

Abstract
Talin is a large flexible rod-shaped protein that activates the integrin family of cell adhesion molecules and couples them to cytoskeletal actin. It exists in both globular and extended conformations, and an intramolecular interaction between the N-terminal F3 FERM subdomain and the C-terminal part of the talin rod contributes to an autoinhibited form of the molecule. Here, we report the solution structure of the primary F3 binding domain within the C-terminal region of the talin rod and use intermolecular nuclear Overhauser effects to determine the structure of the complex. The rod domain (residues 1655–1822) is an amphipathic five-helix bundle; Tyr-377 of F3 docks into a hydrophobic pocket at one end of the bundle, whereas a basic loop in F3 (residues 316–326) interacts with a cluster of acidic residues in the middle of helix 4. Mutation of Glu-1770 abolishes binding. The rod domain competes with β3-integrin tails for binding to F3, and the structure of the complex suggests that the rod is also likely to sterically inhibit binding of the FERM domain to the membrane.
The cytoskeletal protein talin has emerged as a key player, both in regulating the affinity of the integrin family of cell adhesion molecules for ligand (1) and in coupling integrins to the actin cytoskeleton (2). Thus, depletion of talin results in defects in integrin activation (3), integrin signaling through focal adhesion kinase, the maintenance of cell spreading, and the assembly of focal adhesions in cultured cells (4). In the whole organism, studies on the single talin gene in worms (5) and flies (6) show that talin is essential for a variety of integrin-mediated events that are crucial for normal embryonic development. In vertebrates, there are two talin genes, and mice carrying a talin1 null allele fail to complete gastrulation (7). Tissue-specific inactivation of talin1 results in an inability to activate integrins in platelets (8, 9), defects in the membrane-cytoskeletal interface in megakaryocytes (10), and disruption of the myotendinous junction in skeletal muscle (11). In contrast, mice homozygous for a talin2 gene trap allele have no phenotype, although the allele may be hypomorphic (12).
Recent structural studies have provided substantial insights into the molecular basis of talin action. Talin is composed of an N-terminal globular head (∼50 kDa) linked to an extended flexible rod (∼220 kDa). The talin head contains a FERM2 domain (made up of F1, F2, and F3 subdomains) preceded by a domain referred to here as F0 (2). Studies by Wegener et al. (30) have shown how the F3 FERM subdomain, which has a phosphotyrosine binding domain fold, interacts with both the canonical NPXY motif and the membrane-proximal helical region of the cytoplasmic tails of integrin β-subunits (13). The latter interaction apparently activates the integrin by disrupting the salt bridge between the integrin α- and β-subunit tails that normally keeps integrins locked in a low affinity state. The observation that the F0 region is also important in integrin activation (14) may be explained by our recent finding that F0 binds, albeit with low affinity, Rap1-GTP,3 a known activator of integrins (15, 16). The talin rod is made up of a series of amphipathic α-helical bundles (17–20) and contains a second integrin binding site (IBS2) (21), numerous binding sites for the cytoskeletal protein vinculin (22), at least two actin binding sites (23), and a C-terminal helix that is required for assembly of talin dimers (20, 24).
Both biochemical (25) and cellular studies (16) suggest that the integrin binding sites in full-length talin are masked, and both phosphatidylinositol 4,5-bisphosphate (PIP2) and Rap1 have been implicated in exposing these sites. It is well established that some members of the FERM domain family of proteins are regulated by a head-tail interaction (26); gel filtration, sedimentation velocity, and electron microscopy studies all show that talin is globular in low salt buffers, although it is more elongated (∼60 nm in length) in high salt (27). By contrast, the talin rod liberated from full-length talin by calpain-II cleavage is elongated in both buffers, indicating that the head is required for talin to adopt a more compact state. Direct evidence for an interaction between the talin head and rod has recently emerged from NMR studies by Goksoy et al. (28), who demonstrated binding of 15N-labeled talin F3 to a talin rod fragment spanning residues 1654–2344, an interaction that was confirmed by surface plasmon resonance (Kd = 0.57 μm) (28). Chemical shift data also showed that this segment of the talin rod partially masked the binding site in F3 for the membraneproximal helix of the β3-integrin tail (28), directly implicating the talin head-rod interaction in regulating the integrin binding activity of talin. Goksoy et al. (28) subdivided the F3 binding site in this rod fragment into two sites with higher affinity (Kd ∼3.6 μm; residues 1654–1848) and lower affinity (Kd ∼78 μm; residues 1984–2344). Here, we define the rod domain boundaries and determine the NMR structure of residues 1655–1822, a five-helix bundle. We further show that this domain binds F3 predominantly via surface-exposed residues on helix 4, with an affinity similar to the high affinity site reported by Goksoy et al. (28). We also report the structure of the complex between F3 and the rod domain and show that the latter masks the known binding site in F3 for the β3-integrin tail and is expected to inhibit the association of the talin FERM domain with the membrane.
EXPERIMENTAL PROCEDURES
Peptide Preparation—The preparation of U-15N-labeled β3-integrin tail was performed as described previously (29).
Expression of Recombinant Talin Polypeptides—The cDNAs encoding murine talin1 residues 309–400 (F3) and 1655–1822 were synthesized by PCR using a mouse talin1 cDNA as template and cloned into the expression vector pet-151TOPO (Invitrogen). Talin polypeptides were expressed in Escherichia coli BL21 STAR (DE3) cultured either in LB for unlabeled protein or in M9 minimal medium for preparation of isotopically labeled samples for NMR. Recombinant His-tagged talin polypeptides were purified by nickel-affinity chromatography following standard procedures. The His tag was removed by cleavage with AcTEV protease (Invitrogen), and the proteins were further purified by anion-exchange (Domain E) or cation-exchange (F3). U-15N-labeled F3 (309–405) and F3 point mutants were made as described previously (30). Protein concentrations were determined using their respective extinction coefficient at 280 nm. Protein concentrations were based on absorption coefficients calculated from the aromatic content according to ProtParam as follows: talin 1655–1822, 3105 m-1cm-1; talin F3, 16,960 m-1cm-1; PIPKIγ peptide, 8480 m-1cm-1; integrin peptide, 8480 m-1cm-1.
NMR Spectroscopy—NMR experiments for the resonance assignment and structure determination of talin 1655–1822 were carried out with 1 mm protein in 20 mm sodium phosphate, pH 6.5, 50 mm NaCl, 2 mm dithiothreitol, 10% (v/v) 2H2O. NMR spectra of all the proteins were obtained at 298 K using Bruker AVANCE DRX 600 or AVANCE DRX 800 spectrometers both equipped with CryoProbes. Proton chemical shifts were referenced to external 2,2-dimethyl-2-silapentane-5-sulfonic acid, and 15N and 13C chemical shifts were referenced indirectly using recommended gyromagnetic ratios (31). Spectra were processed with TopSpin (Bruker Corp.) and analyzed using Analysis (32). Three-dimensional HNCO, HN(CA)CO, HNCA, HN(CO)CA, HNCACB, and HN(CO)CACB experiments were used for the sequential assignment of the backbone NH, N, CO, Cα, and Cβ resonances. Side chain assignments were obtained using three-dimensional HBHA(CO)NH, HBHANH, H(C)CH-TOCSY, and (H)CCH-TOCSY experiments. Aromatic side chain assignments were obtained using 13C-resolved three-dimensional NOESY-HSQC. The resonance assignments of 1655–1822 have been deposited in the BioMagResBank with the accession number 15457.
Experiments to examine the competition between talin 1655–1822 and the β3-integrin tail were performed on a spectrometer equipped with a 500-MHz Oxford Instruments superconducting magnet and GE-Omega computer. Samples were prepared in 50 mm phosphate buffer, pH 6.1, containing 100 mm NaCl, 1 mm dithiothreitol, 5% 2H2O, and Complete protease inhibitors (Roche Applied Science). Data were processed using NMRPipe (33), and spectra were visualized using SPARKY. Spectra were referenced as described above (31). The 1H and 15N resonances of U-15N-labeled β3-integrin tail were assigned previously (29).
Structure Calculations—Distance restraints were obtained from the following experiments: three-dimensional 15N-edited NOESY-HSQC (800 MHz, 100 ms), 13C-edited NOESY-HSQC (800 MHz, 100 ms), and 13C-edited NOESY-HSQC (800 MHz, 80 ms) on aromatics. All NOESY peaks were picked semiautomatically in Analysis with noise and artifact peaks removed manually. Cross-peak intensities were used to evaluate target distances. Dihedral restraints (Φ/Ψ) were obtained from the TALOS data base (34). Hydrogen-bond restraints within secondary structure elements identified from initial rounds of structure calculation were incorporated based on the temperature dependence of NMR chemical shifts (35) using a series of 1H,15N HSQC spectra collected from 15 to 35 °C. Initial models were generated with CYANA using the CANDID (36) method for NOESY cross-peak assignment and calibration. These models were used as initial structures in structure calculations by Aria (37). The acceptance tolerances in the standard protocol of Aria 1.2 were modified to set violation tolerances to 5.0, 2.0, 1.0, 0.5, 2.0, 0.5, and 0.1 Å for iterations 2–8, respectively, with iteration 1 containing the initial models. Any cross-peaks rejected by Aria were checked manually, and those found to be reliable were added to the calculation. 200 structures were calculated at each iteration, the 20 lowest energy structures retained and 10 used for final restraint analysis. The 30 lowest energy structures from iteration 8 were further refined in the presence of explicit water molecules. Molecular models were generated using PyMOL (38). The structural statistics for each domain are presented in Table 1. The set of 20 lowest energy structures has been submitted to the Protein Data Bank with the accession number 2KBB.
TABLE 1.
Solution structure determination of talin 1655–1822
| Restraints | |
| Unique/Ambiguous NOEs | 4488/520 |
| Intraresidue | 1707/116 |
| Sequential | 948/109 |
| Short range (1 < [i – j] < 5) | 970/145 |
| Long range ([i – j] > 4) | 863/150 |
| ϕ/ψ dihedral anglesa | 257 |
| Energies (kcal mol–1)b | |
| Total | –6688.29 ± 86.70 |
| Van Der Waals | –1626.14 ± 10.38 |
| NOE | 33.50 ± 5.38 |
| r.m.s. deviationsb | |
| NOEs (Å) (no violations > 0.5 Å) | 0.011 ± 0.001 |
| Dihedral restraints (°) (no violations > 5°) | 0.39 ± 0.03 |
| Bonds (Å) | 0.0017 ± 0.0001 |
| Angles (°) | 0.34 ± 0.01 |
| Impropers (°) | 0.244 ± 0.01 |
| Ramachandran map analysisc | |
| Allowed regions | 94.1% |
| Additional allowed regions | 5.2% |
| Generously allowed regions | 0.4% |
| Disallowed regions | 0.3% |
| Pairwise r.m.s. differences (Å)d | |
| Residues 1662–1820 | 0.78 (1.18) |
| Secondary structure | 0.33 (0.76) |
From chemical shifts using Talos
Calculated in ARIA 1.2 for the 20 lowest energy structures refined in water. r.m.s., root mean square
Obtained using PROCHECK-NMR
For backbone atoms; value for all heavy atoms in brackets. r.m.s., root mean square
NMR Titrations—All titrations were carried out in 20 mm phosphate, pH 6.5, 50 mm NaCl, 2 mm dithiothreitol using 1H,15N HSQC. Weighted combined 1H and 15N amide secondary shifts (Δ(H,N)) were calculated using the equation
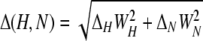 |
(Eq. 1) |
where WH and WN are weighting factors for the 1H and 15N amide shifts, respectively (WH = 1 and WH = 0.15) (39), and Δ= δbound – δfree. Dissociation constants (Kd) were determined by fitting the changes in secondary shift with concentration to the following equation
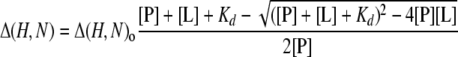 |
(Eq. 2) |
where Δ(H,N) is the weighted secondary shift, Δ(H,N)0 is the shift at saturation, and [P] and [L] are the protein and ligand concentrations, respectively. Data from peaks with the largest shift changes were fitted to this equation using Analysis. Competition experiments were conducted by acquiring 1H,15N HSQC spectra of 50 μm β3-tail in the presence of 0.25 mm talin F3 domain, 1 mm talin 1655–1822, or both. Weighted combined 1H and 15N amide shifts (Δ(H,N)) were calculated as above.
Molecular Docking Calculations—Docking of talin 1655–1822 to F3 was performed with the software HADDOCK2 (40, 41). The starting structures were model 1 of the F3 complex with the β3/PIPKIγ-chimeric peptide (Protein Data Bank (PDB) ID code 2H7D) (30) and the lowest energy NMR structure of talin 1655–1822. Mutagenesis data, chemical shift mapping, and surface accessibility data were used to define the active residues according to HADDOCK definitions. The well defined filtered NOEs between Tyr-377 in F3 and Leu-1680 and Val-1683 of talin 1655–1822 were also used in the calculation. In all, 1000 initial complex structures were generated by rigid body energy minimization, and the best 200 by total energy were selected for torsion angle dynamics and subsequent Cartesian dynamics in an explicit water solvent. Default scaling for energy terms is as described previously (40).
RESULTS
Mapping the Domain Boundaries of the High Affinity F3 Binding Site in the Talin Rod—A fragment of the talin rod, spanning residues 1654–2344, has been shown to contain two non-overlapping binding sites for the talin F3 FERM subdomain (28). To further investigate the molecular basis of the interaction, we first determined the domain boundaries of the relevant section of the talin rod, which is made up of a total of 62 α-helices (Fig. 1A) arranged into ∼13 compact bundles or domains. We have previously established that the most C-terminal bundle (residues 2300–2482), which contains an actin binding site (20), is preceded by two five-helix domains (residues 1974–2140 and 2137–2294) that together make up the integrin binding site referred to as IBS2 (21). Working toward the N terminus, the next domain, which contains a vinculin binding site (VBS3), was thought to be a four-helix bundle (residues 1843–1973) (19), but we have recently found that it is more stable as a five-helix bundle (residues 1815–1973).4 This domain overlaps, by one helix, the F3 binding fragment identified by Goksoy et al. (28) (residues 1654–1848). To establish whether the preceding domain is also a five-helix bundle, we expressed residues 1655–1822, a region that is highly conserved in both talin1 and talin2 and across species (Fig. 1B). The protein was soluble, and its melting temperature, as determined by CD, was 65 °C, providing evidence for a stable fold. Moreover, the 1H,15N HSQC spectrum showed good dispersion indicative of a well folded domain and with peak line widths consistent with a monomeric state.
FIGURE 1.

Structure of the talin 1655–1822 rod domain. A, schematic representation of the domain structure of talin indicating the relative position of the ligand binding sites and the boundaries of the various head and rod domains. B, sequence alignment of human talin1 residues 1655–1822 with the corresponding regions of other talins. C, superimposition of the 20 lowest energy structures consistent with the NMR data. Only the structured region of 1655–1822 is shown, not the disordered N terminus. D, ribbon drawing of a representative low energy structure showing the overall topology of the five-helix bundle. E, map of the surface charge of the domain.
Structure of Talin Residues 1655–1822—The solution structure of talin 1655–1822 was calculated from 5502 distance, and 257 dihedral angle restraints were determined using 13C,15N-labeled protein. The structure consists of five antiparallel amphipathic α-helices forming a bundle stabilized by hydrophobic interactions (Fig. 1, C and D). The loops between helices 1 and 2 and helices 4 and 5 are relatively long (9–10 residues), whereas those between helices 2 and 3 and 3 and 4 are shorter (4–5 residues). Two of the helices contain proline residues (Pro-1715 in helix 2 and Pro-1740 in helix 3) that introduce kinks in the helices; interestingly, these prolines are conserved among talin sequences (Fig. 1B). The topology of the bundle is similar to that seen in talin 482–655 (17) and the two bundles in talin 1974–2293, which make up IBS2 (21). The core is almost exclusively hydrophobic with a conserved phenylalanine (Phe-1738) surrounded by the hydrophobic side chains of Leu, Ile, and Val residues. The only non-hydrophobic residue in the core is a conserved threonine, Thr-1765. Unlike other talin bundles (17–20), there is no aromatic residue capping the end of the bundle. An interesting feature of the domain is that there is a hydrophobic patch on helices 2 and 3, including Leu-1698 and Leu-1743, that is masked by the long loop between helices 1 and 2, which contains several hydrophobic residues (including Ile-1693 and Leu-1687) that dock into this patch. It remains to be established whether this hydrophobic patch is a site for domain-domain interaction in the intact rod (17).
Talin 1655–1822 Interacts with the Talin F3 Domain—The interaction between the talin rod 1655–1822 five-helix bundle and the F3 FERM subdomain of the talin head was studied by collecting 1H,15N HSQC spectra of 15N-labeled talin 1655–1822 in the presence of increasing concentrations of unlabeled F3 (Fig. 2A). A number of resonances showed progressive changes in chemical shift; an example is shown in Fig. 2B. Analysis of the chemical shift changes for five residues (1684, 1685, 1768, 1770, and 1772) yields a Kd for the complex of 11 μm (range 9–14 μm), in reasonable agreement with that (3.6 μm) reported for the 1654–1848 fragment (28). The weighted 1H,15N chemical shifts in talin 1655–1822 induced by F3 are shown as a function of residue number in Fig. 2C and are mapped on the structure in Fig. 2, D and E. The binding site for F3 on talin 1655–1822 is fairly extensive but is centered on helix 4 and the C-terminal end of helix 1 of the five-helix bundle (Fig. 2E). The residues involved in the rod binding site are highly conserved across species (Fig. 1B). The results of the reverse experiment, adding increasing concentrations of unlabeled talin 1655–1822 to 15N-labeled F3, are shown in supplemental Fig. S1. The observed shifts are mapped onto a surface representation of F3 in Fig. 2F and are similar to those observed by Goksoy et al. (28) where a larger talin construct (1654–2344) was used.
FIGURE 2.

Mapping the F3 FERM subdomain binding site in talin 1655–1822. A, 1H,15N HSQC spectra of 100 μm 15N-labeled talin 1655–1822 rod domain in the presence of the F3 FERM subdomain at ratios of 1:0 (blue), 1:0.5 (yellow), 1:1 (green), and 1:1.5 (black). B, chemical shift of the resonance of Glu-1770 of talin 1655–1822 as a function of the concentration of F3. The fitted curve of this residue (shown in red) yields a Kd of 9 (± 0.8) μm. C, weighted shift map obtained from the 1H,15N HSQC spectra of talin 1655–1822 on the addition of F3. D and E, residues in talin 1655–1822 that are significantly perturbed following the addition of F3 are highlighted on the talin 1655–1822 structure. The most significant shifts (>0.2 ppm) are shown in dark blue, and the residue numbers are labeled; smaller perturbations (>0.07 ppm) are shown in light blue. F, residues in talin F3 ((30) PDB 2H7D) that are significantly perturbed following the addition of talin 1655–1822 are highlighted on the F3 structure.
Structure of the Complex between Talin 1655–1822 and Talin F3—Models of the complex were obtained using HAD-DOCK (40, 41) on the basis of chemical shift perturbation data and intermolecular NOEs. The latter were measured by collecting two 13C,15NF1-filtered, F3-edited NOESY-HSQC spectra, one of 13C,15N-labeled talin 1655–1822 in complex with the unlabeled talin F3 subdomain, and one of 13C,15N-labeled F3 in complex with unlabeled talin 1655–1822. Each NOESY spectrum contained more than 100 intermolecular cross-peaks, most of which could be assigned unambiguously; the two experiments were in good agreement with NOEs seen from talin 1655–1822 to F3 and vice versa. The intermolecular NOEs map to the ends of helices 1 and 4 of talin 1655–1822, consistent with the chemical shift mapping. For example, the signals of the aromatic ring of Tyr-377 of F3 exhibit large shifts on the addition of talin 1655–1822, and clear intermolecular NOEs are observed between its aromatic ring protons and the protons of Leu-1680, Val-1683, and Met-1759 of talin 1655–1822. The model was calculated based on rigid body docking of the two proteins because careful examination of the intramolecular NOEs showed that the core structures of the two proteins remained essentially unchanged on complex formation. The final model of the complex is shown in Fig. 3A.
FIGURE 3.

Structure of the complex between talin F3 and talin 1655–1822. A, ribbon representation of the model of talin F3 (red) in complex with talin 1655–1822 (pale blue) obtained using the HADDOCK approach. B, close-up of the interface showing Tyr-377 of F3 and its close proximity to Leu-1680, Val-1683, Met-1759, and Leu-1762 of talin 1655–1822. C, close-up of the interaction of the positively charged activation loop of F3 with the negatively charged residues on talin 1655–1822.
The F3 binding site on talin 1655–1822 is a large elongated area (Figs. 2E and 3A) with two main interaction sites. Thus, Tyr-377 of F3 docks into a small hydrophobic pocket on talin 1655–1822 (Fig. 3B), whereas the F3 activation loop (residues 316–326) binds midway down the helical bundle centered around helix 4. The activation loop of F3 has a high content of positively charged residues, including Lys-316, Lys-318, Lys-320, Lys-322, and Lys-324, and is disordered in free F3. However, on binding talin 1655–1822, it becomes ordered, as indicated by the large chemical shift changes of residues in this region (Fig. 2F and supplemental Fig. S1), with a few residues broadening due to exchange between free and the bound states. The structure of the complex shows that the surface on 1655–1822 with which the activation loop interacts has complementary characteristics, with an overall negative charge, and with residues Asp-1763, Glu-1770, Glu-1798, and Glu-1805 all ideally placed to form electrostatic contacts with the basic residues in F3 (Fig. 3C). The side chain resonances of Asp-1763, Glu-1770, and Glu-1805 all broaden markedly upon the addition of F3, supporting their involvement in the interaction.
Mutations of Residues in Talin 1655–1822 That Contact the F3 Domain Markedly Reduce F3 Binding—To further investigate the interface between the two domains, we made a series of point mutants of talin 1655–1822 (S1684D, Q1764A, T1767A, T1767E, E1770A, and Y1777A) designed to disrupt the interaction with F3. These were based on the structure of the complex (Fig. 3A), the chemical shift mapping (Fig. 2C), and the sequence conservation between talins (Fig. 1B). Each mutant was expressed as an 15N-labeled polypeptide, and all mutants were judged to be correctly folded because they had well dispersed NMR spectra that strongly resembled that of the wild-type protein (data not shown). The most striking effect was seen with the E1770A mutant, which dramatically reduced F3 binding; no significant shifts were observed, even with a 10-fold excess of F3 (Fig. 4B and supplemental Fig. S2B). The chemical shifts of Glu-1770 are markedly affected by F3, and it is in an optimal position for an electrostatic contact to a residue in the activation loop of F3; from the structure of the complex, the most likely partner is Lys-318 or Lys-320. Alanine substitution of Gln-1764 and Tyr-1777 on helix 4, which are not in the binding interface, showed little or no effect on F3 binding (Fig. 4D).
FIGURE 4.

Identification of mutants that disrupt the head-tail interaction. A–C, weighted shift map obtained from the 1H,15N HSQC spectra of talin 1655–1822, or mutants thereof, on the addition of talin F3. A, wild-type (WT) talin 1655–1822. B, E1770A talin 1655–1822. C, T1767E talin 1655–1822. D, dissociation constants for the binding of wild-type talin 1655–1822 and various mutants thereof to F3. E, dissociation constants for the binding of wild-type talin 1655–1822 to various F3 FERM subdomain mutants (obtained from analysis of the chemical shift changes of residues 319, 324, 326, 352, 359, 366, 373, and 378 in the 1H,15N HSQC spectra of F3, or mutants thereof, on the addition of talin 1655–1822).
The program NetPhos2.0 (42) predicts that several residues in the 1655–1822 bundle may be phosphorylated, including Thr-1767 and Ser-1684. Threonine 1767 on helix 1 is adjacent to Glu-1770 and may help to orient the side chain of the latter. Mutation of Thr-1767 to glutamate resulted in a large drop in affinity for F3 (20–30-fold based on NMR titration data); although the mutation completely abolished the interaction of F3 with helix 4, residues in helix 1 were still slightly perturbed (Fig. 4C and supplemental Fig. S2C). However, similar effects were seen with the T1767A mutant, so the data do not reflect a clear role for Thr-1767 phosphorylation in regulating the interaction between F3 and the rod domain. Serine 1684 has been reported to be phosphorylated in platelet talin (43) and is in close proximity to the binding pocket for Tyr-377 of F3 (Fig. 3B). However, a S1684D mutant had only very small effects on the interaction with F3 (Fig. 4D), suggesting that phosphorylation of this residue is also not involved in regulation of the interaction.
Mutations of Residues in Talin F3 That Contact the Talin 1655–1822 Domain Markedly Reduce Talin 1655–1822 Binding—A number of mutants of the talin F3 domain were also expressed in 15N-labeled form and assayed for binding to the talin 1655–1822 rod polypeptide by NMR (Fig. 4E). As noted above, Tyr-377 in F3 occupies a hydrophobic binding pocket on the surface of talin 1655–1822, as shown by intermolecular NOEs to residues Leu-1680, Val-1683, and Met-1759. Mutation of Tyr-377 to alanine resulted in an ∼5-fold decrease in binding affinity, consistent with its role in the complex interface. The structure of the talin F3-talin 1655–1822 complex also suggests a salt-bridge interaction between Lys-324 in F3 and Glu-1798 in the talin rod domain. In support of this prediction, Lys-324 shows a substantial chemical shift change upon binding talin 1655–1822 (supplemental Fig. S1), and the K324D mutation resulted in a large decrease in affinity (∼23-fold; Fig. 4E). Goksoy et al. (28) reported that mutation of M319A in F3 greatly (∼140-fold) decreased its affinity for the talin rod, and full-length M319A talin was twice as effective in activating αIIbβ3 expressed in Chinese hamster ovary cells as full-length wild-type talin. The authors concluded that the M319A mutation inhibits the talin head-rod interaction and results in a greater proportion of talin adopting the open, active, conformation (28). However, our data indicate that Met-319 is not directly involved in the talin F3-talin 1655–1822 interface; the side chain resonances do not shift upon complex formation, and no NOEs are observed from Met-319 to talin 1655–1822. Titration of the M319A F3 mutant with talin 1655–1822 showed that this mutation has a negligible effect on binding (Fig. 4E). It is notable that full-length M319A talin is more effective in activating αIIbβ3-integrin than wild-type talin head on its own (28), suggesting that the mutation must have effect(s) other than changing the conformational equilibrium. Indeed, we find that this mutation results in a slight increase in the affinity of the talin head for the β3-integrin cytoplasmic domain (Kd 225 ± 7 μm when compared with 273 ± 6 μm for the wild-type).
Talin 1655–1822 Competes with the Integrin β3-Tail for Binding to F3—Interactions between F3 and the membrane-proximal region of the β3-integrin tail (Fig. 5A) have previously been shown to be unique to talin and necessary for integrin activation (30). The structure of the complex shows that talin 1655–1822 appears to block the binding site in F3 for the membrane-proximal part of the β3-integrin tail, whereas leaving that for the NPXY part largely accessible (Fig. 5B). This is consistent with the results of competition and mutagenesis experiments with a much larger talin 1654–2344 construct and a β3-integrin-PIPKIγ chimeric peptide (28). We undertook experiments to establish whether the single talin domain 1655–1822 does indeed compete with the wild-type β3-integrin tail for binding to the F3 domain. The 1H and 15N resonances of the β3-integrin tail have previously been assigned (29), and the positions of these peaks in a 1H,15N HSQC spectrum could readily be followed on the addition of various talin domains (Fig. 5C). The addition of F3 caused large chemical shift perturbations both in the NPXY region (residues 744–747) and in the more membrane-proximal portions of the β3-tail. In contrast, the addition of talin 1655–1822 only led to very small perturbations, indicating the absence of any specific interaction between the talin rod construct and the β3-integrin tail. When the F3 domain was added in the presence of a 4-fold excess of talin 1655–1822, the chemical shift perturbations of the residues of the β3-integrin tail were substantially smaller than those observed with the F3 domain alone, indicating competition between the β3-integrin tail and talin 1655–1822 for binding to the F3 domain. This decrease in magnitude was global and was not specific to any one region of the integrin tail. As expected, the E1770A talin rod mutant, which binds F3 very weakly if at all, failed to compete with F3 for binding to the β3-integrin tail (Fig. 5C).
FIGURE 5.

The talin 1655–1822 rod domain competes with the β3-integrin tail for binding to the talin F3 FERM subdomain. A, structure of the talin F3-integrin tail complex ((30) PDB 2H7D). Talin F3 is shown in red; the integrin tail is shown in yellow, and the membrane is shown by a black line. B, the F3-talin 1655–1822 complex oriented as in A, the binding site for the integrin tail, and the membrane association site within the activation loop of F3 are masked by the talin rod domain. Talin F3 is shown in red, and talin 1655–1822 is shown in cyan. C, weighted chemical shift maps of 15N-labeled β3-integrin tail upon the addition of talin 1655–1822 (red), talin F3 domain (blue), or both (green). The upper graph shows results for the wild type rod domain, and the lower graph shows results for the E1770A talin rod domain mutant. Gray bars correspond to peaks that broaden severely and could not be tracked.
DISCUSSION
Early studies by Molony et al. (27) showed that talin can exist in both extended and globular forms and that the globular conformation is dependent on an interaction between the talin head and rod domains. It is now apparent from the recent NMR studies of Goksoy et al. (28) that this globular conformation is at least in part due to an interaction between the talin F3 FERM subdomain and residues 1654–2344 of the talin rod (Kd 0.6 μm). Within this region, the authors provided evidence for two F3 binding sites, one of which (residues 1654–1848) had a higher affinity for F3 (Kd 3.6 μm) than the other (residues 1984–2344; Kd ∼78 μm). We have now determined the authentic domain boundaries of the region that forms the high affinity site and determined the solution structure of this domain (residues 1655–1822), as well as a detailed model of its complex with the F3 domain. The second lower affinity site (1984–2344) identified by Goksoy et al. (28) spans several domains whose structures we have previously determined (1974–2139, 2140–2294, 2300–2541), but we have been unable to detect binding of F3 to these individual domains (data not shown). The fact that F3 binds more tightly to the larger construct spanning 1654–2344 (Kd 0.6 μm) suggests that although the primary F3 binding site in the talin rod spans residues 1655–1822 (Kd ∼9 μm), additional lower affinity interactions with other domains in the vicinity might increase the overall affinity of the head-rod interaction. It is also possible that other regions of the talin head might contribute to binding.
We have shown that the F3 binding surface on talin 1655–1822 is large, comprising two main areas (Fig. 3), one involving Tyr-377 of F3 docking into a small hydrophobic pocket at the top of the helical bundle and a second that involves basic residues in the activation loop of F3, which bind to a cluster of negatively charged residues on helix 4 of talin 1655–1822. Mutagenesis suggests that this helix 4 region is the more important in terms of binding energy because the point mutation E1770A leads to a very marked decrease in affinity. However, Tyr-377 may play a role in regulating binding because it is predicted to be a Src phosphorylation site (42). Although mutation of residue Tyr-377 to alanine showed only a modest decrease in affinity, introduction of a negatively charged phosphate group in this position could have a much greater effect given the hydrophobic nature of the binding pocket on talin 1655–1822.
By overlaying the model of the talin F3-talin 1655–1822 complex with that of F3 bound to a chimeric β3-integrin-PIPKIγ peptide (30), it is clear that the talin rod domain occupies a similar position on F3 to the β3-integrin tail (Fig. 5, A and B); the β3-integrin membrane-proximal region occupies a near equivalent position to helices 4 and 5 of talin 1655–1822. This explains why F3 cannot bind the β-integrin tail when it is involved in the head-tail interaction. Although the interaction region for both ligands on F3 is the same, the manner of binding is different; the talin F3-rod interaction is primarily governed by electrostatic interactions, whereas the talin F3-integrin interaction is primarily hydrophobic in nature. However, our data show that the binding site for the membrane-distal NPXY region of the integrin tail in F3 is not masked by the talin 1655–1822 rod domain, in agreement with the conclusions reached by Goksoy et al. (28), although steric clashes between integrin Trp-739 (a vital residue for talin binding) and talin Pro-1756 suggest that binding might be restricted. Significantly, the WVYSPLH sequence at the C terminus of PIPKIγ can bind to this same site in F3 (44, 45), and PIPKIγ has been shown to play a key role in the adhesion-dependent targeting of cytoplasmic talin, which is predicted to be in the autoinhibited form, to the plasma membrane (46, 47). The binding of talin 1655–1822 to F3 may also block the interaction of F3 and other regions of the talin FERM domain with the membrane. Lys-322, which lies in a loop between strands S1 and S2 of F3, is important in integrin activation, and it has been suggested that it interacts with acidic membrane phospholipids stabilizing the integrin-talin complex (30). The structure of the F3-rod domain complex indicates that this interaction will be blocked. Similarly, the basic surface on the F2 FERM subdomain may also interact with acidic membrane phospholipids, orientating the FERM domain at the membrane interface and facilitating integrin binding. On the basis of the structure of the F2,F3 double domain (48), the binding of the rod domain to F3 would be predicted to prevent such an interaction.
In conclusion, the talin interdomain complex described here provides a clear structural basis for talin autoinhibition. The focus now turns toward defining the mechanisms that lead to talin activation, and PIPKIγ (46, 47), the GTPase Rap1, and its binding partner Rap1-GTP-interacting adaptor molecule (RIAM) (16, 49) have all been implicated in this process.
Supplementary Material
The atomic coordinates and structure factors (code 2KBB) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported, in whole or in part, by National Institutes of Health Grant U54 GM64346 through the Cell Migration Consortium. This work was also supported by grants from the Wellcome Trust and Cancer Research UK.
The on-line version of this article (available at http://www.jbc.org) contains two supplemental figures.
This article was selected as a Paper of the Week.

Author's Choice—Final version full access.
Footnotes
The abbreviations used are: FERM, four-point-one, ezrin, radixin, moesin; HADDOCK, high ambiguity driven biomolecular docking; HSQC, heteronuclear single quantum coherence; NOESY, nuclear Overhauser enhancement spectroscopy; PIPKI, phosphatidylinositol-4-phosphate 5-kinase type 1.
B. T. Goult, P. R. Elliott, N. Bate, B. Patel, A. R. Gingras, J. G. Grossmann, G. C. K. Roberts, D. R. Critchley, and I. L. Barsukov, manuscript in preparation.
B. T. Goult, A. R. Gingras, N. Bate, G. C. K. Roberts, I. L. Barsukov, and D. R. Critchley, unpublished work.
References
- 1.Calderwood, D. A. (2004) J. Cell Sci. 117 657-666 [DOI] [PubMed] [Google Scholar]
- 2.Critchley, D. R., and Gingras, A. R. (2008) J. Cell Sci. 121 1345-1347 [DOI] [PubMed] [Google Scholar]
- 3.Tadokoro, S., Shattil, S. J., Eto, K., Tai, V., Liddington, R. C., de Pereda, J. M., Ginsberg, M. H., and Calderwood, D. A. (2003) Science 302 103-106 [DOI] [PubMed] [Google Scholar]
- 4.Zhang, X., Jiang, G., Cai, Y., Monkley, S. J., Critchley, D. R., and Sheetz, M. P. (2008) Nat. Cell Biol. [DOI] [PMC free article] [PubMed]
- 5.Cram, E. J., Clark, S. G., and Schwarzbauer, J. E. (2003) J. Cell Sci. 116 3871-3878 [DOI] [PubMed] [Google Scholar]
- 6.Tanentzapf, G., Martin-Bermudo, M. D., Hicks, M. S., and Brown, N. H. (2006) J. Cell Sci. 119 1632-1644 [DOI] [PubMed] [Google Scholar]
- 7.Monkley, S. J., Zhou, X. H., Kinston, S. J., Giblett, S. M., Hemmings, L., Priddle, H., Brown, J. E., Pritchard, C. A., Critchley, D. R., and Fassler, R. (2000) Dev. Dyn. 219 560-574 [DOI] [PubMed] [Google Scholar]
- 8.Nieswandt, B., Moser, M., Pleines, I., Varga-Szabo, D., Monkley, S., Critchley, D., and Fassler, R. (2007) J. Exp. Med. 204 3113-3118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petrich, B. G., Marchese, P., Ruggeri, Z. M., Spiess, S., Weichert, R. A., Ye, F., Tiedt, R., Skoda, R. C., Monkley, S. J., Critchley, D. R., and Ginsberg, M. H. (2007) J. Exp. Med. 204 3103-3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang, Y., Litvinov, R. I., Chen, X., Bach, T. L., Lian, L., Petrich, B. G., Monkley, S. J., Critchley, D. R., Sasaki, T., Birnbaum, M. J., Weisel, J. W., Hartwig, J., and Abrams, C. S. (2008) J. Clin. Investig. 118 812-819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conti, F. J., Felder, A., Monkley, S., Schwander, M., Wood, M. R., Lieber, R., Critchley, D., and Muller, U. (2008) Development (Camb.) 135 2043-2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen, N. T., and Lo, S. H. (2005) Biochem. Biophys. Res. Commun. 337 670-676 [DOI] [PubMed] [Google Scholar]
- 13.Wegener, K. L., and Campbell, I. D. (2008) Mol. Membr. Biol. 25 376-387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bouaouina, M., Lad, Y., and Calderwood, D. A. (2008) J. Biol. Chem. 283 6118-6125 [DOI] [PubMed] [Google Scholar]
- 15.Bos, J. L. (2005) Curr. Opin. Cell Biol. 17 123-128 [DOI] [PubMed] [Google Scholar]
- 16.Han, J., Lim, C. J., Watanabe, N., Soriani, A., Ratnikov, B., Calderwood, D. A., Puzon-McLaughlin, W., Lafuente, E. M., Boussiotis, V. A., Shattil, S. J., and Ginsberg, M. H. (2006) Curr. Biol. 16 1796-1806 [DOI] [PubMed] [Google Scholar]
- 17.Papagrigoriou, E., Gingras, A. R., Barsukov, I. L., Bate, N., Fillingham, I. J., Patel, B., Frank, R., Ziegler, W. H., Roberts, G. C., Critchley, D. R., and Emsley, J. (2004) EMBO J. 23 2942-2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fillingham, I., Gingras, A. R., Papagrigoriou, E., Patel, B., Emsley, J., Critchley, D. R., Roberts, G. C., and Barsukov, I. L. (2005) Structure (Camb.) 13 65-74 [DOI] [PubMed] [Google Scholar]
- 19.Gingras, A. R., Vogel, K. P., Steinhoff, H. J., Ziegler, W. H., Patel, B., