

Abstract
Neurotensin is a peptide known to mimic the actions of antipsychotics, but little is known about how it affects synaptic transmission in the striatum, the major input nucleus of the basal ganglia. In this study we measured the effects of neurotensin on EPSCs from medium spiny projection neurons in the sensorimotor striatum, a region implicated in habit formation and control of motor sequences. We found that bath-applied neurotensin reduced glutamate release from presynaptic terminals, and that this effect required retrograde endocannabinoid signaling, as it was prevented by the CB1 cannabinoid receptor antagonist AM251. Neurotensin-mediated inhibition of striatal EPSCs was also blocked by antagonists of D2-like dopamine receptors and group I metabotropic glutmate receptors, as well as by intracellular calcium chelation and phospholipase C inhibition. These results suggest that neurotensin can indirectly engage an endocannabinoid-mediated negative feedback signal to control glutamatergic input to the basal ganglia.
Keywords: neurotensin, synaptic plasticity, neuropeptides, dopamine, glutamate, basal ganglia, striatum, endocannabinoid, CB1
Introduction
The dorsal striatum (neostriatum or caudate-putamen) is a gateway to the basal ganglia. The dorsolateral striatum, also known as the sensorimotor striatum, receives projections from primary sensory and motor cortices as well as motor thalamic nuclei, and sends projections to downstream basal ganglia structures that eventually influence the control of cortical and brainstem motor systems (Nauta, 1989). Recent studies have shown that this structure plays a critical role in habit formation and motor sequencing (Devan and White, 1999; Jog et al., 1999; Yin et al., 2004; Yin and Knowlton, 2006).
Neurotensin (NT), a 13-amino acid peptide found throughout the mammalian brain, is known to modulate dorsal striatal function (Merchant et al., 1992; Merchant and Dorsa, 1993; Merchant et al., 1994; Dobner et al., 2001; Dobner et al., 2003; Caceda et al., 2006). NT is closely associated with dopaminergic pathways to the striatum (Schotte et al., 1988); NT mRNA and NT receptors are found in dopaminergic neurons and striatal medium spiny neurons (MSNs) (Sugimoto and Mizuno, 1987). NT has also been proposed as an endogenous antipsychotic, because drugs like the typical antipsychotic haloperidol, a D2-like dopamine receptor antagonist, can enhance the expression of NT in the striatum (Caceda et al., 2006); and the ability of haloperidol to increase Fos expression in the dorsolateral striatum is also markedly attenuated by genetic deletion of NT (Dobner et al., 2001).
Of the known NT receptors, NTS1 and NTS2 are coupled to G proteins. NTS1, also known as the high-affinity NT receptor, is usually considered the major target of NT action in the striatum (Boudin et al., 1996; Caceda et al., 2006). However, it is not clear what the effects of NT are on synaptic transmission in the striatum. To understand the functions of NT that may contribute to its antipsychotic actions, it would be helpful to know more about the impact of this neuropeptide on striatal physiology. To this end, we measured the effects of bath-applied NT on excitatory synaptic transmission in MSNs from the dorsolateral striatum.
Materials and methods
All experiments were performed in accordance with NIAAA ACUC and NIH animal care guidelines.
Brain Slice Preparation
Brain slices were prepared from postnatal day 15–19 Sprague-Dawley rats (Gerdeman and Lovinger, 2001). The rats were transcardially perfused with ice-cold modified artificial cerebrospinal fluid (aCSF) containing (in mM): 194 sucrose, 30 NaCl, 4.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 glucose; they were then decapitated, and their brains transferred rapidly to the modified aCSF (pH set at 7.4 by aeration with 95% O2/5% CO2). Coronal sections (350 μm thick) were cut in ice-cold modified aCSF using an Integraslice 7550 (Campden instruments, UK). Slices were transferred immediately to a nylon net submerged in normal aCSF containing (in mM): 124 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 D-glucose. Normal aCSF was maintained at pH 7.4 by bubbling with 95% O2/5% CO2 at room temperature (19–22°C). Following at least 1 hr of incubation at room temperature, hemi-slices were transferred to a recording chamber, submerged in normal ACSF. For all experiments, the temperature of the bath was maintained at 28–31°C stable within +/− 1°C during any given experiment.
Whole-Cell Voltage-Clamp Recording
Whole-cell recordings from MSNs were performed as previously described (Gerdeman, 2002). Pipettes were pulled from borosilicate glass on a Flaming-Brown micropipette puller (Novato, CA). Test stimuli (2 pulses 50 ms apart) were delivered via a Master-8 stimulator (A.M.P.I., Jerusalem, Israel) every 20 seconds through a bipolar twisted tungsten wire placed in the dorsolateral striatum or in the white matter adjacent to it. Pipette resistance ranged from 2.5–4.5 MΩ, when filled with an internal solution containing (in mM): 120 cesium methane sulfonate, 5 NaCl, 10 tetraethylammonium chloride, 10 HEPES, 4 lidocaine N-ethyl bromide, 1.1 EGTA, 4 Mg-ATP, and 0.3 Na-GTP, pH adjusted to 7.2 with CsOH, and osmolarity set to 298 mOsm with sucrose. The calcium chelator BAPTA (20 mM) was added to the internal for the experiments designed to lower intracellular calcium concentration.
