

SUMMARY
RanBP2 is a nucleoporin with SUMO E3 ligase activity that functions in both nucleocytoplasmic transport and mitosis. However, the biological relevance of RanBP2 and the in vivo targets of its E3 ligase activity are unknown. Here we show that animals with low amounts of RanBP2 develop severe aneuploidy in the absence of overt transport defects. The main chromosome segregation defect in cells from these mice is anaphase-bridge formation. Topoisomerase IIα (Topo IIα), which decatenates sister centromeres prior to anaphase onset to prevent bridges, fails to accumulate at inner centromeres when RanBP2 levels are low. We find that RanBP2 sumoylates Topo IIα in mitosis and that this modification is required for its proper localization to inner centromeres. Furthermore, mice with low amounts of RanBP2 are highly sensitive to tumor formation. Together, these data identify RanBP2 as a chromosomal instability gene that regulates Topo IIα by sumoylation and suppresses tumorigenesis.
INTRODUCTION
Most human cancers have an abnormal chromosome content, a condition known as aneuploidy. However, the molecular defects underlying the development of aneuploidy and its role in tumorigenesis remain poorly understood (Michor et al., 2005). Deciphering the molecular networks that regulate the proper segregation of chromosomes in mitosis is essential to understanding the mechanisms that can cause chromosomal instability and their role in cancer development. In Saccharomyces cerevisiae, more than 100 genes are known to cause chromosomal instability when defective (Kolodner et al., 2002; Nasmyth, 2002). These genes function in a wide variety of mitotic processes, including chromosome condensation, sister chromatid cohesion, kinetochore assembly, spindle formation, and spindle assembly checkpoint control. Even more genes are expected to contribute to chromosomal stability in humans, but only a limited number have been identified to date (Michor et al., 2005).
Several proteins that mediate transport of macromolecules into and out of the nucleus through nuclear pores have recently been implicated in mitosis. One of these proteins is RanBP2 (or Nup358), a very large multidomain nuclear pore complex protein that exerts SUMO E3 ligase activity in vitro (Matunis and Pickart, 2005; Pichler et al., 2002; Wu et al., 1995). In interphase cells, RanBP2 is localized at the cytoplasmic face of the NPC, where it forms a stable subcomplex with Ran GTPase-activating protein 1 (RanGAP1) and the SUMO E2-conjugating enzyme Ubc9. Only RanGAP1 conjugated to small ubiquitin-like modifier 1 (SUMO1) can interact with RanBP2 (Mahajan et al., 1997; Matunis et al., 1996). Depletion of RanBP2 has no measurable effect on import of proteins into the nucleus but slightly reduces export of mRNA and NES-containing proteins from the nucleus, suggesting that RanBP2 functions as a facilitator of macromolecular export (Bernad et al., 2004; Hutten and Kehlenbach, 2006). At the onset of mitosis when the nuclear envelope (NE) disintegrates and NPCs disassemble, RanBP2-RanGAP1-SUMO1-Ubc9 subcomplexes disperse into the mitotic cytosol and accumulate at the plus ends of free spindle microtubules and at kinetochores of chromosomes that have been captured by spindle microtubules (Joseph et al., 2002, 2004). Kinetochore targeting of RanBP2-RanGAP1-SUMO1-Ubc9 is dependent on the nuclear export receptor Crm1 (Arnaoutov et al., 2005). In HeLa and RGG cells, depletion of RanBP2 causes various mitotic abnormalities, including misalignment of chromosome in metaphase, mislocalization of several kinetochore-associated proteins, and formation of multipolar spindles (Joseph et al., 2004; Salina et al., 2003).
The critical biological functions of the mammalian RanBP2 protein have yet to be elucidated. Furthermore, although RanBP2 has SUMO E3 ligase activity in vitro, it remains unknown whether RanBP2 acts as a SUMO-ligating enzyme in vivo. To address these issues, we bypassed the problem of embryonic lethality of RanBP2 null mice (Aslanukov et al., 2006) by generating a series of mutant mice in which the dose of RanBP2 is reduced in graded fashion. We report here that mice with low amounts of RanBP2 are viable and overtly indistinguishable from wild-type mice. We show that there is an inverse correlation between RanBP2 level of expression and chromosome number instability. The prime mitotic defect associated with RanBP2 insufficiency is formation of chromatin bridges in anaphase, an abnormality linked to impaired Topo IIα-mediated decatenation of sister chromatids at anaphase (Clarke et al., 1993). We show that RanBP2 binds to and regulates the sumoylation and localization of Topo IIα in mitosis. We further show that mice expressing RanBP2 below a threshold level are prone to spontaneous and carcinogen-induced tumorigenesis.
RESULTS
Generation of Mutant Mice with Low Amounts of RanBP2
We created a series of mice in which expression of RanBP2 is reduced in a graded fashion from normal to zero by the use of various combinations of wild-type (RanBP2+), hypomorphic (RanBP2H), and knockout (RanBP2−) alleles. The RanBP2H allele was generated by inserting a neomycin resistance cassette into the third intron of the RanBP2 gene via homologous recombination (Figures 1A and 1B). The RanBP2− allele was established by removing exon 3 from the RanBP2H allele via Cre-mediated recombination (Figures 1A and 1B). As previously described (Aslanukov et al., 2006), RanBP2−/− mice died during embryogenesis. Death occurred prior to day 13.5 of development (data not shown). In contrast, RanBP2−/H and RanBP2H/H mice were viable and overtly indistinguishable from RanBP2+/−, RanBP2+/H, and RanBP2+/+ mice. Western blot analysis of mouse embryonic fibroblast (MEF) lysates revealed that RanBP2+/H, RanBP2+/−, RanBP2H/H, and RanBP2−/H cells contained ~90%, 44%, 31%, and 26%, respectively, of the RanBP2 protein level present in RanBP2+/+ MEFs (Figures 1C and S1).
Figure 1. Generation of Mice with Graded Reduction in RanBP2 Dosage.
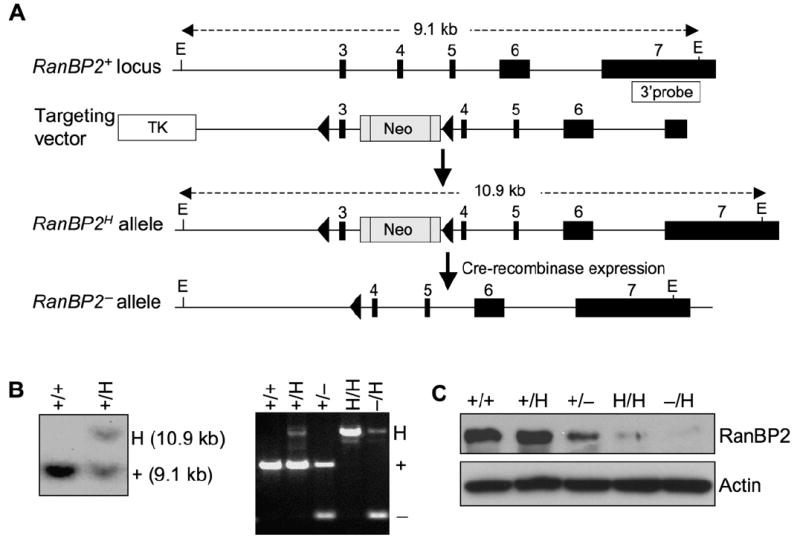
(A) Schematic representation of the RanBP2 gene targeting strategy. The relevant portion of the RanBP2 locus, the targeting vector with loxP (triangles), the hypomorphic and knockout alleles, and the EcoRV restriction sites (E) and the probe used for Southern blotting are indicated.
(B) (left) Southern blot analysis of RanBP2+/H and RanBP2+/+ mice. (Right) PCR-based genotype analysis of RanBP2 mutant mice.
(C) Western blot analysis of MEFs isolated from mice carrying the indicated RanBP2 alleles using anti-RanBP2 antibody. Actin was used as a loading control.
RanBP2−/H Cells Have No Overt Transport-Related Defects
Next, we investigated whether nucleocytoplasmic transport might be impaired in RanBP2−/H cells. In situ hybridization with an oligo(dT)50-mer probe revealed that the intracellular distribution of polyadenylated mRNA was indistinguishable between RanBP2+/+ and RanBP2−/H MEFs, indicating that nuclear export of bulk mRNA was not affected by decreased RanBP2 expression (Figure S2A). Furthermore, NLS-mediated protein import and NES-mediated protein export were both equally efficient in RanBP2+/+ and RanBP2−/H MEFs as measured using established in vivo transport assays (Figures S2B–S2E). Collectively, these results suggest that reduction of RanBP2 protein levels to about a quarter of normal levels has no overt impact on nucleocytoplasmic trafficking.
