphocytes described herein, like all gene therapy procedures, carry potential risks. Lentiviral vector integration or, conceivably, off-target effects resulting from ZFN-mediated genomic modification, are associated with the risk of insertional mutagenesis. In fact, such events have been observed. In a trial of X-linked severe combined immunodeficiency [79], retroviral vector-mediated gene transfer into autologous CD34 + bone marrow cells resulted in a leukemia-like proliferative disorder in three of the study subjects. This outcome was the result of vector integration-mediated upregulation of transcription of the proto-oncogene LMO-2 [80]. However, subsequent studies, including those performed by our collaborator at the University of Pennsylvania, Dr. Bushman, revealed that lentiviral and retroviral vectors have different preferred integration sites, with retroviral vectors favoring integration near transcription start sites [81]. Dr. Bushman has analyzed the integration sites of lentiviral vectors in our first trial, as part of the accompanying safety analysis [37]. Furthermore, recent experiments have shown that mature T cells are more resistant to oncogenic transformation that the hematopoietic stem cells used in the SCID-X1 trial described above [82]. However, we are keenly aware of the possibility of such an event. In response to FDA guidelines [83], our mouse modeling/biotoxicity group evaluates the carcinogenicity or toxicity of engineered human T cells in studies designed in close collaboration with the FDA.
There are additional safety concerns that accompany the in vivo use of engineered human T cells. For example, in T-body immunotherapy, off-target cross-reactivity with non-malignant tissues has been reported. In the trial described above [55], the use of T-bodies to treat metastatic renal cell cancer resulted in unpredicted hepatic toxicity, presumably due to previously undetected expression of the target antigen CAIX in the biliary tract. One strategy to control these off-target effects is to separate the T cell signaling domains on two different T-bodies, i.e., one T-body would contain the CD3ζ signaling domain, while a T-body with different specificity would contain the costimulatory signaling domains, such as CD28 or 4–1BB. Such a strategy would reproduce the physiological signal 1 and 2 checkpoints of T cell activation. Installing a suicide construct within the engineered T cells would provide an additional layer of safety and control. We are currently evaluating the feasibility of suicide gene therapy using the TMPK gene [84]. TMPK catalyzes the rate-limiting step in the conversion of AZT to the toxic intracellular AZT-TP form. Enhanced, minimally modified versions of TMPK possess up to 200-fold enhanced activity [85]. Thus, expression of the enhanced form of TMPK within engineered cells would render them exceedingly sensitive to systemically administered AZT.
TCR-transfer gene therapy also carries unique safety concerns with respect to autoimmune responses engendered by dual-specific T cells. Conceivably autoimmunity could result from one of the following: (1) formation of self-reactive TCR heterodimers by mispairing of the endogenous and transgene TCR chains, (2) breaking of tolerance by activation of tolerized T cells by the transgene TCR, or (3) alloresponses due to partial MHC mismatch between the recipient of the therapy and the host from which the TCR was derived. While these are issues that require careful consideration, our own preclinical results and to some extent clinical if one takes into account the first human trial [68] do not show evidence of adverse effects due to any of these mechanisms.
Closing remarks
The adoptive immunotherapy field is advancing by a tried-and-true method: learning from disappointments and moving forward. Our ability to fully realize the therapeutic potential of adoptive T cell therapy is tied to a more complete understanding of how human T cells receive signals, kill targets, and modulate effective immune responses. Our goal is to perform lab-based experiments that provide insight into how primary T cells function in a manner that will facilitate and enable adoptive T cell therapy clinical trials. Our ability to efficiently modify (and expand) T cells ex vivo provides the opportunity to deliver sufficient immune firepower where it has heretofore been lacking. Sustained transgene expression, coupled with enhanced in vivo engraftment capability, will move adoptive immunotherapy into a realm where long-term therapeutic benefits are the norm rather than the exception.