The osmolarity of the external solution (normal aCSF) was adjusted to 310–315 mOsm with sucrose, and 50 μM picrotoxin was added to the solution to block GABAA receptor-mediated currents. Recordings were made from MSNs (soma diameter 10–15 μm) identified visually with the aid of differential interference contrast (DIC)-enhanced visual guidance. Cells were voltage-clamped at −60 to −70 mV throughout the experiments, and the stimulus intensity was set to the level at which EPSC amplitude was 200–600 pA (0.3–1.6 mA, 0.04–0.12 ms). We analyzed only recordings with series resistance <25 MΩ (usually ranging from 9 to 20 MΩ). The series resistance was not compensated, and if it changed by more than 20% during the course of an experiment, the cell was discarded.
Synaptic currents were recorded with an Axopatch 1D amplifier (Axon Instruments, Foster City, CA), filtered at 5 kHz, digitized at 10 kHz, and stored on a Dell microcomputer (Round Rock, TX). EPSC amplitudes were measured using peak detection software in pCLAMP8 (Union City, CA).
After initiation of the whole-cell recording, we started synaptic stimulation immediately, and began the experiment once a stable baseline was observed, usually after 5 to 10 minutes. Then, after recording for approximately 3 minutes, NT was bath applied for approximately 7 minutes, followed by a 15-minute washout with normal aCSF. AM251 was first dissolved in DMSO, and the stock was added to the normal ACSF before experiments each day at a final DMSO concentration of 0.02% along with 0.1% BSA. Sulpiride, CPCCOEt and MPEP were first dissolved in DMSO, with a final DMSO concentration of 0.02% in ACSF. SR 142948 was first dissolved in DMSO, with a final DMSO concentration of 0.01% in ACSF. In most of the experiments examining receptor antagonist actions, the antagonist was present throughout the recording. The only exception was application of AM251 following NT application.
NT, sulpiride, MPEP, CPCCOEt, AM251, and BAPTA were purchased from Sigma (St. Louis, MO). U73122 and SR 142948 were purchased from Tocris (Bristol, UK).
Results
Neurotensin reduces evoked EPSC amplitude in medium spiny neurons
For each group, planned comparisons using paired t-tests were made between baseline amplitude of the first EPSC (average of 10 traces) and EPSC amplitude immediately after washout (average of 10 traces). As shown in Figure 1A, bath application of 0. 5 μM NT did not affect EPSCs when the stimulation electrode was placed in the white matter (100±7% of baseline, p>0.05), but caused a significant reduction in EPSC amplitude when stimulation was applied within in the dorsolateral striatum (70±7% of baseline, p<0.05). To see whether a higher dose of NT can reduce EPSCs evoked by white matter stimulation, we also applied 1 μM NT, but no significant inhibition was observed (85±8% of baseline, p>0.05; n=6, data not shown). On the basis of these observations, we performed all other experiments with the stimulating electrode placed in the striatum.
Figure 1.

Bath application of NT reduced amplitude of EPSCs recorded from MSNs in the dorsolateral striatum. A) NT did not affect EPSC amplitude when the stimulating electrode was placed in the white matter just above the dorsolateral striatum, but a significant inhibition was observed after NT application when the stimulating electrode was placed in the striatum. B) A replication of the results using intrastriatal stimulation with different doses of NT, showing the amplitude of the first EPSC for each group.
NT dose-dependently reduced the amplitude of EPSCs recorded from the striatum (Figure 1B; 10 nM, 93±5% of baseline, p>0.05; 0.1 μM, 85±7% of baseline, p>0.05; 0.5 μM, 77±5% of baseline, p<0.05; 1 μM, 80±5% of baseline, p<0.05). Because 0.5 μM was found to be the most effective dose in inhibiting the EPSCs, we used this dose for subsequent experiments.
We found that 0.5 μM NT significantly reduced EPSC amplitude (Figure 2A; 77±5% of baseline, p<0.05) while increasing paired-pulse ratio (PPR, second EPSC divided by first EPSC; baseline PPR = 0.97±0.06, after NT PPR = 1.10±0.04, p<0.05). The effect of NT on EPSCs did not appear to recover during the subsequent 15 min wash with aCSF. Application of NT did not alter the holding current at −70 mV, and did not alter input resistance measured from the steady-state component of 10 mV hyperpolarizing pulses (data not shown). In the presence of the potent NTS1 antagonist SR 142948 (Figure 2B), NT did not reduce EPSC amplitude (91±5% of baseline, p>0.05; baseline PPR = 0.97±0.08, after NT PPR = 0.99±0.08), showing that the effects of NT on glutamatergic transmission are due to the activation of NTS1 receptors (Gully et al., 1997).
Figure 2.

A) The results using intrastriatal stimulation with 0.5 µM NT, the most effective dose in inhibiting striatal EPSCs, showing the changes in the first and second of the paired EPSCs. The two pulses were 50 ms apart, and given every 20 s. Note the relatively larger inhibition of the first EPSC of the pair. B) In the presence of the NTS1 antagonist SR 142948, NT failed to alter EPSC amplitude or PPR.
NT-mediated reduction of EPSC amplitude requires activation of D2 and group I metabotropic glutamate receptors
The increase in PPR during NT-mediated inhibition of EPSCs suggests that the neuropeptide produces a decrease in release probability. However, NT receptors are known to activate Gq-coupled G-proteins, a signaling system not usually involved in direct presynaptic inhibiton. We therefore considered the possibility that NT may work indirectly, possibly via interactions with dopamine and glutamate, the two predominant neurotransmitters in the striatum. It is well known that D2 receptor activation can produce transient presynaptic depression in striatum and also participate in induction of presynaptically-expressed long-term depression (LTD) in this brain region (Kreitzer and Malenka, 2005; Yin et al., 2006). Furthermore, both of these forms of presynaptic depression require the activation of postsynaptic group I mGluRs (Calabresi et al., 1992). To test whether NT-mediated inhibition also requires the activation of these receptors, we measured the effects of NT application in the presence of selective antagonists of D2 receptors and mGluRs.
No EPSC changes were observed after NT application in the presence of the selective D2 receptor antagonist sulpiride in the bath (Figure 3A; 100±11% of baseline, P>0.05; baseline PPR = 1.17±0.05, after NT PPR = 1.24±0.06, p>0.05). But when sulpiride was applied after NT, no reversal of the NT-mediated depression was observed (Figure 3B; 10 min after sulpiride application, 60±8% of baseline, P<0.05). These findings indicate that D2 actiavation is necessary for the initiation of NT-induced synaptic depression, but sustained activation of D2 receptors is not necessary to maintain this depression.
Figure 3.
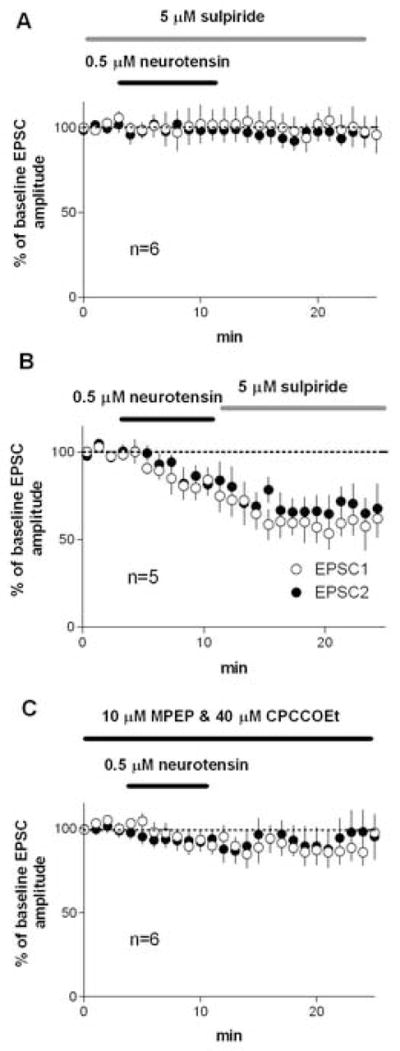
NT-induced inhibition of EPSCs in the dorsolateral striatum was blocked by the D2 receptor antagonist sulpiride and by group I mGluR antagonists. A) Experiment performed in the presence of sulpiride, showing that NT failed to reduce EPSC amplitude. B) Experiment performed with sulpiride applied after NT application, showing that sulpiride applied after the onset of inhibition failed to reverse it. C) Experiment performed in the presence of CPCCOEt (mGluR 1 antagonist) and MPEP (mGluR 5 antagonist), showing that NT failed to reduce EPSC amplitude.
Likewise, synaptic depression and the change in PPR were blocked in the presence of a cocktail of group I mGluR antagonists—the mGluR 1 antagonist CPCCOEt and the mGluR 5 antagonist MPEP (Figure 2B; 91±7% of baseline, P>0.05; baseline PPR = 0.98±0.03, after NT PPR = 1.01±0.05, p>0.05). Hence the NT-induced synaptic depression appears to involve dopaminergic and glutamatergic transmission.
NT-mediated reduction of glutamate release requires CB1 receptor activation, a rise in intracellular calcium, and PLC activation
In the dorsolateral striatum, CB1 cannabinoid receptors are found on the glutamatergic terminals synapsing on MSNs, and activation of these receptors can reduce glutamate release (Gerdeman and Lovinger, 2001). The D2- and mGluR-mediated forms of synaptic depression described in previous studies (Kreitzer and Malenka, 2005; Yin and Lovinger, 2006) are mediated by postsynaptic endocannabinoid release leading to the activation of presynaptic CB1 receptors. To determine if a similar retrograde signaling mechanism is involved in NT-mediated synaptic depression we examined the effect of CB1 blockade on NT actions. In the presence of the selective CB1 antagonist AM251, NT did not reduce EPSC amplitude (Figure 4A; 96±3% of baseline, p>0.05), or cause any change in PPR (baseline PPR = 1.14±0.07, after NT PPR =1.13±0.08).
Figure 4.

NT-induced inhibition of EPSCs in the dorsolateral striatum was blocked and reversed by CB1 receptor antagonism. A) Experiment performed in the continuous present of the CB1 antagonist AM251, NT failed to reduce EPSC amplitude. B) Experiment in which AM251 was applied just after the end of the NT application, showing that the NT-induced inhibition of EPSCs was reversed by bath application of AM251.
Since NT-induced inhibition did not reverse during extended washout, NT might induce a form of LTD similar to that induced by high frequency synaptic activation. However, striatal LTD, once established, is resistant to CB1 blockade (Ronesi et al. 2004). To determine if the CB1 antagonist would reverse established NT-induced synaptic depression, we applied AM251 at the beginning of the post-NT washout period (after 7 min of NT application). The reduction in EPSC amplitude as well as the increased PPR were partially, but not completely reversed by AM251 applied after NT treatment (Figure 4B; after NT EPSC amplitude = 54±6% of baseline, after AM251 EPSC amplitude = 83±3%; baseline PPR=1.22±0.12; after NT PPR = 1.49±0.14; after AM251 PPR = 1.23±0.01). This finding suggests that the lack of reversibility of NT-induced depression stems mainly from continued CB1 activation, perhaps due to a lack of washout of the peptide or continued endocannabinoid production, and thus it does not resemble LTD. However, there is a small component of remaining depression even after AM251 treatment, suggesting that NT may induce a small amount of LTD.
The production and release of endocannabinoids have been shown to depend on a rise in intracellular calcium (Piomelli, 2003), and chelating intracellular calcium also prevents D2/mGluR-mediated synaptic depression in striatum (Yin et al., 2006b). As shown in Figure 5A, the NT-induced reduction of EPSC amplitude was blocked when 20 mM BAPTA was loaded into the postsynaptic medium spiny neuron with the patch pipette (EPSC amplitude = 102±5% of baseline, p>0.05; baseline PPR = 1.06±0.04, after NT PPR = 1.06±0.04). Similarly, NT-mediation inhibition was also blocked by intracellular loading of the PLC inhibitor U73122 (EPSC amplitude = 98±9% of baseline, p>0.05; baseline PPR = 1.19±0.08, after NT PPR = 1.15±0.06, p>0.05).
Figure 5.
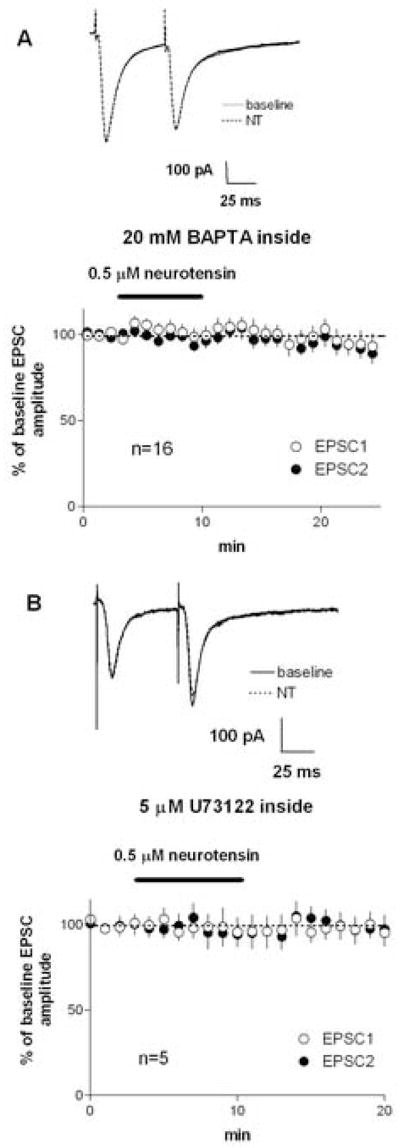
NT-induced inhibition of EPSCs in the dorsolateral striatum requires a rise in intracellular calcium and the activation of the PLC pathway. A) When the fast calcium chelator BAPTA was loaded into the postsynaptic cell via the patch pipette, no inhibition of EPSCs was observed after NT application. B) When the PLC inhibitor U73122 was loaded into the postsynaptic cell via the patch pipette, no inhibition of EPSCs was observed after NT application.
Discussion
We observed that NT inhibits glutamatergic transmission in the dorsolateral striatum, probably by reducing presynaptic glutamate release, since the observed reduction in EPSC amplitude was accompanied by an increase in PPR, an indication of reduced presynaptic release probability (Figure 2A). This is a new and somewhat surprising observation, as most previous neurochemical studies found that NT either increases the release of transmitters like dopamine and glutamate in the striatum (Okuma et al., 1983; Battaini et al., 1986; Hetier et al., 1988; Ferraro et al., 1995; Ferraro et al., 1998; Diaz-Cabiale et al., 2002; Matsuyama et al., 2002; Matsuyama et al., 2003) or increases the excitability of the postsynaptic cell in substantia nigra and medial septum/diagonal band of Broca (Mercuri et al., 1993; Matthews, 1999). By blocking the NT-induced inhibition of striatal EPSCs with the NTS1 receptor antagonist SR 142948 (Gully et al., 1997), we showed that this effect on glutamatergic transmission depends on the activation of NTS1 receptors. Most interestingly, we found that NT-induced inhibition of EPSCs requires retrograde signaling by endocannabinoids, because blockade of CB1 receptors, which are located on the presynaptic terminals, prevented the effect (Figure 4A).