RanBP2 can sumoylate RanGAP1 in vitro, but whether it does so in vivo is unclear (Pichler et al., 2002). RanBP2 has further been proposed to protect SUMO1-modified RanGAP1 from de-sumoylation by SUMO isopeptidases such as SENP2 (Zhang et al., 2002). Western blot analysis of RanBP2+/+ and RanBP2−/H MEF lysates for SUMO1 showed that SUMO1-RanGAP1 levels remained constant in cells with reduced RanBP2 (Figure S2F). The same holds true for other SUMO1-conjugated proteins and SUMO2/3-conjugated proteins level (Figure S2G), suggesting that hypomorphism for the SUMO E3 ligase RanBP2 does not affect the global patterns of SUMO modification in MEFs.
Mice and MEFs with Low Amounts of RanBP2 Develop Severe Aneuploidy
To determine whether RanBP2 insufficiency leads to chromosomal instability in the context of an adult mouse, we collected splenocytes from RanBP2+/+, RanBP2+/H, RanBP2+/−, RanBP2H/H, and RanBP2−/H mice at 5 months of age and performed karyotype analyses. Chromosome counts showed that 0% of RanBP2+/+ and RanBP2+/H splenocytes were aneuploid (Table 1A). In contrast, splenocytes from RanBP2+/−, RanBP2H/H, and RanBP2−/H mice had 1%, 5%, and 15% incidence of aneuploidy, respectively, revealing an inverse correlation between the level of RanBP2 protein and the percentage of aneuploidy in this cell type. Moreover, the range of abnormal chromosome numbers broadened substantially with decreasing expression of RanBP2 protein. The extent of aneuploidy was also determined in MEFs with graded reduction in RanBP2 expression. As shown in Table 1B, in passage 5 MEFs, aneuploidy increased with declining RanBP2 expression levels. These data establish that RanBP2 is a protein that acts to prevent chromosome missegregation and accumulation of aneuploid cells.
Table 1.
Gradual Reduction of RanBP2 Causes Progressively More Aneuploidy in Splenocytes and MEFs
| A | Mouse Age (n) | Mitotic Figures Inspected | Percent Aneuploid Figures (SD) | Karyotypes with Indicated Chromosome Number | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mouse Genotype | 35 | 36 | 37 | 38 | 39 | 40 | 41 | 42 | 43 | |||
| RanBP2+/+ | 5 mo (4) | 200 | 0 (0) | 200 | ||||||||
| RanBP2+/H | 5 mo (3) | 150 | 0 (0) | 150 | ||||||||
| RanBP2+/− | 5 mo (3) | 150 | 1 (0) | 2 | 148 | |||||||
| RanBP2H/H | 5 mo (4) | 200 | 5 (1) | 1 | 6 | 190 | 2 | 1 | ||||
| RanBP2−/H | 5 mo (4) | 200 | 15 (3) | 1 | 1 | 4 | 2 | 12 | 170 | 9 | 0 | 1 |
| B | Mitotic Figures Inspected | Percent Aneuploid Figures (SD) | Karyotypes with Indicated Chromosome Number | |||||||||
| Mitotic MEF Genotype (n) | 37 | 38 | 39 | 40 | 41 | 42 | 43 | |||||
| RanBP2+/+ (3) | 150 | 9 (2) | 4 | 137 | 9 | |||||||
| RanBP2+/H (3) | 150 | 11 (2) | 5 | 133 | 8 | 2 | 2 | |||||
| RanBP2+/− (3) | 150 | 13 (2) | 4 | 0 | 3 | 131 | 10 | 0 | 2 | |||
| RanBP2H/H (3) | 150 | 26 (2) | 2 | 6 | 11 | 111 | 10 | 5 | 5 | |||
| RanBP2−/H (3) | 150 | 33 (2) | 3 | 7 | 8 | 100 | 18 | 10 | 4 | |||
(A) Analysis of numerical chromosomal abnormalities in splenocytes from 5-month-old mice of the indicated genotypes.
(B) Same analysis as in (A) for P5 MEFs of the indicated genotypes.
RanBP2 Insufficiency Induces Anaphase Bridges
To determine the nature of the chromosome missegregation defects underlying the aneuploidy observed in cells with low levels of RanBP2, the chromosome movements of RanBP2 mutant MEFs were followed by timelapse live microscopy. To visualize DNA, MEFs were transduced with a retrovirus expressing YFP-tagged histone 2B. We found that 9% of RanBP2−/H and RanBP2H/H cells failed to progress from metaphase to anaphase, a defect observed in only 2% of RanBP2+/−, RanBP2+/H, and wild-type MEFs (data not shown). Of the RanBP2−/H and RanBP2H/H cells that were able to progress into anaphase, 30% and 28% formed chromatin bridges, respectively (Figures 2A–2C). Most cells had one or two bridges, while a small fraction of cells had more than two (data not shown). These bridges resolved in telophase or during cytokinesis (Figure 2A). The percentage of chromatin bridges was much lower in RanBP2+/− and RanBP2+/H anaphases (16% and 12%, respectively) but still significantly higher than in RanBP2+/+ anaphases (7%) (Figure 2C). In addition, a small but statistically significant increase of centrophilic chromosomes was observed in RanBP2−/H and RanBP2H/H MEFs in comparison to RanBP2+/+ MEFs (Figure 2C). There was a small increase of lagging chromosomes in these MEFs that did not reach statistical significance (Figure 2C). Together, the above findings suggest that anaphase-bridge formation is the main chromosome segregation defect in RanBP2-insufficient cells.
Figure 2. Cells with Low Levels of RanBP2 Form Chromosome Bridges in Anaphase.
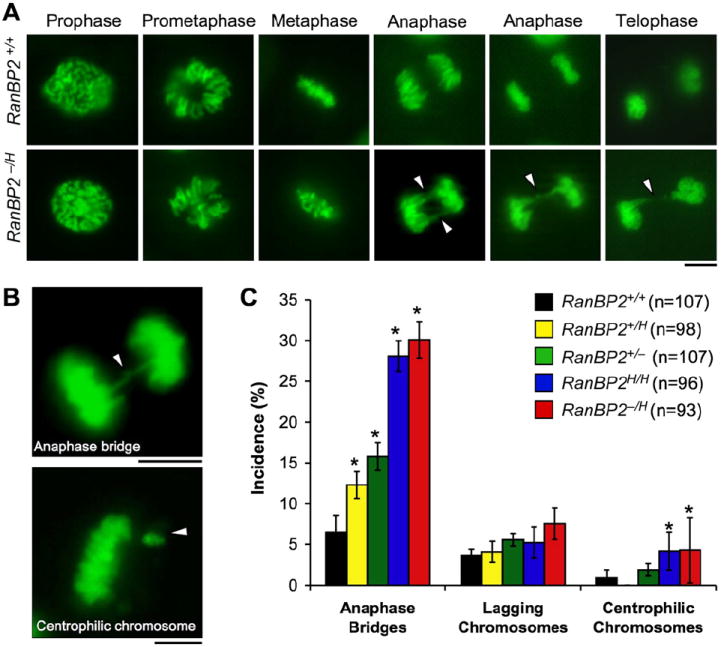
(A) Chromosome dynamics and segregation of RanBP2+/+ and RanBP2−/H MEFs expressing H2B-YFP were observed by timelapse microscopy. Representative images of each mitotic stage are shown. Arrowheads mark chromatin bridges in RanBP2−/H MEFs. Bar = 10 μm.
(B) High-magnification images of RanBP2−/H MEFs with indicated chromosome segregation defects. Bar = 10 μm.
(C) Quantification of the chromosome segregation defects observed by live-cell imaging of MEFs of the indicated genotypes. n = total number of mitotic cells analyzed from at least three independent MEF lines. Error bars indicate SEM, *p < 0.05 versus wild-type cells (Chi-square test).