References
- 1.Heslop HE, Perez M, Benaim E, Rochester R, Brenner MK, Rooney CM. Transfer of EBV-specific CTL to prevent EBV lymphoma post bone marrow transplant. J Clin Apher. 1999;14:154–6. doi: 10.1002/(sici)1098-1101(1999)14:3<154::aid-jca9>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 2.Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333:1038–44. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 3.Porter DL, Levine BL, Bunin N, Stadtmauer EA, Luger SM, Goldstein S, et al. A phase 1 trial of donor lymphocyte infusions expanded and activated ex vivo via CD3/CD28 costimulation. Blood. 2006;107:1325–31. doi: 10.1182/blood-2005-08-3373. [DOI] [PubMed] [Google Scholar]
- 4.Deeks SG, Walker BD. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity. 2007;27:406–16. doi: 10.1016/j.immuni.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proceedings of the National Academy of Sciences. 2005;102:18538–43. doi: 10.1073/pnas.0509182102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T Cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–13. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 7.Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998;58:3491–4. [PubMed] [Google Scholar]
- 8.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 9.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–87. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Harrington LE, Mangan PR, Weaver CT. Expanding the effector CD4 T-cell repertoire: the Th17 lineage. Curr Opin Immunol. 2006;18:349–56. doi: 10.1016/j.coi.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 11.Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. 2006;211:214–24. doi: 10.1111/j.0105-2896.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–12. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 13.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu ZY, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8(+) T cells. J Clin Invest. 2005;115:1616–26. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levine BL. T lymphocyte engineering ex vivo for cancer and infectious disease. Expert Opin Biol Ther. 2008;8:475–89. doi: 10.1517/14712598.8.4.475. [DOI] [PubMed] [Google Scholar]
- 15.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–71. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, et al. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol. 2004;22:589–94. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- 17.Donnelly ML, Luke G, Mehrotra A, Li X, Hughes LE, Gani D, et al. Analysis of the aphthovirus 2A/2B polyprotein ‘cleavage’ mechanism indicates not a proteolytic reaction, but a novel translational effect: a putative ribosomal ‘skip’. J Gen Virol. 2001;82:1013–25. doi: 10.1099/0022-1317-82-5-1013. [DOI] [PubMed] [Google Scholar]
- 18.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174:6477–89. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 19.Akbari O, Freeman GJ, Meyer EH, Greenfield EA, Chang TT, Sharpe AH, et al. Antigen-specific regulatory T cells develop via the ICOS-ICOS-ligand pathway and inhibit allergen-induced airway hyperreactivity. Nat Med. 2002;8:1024–32. doi: 10.1038/nm745. [DOI] [PubMed] [Google Scholar]
- 20.Viola A, Schroeder S, Sakakibara Y, Lanzavecchia A. T lymphocyte costimulation mediated by reorganization of membrane microdomains. Science. 1999;283:680–2. doi: 10.1126/science.283.5402.680. [DOI] [PubMed] [Google Scholar]
- 21.Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273:104–6. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- 22.Wulfing C, Davis MM. A receptor/cytoskeletal movement triggered by costimulation during T cell activation. Science. 1998;282:2266–9. doi: 10.1126/science.282.5397.2266. [DOI] [PubMed] [Google Scholar]
- 23.Abraham RT, Weiss A. Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat Rev Immunol. 2004;4:301–8. doi: 10.1038/nri1330. [DOI] [PubMed] [Google Scholar]
- 24.Pages F, Ragueneau M, Rottapel R, Truneh A, Nunes J, Imbert J, et al. Binding of phosphatidylinositol-3-OH kinase to CD28 is required for T-cell signalling. Nature. 1994;369:327–9. doi: 10.1038/369327a0. [DOI] [PubMed] [Google Scholar]
- 25.Raab M, Pfister S, Rudd CE. CD28 signaling via VAV/SLP-76 adaptors: regulation of cytokine transcription independent of TCR ligation. Immunity. 2001;15:921–33. doi: 10.1016/s1074-7613(01)00248-5. [DOI] [PubMed] [Google Scholar]
