

Abstract
Integrins are a large family of cell adhesion receptors involved in a variety of cellular functions. To study their roles at central synapses, we used two cre recombinase lines to delete the Itgb1 β1 integrin gene in forebrain excitatory neurons at different developmental stages. Removal of the β1 integrins at an embryonic stage resulted in severe cortical lamination defects without affecting the cellular organization of pyramidal neurons in the CA3 and CA1 regions of the hippocampus. Whereas the hippocampal neurons underwent normal dendritic and synaptic differentiation, the adult synapses exhibited deficits in responses to high-frequency stimulation (HFS), as well as in long-term potentiation (LTP). Deletion of β1 integrin at a later postnatal stage also impaired LTP but not synaptic responses to HFS. Thus, the β1-class integrins appear to play distinct roles at different stages of synaptic development, critical for the proper maturation of readily releasable pool of vesicles during early development but essential for LTP throughout adult life.
Keywords: β1 integrin, cell adhesion molecule, hippocampus, synaptic maturation, synaptic vesicle pool, long-term potentiation
Introduction
Synaptic development in the brain requires the coordinate assembly of the presynaptic vesicle secretion machinery and the postsynaptic neurotransmitter reception apparatus at juxtaposed locations between neuronal axons and dendrites. On the presynaptic side, it depends on the proper assembly of different synaptic vesicle pools at the active zone (Rosenmund and Stevens, 1996; Schneggenburger et al., 1999; Wu and Borst, 1999; Schikorski and Stevens, 2001; Mozhayeva et al., 2002; Taschenberger et al., 2002). On the postsynaptic side, it requires the precise organization of different classes of neurotransmitter receptors at the postsynaptic density (Kennedy, 2000; Lee et al., 2004). The coordinate assembly of the synaptic structures involves bidirectional communication mediated by cell surface molecules. For example, interactions between neuroligin and neurexin as well as between members of the Ig superfamily of cell adhesion molecules are involved in triggering differentiation on both sides of the synapse (Scheiffele et al., 2000; Biederer et al., 2002; Yamagata et al., 2002; Shen and Bargmann, 2003; Graf et al., 2004; Chih et al., 2005). The cadherin family of cell adhesion molecules is instead localized to the perisynaptic zone in which it may regulate the organization of the presynaptic membrane as well as synaptic vesicles (Fannon and Colman, 1996; Uchida et al., 1996; Togashi et al., 2002; Bamji et al., 2003).
The integrins are a family of heterodimeric cell adhesion receptors that mediate interactions between cells and between cells and the extracellular matrix (Reichardt and Tomaselli, 1991; Hynes, 2002). In the nervous system, they play important roles in basal lamina assembly and neuronal migration (Galileo et al., 1992; Anton et al., 1999; Graus-Porta et al., 2001; Sanada et al., 2004; Schmid et al., 2004), axonal growth and dendritic development (Tomaselli et al., 1993; DeFreitas et al., 1995; Varnum-Finney et al., 1995; Weaver et al., 1995; Baum and Garriga, 1997; Hoang and Chiba, 1998; Pasterkamp et al., 2003; Moresco et al., 2005; Marrs et al., 2006), neuromuscular junction (NMJ) development and function (Chen and Grinnell, 1995; Martin and Sanes, 1997; Beumer et al., 1999; Rohrbough et al., 2000; Schwander et al., 2004), dendritic spine plasticity (Kramar et al., 2006; Shi and Ethell, 2006), and learning and memory (Grotewiel et al., 1998; Chan et al., 2003; Chan et al., 2006). At central synapses, pharmacological studies have indicated that RGD (Arg-Gly-Asp)-binding integrins play an acute modulatory role in hippocampal long-term potentiation (LTP) (Bahr et al., 1997; Staubli et al., 1998; Kramar et al., 2003; Nagy et al., 2006). Pharmacological and genetic studies have also implicated several β1-binding α-class integrins in the induction and/or consolidation of hippocampal LTP (Chun et al., 2001; Kramar et al., 2002; Chan et al., 2003). More recent studies have also directly implicated the β1 integrins in hippocampal LTP (Chan et al., 2006; Kramar et al., 2006). In addition, studies in neuronal cultures have shown that the integrins may be involved in mediating the enhancement of excitatory synaptogenesis by astrocytes as well as in the functional maturation of hippocampal synapses (Chavis and Westbrook, 2001; Hama et al., 2004). These data suggest that the integrins may play roles in both the development and the function of central synapses. In this article, we used a genetic approach of temporally specific gene deletion to determine the in vivo functions of the β1-class integrins at central synapses at different stages of development.
Materials and Methods
Mouse strains.
The β1 integrin null and conditional alleles were generated as described previously (Stephens et al., 1995; Graus-Porta et al., 2001). The emx1 (empty spiracles homolog 1)–cre and calcium/calmodulin-dependent kinase IIα (CaMKIIα)-cre T29–1 lines were reported by Gorski et al. (2002) and Tsien et al. (Tsien et al., 1996; Sonner et al., 2005), respectively. The cre transgenes were introduced into mice heterozygous for the β1 integrin null and conditional alleles for phenotypic analyses and the heterozygotes alone were used as controls. Mouse colonies were maintained in a mixed 129 and C57BL/6 background. The Rosa26 cre reporter line was described by Soriano (1999). Animal use was in accordance with institutional guidelines.
Primary antibodies.
The following primary antibodies were used at the respective dilutions: anti-neuronal-specific nuclear protein (NeuN) monoclonal (1:500; Chemicon, Temecula, CA), rabbit anti-β-galactosidase (1:500; ICN Cappel, Aurora, OH), rabbit anti-GAD67 (1:1000; Chemicon), anti-β-galactosidase monoclonal (1:200; Promega, Madison, WI), rat anti-β1 integrin monoclonal MAB1997 (20 μg/ml; Chemicon), hamster anti-β1 integrin monoclonal Ha2/11 (7.1 μg/ml; D. M. Kelly, Harvard Medical School, Boston, MA), rabbit anti-ER81 (ETS transcription factor) (1:5000; T. Jessell, Columbia University, New York, NY), anti-bromodeoxyuridine (BrdU) monoclonal (1:400; BD Biosciences, San Jose, CA), rabbit anti-Otx-1 (orthodenticle homolog 1) (1:100; S. McConnelll, Stanford University, Stanford, CA), anti-microtubule-associated protein 2 (MAP2) monoclonal (1:200; Sigma, St. Louis, MO), anti-synaptotagmin monoclonal (clone ASV48, 8 μg/ml), anti-synaptophysin monoclonal (clone SY38, 1:20; Chemicon), rabbit anti-glutamate receptor subunit 1 (GluR1) (1:200; Upstate Biotechnology, Lake Placid, NY), rabbit anti-NR2A (1:500; Upstate Biotechnology), and anti-GAD65 monoclonal (clone GAD-6, 1:1000; Roche, Indianapolis, IN).
Histology and immunohistochemistry.
Brains were dissected from mice anesthetized and perfused with 4% paraformaldehyde in PBS and postfixed for either a few hours or overnight. Vibratome sections of brain tissues were processed for Nissl staining according to standard protocol. For immunohistochemistry, after quenching with 10% methanol plus 3% hydrogen peroxide, sections were blocked with 10% goat serum, 2.5% BSA, 1% glycine, and 0.4% Triton X-100 (except in the case of anti-GAD67 when no detergent was included as recommended by the manufacturer). Primary antibodies were incubated overnight at 4°C, followed by incubations with biotinylated anti-mouse secondary antibodies for 1 h and the ABC solution for an additional 1 h at room temperature (Vector Laboratories, Burlingame, CA). The sections were then processed for diaminobenzidine staining and mounted on glass slides. For immunofluorescent staining, either FITC or cyanine 3-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) were used, and incubation was performed for ∼4 h at room temperature or overnight at 4°C. The sections were then mounted with ProLong Antifade medium (Invitrogen, Carlsbad, CA) and used for image collection under a Bio-Rad (Hercules, CA) MRC 1024 laser scanning confocal microscope. For modified Golgi–Cox staining, an FD Rapid GolgiStain kit was used, and the procedure was performed according to the instructions of the manufacturer (FD NeuroTechnologies, Ellicott City, MD).
Slice electrophysiology.
Recordings from transverse hippocampal slices (400 μm), prepared from male adult mice (postnatal days 42–60) of either wild-type or β1 integrin mutants (both emx1–cre and CaMKIIα–cre targeted) were performed as described previously (Pozzo-Miller et al., 1999). The slices were maintained in an interface chamber for both recovery (>2 h) and recording. Perfusion medium [artificial CSF (ACSF), 34°C] contained the following (in mm): 124 NaCl, 3.0 KCl, 2.5 CaCl2, 1.5 MgCl2, 26 NaHCO3, 1.25 KH2PO4, 10 glucose, and 2 ascorbic acid, pH 7.4. The perfusion rate was 15 ml/h. The field EPSPs (fEPSPs) were evoked in the CA1 stratum radiatum by stimulating the Schaffer–commissural field with twisted bipolar nichrome electrodes and were recorded with ACSF-filled glass pipettes (<5 MΩ) using an Axoclamp-2B amplifier (Molecular Devices, Palo Alto, CA). Test stimuli consisted of monophasic 200 μs pulses of constant current delivered by stimulus isolation units. Basal synaptic transmission was monitored by alternating, low-frequency stimulation (LFS) (every 30 s) of two separate pathways via two stimulating electrodes (S1 and S2) positioned on both sides of the recording. Only slices exhibiting fEPSPs of 2–3 mV in amplitude without superimposed population spikes were used. Stimulus intensity was adjusted to evoked fEPSPs of ∼1.3 mV. All analyses were performed after stable baseline was established for at least 30 min. fEPSPs were digitized (10 kHz), filtered at 3 kHz (eight-pole Bessel filter) using acquisition system pClamp6, stored on magnetic media, and analyzed off-line using analysis system Clampfit 8.0 (Molecular Devices) and Microsoft (Redmond, WA) Excel visual basic programming. The statistics was conducted using number of slices (n) pooled from multiple animals (N).