To examine whether chromatin-bridge formation caused structural chromosome damage, spectral karyotype (SKY) analysis was performed on RanBP2−/H and RanBP2+/+ MEFs. Although this analysis confirmed that RanBP2−/H cells had increased numerical chromosome instability, there was no evidence for increased breakage or fusion of chromosome arms (Figure S3A). Giemsa-stained metaphase spreads from splenocytes and MEFs with various levels of RanBP2 were also inspected for structural chromosomal abnormalities. Again, there was no evidence for increased chromosome breakage or end-to-end fusion in RanBP2 mutant cells, although a small but significant increase in the incidence of chromosome gaps was observed in RanBP2H/H and RanBP2−/H splenocytes and MEFs (Figures S3B–S3D). These data suggest that chromosome-bridge formation causes aneuploidy in the absence of extensive structural chromosome damage. However, the possibility that MEFs with structural damage have reduced viability and thus go unnoticed cannot be excluded.
Previous studies of HeLa cells have documented that depletion of RanBP2 by RNA interference perturbs mitotic spindle formation and loading of certain mitotic checkpoint proteins onto kinetochores in early mitosis (Joseph et al., 2004; Salina et al., 2003). However, no such mitotic defects were detectable in RanBP2 hypomorphic MEFs (Figure S4).
Topo IIα Targeting to Inner Centromeres in Mitosis is Dependent on RanBP2
Formation of chromatin bridges in anaphase is a distinctive feature of cells in which DNA decatenation is impaired by mutation or chemical inhibition of Topo IIα (Clarke et al., 1993). Topo IIα is active in S phase and at the metaphase-anaphase transition, where it disentangles sister centromeres to enable their separation in anaphase (Bhat et al., 1996). To examine whether ana-phase-bridge formation in RanBP2 hypomorphic MEFs might be due to Topo IIα insufficiency, we examined lysates from asynchronous MEF cultures of various RanBP2 genotypes for Topo IIα expression by immunoblotting. As shown in Figure 3A, Topo IIα expression levels were independent of RanBP2 status. RanBP2+/+ and RanBP2−/H MEFs that were arrested in mitosis by nocodazole also contained similar amounts of Topo IIα (Figure 3B).
Figure 3. RanBP2 Binds to Topo IIα in Mitosis and Is Essential for Its Accumulation at Inner Centromeres.
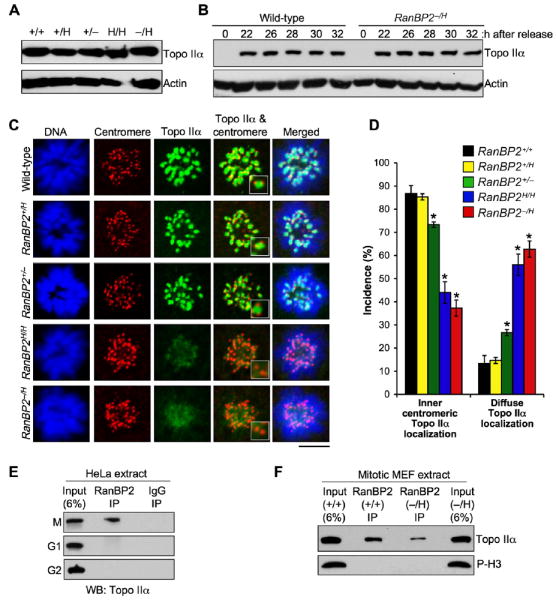
(A) Western blots of asynchronous MEF lysates probed with antibody to Topo IIα. Actin served as a loading control.
(B) Western blots of synchronized RanBP2+/+ and RanBP2−/H MEF lysates. MEFs were synchronized in G0 by serum starvation and then released for the indicated durations in serum-containing medium. Nocodazole was added 23 hr after cells were released. Blots were probed for Topo IIα. Actin was used as loading control.
(C) Immunolocalization of Topo IIα in MEFs with various levels of RanBP2 during prometaphase. Centromeres were visualized with ACA antibody. DNA was stained with Hoechst. Magnified images of the centromeric and inner centromeric regions are shown in the insets. Bar = 10 μm.
(D) Quantification of prometaphases with inner centromeric versus diffuse localization of Topo IIα. Seventy-five prometaphases were analyzed per genotype (three independent MEF lines were analyzed per genotype). Error bars indicate SEM, *p < 0.05 versus wild-type cells (Chi-square test).
(E) Immunoblots of mitotic (M), G1, or G2 HeLa extracts subjected to immunoprecipitation with RanBP2 antibody and analyzed for coprecipitation of Topo IIα.
(F) Immunoblots of mitotic extracts from RanBP2+/+ and RanBP2−/H MEFs subjected to immunoprecipitation with RanBP2 antibody and analyzed for coimmunoprecipitation of Topo IIα. Phosphohistone H3 (P-H3) signals indicate that similar amounts of mitotic cells were present in the lysates used for immunoprecipitation.
Next, we hypothesized that RanBP2 deficiency might cause anaphase-bridge formation by interfering with proper localization of Topo IIα to inner centromeres of mitotic chromosomes. To test for this possibility, RanBP2+/+ and RanBP2−/H MEFs were arrested in prometaphase with monastrol, an inhibitor of the mitotic kinesin Eg5, and immunostained with anti-Topo IIα and anti-centromeric antibodies (ACA). We found that Topo IIα concentrated at inner centromeres in most RanBP2+/+, RanBP2+/H, and RanBP2+/− cells (Figures 3C, 3D, and S5). In contrast, the majority of RanBP2H/H and RanBP2−/H MEFs failed to accumulate Topo IIα at inner centromeres. These results demonstrate that proper targeting of Topo IIα to inner centromeres of mitotic chromosomes is dependent on RanBP2 and suggest that anaphase-bridge formation in RanBP2 hypomorphic cells is caused by Topo IIα mislocalization.
RanBP2 Binds to Topo IIα in Mitosis
Because Topo IIα localization in mitosis is RanBP2 dependent, we suspected that the two proteins might form a complex. To investigate this, RanBP2 was immunoprecipitated from interphase and mitotic HeLa cell extracts and examined for Topo IIα coprecipitation. As shown in Figure 3E, about 4%–5% of the cellular Topo IIα pool coimmunoprecipitated with RanBP2 from mitotic extracts but not from G1 or G2 extracts. In the reverse experiment, RanBP2 immunoprecipitated with Topo IIα from mitotic extracts but again not from interphase extracts (data not shown). To determine whether the abundance of RanBP2-Topo IIα complexes in mitosis was affected by RanBP2 insufficiency, mitotic extracts from RanBP2−/H and RanBP2+/+ MEFs were subjected to coimmunoprecipitation with anti-RanBP2 antibodies. Although Topo IIα coprecipitated with RanBP2 from RanBP2−/H cell extracts, the amount of protein coprecipitated from RanBP2+/+ cell extracts was considerably greater (Figure 3F). These data suggest that RanBP2 and Topo IIα form a complex in mitosis and that RanBP2-insufficient cells have reduced amounts of this complex. RanBP2 does not colocalize with Topo IIα at inner centromeres (data not shown). Both RanBP2 and Topo IIα, however, are present in the mitotic cytosol, suggesting that interaction between them takes place in this subcellular compartment.
The RanBP2 SUMO E3 Ligase Domain Regulates Proper Chromosome Segregation and Targeting of Topo IIα to Inner Centromeres
In S. cerevisiae, SUMO modification of topoisomerase II (Top2p), which is catalyzed by the E3 ligases Siz1p and Siz2p, targets the protein to the pericentromeric regions of mitotic chromosomes (Takahashi et al., 2006). Studies in Xenopus egg extracts and human cells suggest that Topo IIα is also subjected to sumoylation in higher eukaryotes (Azuma et al., 2003, 2005; Mao et al., 2000). Since RanBP2 is a nucleoporin with SUMO E3 ligase activity (Pichler et al., 2002), we speculated that this activity might be required for accumulation of Topo IIα at inner centromeres and proper separation of sister chromatids. To test this hypothesis, we expressed influenza hemagglutinin (HA) epitope-tagged RanBP2(2553–2838), a 33 kDa fragment spanning the RanBP2 SUMO E3 ligase domain (Pichler et al., 2004) (Figure 4A), in RanBP2−/H MEFs and measured its ability to correct ana-phase-bridge formation and Topo IIα localization. Three catalytic mutants of the SUMO E3 ligase domain, HA-RanBP2(2553–2838)L2651A,L2653A, HA-RanBP2(2553–2838)F2657A,F2658A, and HA-RanBP2(2553–2838)L2651A,L2653A,F2657A,F2658A, and two regions flanking the SUMO E3 ligase domain, HA-RanBP2(1950–2553) and HA-RanBP2(2839–3224), were also expressed in RanBP2−/H cells as controls (Figure 4A). Western blotting for HA revealed that all RanBP2 fragments were expressed at similar levels (Figure S6). Consistent with our earlier results, 28% of RanBP2−/H MEFs expressing empty vector formed anaphase bridges (Figure 4B). A similarly high percentage of anaphase bridges was observed in RanBP2−/H MEFs expressing the catalytic mutants or the flanking regions of the RanBP2 SUMO E3 ligase domain. By contrast, only 11% of RanBP2−/H MEFs expressing the fully functional RanBP2 SUMO E3 ligase domain (2553–2838) formed anaphase bridges, a percentage that was similar to that recorded for RanBP2+/+ cells expressing the empty vector. Ectopic expression of the HA-RanBP2(2553–2838) had no overt effect on global sumoylation patterns as measured by western blotting (data not shown). Importantly, overexpression of PIASy, a SUMO E3 ligase required for sumoylation of Topo IIα in Xenopus egg extracts (Azuma et al., 2003, 2005), did not correct anaphase-bridge formation (Figures 4B and S6).
Figure 4. Expression of the RanBP2 SUMO E3 Ligase Domain in RanBP2-Insufficient Cells Prevents Anaphase-Bridge Formation and Topo IIα Mislocalization.
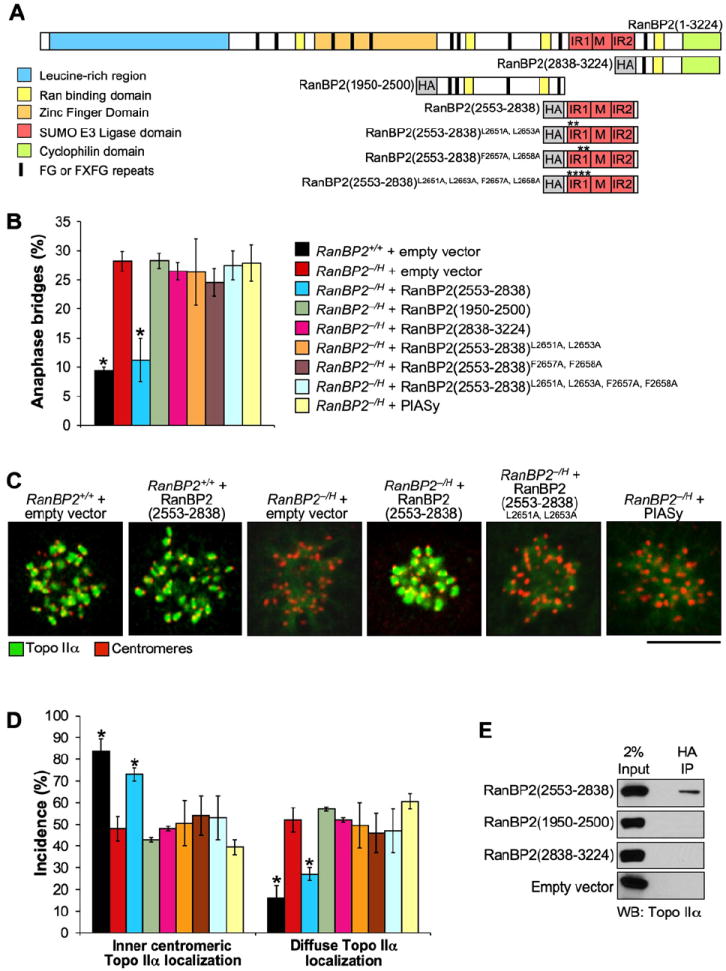
(A) Overview of the HA-tagged mutant RanBP2 proteins used in correction experiments.
(B) Incidence of chromatin bridges in RanBP2−/H MEFs expressing the proteins indicated in (A) or HA-tagged PIASy. Three independent MEF lines were evaluated per genotype (~25–40 anaphases per line). Error bars indicate SEM, *p < 0.05 versus RanBP2−/H MEFs carrying empty expression vector (Chi-square test).
(C) Immunolocalization of Topo IIα (green) in monastrol-treated RanBP2−/H MEFs expressing the indicated proteins. Centromeres were visualized with human ACA antibody (red). Bar = 10 μm.
(D) Quantification of prometaphases with inner centromeric versus diffuse Topo IIα localization. Seventy-five prometaphases were analyzed per genotype (three independent MEF lines were analyzed per genotype). Error bars indicate SEM. Chart legend is as in (B). *p < 0.05 versus RanBP2−/H cells carrying empty expression vectors (Chi-square test).
(E) Western blot analysis of HA immunoprecipitates from mitotic extracts of MEF cells expressing the indicated HA-RanBP2 fragments. Blots were probed for Topo IIα.
Complementary immunostaining experiments with antibodies to Topo IIα and centromeres revealed that the expression of RanBP2 SUMO E3 ligase domain in RanBP2−/H cells greatly improved the targeting of Topo IIα to inner centromeres of mitotic chromosomes (Figures 4C and 4D). The percentage of cells with diffuse Topo IIα localization was reduced to only 25% in RanBP2−/H cells expressing RanBP2(2553–2838) compared to 50% in RanBP2−/H cells expressing the empty vector. No such improvement was observed when catalytic mutants or regions flanking the RanBP2 SUMO E3 ligase domain or PIASy were overexpressed (Figures 4C and 4D). Together these data establish a specific requirement for RanBP2 SUMO E3 ligase activity in targeting Topo IIα protein to inner centromeres in early mitosis to allow for decatenation of sister centromeres prior to anaphase onset.
To investigate whether the SUMO E3 ligase domain might exert its corrective effects by targeting Topo IIα protein, we determined whether it formed a complex with Topo IIα in mitosis. To this end, we expressed HA-RanBP2(2553–2838) in RanBP2+/+ MEFs and precipitated it with an antibody against HA from mitotic extracts. As shown in Figure 4E, Topo IIα indeed coimmunoprecipitated with HA-RanBP2(2553–2838) from these extracts. In contrast, Topo IIα failed to coimmunoprecipitate with HA-RanBP2(1950–2553) and HA-RanBP2(2839–3224). Thus, the SUMO E3 ligase domain of RanBP2 is sufficient for complex formation with Topo IIα in vivo.
RanBP2 Promotes Sumoylation of Topo IIα In Vitro and In Vivo
Next, we used an in vitro sumoylation assay to address whether Topo IIα is a substrate for SUMO modification by RanBP2. We incubated full-length purified human Topo IIα with recombinant SUMO E1, Ubc9, His-HA-RanBP2(2553–2838), SUMO1, and ATP for 1 hr. Topo IIα sumoylation was then analyzed by immunoblotting for Topo IIα. As shown in Figure 5A, Topo IIα was efficiently sumoylated by His-HA-RanBP2(2553–2838). No such modification occurred in the absence of His-HA-RanBP2(2553–2838) or ATP, or when catalytic mutant His-HA-RanBP2(2553–2838)L2651A,L2653A was used instead of His-HA-RanBP2(2553–2838). We found that Topo IIα was also efficiently sumoylated by His-HA-RanBP2(2553–2838) when SUMO1 was replaced by SUMO2 or SUMO3 (Figure S7A). Collectively, these data demonstrate that Topo IIα is a substrate of RanBP2 for SUMO modification in vitro.
Figure 5. RanBP2 Sumoylates Topo IIα in Mitosis.

(A) In vitro sumoylation of Topo IIα by the RanBP2 E3 ligase domain. Twenty nanograms of recombinant human Topo IIα was tested for SUMO1 modification in the presence or absence of 100 ng wild-type (His-HA-RanBP2[2553–2838]) or mutant (His-HA-RanBP2[2553–2838]L2651A,L2653A) RanBP2 SUMO E3 ligase domain, 110 ng E1, 50 ng Ubc9, 5 μg SUMO1, and 1 mM ATP. Samples were incubated at 37°C for 1 hr and then analyzed by western blotting with antibody to Topo IIα.
(B) In vivo sumoylation of Topo IIα by RanBP2. Mitotic shake-off lysates from RanBP2+/+ and RanBP2−/H MEFs containing the indicated expression vectors were prepared and analyzed by western blotting for Topo IIα and HA. P-H3 and actin antibodies were used to verify that similar amounts of mitotic cells and protein were present in lysates used.
(C) RanBP2 sumoylation of Topo IIα under physiological conditions. Mitotic shake-off lysates were prepared from RanBP2+/+ and RanBP2−/H MEFs and analyzed by western blotting for Topo IIα. P-H3 and actin antibodies were used to ensure that lysates contained similar amounts of mitotic cells and protein.
(D) Localization of SUMO1-Topo IIα-EGFP in RanBP2−/H MEFs during prometaphase. (Left) Confocal image of a prometaphase expressing SUMO1-Topo IIα-EGFP. Centromeres were visualized by immunostaining with ACA antibody. Bar = 5 μm. (Right) Quantification of cells with inner centromeric versus diffuse localization of SUMO1-Topo IIα-EGFP during prometaphase. Two independent RanBP2−/H MEF lines were analyzed (50 prometaphases per line). Error bars indicate SEM, *p < 0.05 versus untransfected RanBP2−/H MEFs (paired t test). Localization of Topo IIα in untransfected RanBP2−/H MEFs was determined by immunostaining for Topo IIα and centromeres as in Figure 3C.
(E) Incidence of chromatin bridges in RanBP2−/H MEFs in the absence or presence of SUMO1-Topo IIα-EGFP. Two independent RanBP2−/H MEF lines were analyzed (50 prometaphases per line). Error bars indicate SEM, *p < 0.05 versus RanBP2−/H untransfected MEFs (paired t test).
(F) Immunolocalization of Topo IIα in PIASy−/− MEFs during prometaphase. (Left) Confocal image of a PIASy−/− MEF immunostained for Topo IIα and centromeres. Bar = 5 μm. (Right) Quantification of PIASy+/+ and PIASy−/− prometaphases with inner centromeric versus diffuse localization of Topo IIα. Three independent MEF lines were analyzed (~25–50 prometaphases per line). Error bars indicate SEM.
(G) Incidence of chromatin bridges in PIASy−/− MEFs examined by live-cell imaging. Three independent PIASy+/+ and PIASy−/− MEF lines were analyzed (~25–40 anaphases per line). Error bars indicate SEM.
(H) Sumoylation of endogenous Topo IIα in PIASy−/− MEFs. Experimental details were as in (C).
(I) Proposed model for the role of RanBP2 in sister-chromosome separation. In mitosis, RanBP2 (in conjunction with Ubc9) binds to and sumoylates Topo IIα in the mitotic cytosol. This modification serves as a signal for Topo IIα targeting to inner centromeres, where it functions to decatenate sister centromeres, thus allowing for proper separation of sister chromosomes in anaphase. As SUMO-modified Topo IIα represents only a small fraction of the total Topo IIα pool (Figure 5C), SUMO conjugation may only be required for initial targeting of Topo IIα to centromeres but not for maintenance of centromeric localization.
If RanBP2 indeed functions to sumoylate Topo IIα in mitosis, one would expect mitotic RanBP2−/H MEFs to have reduced levels of SUMO-conjugated Topo IIα. To test whether this is the case, we stably expressed HA-tagged SUMO1 in RanBP2+/+ and RanBP2−/H MEFs (Figure 5B), harvested mitotic cells by mitotic shake-off, and analyzed lysates prepared from these cells for the presence of SUMO-conjugated Topo IIα by western blotting for Topo IIα. In addition to unmodified Topo IIα, RanBP2+/+ MEFs possessed a higher-molecular-weight SUMO1-conjugated Topo IIα band (Figure 5B). The presence of HA-SUMO1 in the upper band was confirmed by anti-HA immunoblotting. SUMO1-conjugated Topo IIα was not detectable in RanBP2−/H MEFs. Importantly, expression of HA-RanBP2(2553–2838), but not HA-RanBP2(2553–2838)L2651A,L2653A, was able to restore sumoylation of Topo IIα in these MEFs. To determine whether Topo IIα sumoylation was impaired under physiological conditions, lysates from mitotic RanBP2+/+ and RanBP2−/H MEFs were analyzed for the presence of SUMO-Topo IIα with anti-Topo IIα antibody. As shown in Figure 5C, SUMO-modified Topo IIα was detectable in RanBP2+/+ lysates but not in RanBP2−/H lysates. As expected, SUMO-modified Topo IIα was not observed when RanBP2+/+ MEFs were lysed in the absence of the isopeptidase inhibitor N-ethylmaleimide (NEM; see Figure S7B). The observation that RanBP2 hypomorphism leads to decreased sumoylation of Topo IIα in mitosis suggests that Topo IIα is an in vivo substrate of RanBP2 during this cell-cycle stage.
To further test whether Topo IIα is the relevant target of RanBP2 SUMO E3 ligase activity in mitosis, we expressed a Topo IIα construct encoding a nonhydrolyzable SUMO1 in linear N-terminal fusion with Topo IIα-EGFP in RanBP2−/H MEFs and determined its ability to restore Topo IIα localization and efficient separation of sister chromatids in these cells. SUMO1-Topo IIα-EGFP fusion protein indeed targeted with high efficiency to inner centromeres of RanBP2 hypomorphic MEFs in mitosis (Figure 5D) and caused a dramatic reduction in ana-phase-bridge formation in these cells (Figure 5E). These data support the idea that the mitotic defects seen in RanBP2 hypomorphic cells are due to impaired SUMO modification of Topo IIα.
Depletion of PIASy in Xenopus egg extracts or HeLa cells has been shown to inhibit Topo IIα targeting to inner centromeres and block chromosome segregation (Azuma et al., 2003, 2005; Diaz-Martinez et al., 2006), suggesting that RanBP2 and PIASy E3 ligases might have overlapping functions in mitosis. To investigate this, we analyzed the subcellular localization of Topo IIα in MEFs derived from PIASy−/− mice (Roth et al., 2004). As shown in Figure 5F, PIASy loss had no impact on Topo IIα targeting to inner centromeres in mitosis. MEFs from an independently generated PIASy−/− mouse strain yielded the same results (Wong et al., 2004) (data not shown). Furthermore, anaphase-bridge frequencies and aneuploidy were not significantly elevated in PIASy−/− MEFs (Figures 5G and S8). Western blot analysis of mitotic lysates from PIASy+/+ and PIASy−/− MEFs for Topo IIα revealed that SUMO modification of Topo IIα was unperturbed by the deletion of PIASy (Figure 5H). These data demonstrate that, at least in MEFs, RanBP2 rather than PIASy regulates accumulation of Topo IIα at inner centromeres and proper chromosome segregation.
RanBP2 Insufficient Mice Are Prone to Carcinogen-Induced and Spontaneous Tumors
As RanBP2 insufficiency leads to chromosome number instability, a condition that has been linked to cancer development, we sought to determine whether mice with reduced RanBP2 expression are prone to tumorigenesis. To this end, we performed a tumor bioassay with 7,12-dimethylbenz[a]anthracene (DMBA), a carcinogen that predisposes wild-type mice to lung tumors, skin tumors, and thymic lymphomas when applied to the skin (Serrano et al., 1996). Pups from RanBP2H/H × RanBP2−/H and RanBP2+/− × RanBP2+/H intercrosses were given a single application of 50 μl 0.5% DMBA to the dorsal surface on postnatal day 5. Treated animals were then monitored for development of overt tumors over a period of 5 months. The incidence of skin tumors was dramatically increased in RanBP2H/H and RanBP2−/H mice compared to RanBP2+/+, RanBP2+/H, and RanBP2+/− mice (Figures 6A and 6B). Seventy-seven percent of RanBP2−/H and 60% of RanBP2H/H mice developed skin tumors compared to 31% of RanBP2+/−, 24% of RanBP2+/H, and 13% of wild-type mice. Moreover, the latency with which skin tumors formed was much shorter in RanBP2H/H and RanBP2−/H mice than in RanBP2+/+, RanBP2+/H, and RanBP2+/− mice (Figure 6B). Compared with RanBP2+/+ mice, the skin tumor burden was 16- and 23-fold increased in RanBP2H/H and RanBP2−/H mice, respectively (Figure 6C). In RanBP2+/− and RanBP2+/H mice this increase was only 3-fold. At 5 months of age, the animals were sacrificed and their internal organs were screened for tumors. Lung tumors were identified in 58% of RanBP2+/+ mice (Figures 6D and 6E). Small increases in the incidence of lung tumorigenesis were observed in RanBP2+/− and RanBP2+/H mice, but they were not statistically significant. In contrast, the increases in lung tumor incidence were much larger in RanBP2H/H and RanBP2−/H animals, with 95% of RanBP2H/H and 100% of RanBP2−/H animals having this tumor type. Similarly, the lung tumor burden in RanBP2H/H and RanBP2−/H mice was considerably higher than in RanBP2+/−, RanBP2+/H, and RanBP2+/+ mice, with RanBP2H/H and RanBP2−/H animals developing on average 17 and 14 tumors per mouse, respectively, compared to three or less in the other genotypes (Figure 6F).
Figure 6. RanBP2 Suppresses Spontaneous and DMBA-Induced Tumorigenesis.
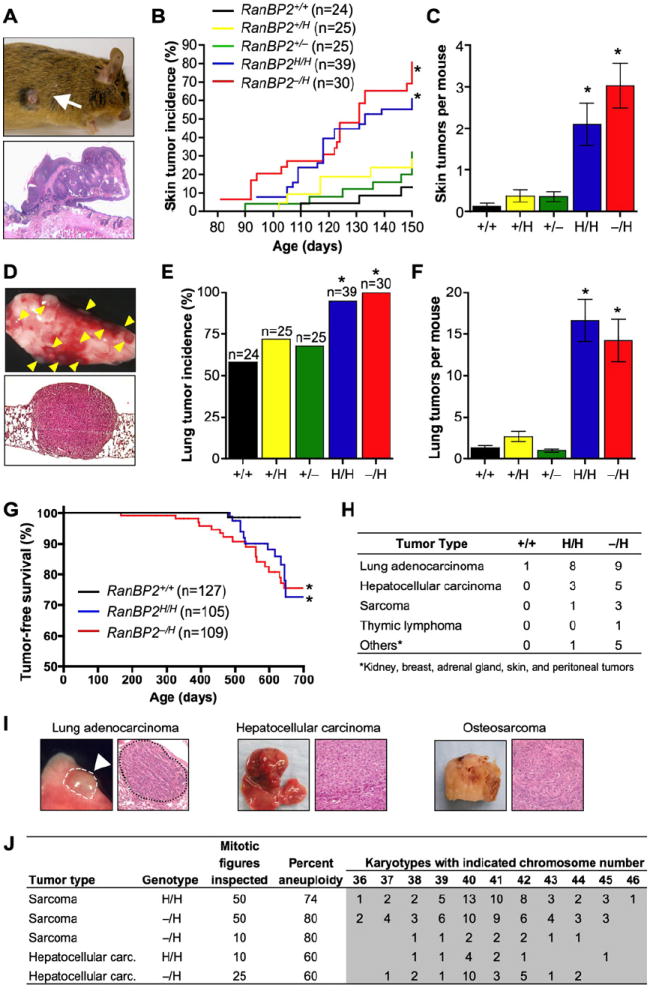
(A) Gross appearance (top) and histology (bottom) of skin tumor of a DMBA-treated RanBP2−/H mouse.
(B) Skin tumor incidence of DMBA-treated mice of the indicated genotypes. *p < 0.05 versus wild-type mice (Log rank test).
(C) Skin tumor burden of DMBA-treated mice of the indicated genotypes. Error bars indicate SEM, *p < 0.05 versus wild-type mice (Mann Whitney test).
(D) Gross appearance (top) and histology (bottom) of lung tumors of a DMBA-treated RanBP2−/H mouse.
(E) Lung tumor incidence of DMBA-treated mice of the indicated genotypes. Error bars indicate SEM, *p < 0.05 versus wild-type mice (Chi-square test).
(F) Lung tumor burden of DMBA-treated mice of the indicated genotypes. Error bars indicate SEM, *p < 0.05 versus wild-type mice (Mann Whitney test).
(G) Tumor-free survival of RanBP2+/+, RanBP2H/H, and RanBP2−/H mice. *p < 0.01 versus wild-type mice (Log rank test).
(H) Tumor spectrum of mice with various RanBP2 genotypes.
(I) Gross images and histological analysis of representative spontaneous tumors from RanBP2−/H mice.
(J) Chromosome counts on primary tumors of RanBP2H/H and RanBP2−/H mice.
Furthermore, to determine if RanBP2 insufficiency promotes spontaneous tumorigenesis, cohorts of RanBP2+/+, RanBP2H/H, and RanBP2−/H mice were established and monitored biweekly for signs of overt tumors or ill health for up to 2 years. Moribund animals were sacrificed and screened for tumors. Tumors were collected and embedded in paraffin for histopathology. RanBP2−/H and RanBP2H/H mice formed tumors with increased incidence and shorter latency than RanBP2+/+ mice (Figure 6G). Lung adenocarcinomas were the most prevalent tumor type in RanBP2−/H and RanBP2H/H mice (Figures 6H and 6I). Other frequently observed tumor types in these mice were hepatocellular carcinomas and sarcomas. Chromosome counts and interphase fluorescence in situ hybridization (FISH) using probes for chromosomes 4, 7, 9, and 12 demonstrated that tumors of all three types consistently displayed severe aneuploidy (Figures 6J and S9). Thus, when RanBP2 expression drops below a critical threshold level, mice become prone to spontaneous and carcinogen-induced tumorigenesis. Furthermore, these studies establish RanBP2 as a protein with tumor-suppressive activity and reveal a correlation between aneuploidy and tumorigenesis.
DISCUSSION
RanBP2 has been implicated in both nucleocytoplasmic transport and mitosis and is unique among nucleoporins in that it is a SUMO E3 ligase. However, the critical biological functions of RanBP2 and the in vivo targets of its ligase activity have been unknown. Progress in understanding the physiological functions of mammalian nucleoporins has been hampered by the fact that most loss-of-function mutations disrupt early embryogenesis so severely that the embryo dies. One way around this problem is to study hypomorphic mutant mice. Using this approach, we established a requirement for RanBP2 in maintenance of chromosome number stability and suppression of tumorigenesis. The observation that the mitotic phenotype caused by RanBP2 hypomorphism is reminiscent of Topo IIα inhibition led to the discovery that RanBP2 regulation of chromosome segregation fidelity is mediated, at least in part, by the sumoylation of Topo IIα in mitosis.
RanBP2 Sumoylates Topo IIα to Mediate Sister-Chromatid Separation
Siz1p and Siz2p, two proteins that provide the major SUMO (Smt3p) E3 ligase activity in budding yeast, execute SUMO modification of Top2p in mitosis (Takahashi et al., 2006). This modification seems to function as a signal for Top2p targeting to pericentromeric regions of mitotic chromosomes. Seven observations reported here suggest that, in mammals, RanBP2 catalyzes the SUMO modification of Topo IIα in mitosis to direct this protein to inner centromeres for accurate chromosome separation prior to anaphase onset (Figure 5I). First, catalytic inhibition of Topo IIα causes formation of chromatin bridges in anaphase due to incomplete DNA decatenation (Clarke et al., 1993). Ana-phase-bridge formation is the primary phenotype of RanBP2 hypomorphic cells. Second, the RanBP2 hypomorphic cells fail to accumulate Topo IIα at inner centromeres of mitotic chromosomes at high incidence. Third, ectopic expression of the RanBP2 SUMO E3 ligase domain in RanBP2 hypomorphic cells by itself is sufficient to restore proper localization of Topo IIα to inner centromeres and prevent anaphase-bridge formation. These corrective effects require SUMO conjugation, as they were not observed in RanBP2 hypomorphic cells expressing catalytic mutants of the RanBP2 E3 ligase domain. Unlike Topo IIα, the RanBP2 SUMO E3 ligase domain and full-length RanBP2 do not concentrate at inner centromeres of mitotic chromosomes, suggesting that Topo IIα is sumoylated by RanBP2 in the mitotic cytosol and, from there, translocates to inner centromeres of duplicated chromosomes without RanBP2 (Figure 5I). Fourth, RanBP2 forms a complex with Topo IIα specifically in mitosis. Importantly, the abundance of RanBP2-Topo IIα complexes is substantially reduced in RanBP2 hypomorphic cells. Fifth, the RanBP2 SUMO E3 ligase domain, which is sufficient for Topo IIα binding, efficiently catalyzes SUMO conjugation of Topo IIα in an in vitro assay using recombinant proteins. We show that Topo IIα can be modified by all three SUMO species in this assay. Sixth, RanBP2 hypomorphic cells have substantially lower levels of SUMO-modified Topo IIα than cells expressing a full complement of RanBP2. Furthermore, ectopic expression of the RanBP2 SUMO E3 ligase domain in RanBP2 hypomorphic cells is sufficient to restore SUMO conjugation of Topo IIα in mitosis. Seventh, ectopically expressed SUMO1-Topo IIα fusion protein corrects both Topo IIα mislocalization and anaphase-bridge formation in RanBP2 hypomorphic MEFs. As expression of the catalytically active SUMO E3 ligase domain has similar corrective effects in these cells, it is reasonable to conclude that Topo IIα is the relevant substrate of RanBP2 in mitosis.