- 26.Shapiro VS, Truitt KE, Imboden JB, Weiss A. CD28 mediates transcriptional upregulation of the inter-leukin-2 (IL-2) promoter through a composite element containing the CD28RE and NF-IL-2B AP-1 sites. Mol Cell Biol. 1997;17:4051–8. doi: 10.1128/mcb.17.7.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vandenberghe P, Freeman GJ, Nadler LM, Fletcher MC, Kamoun M, Turka LA, et al. Antibody and B7/BB1-mediated ligation of the CD28 receptor induces tyrosine phosphorylation in human T cells. J Exp Med. 1992;175:951–60. doi: 10.1084/jem.175.4.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–77. doi: 10.1016/s1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- 29.Parry RV, Rumbley CA, Vandenberghe LH, June CH, Riley JL. CD28 and inducible costimulatory protein Src homology 2 binding domains show distinct regulation of phosphatidylinositol 3-Kinase, Bcl-xL, and IL-2 expression in primary human CD4 T lymphocytes. J Immunol. 2003;171:166–74. doi: 10.4049/jimmunol.171.1.166. [DOI] [PubMed] [Google Scholar]
- 30.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173:945–54. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- 31.Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kovacs B, Parry RV, Ma Z, Fan E, Shivers DK, Freiberg BA, et al. Ligation of CD28 by its natural ligand CD86 in the absence of TCR stimulation induces lipid raft polarization in human CD4 T cells. J Immunol. 2005;175:7848–54. doi: 10.4049/jimmunol.175.12.7848. [DOI] [PubMed] [Google Scholar]
- 33.Chemnitz JM, Lanfranco AR, Braunstein I, Riley JL. B and T lymphocyte attenuator-mediated signal transduction provides a potent inhibitory signal to primary human CD4 T cells that can be initiated by multiple phosphotyrosine motifs. J Immunol. 2006;176:6603–14. doi: 10.4049/jimmunol.176.11.6603. [DOI] [PubMed] [Google Scholar]
- 34.Bowen DL, Lane HC, Fauci AS. Immunopathogenesis of the acquired immunodeficiency syndrome. Ann Intern Med. 1985;103:704–9. doi: 10.7326/0003-4819-103-5-704. [DOI] [PubMed] [Google Scholar]
- 35.Carroll RG, Riley JL, Levine BL, Feng Y, Kaushal S, Ritchey DW, et al. Differential regulation of HIV-1 fusion cofactor expression by CD28 costimulation of CD4+ T cells. Science. 1997;276:273–6. doi: 10.1126/science.276.5310.273. [DOI] [PubMed] [Google Scholar]
- 36.Levine BL, Mosca JD, Riley JL, Carroll RG, Vahey MT, Jagodzinski LL, et al. Antiviral effect and ex vivo CD4 + T Cell proliferation in HIV-positive patients as a result of CD28 costimulation. Science. 1996;272:1939–43. doi: 10.1126/science.272.5270.1939. [DOI] [PubMed] [Google Scholar]
- 37.Levine BL, Humeau LM, Boyer J, MacGregor RR, Rebello T, Lu X, et al. Gene transfer in humans using a conditionally replicating lentiviral vector. Proceedings of the National Academy of Sciences. 2006;103:17372–7. doi: 10.1073/pnas.0608138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Humeau LM, Binder GK, Lu X, Slepushkin V, Merling R, Echeagaray P, et al. Efficient lentiviral vector-mediated control of HIV-1 replication in CD4 lymphocytes from diverse HIV+ infected patients grouped according to CD4 count and viral load. Mol Ther. 2004;9:902–13. doi: 10.1016/j.ymthe.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 39.Stremlau M, Perron M, Welikala S, Sodroski J. Species-specific variation in the B30.2 (SPRY) domain of TRIM5{alpha} determines the potency of human immunodeficiency virus restriction. J Virol. 2005;79:3139–45. doi: 10.1128/JVI.79.5.3139-3145.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. The cytoplasmic body component TRIM5 [alpha] restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–53. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 41.Richardson MW, Carroll RG, Stremlau M, Korokhov N, Humeau LM, Silvestri G, et al. Mode of transmission affects the sensitivity of HIV-1 to restriction by Rhesus TRIM5α. J Virol. 2008 doi: 10.1128/JVI.01046-08. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lederman MM, Veazey RS, Offord R, Mosier DE, Dufour J, Mefford M, et al. Prevention of vaginal SHIV transmission in Rhesus Macaques through inhibition of CCR5. Science. 2004;306:485–7. doi: 10.1126/science.1099288. [DOI] [PubMed] [Google Scholar]