Input–output curves were obtained by stimulating the CA1 synapses by single pulses of increasing intensities and plotting the fiber volley against the slopes of fEPSPs. For paired-pulse facilitation (PPF), a twin pulse of different interpulse interval (IPI) (10–100 ms) was applied to the CA1 synapses. The ratios of second fEPSP to first fEPSP slope were plotted against interpulse intervals. Two stimulus protocols were used to study different pools of synaptic vesicles: high-frequency stimulation (HFS) (40 stimuli at 100 Hz) and prolonged repetitive stimulation (80 stimuli at 14 Hz) in the presence of the NMDA receptor antagonist d(−)-2 amino-5-phosphonovaleric acid (APV) (100 μm). LTP was induced by tetanic stimulation (100 Hz, 2 trains, 1 s each, spaced by 20 s). NMDA receptor-mediated fEPSPs were isolated pharmacologically using Mg2+-free ACSF containing the AMPA receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione disodium (CNQX) (10 μm). At the end of recordings, APV was applied to confirm that they were NMDA-mediated synaptic responses.
Whole-cell recordings were made from CA1 pyramidal neurons of hippocampal slices using the blind patch method. Slices were maintained in a submerged chamber for both recovery (1 h) at room temperature and recordings at 30°C. Recording electrodes (5–8 MΩ) were filled with intracellular solution containing the following (in mm): 130 K-gluconate, 10 HEPES, 2 Mg2-ATP, 0.5 Na3-GTP, 11 EGTA, 10 phosphocreatine, and 5 QX-314 [2(triethylamino)-N-(2,6-dimethylphenyl) acetamine], pH 7.2 (saturated with 95%O2, 5% CO2; 290 mOsm). Perfusion rate was 1.5 ml/min. EPSCs were evoked at a rate of 0.1 Hz by a stimulating electrode placed in the stratum radiatum. Access resistance was monitored throughout experiments and ranged from 10 to 25 MΩ. Data was discarded when access resistance changed by >20% during an experiment. To isolate NMDA-mediated EPSCs, neurons were voltage clamped at −40 mV in the presence of CNQX (20 μm) and the GABAA receptor antagonist bicuculline methiodide (10 μm). Signals were recorded using an Axopatch 200B, filtered at 5 kHz, and digitized at 10 kHz.
Results
Deletion of β1 integrin from the forebrain excitatory neurons using emx1–cre
The integrins are heterodimeric membrane proteins that consist of an α and a β subunit. They include two major classes, one with a common β1 subunit and the other with a common αv subunit. In the nervous system, both classes of integrins are widely expressed during development and in the adult, in regions including the cerebral cortex and the hippocampus (Pinkstaff et al., 1998, 1999). To study the in vivo function of β1 integrins in the nervous system, we used a conditional allele of the β1 integrin gene in which the first coding exon was flanked by a pair of loxP sites (Graus-Porta et al., 2001). In previous experiments, this conditional allele was introduced into a cre line whose expression is driven by the neuronal enhancer element of the nestin gene promoter. This resulted in deletion of the β1 integrin gene throughout much of the nervous system and led to severe developmental defects in cerebral cortical lamination and cerebellar foliation (Graus-Porta et al., 2001; Blaess et al., 2004). To study more specifically the functions of β1-class integrins in excitatory neurons, we used another cre line whose expression is driven by the emx1 gene promoter by virtue of an internal ribosome entry site element engineered into the emx1 locus (Gorski et al., 2002). In contrast to the nestin promoter-regulated cre, the expression of cre in this strain is restricted to excitatory neurons and glial cells and their precursors derived from the dorsal forebrain neuroepithelium but is absent from the inhibitory interneurons, which originate from a different source in the ventral pallium. When crossed to a Rosa26 reporter line (Soriano, 1999), the emx1–cre induces expression of the lacZ reporter gene in excitatory neurons in both the cerebral cortex (data not shown) and the hippocampus (Fig. 1a–c), whereas no detectable level of β-galactosidase immunoreactivity was observed in inhibitory interneurons (Fig. 1d–f), confirming previous results (Gorski et al., 2002). This restriction of recombination to the excitatory neurons and the glia has allowed us to study the cell-autonomous requirement for β1 integrins in the development and function of excitatory synapses, particularly in the hippocampus.
Figure 1.
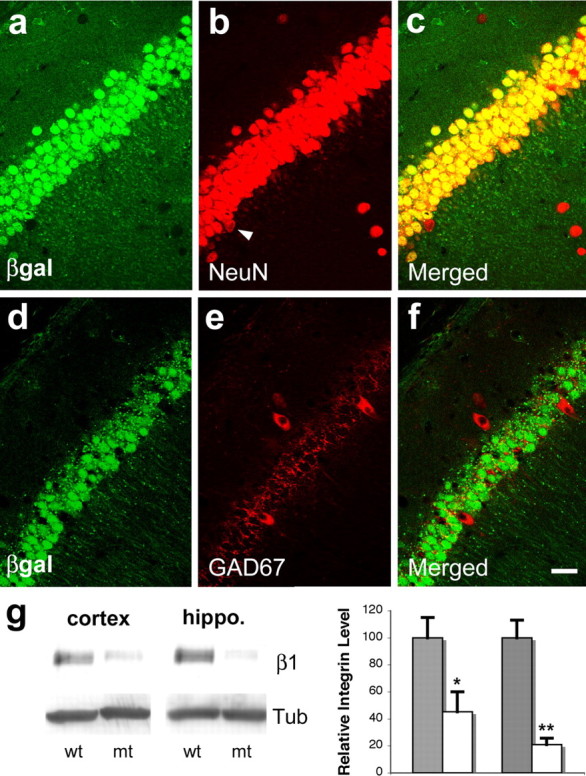
emx1–cre induces recombination in excitatory but not inhibitory neurons in the hippocampus. a–c, Confocal micrographs of neurons in the CA1 region of the hippocampus in adult mice harboring the Rosa26 reporter and the emx1–cre transgenes. Neurons were stained for β-galactosidase (βgal, green) and the pan-neuronal marker NeuN (red). Notice that the vast majority of the neurons are positive for β-galactosidase except for a few presumable inhibitory neurons (arrowhead in b). d–f, Confocal micrographs of neurons from the same region stained for β-galactosidase (green) and the inhibitory neuron marker GAD67 (red). Notice the absence of β-galactosidase in these neurons. The background staining in e was probably attributable to the absence of detergent in the procedure. g, Western blot analysis of β1 integrin expression in the cerebral cortex and hippocampus (hippo.) of wild-type (wt) and mutant (mt) animals. Relative expression levels of β1 integrin from three postnatal day 2 animals of each genotype (wt, gray columns; mt, white columns) were quantified against β tubulin (tub) (left, cortex; right, hippocampus). Error bars depict SEM. *p < 0.05; **p < 0.01 (t test). Scale bar, 50 μm for a–f.
Defective cortical but normal hippocampal lamination in β1 integrin/emx1–cre mouse
To obtain β1 integrin mutants, we made use of a null allele of β1 integrin gene (Stephens et al., 1995) and generated heterozygous animals with one null allele and one conditional allele. This ensures the efficiency of deleting the β1 integrin gene because only one allele needs to be recombined in each cell in the presence of the Cre recombinase. We confirmed the deletion of β1 integrin gene by examining its protein expression. We found that, at late embryonic stage, no β1 integrin immunostaining was detected in the radial glia or neurons in the developing cerebral cortex or hippocampus (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Western blot analysis further confirmed the loss of β1 integrin protein from the cerebral cortex and hippocampus (Fig. 1g). The emx1–cre mutants were born at a normal Mendelian ratio but showed decreased postnatal viability, presumably as a result of seizures caused by cortical lamination defects (see below). Nonetheless, a significant number of these animals survived to adulthood, permitting both anatomical and electrophysiological analyses at the adult stage.
Nissl staining of the cerebral cortices of these mutant animals indicated that there were severe neuronal layering defects. In control animals, the cortex is normally organized into distinct layers with the cell-sparse layer I positioned closest to the pial surface (Fig. 2a,c). In mutant animals, however, no distinct cortical layers were obvious except for layer I, whose border with the other cortical layers was also severely distorted (Fig. 2b,d). Neuronal birthdating experiments further revealed that, in some areas, neurons normally destined for the deeper layers had apparently invaded the superficial layers, whereas in other areas neurons normally destined for the superficial layers were trapped in the deeper layers (Fig. 3a–d). These lamination defects were also confirmed by staining for layer specific markers such as ER81 and Otx-1 (Fig. 3, e,f and g,h, respectively).
Figure 2.
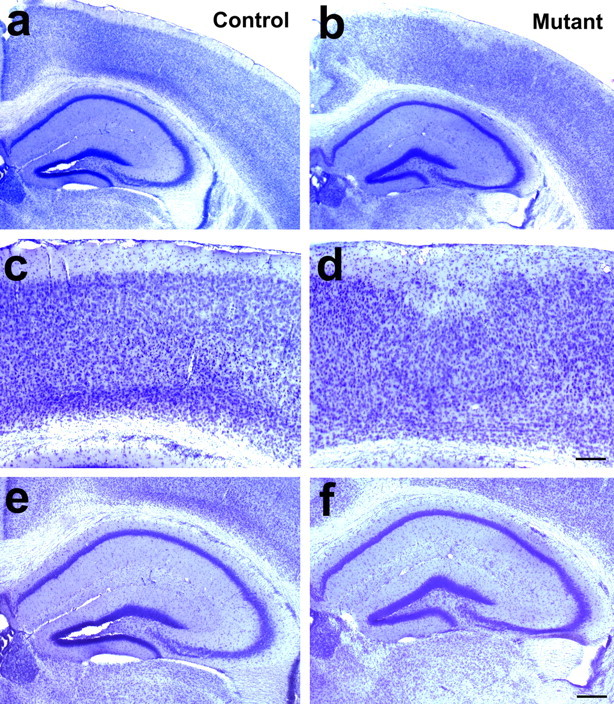
Severe cortical lamination defects but normal hippocampal morphology in the β1 integrin/emx1–cre mice. a, b, Low-magnification micrographs of Nissl-stained sections of the dorsal forebrain region in the control (a) and mutant (b) animals. c, d, Close-up views of the cortical regions in a and b, showing the lamination defects in the mutant (d) compared with the control (c). e, f, Close-up views of the hippocampal formation. No obvious difference in morphology was observed between the control (e) and the mutant (f). Scale bars: c–f, 200 μm.
Figure 3.
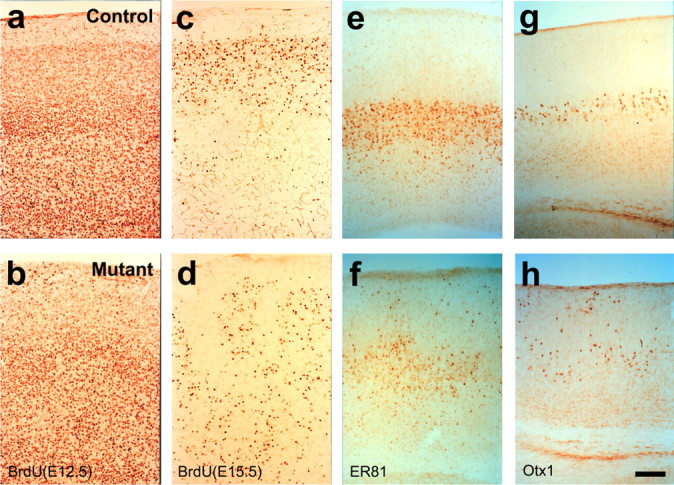
Neuronal displacement in the cerebral cortex of the β1 integrin/emx1–cre mice. a–d, Neuronal birthdating using BrdU incorporation. Neurons destined for the deeper layers in the control cortex (a), which were heavily labeled by BrdU injection at embryonic day 12.5 (E12.5), were displaced into more superficial layers in the mutant cortex (b). Neurons destined for superficial layers in the control cortex (c), labeled by BrdU injection at E15.5, were found trapped in deeper layers in the mutant cortex (d). e–h, Stainings of deep layer neuronal markers ER81 (e, f) and Otx-1 (g, h) in control (e, g) and mutant (f, h) cortices show displacement of neurons expressing these proteins in the mutant. Scale bar, 200 μm.
In contrast to the cerebral cortex, we observed a relatively normal organization of the neuronal layer in the hippocampal formation. In both the CA1 and CA3 regions of the mutants, the pyramidal neuron cell bodies were tightly packed in a single layer in their normal location (Fig. 2, compare f, e), although occasional defects in the organization of granule cells in the dentate gyrus were observed (data not shown), as has been reported after deletion with nestin–cre (Forster et al., 2002). This suggests that the β1 integrins are not essential for the migration of pyramidal neurons to their normal location in the hippocampus. In addition, because the dendritic fields of the CA1 and CA3 neurons also appeared to be of relatively normal size (Figs. 2e,f, 4) and their thickness was comparable between the control and the mutant (p > 0.7), this suggests that the overall differentiation of these neurons was not compromised. This thus allowed us to investigate the potential role(s) of the β1 integrins in the differentiation and function of the hippocampal pyramidal neurons in a relatively normal in vivo cellular environment.
Figure 4.
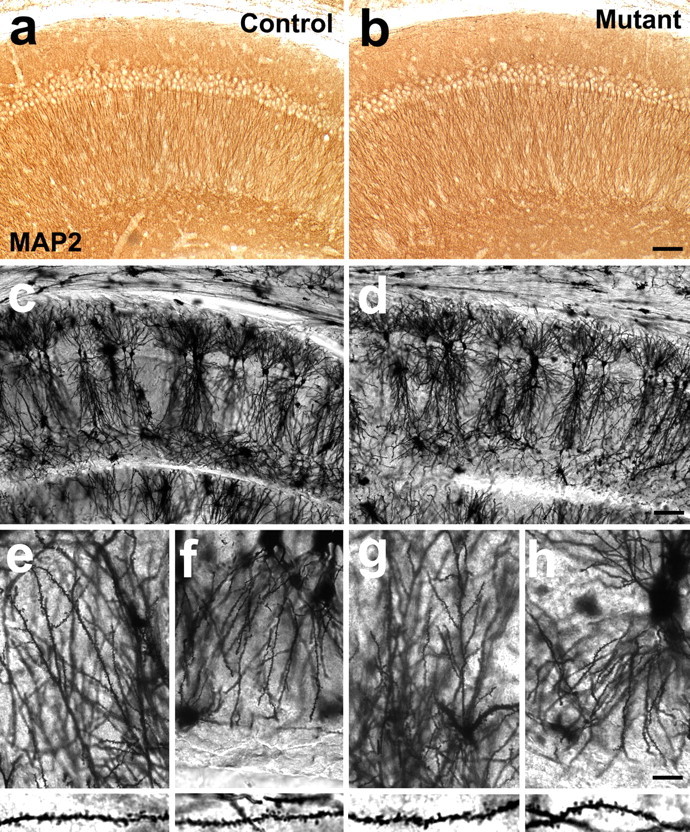
Normal dendritic differentiation in the hippocampal region of the β1 integrin/emx1–cre mice. a, b, Micrographs of staining for the dendritic marker MAP2 in the hippocampal CA1 region of control (a) and mutant (b) animals. c–h, Micrographs of Golgi-impregnated pyramidal neurons in the hippocampal CA1 region of control (c, e, f) and mutant (d, g, h) mice. Normal overall apical and basal dendritic morphology was observed at a low magnification (c, d). Normal dendritic spines were also observed along both the apical (e, g) and the basal (f, h) dendrites at a higher magnification. Scale bars: a–d, 50 μm; e–h, 25 μm.
Normal overall differentiation of dendrites and synapses in β1 integrin/emx1–cre hippocampus
In developing hippocampal neurons in culture, the β1 integrins are localized to the soma as well as the processes of the cells (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). To investigate whether the β1 integrins play a role in the differentiation of hippocampal neuronal processes in vivo, we first stained for the expression of the dendritic marker MAP2. We found that the pyramidal neurons in the CA1 region of the mutant hippocampus had relatively normal dendritic architecture (Fig. 4a,b). The mutant dendrites extended normally into the dendritic field and had similar intensity of MAP2 staining compared with those from the control. Similar patterns were also observed in the CA3 region of the hippocampus (data not shown). Together with the relatively normal size of the dendritic fields (Fig. 2g), these results indicate that the overall differentiation of pyramidal neuron dendrites was not affected by the loss of β1 integrins. To further examine the dendritic morphology of these neurons, we performed Golgi staining on these brains. Consistent with the observations on MAP2-stained neurons, we found that the impregnated mutant CA1 pyramidal neurons also had relatively normal morphologies with well developed apical and basal dendritic branches (Fig. 4c,d). Closer examination revealed that, not only did the dendrites have relatively normal morphologies (Fig. 4e–h, top), they were also decorated by regularly spaced dendritic spines at an apparently normal density (Fig. 4e–h, bottom). Thus, the β1 integrins do not seem to be required for the normal overall differentiation of pyramidal neuron dendrites and spines in the hippocampus.
To determine whether the β1 integrins regulate synaptic differentiation in vivo, we next examined the expression pattern of a number of synaptic markers in the hippocampus. We first examined the expression of synaptotagmin, a marker of all presynaptic terminals, in the hippocampus by immunohistochemistry. We observed a relatively normal pattern and intensity of synaptotagmin staining in the CA1 region of the mutant hippocampus (Fig. 5a,b). Similar results were also observed in the CA3 region of the hippocampus (data not shown). Staining of synaptophysin, another protein present in all presynaptic terminals, also revealed similar results. At high resolution, the synaptophysin-positive puncta showed similar densities and intensities in the CA1 dendritic fields of the control and mutant animals (Fig. 5c,d). To further investigate synaptic differentiation, we next used antibodies specific to the postsynaptic compartments of excitatory synapses. We found that the mutant hippocampus had a relatively normal density and intensity of puncta positive for the AMPA subclass of glutamate receptor subunit GluR1 (Fig. 5e,f). Similar results were also obtained for the NMDA subclass of glutamate receptor subunit NR2A (data not shown). To investigate the differentiation of inhibitory synapses, we examined the staining of GAD65 in the axonal terminals of inhibitory neurons and observed relatively normal pattern of expression in the mutants (Fig. 5g,h). Thus, these results suggest that the overall differentiation of excitatory synapses in the hippocampus was not perturbed by the loss of β1 integrins in excitatory neurons and glial cells.
Figure 5.
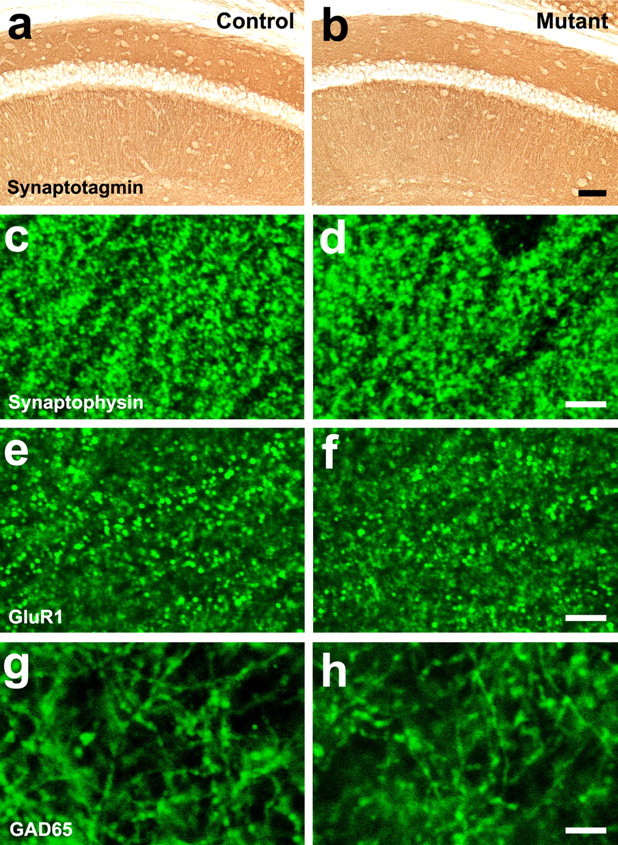
Normal synaptic differentiation in the hippocampus of the β1 integrin/emx1–cre mice. a, b, Micrographs of synaptotagmin staining in the hippocampal CA1 region of control (a) and mutant (b) animals. c–h, Confocal micrographs of immunofluorescent stainings for the pan-synaptic marker synaptophysin (c, d), the excitatory synapse-specific marker GluR1 (e, f), and the inhibitory synapse-specific presynaptic marker GAD65 (g, h) in the apical dendritic field of the CA1 region of control (c, e, g) and mutant (d, f, h) animals. No obvious differences were observed between the controls and the mutants. Scale bars: a, b, 50 μm; c–h, 5 μm.
Impairment of LTP but not basal transmission in β1 integrin/emx1–cre hippocampus
To determine whether the loss of β1-class integrins from excitatory neurons during early development affects the function of adult hippocampal synapses, we examined the electrophysiological properties of the Schaffer collateral–CA1 synapses in hippocampal slices derived from emx1–cre-targeted animals. Recording of fEPSPs from hippocampal slices indicated that basal synaptic transmission, as reflected by the input–output curve, was normal at the mutant synapses (Fig. 6a). No significant difference was observed in fEPSP slopes induced by a wide range of stimulation intensities (Kolmogorov–Smirnov test, p > 0.5). Thus, the overall efficacy at CA1 synapses was similar between the mutant and the control.
Figure 6.

Electrophysiological defects at the Schaffer collateral–CA1 synapses in the β1 integrin/emx1–cre mice. Field EPSPs were recorded at CA1 synapses by stimulating the Schaffer collaterals. For recordings of all types of plasticity, the stimulation intensity was adjusted to evoke fEPSPs of ∼1.3 mV. a, Input–output curves for basal synaptic transmission. fEPSP slopes were plotted against the fiber volley amplitude. Wild type, N = 5 animals, n = 9 slices; mutant, N = 4 animals, n = 6 slices. In this and all other electrophysiology figures, open circles indicate wild type (WT) and filled triangles indicate mutant (MT). Data from multiple slices of the same genotype are pooled and presented as mean ± SEM (b) LTP. fEPSP slopes were plotted against time (in minutes) before and after tetanic stimulation (two times for 100 Hz trains, 1 s each, interval 20 s). Wild type, N = 7 animals, n = 14 slices; mutant, N = 5 animals, n = 9 slices. Representative traces before and after LTP are shown as insets. c, PPF. The percentage of increase in the second fEPSP slope over the first one was calculated, and mean values were plotted against different IPIs (10–100 ms). Wild type, N = 6 animals, n = 13 slices; mutant, N = 5 animals, n = 14 slices. d, Synaptic responses to HFP (100 Hz, 40 pulses). In d and e, the recordings were performed in the presence of APV to block the NMDA receptor. fEPSP slopes were plotted against stimulus number. Wild type, N = 5 animals, n = 20 slices; mutant, N = 5 animals, n = 15 slices. e, Synaptic responses to LFS (14 Hz). fEPSP slopes were plotted against stimulus number. Wild type, N = 4 animals, n = 18 slices; mutant, N = 4 animals, n = 20 slices. f, NMDA fEPSPs. NMDA-fEPSPs were isolated pharmacologically in Mg2+-free ACSF containing 10 μm CNQX. The mean slopes of fEPSPs are plotted against the amplitudes of fiber volley. Wild type, N = 3 animals, 12 slices; mutant, N = 3 animals, 18 slices.
We next examined the role of β1 integrins in long-term synaptic plasticity. emx1–cre-targeted mutants exhibited significant defects in LTP (Fig. 6b). In wild-type slices, tetanic stimulation (two times for 100 Hz, 1 s) induced robust LTP that lasted for at least 60 min. The fEPSP slope was 36.9 ± 7.4% above baseline level 60 min after the tetanus. In mutant slices, the same stimulation protocol induced synaptic potentiation that declined after the initial 10 min. The fEPSP slope at the end of the experiments was 11.9 ± 10.6% above baseline, significantly smaller than in the wild-type control (p < 0.01). These results are consistent with previous data implicating RGD-binding integrins in the expression of LTP (Bahr et al., 1997; Staubli et al., 1998; Kramar et al., 2003; Nagy et al., 2006). They are also consistent with studies that implicate several β1-binding α-class integrins in hippocampal LTP (Chun et al., 2001; Kramar et al., 2002; Chan et al., 2003). This suggests that the β1 integrins may be a major class of integrins involved in LTP at the hippocampal synapses. Because emx1–cre induces β1 integrin deletion in excitatory but not inhibitory neurons, our results further argue that the β1 integrins are most likely required in the excitatory neurons for mediating their function in LTP, although they do not exclude the possibility that the presence of β1 integrin may also be required in glial cells.
Presynaptic and postsynaptic changes as consequences of β1 integrin deletion
The LTP deficits we observed in the β1 integrin mutants could be attributable to impairments in presynaptic neurotransmitter release, postsynaptic responsiveness, or both. To probe presynaptic function, we first analyzed PPF, measured as the ratio of fEPSP slopes in response to two successive stimulation pulses at various IPIs. Although no change was observed in PPF at most IPIs, the ratio of fEPSP slopes increased when the IPI became very short (10 ms; p < 0.05) (Fig. 6c). These results raise the possibility that the β1 integrins may alter synaptic response to HFS.
To test this idea directly, we recorded the responses of the CA1 synapses to a brief train of HFS (100 Hz, 40 pulses) in the presence of the NMDA receptor antagonist APV (100 μm). After an initial increase, the fEPSP slopes exhibited a continuous decline over time in wild-type slices (Fig. 6d). Consistent with the increase in PPF at the IPI of 10 ms, the response to the second stimulus in the train was increased in the mutant slices. To better illustrate the differences in the decay phase of HFS response between wild-type and mutant slices, we plotted all of the data normalized to the response to the second stimulus (Fig. 6d). The fEPSP slope recorded from the mutant synapses declined more rapidly than that from the control synapses (Kolmogorov–Smirnov test, p < 0.05). The rate constant (τ), obtained by fitting the decay phase of the plots with an exponential equation, was 20.0 ± 3.9 stimuli for the control but 10.4 ± 1.2 stimuli for the mutant synapses (p < 0.02). To exclude the possibility that repetitive stimulation may recruit inhibition in the mutant slices, we performed the same experiment in the presence of the GABAA antagonist bicuculline (10 μm). The CA3 regions of the hippocampal slices were removed to prevent excessive excitation and afterdischarges induced by bicuculline. Under these conditions, synaptic responses to HFS were still impaired in β1 integrin mutants (supplemental Fig. 3a, available at www.jneurosci.org as supplemental material). The brief HFS is effective in depleting the readily releasable pool (RRP) of synaptic vesicles but is too fast to affect the reserve pool of vesicles. Thus, a faster decline of response to HFS implies a smaller RRP at mutant synapses.
The increase in PPF at a short IPI observed at β1 integrin mutant synapses could be explained by a decrease in probability of transmitter release (Pr). To directly investigate potential changes in Pr, we performed whole-cell recording of NMDA currents in the presence of MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate] (5 μm), an irreversible, open channel blocker for NMDA receptor. The NMDA currents were pharmacologically isolated by blocking AMPA and GABAA receptors with CNQX (20 μm) and bicuculline, respectively. The currents were monitored at a holding potential of −40 mV once every 10 s, with the amplitude set to ∼100 pA. After a stable baseline was obtained, stimulation was paused for 5 min when MK-801 was perfused into the slices. When stimulation was resumed in the presence of MK-801, the amplitude of NMDA currents progressively declined (Fig. 7a,b). Scaling of the current at the end of experiment (Fig. 7a, #3) superimposed with that at the beginning (Fig. 7a, #2), suggesting that the effective concentration of MK-801 remained constant during the course of the experiments (Fig. 7c). The time constant of the progressive decline of NMDA currents is inversely related to the Pr (Hessler et al., 1993; Rosenmund et al., 1993). The decay of the averaged responses appeared significantly slower in the mutant compared with wild-type slices (Fig. 7a,b), suggesting a lower Pr in the mutant. A double-exponential fit of the decline revealed a fast and a slow component. The fast decay constant was significantly slower in β1 integrin mutants compared with wild-type animals (21.9 ± 2.5 vs 7.8 ± 0.9 stimuli; p < 0.01), whereas the slow time constant was not statistically different between genotypes (46.2 ± 5.06 vs 52.6 ± 5.7 stimuli; p > 0.1) (Fig. 7d). An alternative measure is the number of stimuli to attain 50% block of the initial EPSC amplitude. This number for the mutants was significantly higher, with a mean of 21.6 ± 3.8 stimuli compared with that of the wild types, which had a mean of 10.1 ± 1.7 stimuli (p < 0.05) (Fig. 7d). Collectively, these data suggest that the Pr is reduced in mice lacking β1 integrin.
Figure 7.

Reduction in Pr in β1 integrin/emx1–cre mice. The NMDA currents (NMDA EPSCs) were pharmacologically isolated by applying CNQX and bicuculline and monitored by whole-cell recording once every 10 s (Vh of −40 mV). After perfusion of MK-801, the amplitude of NMDA currents progressively declined. a, Example of MK-801-induced blockade of NMDA EPSCs in wild-type and β1 integrin/emx1–cre mice. b, Average decay profile of NMDA currents for wild-type and β1 integrin/emx1–cre mice. The amplitudes of NMDA EPSCs were normalized to that of the first EPSC, and the plots were fitted by a double-exponential function. Wild type, n = 6; mutant, n = 6. c, Averaged NMDA EPSCs during baseline (1 in a) and in the presence of MK-801 (2 and 3 in a). Adjacent are the EPSCs of 2 and 3 scaled to that of 1. d, Summary graphs depicting the fast and slow time constants and the time to reach half-maximal EPSC amplitude for β1 integrin/emx1–cre mice and age-matched wild-type littermates. WT, White columns; MT, filled columns. *p < 0.05.