Previous studies in Xenopus egg extracts have implicated PIASy in the sumoylation of Topo IIα (Azuma et al., 2003, 2005). This combined with the observation that depletion of PIASy from these extracts results in metaphase arrest led to speculation that sumoylation of Topo IIα is essential for sister-chromatid separation in mitosis. The observation that siRNA-mediated depletion of PIASy from HeLa cells interferes with targeting of Topo IIα and blocks separation of duplicated chromosomes further supported this idea (Diaz-Martinez et al., 2006). However, here we demonstrate that Topo IIα sumoylation, Topo IIα accumulation at inner centromeres, and proper separation of sister chromosomes are unperturbed in PIASy−/− MEFs. Consistent with this, PIASy−/− mice are viable, fertile, and devoid of any overt phenotypes (Roth et al., 2004; Wong et al., 2004). Furthermore, PIASy overexpression was found to be unable to correct Topo IIα mislocalization and chromosome missegregation in RanBP2 hypomorphic MEFs. One interpretation is that the role of RanBP2 and PIASy in Topo IIα sumoylation is species, cell type, and/or transformation status dependent. It is also possible that off-target effects of siRNA oligos drive aberrant Topo IIα localization and chromosome segregation in the PIASy-depleted HeLa cells. Furthermore, it is unknown whether depletion of PIASy in HeLa cells correlates with impaired SUMO modification of Topo IIα.
RanBP2 in Chromosomal Instability and Tumorigenesis
Our finding that RanBP2 hypomorphic mice are highly susceptible to carcinogen-induced tumors and prone to various spontaneous tumors reveals a novel and important role for RanBP2 in suppression of tumorigenesis. What could be the tumor-suppressive function(s) of RanBP2? Given that RanBP2 hypomorphism induces aneuploidy and that the incidence of aneuploidy is high in human cancers, it is tempting to speculate that RanBP2 exerts its tumor-suppressive effect by ensuring accurate segregation of duplicated chromosomes in mitosis. The finding that aneuploidy is high in tumors from RanBP2 hypomorphic mice supports this notion. Yet, whether aneuploidy is causally implicated in cancer development has been a subject of intense investigation and debate. Recent studies of mutant mouse strains with defects in mitotic checkpoint genes that increase the incidence of missegregation of whole chromosomes suggest that aneuploidy indeed has the ability to promote tumorigenesis (Weaver and Cleveland, 2006). Structural chromosome defects have also been implicated in tumor development. Despite having high rates of chromatin-bridge formation, RanBP2 hypomorphic MEFs showed no evidence of increased gross chromosomal aberrations. However, this does not exclude the possibility that structural chromosome damage from improper decatenation contributes to tumor development in RanBP2 mutant mice. For instance, it is possible that transformed cells may be more resistant to cell death induced by structural damage than nontransformed cells.
Although RanBP2 is a well-established component of the mammalian NPC, cells that are hypomorphic for this protein displayed no overt defects in NLS-dependent protein import, NES-dependent protein export, or mRNA export. These findings suggest that tumor development in RanBP2 hypomorphic mice is unlikely due to impairments in some of the major transport pathways. However, it cannot be excluded that specific NLS- or NES-containing proteins that are important for controlled cell proliferation or induction of apoptosis are expressed at inappropriate levels or unable to reach their intracellular sites of action in RanBP2 hypomorphic mice. Moreover, only those transport pathways that were functionally impaired by RanBP2 depletion in HeLa cells were included in our analysis (Bernad et al., 2004; Hutten and Kehlenbach, 2006). Thus, it is possible that certain transport pathways are not optimally functioning in RanBP2 hypomorphic cells.
To explore whether RanBP2 might have a role in human cancer, we compared the relative expression of RanBP2 in normal tissue versus tumors using human gene expression data from the Oncomine database (http://www.oncomine.com). RanBP2 hypomorphic mice are particularly sensitive to spontaneous and carcinogen-induced lung tumors. Consistent with these data, two independent studies revealed that RanBP2 transcript levels are substantially reduced in human non-small cell lung cancers (Beer et al., 2002; Garber et al., 2001). These findings, along with data showing that RanBP2 expression is frequently downregulated in human lung cancer cell lines and primary lung tumor samples (D. Baker and J.M.v.D., unpublished data), suggest that RanBP2 downregulation is a frequent event in human lung tumorigenesis.
EXPERIMENTAL PROCEDURES
Generation of Hypomorphic and Knockout Mice and Tumor Susceptibility Studies
The gene targeting procedure used to generate the hypomorphic RanBP2 allele (H) was as previously described (Dawlaty and van Deursen, 2006). Correctly targeted ES cell clones were injected into blastocysts and RanBP2+/H offspring were obtained from the resulting chimeras through standard procedures. RanBP2+/− mice were established by Cre-mediated excision of RanBP2 exon 3 in the female germline by the use of MMTV-Cre transgenic mice (this excision causes out-of-frame fusion of exons 2 and 4). All mice were maintained on a 129Sv/E × C57BL/6 genetic background. Mice in tumor susceptibility studies were monitored daily. Moribund mice were killed and their major organs screened for overt tumors. Tumors were processed for histopathology by standard procedures. Prism software was used for the generation of tumor-free survival curves and for statistical analyses. DMBA treatment was as described (Serrano et al., 1996).
Western Blotting and Coimmunoprecipitation
Western blot analyses and coimmunoprecipitations were essentially done as described (Kasper et al., 1999). Lysis buffer for detection of SUMO-modified Topo IIα by western blotting consisted of 50 mM Tris HCl (pH 7.5), 150 mM NaCl, 0.1% NP40, 0.5% SDS, protease inhibitor cocktail (Roche, Cat. #11-836-170-001), and 20 mM NEM. Samples were kept on ice for 15 min and vortexed at ~2 min intervals and then boiled in Laemmli buffer. Antibodies were rabbit anti-hRanBP2(2500–3224) and rabbit anti-hRanBP2(2550–2837) (Joseph et al., 2004); rabbit anti-Topo IIα (Topogen); mouse anti-β-actin (AC-151, Sigma); rat anti-HA (3F10; Roche); and mouse anti-phosphohistone H3 (Ser10) (Upstate).
Indirect Immunofluorescence and Live-Cell Imaging
MEFs were cultured on 10-well glass slides for 24 hr, arrested in prometaphase with 100 μM monastrol for 3 hr, and fixed with 3% paraformaldehyde in PBS for 12 min. Antibody incubations were as described (Kasper et al., 1999). Antibodies were rabbit anti-Topo IIα (Topogen); human anti-centromeric antibody (Antibodies Incorporated); rabbit anti-Mad1 (Dr. T. Yen); and rabbit anti-Cenp-E (Dr. D. Cleveland). Live-cell imaging was performed as described (Jeganathan et al., 2005).
In Vitro Sumoylation Assays
Sumoylation reactions were carried out in a total volume of 20 ml. Reactions contained 50 mM HEPES (pH 8), 100 mM NaCl, 1 mM DTT, 1 mM Mg-ATP, 50 nM E1 (110 ng), 25 μM SUMO1, 2, or 3 (5 μg), 125 nM Ubc9 (50 ng), 100 ng recombinant His-HA-RanBP2(2553–2838) or His-HA-RanBP2(2553–2838)L2651A,L2653A, and 20 ng of purified full-length Topo IIα (Topogen). Reactions were performed at 37°C for 1 hr, stopped with equal volume of 2×Laemmli buffer, resolved by SDS-PAGE on 5% Tris-HCl gels, and analyzed by immunoblotting for Topo IIα. Recombinant E1, Ubc9, and SUMO1–3 were from Boston Biochem (SUMO1 conjugation kit; Cat. #K-710). To produce recombinant RanBP2 His-HA-RanBP2(2553–2838) and His-HA-RanBP2(2553–2838)L2651A,L2653A, their corresponding cDNA constructs were cloned into BamH1 and Xho1 sites of pET28a(+) (Novagen). Proteins tagged with His were expressed in BL21 (DE3) bacteria at 15°C and purified from bacterial lysates with Ni2+ agarose.
Supplementary Material
Acknowledgments
We thank Darren Baker, Fang Jin, Mike Thompson, and Mike Zimmer for assistance, staff on the Mayo Clinic Cytogenetics Shared Resource for FISH analysis, members of the van Deursen lab for helpful discussions, and Drs. Mike Matunis, Frauke Melchior, Nabeel Yaseen, Jan Ellenberg, Mary Dasso, Tim Yen, and Don Cleveland for reagents. This work was supported by NIH grant RO1-CA077262.