It is well-established that the production and release of endocannabinoids can be stimulated by a rise of intracellular calcium (Piomelli, 2003; Hashimotodani et al., 2005). We therefore tested whether chelating intracellular calcium with BAPTA has any effect on NT-mediated inhibition, and found that this manipulation prevented the observed reduction in glutamatergic transmission (Figure 5A). A rise in intracellular calcium is therefore necessary for the NT-induced inhibition of EPSCs, suggesting that the postsynaptic MSN is the site of endocannabinoid synthesis and release. This finding also indicates that the postsynaptic neuron is the most likely locus of NTS1-stimulated endocannabinoid production. Likewise, PLC has been shown to be a critical enzyme involved in 2-AG synthesis (Piomelli, 2003) and can also be involved in AEA synthesis(Liu et al., 2006), and we found that the PLC inhibitor U73122 loaded into the postsynaptic neuron also blocked the NT-mediated inhibition of EPSCs (Figure 5B).
We also examined the role of the two major striatal neurotransmitters, dopamine and glutamate, in NTS-stimulated endocannabinoid signaling. It has been reported that activation of striatal D2 receptors and group I mGluRs can both result in endocannabinoid production and release (Giuffrida et al., 1999; Maejima et al., 2001; Piomelli, 2003; Kushmerick et al., 2004; Maejima et al., 2005; Yin and Lovinger, 2006). Furthermore, NT has been shown to increase DA release and activate dopamine-dependent signaling pathways in striatal slices(Okuma et al., 1983; Battaini et al., 1986; Hetier et al., 1988; Matsuyama et al., 2002). We found that blockade of group I mGluRs and D2 receptors prevented the NT-induced inhibition of EPSCs in the dorsolateral striatum (Figure 3). These receptors can potentially act downstream of NT, but we cannot yet rule out additive or synergistic effects of the drugs (e.g. on postsynaptic endocannabinoid production).
Possible mechanisms for NT-induced inhibition of EPSCs
As NT is often thought to increase glutamate release in the striatum (Matsuyama et al., 2003), our observation that it inhibits striatal EPSCs via retrograde endocannabinoid signaling is surprising. There are three possible mechanisms for the observed effect: 1) enhanced glutamate release activating extrasynaptically located group I mGluRs; 2) direct activation of the Gq-coupled NTS1 on dendrites and soma of MSNs, with a consequent increase in intracellular calcium; and 3) enhanced dopamine release activating postsynaptic D2 receptors.
First, as shown by previous studies, simply by enhancing glutamate release from cortical and thalamic afferents NT could activate postsynaptically located mGluRs, which have been implicated in endocannabinoid production and release (Giuffrida et al., 1999; Maejima et al., 2001; Yin and Lovinger, 2006). When coupled to the activation of postsynaptic D2 receptors (which is likely given dopamine release likely caused by stimulation of dopaminergic terminals by the intrastriatally placed stimulating electrode used in our experiments), this could trigger the retrograde endocannabinoid signal. This possibility is attractive in view of the fact that NTS1 receptors are abundant on presynaptic terminals in the striatum (Boudin et al., 1996). However, if there is indeed increased glutamate release we should be able to observe it directly as a postsynaptic ionotropic receptor-mediated current or an increase in EPSC amplitude. Indeed, when we blocked the group I mGluRs or applied high intracellular BAPTA, thus eliminating retrograde signaling by endocannabinoids, the potentiating effect of NT on presynaptic release should have been observed. As we failed to observe consistent potentiation of glutamate release under such conditions, no direct evidence is provided by our data in support of this possible mechanism.
Secondly, it is possible that NT acts on somatodendritic NTS1 receptors on the MSNs themselves. Because this receptor is Gq-coupled, and known to activate the Phospholipase C pathway (Hermans et al., 1992; Trudeau, 2000; Belmeguenai et al., 2003), it could directly lead to endocannbinoid production and release (Piomelli, 2003). Thus NTS1 could be similar to group I mGluRs, which are also Gq-coupled, the activation of which is known to result in endocannabinoid production and release. While this is certainly an intriguing possibility, it is less plausible because NTS1 receptors are predominantly found on axon terminals in the striatum, though this issue has not been examined thoroughly (Delle Donne et al., 2004). Furthermore, we did not observe any evidence that NT is capable of activating endocannabinoid signaling in the absence of dopamine and glutamate actions, casting some doubt on the idea that NT activation of postsynaptic Gq-coupled receptors plays any direct role in synaptic depression. Still, this hypothesis cannot be ruled out at present.
Finally, since NTS1 and D2 receptors are colocalized within single axon terminals, it is possible that NT simply potentiates dopamine release by antagonizing the function of presynaptic D2 autoreceptors, as has previously been suggested (Chapman et al., 1992; Diaz-Cabiale et al., 2002; Delle Donne et al., 2004). Indeed, it is well documented that NT stimulates dopamine release in striatal slices(Okuma et al., 1983; Battaini et al., 1986; Hetier et al., 1988). One interesting observation from the current study is the difference between white matter and striatal stimulation electrode placements. When the stimulating electrode was placed in the white matter just above the striatum, no effect of NT was observed, but when it was placed in the striatum, NT reduced glutamatergic transmission (Figure 1A). Compared with white matter stimulation, intrastriatal stimulation is more likely to activate dopaminergic fibers directly, or at least to cause more dopamine release. Thus NT may only exert its effects on dopamine release (e.g. potentiate DA release) when dopaminergic terminals are already being activated by synaptic stimulation.
What, then, is the consequence of enhanced dopamine release? Recent work has shown that activation of D2 receptors with the selective agonist quinpirole, combined with sufficient synaptically evoked glutamate release can trigger retrograde signaling by endocannabinoids (Yin and Lovinger, 2006). As already mentioned, LTD in the dorsolateral striatum also requires retrograde endocannabinoid signaling (Gerdeman et al., 2002), but since NT-induced reduction in striatal EPSCs is nearly completely reversed by the CB1 antagonist AM251 (Figure 3B), it is more similar to the inhibition of corticostriatal EPSCs by the D2 receptor agonist (Yin and Lovinger, 2006). However, a small amount of inhibition persisted even after prolonged AM251 application, suggesting that NT might induce LTD at a small number of synapses. This possibility can be investigated in future studies. It should be noted that in this experiment AM251 application began several minutes after the onset of NT-induced inhibition, a time point at which LTD would be CB1-independent (Ronesi et al., 2004). Interestingly, quinpirole-mediated inhibition of EPSCs was only observed when the stimulating electrode was placed in the cortex, because striatal stimulation could activate D2 receptors by stimulating local dopamine release even in the absence of quinpirole. By contrast, in the present study, striatal, rather than cortical, stimulation was needed to observe the effects of NT, suggesting that dopamine release is a necessary condition for inhibitory effect of NT on striatal EPSCs. Moreover, since the D2 receptor antagonist sulpiride blocked NT-mediated inhibition, any NT effect on dopamine release would likely involve activation of the same pool of D2 receptors involved in D2 agonist-induced synaptic depression. Interestingly, sulpiride could not reverse NT-induced synaptic depression once it had been established, in contrast to the actions of AM251. This finding indicates that sustained endocannabinoid release and CB1 activation does not require continued D2 activation.
The data from our study cannot rule out any of the three possibilities listed above; nor, it should be stressed, are these possibilities mutually exclusive. In particular, we can suggest the following combination—potentiated dopamine release by NT activating on presynaptically located NTS1 receptors, simultaneously activating postsynaptically-located D2 receptors, combined with the activation of postynaptically located NTS1 receptors and consequent rise in intracellular calcium through Gq-coupled pathways. As already mentioned, such a mechanism is in fact quite similar to that previously found to underlie inhibition of glutamatergic transmission during coincident activation of group I mGluRs and D2 receptors in the dorsolateral striatum (Yin and Lovinger, 2006). It is even possible that presynaptic NT receptors are involved in the initial, D2-dependent phase of synaptic depression, while postsynaptic NTS1 receptors contribute to maintenance of endocannabinoid actions at later time points when inhibition becomes independent of D2 receptors.
In any case, we have discovered a new role for NT in inhibiting excitatory transmission in the striatum, and regardless of the precise mechanisms underlying this effect, it is clear that the role of NT in modulating striatal activity is more counterintuitive than previously work measuring immediate early gene expression has suggested (Dobner et al., 2003; Caceda et al., 2006). If, for example, increased dopamine release acting on D2 receptors turns out to be responsible for NT-mediated inhibition of EPSCs in the dorsolateral striatum, then the control of glutamatergic inputs to the striatum, rather than the reduction of dopaminergic transmission, would appear to be a relevant mechanism for NT’s antipsychotic actions. This intriguing possibility clearly awaits further investigation.
Acknowledgments
This research was supported by the Division of Intramural Clinical and Basic Research of the NIH, NIAAA. We would like to thank Dr. Margaret Davis for helpful discussion of the data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Battaini F, Govoni S, Di Giovine S, Trabucchi M. Neurotensin effect on dopamine release and calcium transport in rat striatum: interactions with diphenylalkylamine calcium antagonists. Naunyn Schmiedebergs Arch Pharmacol. 1986;332:267–270. doi: 10.1007/BF00504865. [DOI] [PubMed] [Google Scholar]
- Belmeguenai A, Desrues L, Leprince J, Vaudry H, Tonon MC, Louiset E. Neurotensin stimulates both calcium mobilization from inositol trisphosphate-sensitive intracellular stores and calcium influx through membrane channels in frog pituitary melanotrophs. Endocrinology. 2003;144:5556–5567. doi: 10.1210/en.2003-0176. [DOI] [PubMed] [Google Scholar]