- 43.Mosier DE, Picchio GR, Gulizia RJ, Sabbe R, Poignard P, Picard L, et al. Highly potent RANTES analogues either prevent CCR5-using human immunodeficiency virus type 1áInfection in vivo or rapidly select for CXCR4-using variants. J Virol. 1999;73:3544–50. doi: 10.1128/jvi.73.5.3544-3550.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Santiago Y, Chan E, Liu PQ, Orlando S, Zhang L, Urnov FD, et al. From the cover: targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proceedings of the National Academy of Sciences. 2008;105:5809–14. doi: 10.1073/pnas.0800940105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, et al. Establishment of HIV-1 resistance in CD4(+) T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;7:808–16. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, et al. T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science. 1995;270:475–80. doi: 10.1126/science.270.5235.475. [DOI] [PubMed] [Google Scholar]
- 47.Mitsuyasu RT, Anton PA, Deeks SG, Scadden DT, Connick E, Downs MT, et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood. 2000;96:785–93. [PubMed] [Google Scholar]
- 48.Cooper LJ, Al-Kadhimi Z, DiGiusto D, Kalos M, Colcher D, Raubitschek A, et al. Development and application of CD19-specific T cells for adoptive immunotherapy of B cell malignancies. Blood Cells Mol Dis. 2004;33:83–9. doi: 10.1016/j.bcmd.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 49.Eshhar Z, Waks T, Bendavid A, Schindler DG. Functional expression of chimeric receptor genes in human T cells. J Immunol Methods. 2001;248:67–76. doi: 10.1016/s0022-1759(00)00343-4. [DOI] [PubMed] [Google Scholar]
- 50.Gross G, Gorochov G, Waks T, Eshhar Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant Proc. 1989;21:127–30. [PubMed] [Google Scholar]
- 51.Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 52.Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112:2261–71. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15:825–33. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 54.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–15. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20–e2. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 56.Riley JL, June CH. The CD28 family: a T-cell rheostat for therapeutic control of T-cell activation. Blood. 2005;105:13–21. doi: 10.1182/blood-2004-04-1596. [DOI] [PubMed] [Google Scholar]
- 57.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCR[zeta]/CD28 receptor. Nat Biotech. 2002;20:70–5. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 58.Maric M, Zheng P, Sarma S, Guo Y, Liu Y. Maturation of cytotoxic T lymphocytes against a B7-transfected nonmetastatic tumor: a critical role for costimulation by B7 on both tumor and host antigen-presenting cells. Cancer Res. 1998;58:3376–84. [PubMed] [Google Scholar]
- 59.Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, et al. CD28 Costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995–1004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 60.Altvater B, Pscherer S, Landmeier S, Niggemeier V, Juergens H, Vormoor J, et al. CD28 co-stimulation via tumour-specific chimaeric receptors induces an incomplete activation response in Epstein-Barr virus-specific effector memory T cells. Clin Exp Immunol. 2006;144:447–57. doi: 10.1111/j.1365-2249.2006.03095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Riley JL, Mao M, Kobayashi S, Biery M, Burchard J, Cavet G, et al. Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proceedings of the National Academy of Sciences. 2002;99:11790–5. doi: 10.1073/pnas.162359999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maus MV, Thomas AK, Leonard DGB, Allman D, Addya K, Schlienger K, et al. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4–1BB. Nat Biotech. 2002;20:143–8. doi: 10.1038/nbt0202-143. [DOI] [PubMed] [Google Scholar]
- 63.Nadler LM, Anderson KC, Marti G, Bates M, Park E, Daley JF, et al. B4, a human B lymphocyte-associated antigen expressed on normal, mitogen-activated, and malignant B lymphocytes. J Immunol. 1983;131:244–50. [PubMed] [Google Scholar]
- 64.Hassan R, Bera T, Pastan I, Mesothelin A. New target for immunotherapy. Clin Cancer Res. 2004;10:3937–42. doi: 10.1158/1078-0432.CCR-03-0801. [DOI] [PubMed] [Google Scholar]
- 65.Rudolf D, Silberzahn T, Walter S, Maurer D, Engelhard J, Wernet D, et al. Potent costimulation of human CD8 T cells by anti-4–1BB and anti-CD28 on synthetic artificial antigen presenting cells. Cancer Immunol Immunother. 2008;57:175–83. doi: 10.1007/s00262-007-0360-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang H, Snyder KM, Suhoski MM, Maus MV, Kapoor V, June CH, et al. 4–1BB is superior to CD28 costimulation for generating CD8+ cytotoxic lymphocytes for adoptive immunotherapy. J Immunol. 2007;179:4910–18. doi: 10.4049/jimmunol.179.7.4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Clay TM, Custer MC, Sachs J, Hwu P, Rosenberg SA, Nishimura MI. Efficient transfer of a tumor antigen-reactive TCR to human peripheral blood lymphocytes confers anti-tumor reactivity. J Immunol. 1999;163:507–13. [PubMed] [Google Scholar]