Synaptic vesicles in the nerve terminals undergo a cycling process in which the depleted docked vesicles are replenished by the reserved pool of vesicles and/or through endocytosis (Dobrunz and Stevens, 1997; Pyle et al., 2000; Richards et al., 2000). When a prolonged, LFS is applied, the depletion is usually faster than the replenishment, leading to a gradual decline of EPSP slopes, or synaptic depression (Geppert et al., 1994, 1997). To investigate potential effects of β1 integrin deletion on the reserved pool of synaptic vesicles, we examined the synaptic responses to LFS (14 Hz), again in the presence of APV (Fig. 6e). No significant difference was observed initially, but, as the stimulus number further increased, the fEPSP slope of the mutant synapses decreased more rapidly (Fig. 6e). Statistical analysis revealed a significantly faster decline of synaptic responses to LFS in the mutant slices (Kolmogorov–Smirnov test, p < 0.05), suggesting a smaller size of the reserved vesicle pool and/or defects in its mobilization in neurons lacking integrin β1. The same experiment was repeated in the presence of bicuculline, and the mutant slices still showed an impaired response compared with the wild-type slices (supplemental Fig. 3b, available at www.jneurosci.org as supplemental material). Thus, the differences in LFS responses observed between genotypes are unlikely to be attributable to the differential recruitment of the inhibitory network after repetitive stimulation.
The deficits in LFS response raised the possibility that the impairment in LTP maintenance may be attributable to accumulating vesicular depletion when LTP was monitored by stimulation at 0.033 Hz. We addressed this by performing experiments in which stimulation was interrupted after LTP induction. After tetanic stimulation, the resulting LTP was measured for a period of 10 min, after which, monitoring stimulation was stopped for 10 min. After resuming stimulation, the field responses remained potentiated in wild-type slices with a mean fEPSP slope of 150 ± 12% (supplemental Fig. 3c, available at www.jneurosci.org as supplemental material). In contrast, the responses recorded from mutant slices decayed back to baseline to a mean value of 107 ± 10%, indicating that the failure in LTP maintenance at β1 integrin-deficient synapses was not the result of vesicle depletion.
To determine whether deletion of integrin β1 could also elicit postsynaptic changes, we measured the input–output curves of NMDA receptor-mediated field EPSPs. Slices were incubated in Mg2+-free ACSF containing CNQX (10 μm) to block AMPA receptors. We found that the NMDA fEPSP slopes of the mutant synapses were consistently lower than those of the control synapses over a range of stimulation intensities (Fig. 6f). Statistical analysis of the two plots indicated that NMDA receptor activity at the mutant synapses is significantly lower compared with the wild-type synapses (Kolmogorov–Smirnov test, p < 0.01). These results are consistent with recent reports in which integrin ligation was found to increase the amplitudes of NMDA receptor-mediated synaptic currents in adult hippocampal slices (Lin et al., 2003; Bernard-Trifilo et al., 2005). The impairment in NMDA receptor function could contribute to the LTP deficits we observed in integrin β1 mutant slices. Together, our results show that the loss of the β1-class integrins during embryonic hippocampal development impacts both the presynaptic and postsynaptic properties of the adult hippocampal excitatory synapses.
Distinct synaptic deficits in β1 integrin/CaMKIIα–cre hippocampus
The presynaptic defects we observed in the emx1–cre mutants suggest that absence of the β1 integrins during synaptic development results in synaptic deficits that persist into adulthood. Conversely, pharmacological studies have implied that the RGD-binding integrins acutely regulate LTP at adult hippocampal synapses. This raises the possibility that the β1 integrins may play different roles during development and in the adult. To test this, we made use of another cre line, CaMKIIα–cre, which induces recombination in the hippocampal pyramidal neurons at a later stage than emx1–cre, at ∼2–4 weeks postnatally (Tsien et al., 1996; Xu et al., 2000; Sonner et al., 2005). When this cre line was used to delete the β1 integrin gene, we found that, as expected, it did not lead to any neuronal lamination defects in either the cerebral cortex (data not shown) or the hippocampal formation (Fig. 8a,b). This is consistent with the fact the cre gene expression in this line comes on postnatally after neuronal migration is completed in both the cerebral cortex and the hippocampus. The dendritic fields of the CA1 and CA3 regions in the CaMKIIα–cre-targeted hippocampus were also of relatively normal size, suggesting normal overall dendritic differentiation. Indeed, staining for the dendritic marker MAP2 showed that the mutant CA1 pyramidal neurons had dendritic processes with similar morphology and MAP2 immunoreactivity to those of control animals (Fig. 8c,d). In addition, the general pattern of staining in the hippocampus for the synaptic marker synaptotagmin was also relatively normal (Fig. 8e,f). Thus, consistent with our results using emx1–cre, the β1 integrins do not seem to be required for the overall dendritic and synaptic differentiation by the CA1 and CA3 pyramidal neurons.
Figure 8.
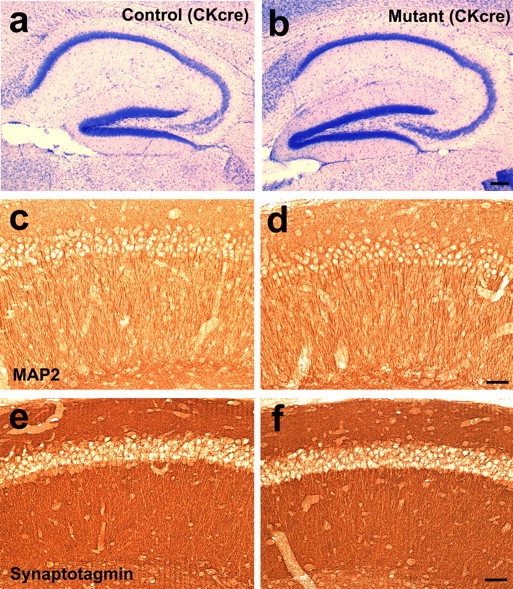
Normal dendritic and synaptic differentiation in the hippocampal region of the β1 integrin/CaMKIIα–cre mice. a, b, Micrographs of Nissl-stained sections of the hippocampal formation in the control (a) and mutant (b) animals. c, d, Micrographs of staining for the dendritic marker MAP2 in the hippocampal CA1 region of control (c) and mutant (d) animals. e, f, Micrographs of synaptotagmin staining in the CA1 region of control (e) and mutant (f) animals. Scale bars: a, b, 200 mm; c–f, 50 mm.
To determine whether the loss of β1-class integrins from mature synapses affects their synaptic physiology, we next performed recording on the Schaffer collateral–CA1 synapses using hippocampal slices from the CaMKIIα–cre animals. We found that, as expected, the basal transmission at these synapses was relatively normal (data not shown). Moreover, the mutant synapses showed similar PPFs over a range of interpulse intervals as the wild-type synapses (ANOVA test, p > 0.05 in all points), indicating a normal sensitivity to calcium and normal Pr (Fig. 9b). Similar to what was observed in the emx1–cre mutants, the synapses of the CaMKIIα–cre mutants also exhibited a significantly faster decline of synaptic response to LFS (Kolmogorov–Smirnov test, p < 0.01) (compare Figs. 9d, 6e). This suggests that the mobilization of the reserved pool of vesicles requires the continuous presence of β1 integrins at the mature hippocampal synapses. However, unlike those in the emx1–cre mutants, the synapses in the CaMKIIα–cre mutants displayed completely normal response to HFS (Kolmogorov–Smirnov test, p > 0.05) (Fig. 9c), suggesting that the size of RRP and the release of vesicles at these synapses were normal. These results suggest that the continuous presence of β1 integrins at the hippocampal synapses is essentially dispensable for the maintenance of their normal presynaptic properties, particularly for the regulated release of RRP vesicles, once the synapses have matured. The defective presynaptic response to HFS we observed in the emx1–cre mutants was thus likely attributable to the loss of the β1-class integrins during synaptic development.
Figure 9.

Electrophysiological defects at the hippocampal synapses in the β1 integrin/CaMKIIα-cre mice. a, LTP. Wild type (WT), N = 5 animals, n = 9 slices; mutant (MT), N = 4 animals, n = 5 slices. Data from multiple slices of the same genotype are pooled and presented as mean ± SEM. Representative traces before and after LTP are shown as insets. b, Paired-pulse facilitation. Wild type, N = 3 animals, n = 6 slices; mutant, N = 3 animals, n = 7 slices. c, Synaptic responses to high-frequency stimulation (100 Hz). Wild type, N = 4 animals, n = 10 slices; mutant, N = 4 animals, n = 14 slices. d, Synaptic responses to low-frequency stimulation (14 Hz). Wild type, N = 3 animals, n = 4 slices; mutant, N = 3 animals, n = 7 slices.
In addition to deficits in their response to LFS, the CaMKIIα–cre mutant synapses exhibited deficits in LTP very similar to those observed in the emx1–cre mutants (Fig. 9a). The fEPSP slope 60 min after the tetanus was 52.1 ± 8.0% for wild-type but 7.4 ± 7.3% for CaMKIIα–cre slices (p < 0.01). Because the β1 integrin gene is deleted in the CaMKIIα–cre mutants only after postnatal week 2–4, these results indicate that the continuous presence of β1-class integrins at the mature hippocampal synapses is required for normal LTP. Thus, the β1 integrins appear to play an active signaling role in the process of LTP expression at the mature hippocampal synapse.
Discussion
The integrins mediates interactions between cells and the extracellular matrix and play critical roles in many developmental processes as well as in the adult organism. In this study, we used a conditional allele of the β1 integrin gene to study the function of the β1-class integrins in the nervous system. We found that the β1 integrins play a pivotal role in the process of cortical neuron layer formation but are essentially dispensable for the normal cellular organization and overall dendritic and synaptic differentiation of pyramidal neurons in the hippocampus. Instead, we found that they play distinct roles in the development and function of the Schaffer collateral–CA1 excitatory synapses. Their presence appears to be required at an early stage for the maturation of the readily releasable pool of vesicles at the presynaptic terminal. In their absence during this period, the synapses become permanently impaired in their release of docked vesicles and consequently in their response to high-frequency stimulation. At mature synapses, their continuous presence is essential for the normal expression of LTP but is mostly dispensable for the normal release of synaptic vesicles from the readily releasable pool. Thus, the β1-class integrins play distinct roles in the development and function of central excitatory synapses.
Integrin function in neurite outgrowth and synaptic differentiation
Through binding the ECM, the integrins provide cells with both a biochemical and a mechanical link to the ECM (Hynes, 2002). This unique property has been postulated to serve a key role in the processes of cell and growth cone migration. Indeed, in Drosophila and Caenorhabditis elegans, integrin mutants exhibit axon pathfinding or fasciculation defects (Baum and Garriga, 1997; Hoang and Chiba, 1998). In vertebrates, in vitro observations have long argued for a role of the integrins in promoting neurite outgrowth on ECM substrates (Tomaselli et al., 1993; DeFreitas et al., 1995; Varnum-Finney et al., 1995; Weaver et al., 1995). They may also mediate the responses of axon growth cones to certain guidance cues such as semaphorin 7A (Pasterkamp et al., 2003). However, data for such roles in vivo have been mostly lacking. Instead, strong evidence argues for a role of integrins in neuronal migration. For example, the α3 and αv integrin receptors seem to regulate distinct aspects of neuronal migration during corticogenesis (Anton et al., 1999; Dulabon et al., 2000; Sanada et al., 2004; Schmid et al., 2004). The β1 integrins are also critical for lamination and foliation of the cerebral and cerebellar cortices (Graus-Porta et al., 2001). We observed similar phenotypes in the layering of the cerebral cortex and the dentate gyrus using emx1–cre-targeted deletion of β1 integrin. Thus, although the in vivo role of integrins in vertebrate axonal guidance remains to be established, these molecules are clearly important for neuronal migration.
The relatively normal cellular organization in the hippocampus of β1 integrin/emx1–cre mutants provides a unique opportunity to investigate its function in synaptic differentiation. Studies in this area have so far been focused primarily on the peripheral NMJ. At the Drosophila NMJ, the integrins have been found to regulate the number of synaptic boutons but play a minimal role in regulating synaptic ultrastructure or miniature EPSP (Beumer et al., 1999, 2002; Rohrbough et al., 2000). At the mouse NMJ, β1 integrin expression in the skeletal muscle but not in motor neurons is required for the proper terminal growth of the axons (Schwander et al., 2004). In the hippocampus, we observed that β1 integrin-deficient pyramidal neurons appeared to express normal overall densities of dendritic spines along the apical and basal dendrites. The overall densities of both excitatory and inhibitory synapses also appear normal. In addition, the basal transmission at the Schaffer collateral–CA1 synapses appears unaffected in β1 integrin/emx1–cre mice. In an independent study using β1 integrin/CaMKIIα–cre mice, however, basal synaptic transmission was found to be reduced, particularly at low-stimulation strengths (Chan et al., 2006). The reason for this discrepancy remains unclear. Regardless, β1 integrins do not seem to regulate overall synaptic density in the hippocampus, nor do they seem to be required for the general differentiation of the synapses.
Integrin function in the developmental maturation of presynaptic physiology
By taking advantage of the temporal difference in cre expression in the emx1–cre and CaMKIIα–cre lines, we identified a specific role of β1 integrins in presynaptic physiology and pinpointed their likely requirement to a time window during early development. We found that two forms of short-term synaptic plasticity, PPF at a short interpulse interval and synaptic response to HFS, are impaired in the emx1–cre mutants but not in the CaMKIIα–cre mutants. Because the only difference between these two cre lines in inducing recombination in pyramidal neurons is a temporal delay for CaMKIIα–cre, our results point strongly to a requirement for β1 integrins at an early stage of development in the proper presynaptic function of adult synapses, although we cannot rule out the possibility that they may act in glial cells of the adult hippocampus to regulate this process.
Two populations of synaptic vesicles have been described at the nerve terminal: the RRP and the reserved pool (Sudhof, 2000). Of them, only the RRP vesicles, equivalent to those morphologically docked at presynaptic membranes, are immediately available for release (Schikorski and Stevens, 1997; Murthy and Stevens, 1999). A brief HFS depletes most vesicles in the RRP but essentially spares the reserved pool and is therefore a good measure of the RRP size. We show that HFS induces a faster decline of fEPSP slopes in the emx1–cre mutant synapses, suggesting that expression of β1 integrins in early development is critical for controlling the number of docked vesicles at adult hippocampal synapses. Additional work using quantitative electron microscopy should provide additional evidence for this proposal.
The increase in PPF at a short IPI at the CA1 synapses of emx1–cre mutants reflects a decrease in Pr. This is supported by our finding using progressive, activity-dependent blockade of the NMDA receptor, a reliable assay for Pr. When NMDA currents are monitored over time in the presence of the open channel blocker MK-801, the amplitudes of the EPSCs gradually decrease at a rate dependent on the Pr. We observed that the mutant synapses exhibited a significantly slower decay in this assay, suggesting that deletion of β1 integrin decreases the Pr. In marked contrast, a previous report showed that application of RGD peptides or an antibody against β3 (but not β1) integrin to cultured hippocampal neurons resulted in an increase in the Pr, measured by the same method. These different observations may reflect a functional difference in the roles of β1 and β3 integrins. Alternatively, they may be attributable to differences between the in vitro cultures analyzed in the previous report and the genetically modified slices used in our study. It is interesting to note that RGD peptides attenuate the increase in transmitter release induced by mechanical stretch or hypertonic solution at the NMJ, suggesting that blockade of RGD-binding integrin function reduces the Pr at the NMJ (Chen and Grinnell, 1995; Kashani et al., 2001).
The alterations in the release kinetics of the RRP vesicles at the β1 integrin-deficient synapses is reminiscent of a recent finding that the level of phosphatidylinositol-4,5-biphosphate at presynaptic sites impacts synaptic vesicle trafficking and the size of RRP (Di Paolo et al., 2004). Interestingly, talin, an integrin cytodomain binding protein, has been found to regulate the activity of phosphatidylinositol kinase type Iγ as well as presynaptic vesicle trafficking (Di Paolo et al., 2002; Ling et al., 2002; Morgan et al., 2004). It would be of interest to explore in the future whether the integrins may be involved in this pathway, perhaps in the recruitment of talin and phosphatidylinositol kinase type Iγ to the synapse.
Integrin function at mature synapses and in learning and memory
At the Drosophila NMJ, the integrins are involved in regulating synaptic transmission and plasticity (Rohrbough et al., 2000). In the mammalian hippocampus, an acute involvement of the RGD-binding integrins in LTP maintenance has also been observed (Bahr et al., 1997; Staubli et al., 1998; Kramar et al., 2003). Several β1-binding α-class integrins, including α3, α5, and α8, have also been implicated in the induction and/or consolidation of hippocampal LTP (Chun et al., 2001; Kramar et al., 2002; Chan et al., 2003). We found that genetic deletion of the β1-class integrins from the excitatory pyramidal neurons using two different cre lines resulted in similar LTP defects both closely resembling those observed in previous pharmacological studies. Because β1 integrin is the known exclusive binding partner for α3, α5, and α8 integrin, the RGD-binding α3β1, α5β1, and α8β1 integrins may thus be the major class of integrins involved in mediating signal transduction at the hippocampal synapses. Because the postnatal deletion of β1 integrin in the CaMKIIα–cre mutants resulted in very similar phenotypes to those after embryonic deletion using emx1–cre, our results also point to a specific postnatal involvement of β1 integrin in LTP. Consistent with our finding, a recent study reported that postnatal deletion of β1 integrin using CaMKIIα–cre resulted in the impairment of LTP (Chan et al., 2006). Interestingly, these mice also exhibit normal PPF and normal response to HFS. These results, together with our finding that PPF and HFS responses are impaired in the emx1–cre mutants, suggest that early expression of β1 integrin is required for some (Pr, vesicle docking) but not other (LTP) functions of the adult synapses.
LTP has long been regarded as a plausible cellular substrate of learning and memory. The requirement for β1 integrins in LTP suggests that they may also be involved in the processes of learning and memory. Consistent with this, mice with reduced expression of the α3, α5, and α8 integrin subunits have been found to display defects in spatial memory (Chan et al., 2003). Expression of the integrin-associated protein gene has also been found to be upregulated during memory formation (Huang et al., 1998), suggesting an orchestrated response of integrin-related pathways during learning and memory. Interestingly, mice with postnatal deletion of β1 integrins exhibit normal hippocampal-dependent spatial and contextual memory but are impaired in working memory (Chan et al., 2006). Thus, future studies that elucidate the signaling cascade triggered by integrin engagement at the synapses may not only clarify the specific role of β1 integrins during LTP but also provide new insight into the processes of learning and memory.
Footnotes
This work was supported, in part, by funds from the Intramural Program of the National Institute of Child Health and Human Development (B.L.) and by National Institutes of Health Grant R01NS19090 (L.F.R.). Z.H. was supported by a postdoctoral fellowship from the Helen Hay Whitney foundation. N.H.W. was supported by fellowships from the Alberta Heritage Foundation for Medical Research and Natural Sciences and Engineering Research Council of Canada. L.F.R. is an investigator of the Howard Hughes Medical Institute. We thank K. Jones, S. Tonegawa, C. Damsky, and P. Soriano for the emx1-cre, CamKIIα-cre, β1 integrin neo (null), and Rosa26 reporter mice, S. McConnell and T. Jessell for antibodies, and members of the Reichardt laboratory for helpful discussions.
References
- Anton ES, Kreidberg JA, Rakic P. Distinct functions of alpha3 and alpha(v) integrin receptors in neuronal migration and laminar organization of the cerebral cortex. Neuron. 1999;22:277–289. doi: 10.1016/s0896-6273(00)81089-2. [DOI] [PubMed] [Google Scholar]
- Bahr BA, Staubli U, Xiao P, Chun D, Ji ZX, Esteban ET, Lynch G. Arg-Gly-Asp-Ser-selective adhesion and the stabilization of long-term potentiation: pharmacological studies and the characterization of a candidate matrix receptor. J Neurosci. 1997;17:1320–1329. doi: 10.1523/JNEUROSCI.17-04-01320.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamji SX, Shimazu K, Kimes N, Huelsken J, Birchmeier W, Lu B, Reichardt LF. Role of beta-catenin in synaptic vesicle localization and presynaptic assembly. Neuron. 2003;40:719–731. doi: 10.1016/s0896-6273(03)00718-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum PD, Garriga G. Neuronal migrations and axon fasciculation are disrupted in ina-1 integrin mutants. Neuron. 1997;19:51–62. doi: 10.1016/s0896-6273(00)80347-5. [DOI] [PubMed] [Google Scholar]
- Bernard-Trifilo JA, Kramar EA, Torp R, Lin CY, Pineda EA, Lynch G, Gall CM. Integrin signaling cascades are operational in adult hippocampal synapses and modulate NMDA receptor physiology. J Neurochem. 2005;93:834–849. doi: 10.1111/j.1471-4159.2005.03062.x. [DOI] [PubMed] [Google Scholar]
- Beumer K, Matthies HJ, Bradshaw A, Broadie K. Integrins regulate DLG/FAS2 via a CaM kinase II-dependent pathway to mediate synapse elaboration and stabilization during postembryonic development. Development. 2002;129:3381–3391. doi: 10.1242/dev.129.14.3381. [DOI] [PubMed] [Google Scholar]
- Beumer KJ, Rohrbough J, Prokop A, Broadie K. A role for PS integrins in morphological growth and synaptic function at the postembryonic neuromuscular junction of Drosophila. Development. 1999;126:5833–5846. doi: 10.1242/dev.126.24.5833. [DOI] [PubMed] [Google Scholar]
- Biederer T, Sara Y, Mozhayeva M, Atasoy D, Liu X, Kavalali ET, Sudhof TC. SynCAM, a synaptic adhesion molecule that drives synapse assembly. Science. 2002;297:1525–1531. doi: 10.1126/science.1072356. [DOI] [PubMed] [Google Scholar]
- Blaess S, Graus-Porta D, Belvindrah R, Radakovits R, Pons S, Littlewood-Evans A, Senften M, Guo H, Li Y, Miner JH, Reichardt LF, Muller U. β1-integrins are critical for cerebellar granule cell precursor proliferation. J Neurosci. 2004;24:3402–3412. doi: 10.1523/JNEUROSCI.5241-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Weeber EJ, Kurup S, Sweatt JD, Davis RL. Integrin requirement for hippocampal synaptic plasticity and spatial memory. J Neurosci. 2003;23:7107–7116. doi: 10.1523/JNEUROSCI.23-18-07107.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Weeber EJ, Zong L, Fuchs E, Sweatt JD, Davis RL. β1-integrins are required for hippocampal AMPA receptor-dependent synaptic transmission, synaptic plasticity, and working memory. J Neurosci. 2006;26:223–232. doi: 10.1523/JNEUROSCI.4110-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavis P, Westbrook G. Integrins mediate functional pre- and postsynaptic maturation at a hippocampal synapse. Nature. 2001;411:317–321. doi: 10.1038/35077101. [DOI] [PubMed] [Google Scholar]
- Chen BM, Grinnell AD. Integrins and modulation of transmitter release from motor nerve terminals by stretch. Science. 1995;269:1578–1580. doi: 10.1126/science.7667637. [DOI] [PubMed] [Google Scholar]
- Chih B, Engelman H, Scheiffele P. Control of excitatory and inhibitory synapse formation by neuroligins. Science. 2005;307:1324–1328. doi: 10.1126/science.1107470. [DOI] [PubMed] [Google Scholar]
- Chun D, Gall CM, Bi X, Lynch G. Evidence that integrins contribute to multiple stages in the consolidation of long term potentiation in rat hippocampus. Neuroscience. 2001;105:815–829. doi: 10.1016/s0306-4522(01)00173-7. [DOI] [PubMed] [Google Scholar]
- DeFreitas MF, Yoshida CK, Frazier WA, Mendrick DL, Kypta RM, Reichardt LF. Identification of integrin alpha 3 beta 1 as a neuronal thrombospondin receptor mediating neurite outgrowth. Neuron. 1995;15:333–343. doi: 10.1016/0896-6273(95)90038-1. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, Pellegrini L, Letinic K, Cestra G, Zoncu R, Voronov S, Chang S, Guo J, Wenk MR, De Camilli P. Recruitment and regulation of phosphatidylinositol phosphate kinase type 1 gamma by the FERM domain of talin. Nature. 2002;420:85–89. doi: 10.1038/nature01147. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, Moskowitz HS, Gipson K, Wenk MR, Voronov S, Obayashi M, Flavell R, Fitzsimonds RM, Ryan TA, De Camilli P. Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature. 2004;431:415–422. doi: 10.1038/nature02896. [DOI] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Dulabon L, Olson EC, Taglienti MG, Eisenhuth S, McGrath B, Walsh CA, Kreidberg JA, Anton ES. Reelin binds alpha3beta1 integrin and inhibits neuronal migration. Neuron. 2000;27:33–44. doi: 10.1016/s0896-6273(00)00007-6. [DOI] [PubMed] [Google Scholar]
- Fannon AM, Colman DR. A model for central synaptic junctional complex formation based on the differential adhesive specificities of the cadherins. Neuron. 1996;17:423–434. doi: 10.1016/s0896-6273(00)80175-0. [DOI] [PubMed] [Google Scholar]
- Forster E, Tielsch A, Saum B, Weiss KH, Johanssen C, Graus-Porta D, Muller U, Frotscher M. Reelin, Disabled 1, and beta 1 integrins are required for the formation of the radial glial scaffold in the hippocampus. Proc Natl Acad Sci USA. 2002;99:13178–13183. doi: 10.1073/pnas.202035899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galileo DS, Majors J, Horwitz AF, Sanes JR. Retrovirally introduced antisense integrin RNA inhibits neuroblast migration in vivo. Neuron. 1992;9:1117–1131. doi: 10.1016/0896-6273(92)90070-t. [DOI] [PubMed] [Google Scholar]
- Geppert M, Bolshakov VY, Siegelbaum SA, Takei K, De Camilli P, Hammer RE, Sudhof TC. The role of Rab3A in neurotransmitter release. Nature. 1994;369:493–497. doi: 10.1038/369493a0. [DOI] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Stevens CF, Sudhof TC. The small GTP-binding protein Rab3A regulates a late step in synaptic vesicle fusion. Nature. 1997;387:810–814. doi: 10.1038/42954. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 2004;119:1013–1026. doi: 10.1016/j.cell.2004.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Muller U. Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 2001;31:367–379. doi: 10.1016/s0896-6273(01)00374-9. [DOI] [PubMed] [Google Scholar]
- Grotewiel MS, Beck CD, Wu KH, Zhu XR, Davis RL. Integrin-mediated short-term memory in Drosophila. Nature. 1998;391:455–460. doi: 10.1038/35079. [DOI] [PubMed] [Google Scholar]
- Hama H, Hara C, Yamaguchi K, Miyawaki A. PKC signaling mediates global enhancement of excitatory synaptogenesis in neurons triggered by local contact with astrocytes. Neuron. 2004;41:405–415. doi: 10.1016/s0896-6273(04)00007-8. [DOI] [PubMed] [Google Scholar]
- Hessler NA, Shirke AM, Malinow R. The probability of transmitter release at a mammalian central synapse. Nature. 1993;366:569–572. doi: 10.1038/366569a0. [DOI] [PubMed] [Google Scholar]
- Hoang B, Chiba A. Genetic analysis on the role of integrin during axon guidance in Drosophila. J Neurosci. 1998;18:7847–7855. doi: 10.1523/JNEUROSCI.18-19-07847.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AM, Wang HL, Tang YP, Lee EH. Expression of integrin-associated protein gene associated with memory formation in rats. J Neurosci. 1998;18:4305–4313. doi: 10.1523/JNEUROSCI.18-11-04305.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Kashani AH, Chen BM, Grinnell AD. Hypertonic enhancement of transmitter release from frog motor nerve terminals: Ca2+ independence and role of integrins. J Physiol (Lond) 2001;530:243–252. doi: 10.1111/j.1469-7793.2001.0243l.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MB. Signal-processing machines at the postsynaptic density. Science. 2000;290:750–754. doi: 10.1126/science.290.5492.750. [DOI] [PubMed] [Google Scholar]
- Kramar EA, Bernard JA, Gall CM, Lynch G. Alpha3 integrin receptors contribute to the consolidation of long-term potentiation. Neuroscience. 2002;110:29–39. doi: 10.1016/s0306-4522(01)00540-1. [DOI] [PubMed] [Google Scholar]
- Kramar EA, Bernard JA, Gall CM, Lynch G. Integrins modulate fast excitatory transmission at hippocampal synapses. J Biol Chem. 2003;278:10722–10730. doi: 10.1074/jbc.M210225200. [DOI] [PubMed] [Google Scholar]
- Kramar EA, Lin B, Rex CS, Gall CM, Lynch G. Integrin-driven actin polymerization consolidates long-term potentiation. Proc Natl Acad Sci USA. 2006;103:5579–5584. doi: 10.1073/pnas.0601354103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Wenk MR, Kim Y, Nairn AC, De Camilli P. Regulation of synaptojanin 1 by cyclin-dependent kinase 5 at synapses. Proc Natl Acad Sci USA. 2004;101:546–551. doi: 10.1073/pnas.0307813100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B, Arai AC, Lynch G, Gall CM. Integrins regulate NMDA receptor-mediated synaptic currents. J Neurophysiol. 2003;89:2874–2878. doi: 10.1152/jn.00783.2002. [DOI] [PubMed] [Google Scholar]