- Boudin H, Pelaprat D, Rostene W, Beaudet A. Cellular distribution of neurotensin receptors in rat brain: immunohistochemical study using an antipeptide antibody against the cloned high affinity receptor. J Comp Neurol. 1996;373:76–89. doi: 10.1002/(SICI)1096-9861(19960909)373:1<76::AID-CNE7>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Caceda R, Kinkead B, Nemeroff CB. Neurotensin: role in psychiatric and neurological diseases. Peptides. 2006;27:2385–2404. doi: 10.1016/j.peptides.2006.04.024. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci. 1992;12:4224–4233. doi: 10.1523/JNEUROSCI.12-11-04224.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman MA, See RE, Bissette G. Neurotensin increases extracellular striatal dopamine levels in vivo. Neuropeptides. 1992;22:175–183. doi: 10.1016/0143-4179(92)90160-x. [DOI] [PubMed] [Google Scholar]
- Delle Donne KT, Chan J, Boudin H, Pelaprat D, Rostene W, Pickel VM. Electron microscopic dual labeling of high-affinity neurotensin and dopamine D2 receptors in the rat nucleus accumbens shell. Synapse. 2004;52:176–187. doi: 10.1002/syn.20018. [DOI] [PubMed] [Google Scholar]
- Devan BD, White NM. Parallel information processing in the dorsal striatum: relation to hippocampal function. J Neurosci. 1999;19:2789–2798. doi: 10.1523/JNEUROSCI.19-07-02789.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Cabiale Z, Fuxe K, Narvaez JA, Finetti S, Antonelli T, Tanganelli S, Ferraro L. Neurotensin-induced modulation of dopamine D2 receptors and their function in rat striatum: counteraction by a NTR1-like receptor antagonist. Neuroreport. 2002;13:763–766. doi: 10.1097/00001756-200205070-00006. [DOI] [PubMed] [Google Scholar]
- Dobner PR, Deutch AY, Fadel J. Neurotensin: dual roles in psychostimulant and antipsychotic drug responses. Life Sci. 2003;73:801–811. doi: 10.1016/s0024-3205(03)00411-9. [DOI] [PubMed] [Google Scholar]
- Dobner PR, Fadel J, Deitemeyer N, Carraway RE, Deutch AY. Neurotensin-deficient mice show altered responses to antipsychotic drugs. Proc Natl Acad Sci U S A. 2001;98:8048–8053. doi: 10.1073/pnas.141042198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro L, Tanganelli S, O’Connor WT, Bianchi C, Ungerstedt U, Fuxe K. Neurotensin increases endogenous glutamate release in the neostriatum of the awake rat. Synapse. 1995;20:362–364. doi: 10.1002/syn.890200409. [DOI] [PubMed] [Google Scholar]
- Ferraro L, Antonelli T, O’Connor WT, Fuxe K, Soubrie P, Tanganelli S. The striatal neurotensin receptor modulates striatal and pallidal glutamate and GABA release: functional evidence for a pallidal glutamate-GABA interaction via the pallidal-subthalamic nucleus loop. J Neurosci. 1998;18:6977–6989. doi: 10.1523/JNEUROSCI.18-17-06977.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman G, Lovinger DM. CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J Neurophysiol. 2001;85:468–471. doi: 10.1152/jn.2001.85.1.468. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoids release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Giuffrida A, Parsons LH, Kerr TM, Rodriguez de Fonseca F, Navarro M, Piomelli D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci. 1999;2:358–363. doi: 10.1038/7268. [DOI] [PubMed] [Google Scholar]
- Gully D, Labeeuw B, Boigegrain R, Oury-Donat F, Bachy A, Poncelet M, Steinberg R, Suaud-Chagny MF, Santucci V, Vita N, Pecceu F, Labbe-Jullie C, Kitabgi P, Soubrie P, Le Fur G, Maffrand JP. Biochemical and pharmacological activities of SR 142948A, a new potent neurotensin receptor antagonist. J Pharmacol Exp Ther. 1997;280:802–812. [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto K, Maejima T, Araishi K, Shin HS, Kano M. Phospholipase Cbeta serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron. 2005;45:257–268. doi: 10.1016/j.neuron.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Hermans E, Maloteaux JM, Octave JN. Phospholipase C activation by neurotensin and neuromedin N in Chinese hamster ovary cells expressing the rat neurotensin receptor. Brain Res Mol Brain Res. 1992;15:332–338. doi: 10.1016/0169-328x(92)90126-v. [DOI] [PubMed] [Google Scholar]
- Hetier E, Boireau A, Dubedat P, Blanchard JC. Neurotensin effects on evoked release of dopamine in slices from striatum, nucleus accumbens and prefrontal cortex in rat. Naunyn Schmiedebergs Arch Pharmacol. 1988;337:13–17. doi: 10.1007/BF00169470. [DOI] [PubMed] [Google Scholar]
- Jog MS, Kubota Y, Connolly CI, Hillegaart V, Graybiel AM. Building neural representations of habits. Science. 1999;286:1745–1749. doi: 10.1126/science.286.5445.1745. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushmerick C, Price GD, Taschenberger H, Puente N, Renden R, Wadiche JI, Duvoisin RM, Grandes P, von Gersdorff H. Retroinhibition of presynaptic Ca2+ currents by endocannabinoids released via postsynaptic mGluR activation at a calyx synapse. J Neurosci. 2004;24:5955–5965. doi: 10.1523/JNEUROSCI.0768-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wang L, Harvey-White J, Osei-Hyiaman D, Razdan R, Gong Q, Chan AC, Zhou Z, Huang BX, Kim HY, Kunos G. A biosynthetic pathway for anandamide. Proc Natl Acad Sci U S A. 2006;103:13345–13350. doi: 10.1073/pnas.0601832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31:463–475. doi: 10.1016/s0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- Maejima T, Oka S, Hashimotodani Y, Ohno-Shosaku T, Aiba A, Wu D, Waku K, Sugiura T, Kano M. Synaptically driven endocannabinoid release requires Ca2+-assisted metabotropic glutamate receptor subtype 1 to phospholipase Cbeta4 signaling cascade in the cerebellum. J Neurosci. 2005;25:6826–6835. doi: 10.1523/JNEUROSCI.0945-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S, Fukui R, Higashi H, Nishi A. Regulation of DARPP-32 Thr75 phosphorylation by neurotensin in neostriatal neurons: involvement of glutamate signalling. Eur J Neurosci. 2003;18:1247–1253. doi: 10.1046/j.1460-9568.2003.02859.x. [DOI] [PubMed] [Google Scholar]
- Matsuyama S, Higashi H, Maeda H, Greengard P, Nishi A. Neurotensin regulates DARPP-32 thr34 phosphorylation in neostriatal neurons by activation of dopamine D1-type receptors. J Neurochem. 2002;81:325–334. doi: 10.1046/j.1471-4159.2002.00822.x. [DOI] [PubMed] [Google Scholar]
- Matthews RT. Neurotensin depolarizes cholinergic and a subset of non-cholinergic septal/diagonal band neurons by stimulating neurotensin-1 receptors. Neuroscience. 1999;94:775–783. doi: 10.1016/s0306-4522(99)00364-4. [DOI] [PubMed] [Google Scholar]
- Merchant KM, Dorsa DM. Differential induction of neurotensin and c-fos gene expression by typical versus atypical antipsychotics. Proc Natl Acad Sci U S A. 1993;90:3447–3451. doi: 10.1073/pnas.90.8.3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant KM, Dobner PR, Dorsa DM. Differential effects of haloperidol and clozapine on neurotensin gene transcription in rat neostriatum. J Neurosci. 1992;12:652–663. doi: 10.1523/JNEUROSCI.12-02-00652.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant KM, Dobie DJ, Filloux FM, Totzke M, Aravagiri M, Dorsa DM. Effects of chronic haloperidol and clozapine treatment on neurotensin and c-fos mRNA in rat neostriatal subregions. J Pharmacol Exp Ther. 1994;271:460–471. [PubMed] [Google Scholar]
- Mercuri NB, Stratta F, Calabresi P, Bernardi G. Neurotensin induces an inward current in rat mesencephalic dopaminergic neurons. Neurosci Lett. 1993;153:192–196. doi: 10.1016/0304-3940(93)90320-k. [DOI] [PubMed] [Google Scholar]
- Nauta WJH. Reciprocal links of the corpus striatum with the cerebral cortex and limbic system: A common substrate for movement and thought? In: Mueller, editor. Neurology and psychiatry: a meeting of minds. Basel: Karger; 1989. pp. 43–63. [Google Scholar]
- Okuma Y, Fukuda Y, Osumi Y. Neurotensin potentiates the potassium-induced release of endogenous dopamine from rat striatal slices. Eur J Pharmacol. 1983;93:27–33. doi: 10.1016/0014-2999(83)90027-4. [DOI] [PubMed] [Google Scholar]
- Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- Ronesi J, Gerdeman GL, Lovinger DM. Disruption of endocannabinoid release and striatal long-term depression by postsynaptic blockade of endocannabinoid membrane transport. J Neurosci. 2004;24:1673–1679. doi: 10.1523/JNEUROSCI.5214-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotte A, Rostene W, Laduron PM. Different subcellular localization of neurotensin-receptor and neurotensin-acceptor sites in the rat brain dopaminergic system. J Neurochem. 1988;50:1026–1031. doi: 10.1111/j.1471-4159.1988.tb10568.x. [DOI] [PubMed] [Google Scholar]
- Sugimoto T, Mizuno N. Neurotensin in projection neurons of the striatum and nucleus accumbens, with reference to coexistence with enkephalin and GABA: an immunohistochemical study in the cat. J Comp Neurol. 1987;257:383–395. doi: 10.1002/cne.902570307. [DOI] [PubMed] [Google Scholar]
- Trudeau LE. Neurotensin regulates intracellular calcium in ventral tegmental area astrocytes: evidence for the involvement of multiple receptors. Neuroscience. 2000;97:293–302. doi: 10.1016/s0306-4522(99)00597-7. [DOI] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ. The role of the basal ganglia in habit formation. Nat Rev Neurosci. 2006;7:464–476. doi: 10.1038/nrn1919. [DOI] [PubMed] [Google Scholar]
- Yin HH, Lovinger DM. Frequency-specific and D2 receptor-mediated inhibition of glutamate release by retrograde endocannabinoid signaling. Proc Natl Acad Sci U S A. 2006;103:8251–8256. doi: 10.1073/pnas.0510797103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ, Balleine BW. Lesions of dorsolateral striatum preserve outcome expectancy but disrupt habit formation in instrumental learning. Eur J Neurosci. 2004;19:181–189. doi: 10.1111/j.1460-9568.2004.03095.x. [DOI] [PubMed] [Google Scholar]
- Yin HH, Davis MI, Ronesi JA, Lovinger DM. The role of protein synthesis in striatal long-term depression. J Neurosci. 2006;26:11811–11820. doi: 10.1523/JNEUROSCI.3196-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]