- 68.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Davis MM, Boniface JJ, Reich Z, Lyons D, Hampl J, Arden B, et al. Ligand recognition by alphabeta T cell receptors. Annu Rev Immunol. 1998;16:523–44. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- 70.Fremont DH, Rees WA, Kozono H. Biophysical studies of T-cell receptors and their ligands. Curr Opin Immunol. 1996;8:93–100. doi: 10.1016/s0952-7915(96)80111-7. [DOI] [PubMed] [Google Scholar]
- 71.Garcia KC, Tallquist MD, Pease LR, Brunmark A, Scott CA, Degano M, et al. Alpha beta T cell receptor interactions with syngeneic and allogeneic ligands: affinity measurements andácrystallization. Proceedings of the National Academy of Sciences. 1997;94:13838–43. doi: 10.1073/pnas.94.25.13838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Derby MA, exander-Miller MA, Tse R, Berzofsky JA. High-avidity CTL exploit two complementary mechanisms to provide better protection against viral infection than low-avidity CTL. J Immunol. 2001;166:1690–7. doi: 10.4049/jimmunol.166.3.1690. [DOI] [PubMed] [Google Scholar]
- 73.Dunn SM, Rizkallah PJ, Baston E, Mahon T, Cameron B, Moysey R, et al. Directed evolution of human T cell receptor CDR2 residues by phage display dramatically enhances affinity for cognate peptide-MHC without increasing apparent cross-reactivity. Protein Sci. 2006;15:710–21. doi: 10.1110/ps.051936406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol. 2005;23:349–54. doi: 10.1038/nbt1070. [DOI] [PubMed] [Google Scholar]
- 75.Molloy PE, Sewell AK, Jakobsen BK. Soluble T cell receptors: novel immunotherapies. Curr Opin Pharmacol. 2005;5:438–43. doi: 10.1016/j.coph.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 76.Varela-Rohena A, Molloy PE, Dunn SM, Li Y, Suhoski MM, Carroll RG, et al. Control of HIV-1 immune escape by CD8 T-cells expressing enhanced T-cell receptor. Nat Med. 2008 doi: 10.1038/nm.1779. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Iversen AKN, Stewart-Jones G, Learn GH, Christie N, Sylvester-Hviid C, Armitage AE, et al. Conflicting selective forces affect T cell receptor contacts in an immunodominant human immunodeficiency virus epitope. Nature Immunol. 2006;7:179–89. doi: 10.1038/ni1298. [DOI] [PubMed] [Google Scholar]
- 78.Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, et al. CD8(+) T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med. 2007;13:46–53. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]
- 79.Hacein-Bey-Abina S, Le DF, Carlier F, Bouneaud C, Hue C, De Villartay JP, et al. Sustained correction of X-linked severe combined immunodeficiency by ex vivo gene therapy. N Engl J Med. 2002;346:1185–93. doi: 10.1056/NEJMoa012616. [DOI] [PubMed] [Google Scholar]
- 80.Hacein-Bey-Abina S, von KC, Schmidt M, Le DF, Wulffraat N, McIntyre E, et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2003;348:255–6. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 81.Mitchell RS, Beitzel BF, Schroder ARW, Shinn P, Chen H, Berry CC, et al. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004;2:e234. doi: 10.1371/journal.pbio.0020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Newrzela S, Cornils K, Li Z, Baum C, Brugman MH, Hartmann M, et al. Resistance of mature T cells to oncogene transformation. Blood. 2008;112:2278–86. doi: 10.1182/blood-2007-12-128751. [DOI] [PubMed] [Google Scholar]
- 83.Manilla P, Rebello T, Afable C, Lu X, Slepushkin V, Humeau LM, et al. Regulatory considerations for novel gene therapy products: a review of the process leading to the first clinical lentiviral vector. Hum Gene Ther. 2005;16:17–25. doi: 10.1089/hum.2005.16.17. [DOI] [PubMed] [Google Scholar]
- 84.Sato T, Neschadim A, Konrad M, Fowler DH, Lavie A, Medin JA. Engineered human tmpk/AZT As a novel enzyme/prodrug axis for suicide gene therapy. Mol Ther. 2007;15:962–70. doi: 10.1038/mt.sj.6300122. [DOI] [PubMed] [Google Scholar]
- 85.Brundiers R, Lavie A, Veit T, Reinstein J, Schlichting I, Ostermann N, et al. Modifying human thymidylate kinase to potentiate azidothymidine activation. J Biol Chem. 1999;274:35289–92. doi: 10.1074/jbc.274.50.35289. [DOI] [PubMed] [Google Scholar]