Footnotes
SUPPLEMENTAL DATA Supplemental Data include Experimental Procedures, References, and nine figures and can be found with this article online at http://www.cell.com/cgi/content/full/133/1/103/DC1/.
References
- Arnaoutov A, Azuma Y, Ribbeck K, Joseph J, Boyarchuk Y, Karpova T, McNally J, Dasso M. Crm1 is a mitotic effector of Ran-GTP in somatic cells. Nat Cell Biol. 2005;7:626–632. doi: 10.1038/ncb1263. [DOI] [PubMed] [Google Scholar]
- Aslanukov A, Bhowmick R, Guruju M, Oswald J, Raz D, Bush RA, Sieving PA, Lu X, Bock CB, Ferreira PA. RanBP2 modulates Cox11 and hexokinase I activities and haploinsufficiency of RanBP2 causes deficits in glucose metabolism. PLoS Genet. 2006;2:e177. doi: 10.1371/journal.pgen.0020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma Y, Arnaoutov A, Dasso M. SUMO-2/3 regulates topoisomerase II in mitosis. J Cell Biol. 2003;163:477–487. doi: 10.1083/jcb.200304088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuma Y, Arnaoutov A, Anan T, Dasso M. PIASy mediates SUMO-2 conjugation of Topoisomerase-II on mitotic chromosomes. EMBO J. 2005;24:2172–2182. doi: 10.1038/sj.emboj.7600700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- Bernad R, van der Velde H, Fornerod M, Pickersgill H. Nup358/RanBP2 attaches to the nuclear pore complex via association with Nup88 and Nup214/CAN and plays a supporting role in CRM1-mediated nuclear protein export. Mol Cell Biol. 2004;24:2373–2384. doi: 10.1128/MCB.24.6.2373-2384.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat MA, Philp AV, Glover DM, Bellen HJ. Chromatid segregation at anaphase requires the barren product, a novel chromosome-associated protein that interacts with Topoisomerase II. Cell. 1996;87:1103–1114. doi: 10.1016/s0092-8674(00)81804-8. [DOI] [PubMed] [Google Scholar]
- Clarke DJ, Johnson RT, Downes CS. Topoisomerase II inhibition prevents anaphase chromatid segregation in mammalian cells independently of the generation of DNA strand breaks. J Cell Sci. 1993;105:563–569. doi: 10.1242/jcs.105.2.563. [DOI] [PubMed] [Google Scholar]
- Dawlaty MM, van Deursen JM. Gene targeting methods for studying nuclear transport factors in mice. Methods. 2006;39:370–378. doi: 10.1016/j.ymeth.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Diaz-Martinez LA, Gimenez-Abian JF, Azuma Y, Guacci V, Gimenez-Martin G, Lanier LM, Clarke DJ. PIASgamma is required for faithful chromosome segregation in human cells. PLoS ONE. 2006;1:e53. doi: 10. 1371/journal.pone.0000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen GD, Perou CM, Whyte RI, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci USA. 2001;98:13784–13789. doi: 10.1073/pnas.241500798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutten S, Kehlenbach RH. Nup214 is required for CRM1-dependent nuclear protein export in vivo. Mol Cell Biol. 2006;26:6772–6785. doi: 10.1128/MCB.00342-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeganathan KB, Malureanu L, van Deursen JM. The Rae1-Nup98 complex prevents aneuploidy by inhibiting securin degradation. Nature. 2005;438:1036–1039. doi: 10.1038/nature04221. [DOI] [PubMed] [Google Scholar]
- Joseph J, Tan SH, Karpova TS, McNally JG, Dasso M. SUMO-1 targets RanGAP1 to kinetochores and mitotic spindles. J Cell Biol. 2002;156:595–602. doi: 10.1083/jcb.200110109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph J, Liu ST, Jablonski SA, Yen TJ, Dasso M. The Ran-GAP1-RanBP2 complex is essential for microtubule-kinetochore interactions in vivo. Curr Biol. 2004;14:611–617. doi: 10.1016/j.cub.2004.03.031. [DOI] [PubMed] [Google Scholar]
- Kasper LH, Brindle PK, Schnabel CA, Pritchard CE, Cleary ML, van Deursen JM. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol Cell Biol. 1999;19:764–776. doi: 10.1128/mcb.19.1.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodner RD, Putnam CD, Myung K. Maintenance of genome stability in Saccharomyces cerevisiae. Science. 2002;297:552–557. doi: 10.1126/science.1075277. [DOI] [PubMed] [Google Scholar]
- Mahajan R, Delphin C, Guan T, Gerace L, Melchior F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell. 1997;88:97–107. doi: 10.1016/s0092-8674(00)81862-0. [DOI] [PubMed] [Google Scholar]
- Mao Y, Desai SD, Liu LF. SUMO-1 conjugation to human DNA topoisomerase II isozymes. J Biol Chem. 2000;275:26066–26073. doi: 10.1074/jbc.M001831200. [DOI] [PubMed] [Google Scholar]
- Matunis MJ, Pickart CM. Beginning at the end with SUMO. Nat Struct Mol Biol. 2005;12:565–566. doi: 10.1038/nsmb0705-565. [DOI] [PubMed] [Google Scholar]
- Matunis MJ, Coutavas E, Blobel G. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein Ran-GAP1 between the cytosol and the nuclear pore complex. J Cell Biol. 1996;135:1457–1470. doi: 10.1083/jcb.135.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michor F, Iwasa Y, Vogelstein B, Lengauer C, Nowak MA. Can chromosomal instability initiate tumorigenesis? Semin Cancer Biol. 2005;15:43–49. doi: 10.1016/j.semcancer.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Nasmyth K. Segregating sister genomes: the molecular biology of chromosome separation. Science. 2002;297:559–565. doi: 10.1126/science.1074757. [DOI] [PubMed] [Google Scholar]
- Pichler A, Gast A, Seeler JS, Dejean A, Melchior F. The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell. 2002;108:109–120. doi: 10.1016/s0092-8674(01)00633-x. [DOI] [PubMed] [Google Scholar]
- Pichler A, Knipscheer P, Saitoh H, Sixma TK, Melchior F. The RanBP2 SUMO E3 ligase is neither HECT- nor RING-type. Nat Struct Mol Biol. 2004;11:984–991. doi: 10.1038/nsmb834. [DOI] [PubMed] [Google Scholar]
- Roth W, Sustmann C, Kieslinger M, Gilmozzi A, Irmer D, Kremmer E, Turck C, Grosschedl R. PIASy-deficient mice display modest defects in IFN and Wnt signaling. J Immunol. 2004;173:6189–6199. doi: 10.4049/jimmunol.173.10.6189. [DOI] [PubMed] [Google Scholar]
- Salina D, Enarson P, Rattner JB, Burke B. Nup358 integrates nuclear envelope breakdown with kinetochore assembly. J Cell Biol. 2003;162:991–1001. doi: 10.1083/jcb.200304080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Yong-Gonzalez V, Kikuchi Y, Strunnikov A. SIZ1/SIZ2 control of chromosome transmission fidelity is mediated by the sumoylation of topoisomerase II. Genetics. 2006;172:783–794. doi: 10.1534/genetics.105.047167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BA, Cleveland DW. Does aneuploidy cause cancer? Curr Opin Cell Biol. 2006;18:658–667. doi: 10.1016/j.ceb.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Wong KA, Kim R, Christofk H, Gao J, Lawson G, Wu H. Protein inhibitor of activated STAT Y (PIASy) and a splice variant lacking exon 6 enhance sumoylation but are not essential for embryogenesis and adult life. Mol Cell Biol. 2004;24:5577–5586. doi: 10.1128/MCB.24.12.5577-5586.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Matunis MJ, Kraemer D, Blobel G, Coutavas E. Nup358, a cytoplasmically exposed nucleoporin with peptide repeats, Ran-GTP binding sites, zinc fingers, a cyclophilin A homologous domain, and a leucine-rich region. J Biol Chem. 1995;270:14209–14213. doi: 10.1074/jbc.270.23.14209. [DOI] [PubMed] [Google Scholar]
- Zhang H, Saitoh H, Matunis MJ. Enzymes of the SUMO modification pathway localize to filaments of the nuclear pore complex. Mol Cell Biol. 2002;22:6498–6508. doi: 10.1128/MCB.22.18.6498-6508.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.