- Ling K, Doughman RL, Firestone AJ, Bunce MW, Anderson RA. Type I gamma phosphatidylinositol phosphate kinase targets and regulates focal adhesions. Nature. 2002;420:89–93. doi: 10.1038/nature01082. [DOI] [PubMed] [Google Scholar]
- Marrs GS, Honda T, Fuller L, Thangavel R, Balsamo J, Lilien J, Dailey ME, Arregui C. Dendritic arbors of developing retinal ganglion cells are stabilized by beta1-integrins. Mol Cell Neurosci. 2006;32:230–241. doi: 10.1016/j.mcn.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Martin PT, Sanes JR. Integrins mediate adhesion to agrin and modulate agrin signaling. Development. 1997;124:3909–3917. doi: 10.1242/dev.124.19.3909. [DOI] [PubMed] [Google Scholar]
- Moresco EM, Donaldson S, Williamson A, Koleske AJ. Integrin-mediated dendrite branch maintenance requires Abelson (Abl) family kinases. J Neurosci. 2005;25:6105–6118. doi: 10.1523/JNEUROSCI.1432-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JR, Di Paolo G, Werner H, Shchedrina VA, Pypaert M, Pieribone VA, De Camilli P. A role for talin in presynaptic function. J Cell Biol. 2004;167:43–50. doi: 10.1083/jcb.200406020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozhayeva MG, Sara Y, Liu X, Kavalali ET. Development of vesicle pools during maturation of hippocampal synapses. J Neurosci. 2002;22:654–665. doi: 10.1523/JNEUROSCI.22-03-00654.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Stevens CF. Reversal of synaptic vesicle docking at central synapses. Nat Neurosci. 1999;2:503–507. doi: 10.1038/9149. [DOI] [PubMed] [Google Scholar]
- Nagy V, Bozdagi O, Matynia A, Balcerzyk M, Okulski P, Dzwonek J, Costa RM, Silva AJ, Kaczmarek L, Huntley GW. Matrix metalloproteinase-9 is required for hippocampal late-phase long-term potentiation and memory. J Neurosci. 2006;26:1923–1934. doi: 10.1523/JNEUROSCI.4359-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasterkamp RJ, Peschon JJ, Spriggs MK, Kolodkin AL. Semaphorin 7A promotes axon outgrowth through integrins and MAPKs. Nature. 2003;424:398–405. doi: 10.1038/nature01790. [DOI] [PubMed] [Google Scholar]
- Pinkstaff JK, Lynch G, Gall CM. Localization and seizure-regulation of integrin beta 1 mRNA in adult rat brain. Brain Res Mol Brain Res. 1998;55:265–276. doi: 10.1016/s0169-328x(98)00007-2. [DOI] [PubMed] [Google Scholar]
- Pinkstaff JK, Detterich J, Lynch G, Gall C. Integrin subunit gene expression is regionally differentiated in adult brain. J Neurosci. 1999;19:1541–1556. doi: 10.1523/JNEUROSCI.19-05-01541.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzo-Miller LD, Gottschalk W, Zhang L, McDermott K, Du J, Gopalakrishnan R, Oho C, Sheng ZH, Lu B. Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knock-out mice. J Neurosci. 1999;19:4972–4983. doi: 10.1523/JNEUROSCI.19-12-04972.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle JL, Kavalali ET, Piedras-Renteria ES, Tsien RW. Rapid reuse of readily releasable pool vesicles at hippocampal synapses. Neuron. 2000;28:221–231. doi: 10.1016/s0896-6273(00)00098-2. [DOI] [PubMed] [Google Scholar]
- Reichardt LF, Tomaselli KJ. Extracellular matrix molecules and their receptors: functions in neural development. Annu Rev Neurosci. 1991;14:531–570. doi: 10.1146/annurev.ne.14.030191.002531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards DA, Guatimosim C, Betz WJ. Two endocytic recycling routes selectively fill two vesicle pools in frog motor nerve terminals. Neuron. 2000;27:551–559. doi: 10.1016/s0896-6273(00)00065-9. [DOI] [PubMed] [Google Scholar]
- Rohrbough J, Grotewiel MS, Davis RL, Broadie K. Integrin-mediated regulation of synaptic morphology, transmission, and plasticity. J Neurosci. 2000;20:6868–6878. doi: 10.1523/JNEUROSCI.20-18-06868.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science. 1993;262:754–757. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- Sanada K, Gupta A, Tsai LH. Disabled-1-regulated adhesion of migrating neurons to radial glial fiber contributes to neuronal positioning during early corticogenesis. Neuron. 2004;42:197–211. doi: 10.1016/s0896-6273(04)00222-3. [DOI] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci. 1997;17:5858–5867. doi: 10.1523/JNEUROSCI.17-15-05858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Morphological correlates of functionally defined synaptic vesicle populations. Nat Neurosci. 2001;4:391–395. doi: 10.1038/86042. [DOI] [PubMed] [Google Scholar]
- Schmid RS, Shelton S, Stanco A, Yokota Y, Kreidberg JA, Anton ES. alpha3beta1 integrin modulates neuronal migration and placement during early stages of cerebral cortical development. Development. 2004;131:6023–6031. doi: 10.1242/dev.01532. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Schwander M, Shirasaki R, Pfaff SL, Muller U. Beta1 integrins in muscle, but not in motor neurons, are required for skeletal muscle innervation. J Neurosci. 2004;24:8181–8191. doi: 10.1523/JNEUROSCI.1345-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Bargmann CI. The immunoglobulin superfamily protein SYG-1 determines the location of specific synapses in Celegans. Cell. 2003;112:619–630. doi: 10.1016/s0092-8674(03)00113-2. [DOI] [PubMed] [Google Scholar]
- Shi Y, Ethell IM. Integrins control dendritic spine plasticity in hippocampal neurons through NMDA receptor and Ca2+/calmodulin-dependent protein kinase II-mediated actin reorganization. J Neurosci. 2006;26:1813–1822. doi: 10.1523/JNEUROSCI.4091-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonner JM, Cascio M, Xing Y, Fanselow MS, Kralic JE, Morrow AL, Korpi ER, Hardy S, Sloat B, Eger EI, II, Homanics GE. Alpha 1 subunit-containing GABA type A receptors in forebrain contribute to the effect of inhaled anesthetics on conditioned fear. Mol Pharmacol. 2005;68:61–68. doi: 10.1124/mol.104.009936. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Staubli U, Chun D, Lynch G. Time-dependent reversal of long-term potentiation by an integrin antagonist. J Neurosci. 1998;18:3460–3469. doi: 10.1523/JNEUROSCI.18-09-03460.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens LE, Sutherland AE, Klimanskaya IV, Andrieux A, Meneses J, Pedersen RA, Damsky CH. Deletion of beta 1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes Dev. 1995;9:1883–1895. doi: 10.1101/gad.9.15.1883. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle revisited. Neuron. 2000;28:317–320. doi: 10.1016/s0896-6273(00)00109-4. [DOI] [PubMed] [Google Scholar]
- Taschenberger H, Leao RM, Rowland KC, Spirou GA, von Gersdorff H. Optimizing synaptic architecture and efficiency for high-frequency transmission. Neuron. 2002;36:1127–1143. doi: 10.1016/s0896-6273(02)01137-6. [DOI] [PubMed] [Google Scholar]
- Togashi H, Abe K, Mizoguchi A, Takaoka K, Chisaka O, Takeichi M. Cadherin regulates dendritic spine morphogenesis. Neuron. 2002;35:77–89. doi: 10.1016/s0896-6273(02)00748-1. [DOI] [PubMed] [Google Scholar]
- Tomaselli KJ, Doherty P, Emmett CJ, Damsky CH, Walsh FS, Reichardt LF. Expression of β1 integrins in sensory neurons of the dorsal root ganglion and their functions in neurite outgrowth on two laminin isoforms. J Neurosci. 1993;13:4880–4888. doi: 10.1523/JNEUROSCI.13-11-04880.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Uchida N, Honjo Y, Johnson KR, Wheelock MJ, Takeichi M. The catenin/cadherin adhesion system is localized in synaptic junctions bordering transmitter release zones. J Cell Biol. 1996;135:767–779. doi: 10.1083/jcb.135.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnum-Finney B, Venstrom K, Muller U, Kypta R, Backus C, Chiquet M, Reichardt LF. The integrin receptor alpha 8 beta 1 mediates interactions of embryonic chick motor and sensory neurons with tenascin-C. Neuron. 1995;14:1213–1222. doi: 10.1016/0896-6273(95)90268-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CD, Yoshida CK, de Curtis I, Reichardt LF. Expression and in vitro function of β1-integrin laminin receptors in the developing avian ciliary ganglion. J Neurosci. 1995;15:5275–5285. doi: 10.1523/JNEUROSCI.15-07-05275.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Borst JG. The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron. 1999;23:821–832. doi: 10.1016/s0896-6273(01)80039-8. [DOI] [PubMed] [Google Scholar]
- Xu B, Zang K, Ruff NL, Zhang YA, McConnell SK, Stryker MP, Reichardt LF. Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron. 2000;26:233–245. doi: 10.1016/s0896-6273(00)81153-8. [DOI] [PubMed] [Google Scholar]
- Yamagata M, Weiner JA, Sanes JR. Sidekicks: synaptic adhesion molecules that promote lamina-specific connectivity in the retina. Cell. 2002;110:649–660. doi: 10.1016/s0092-8674(02)00910-8. [DOI] [PubMed] [Google Scholar]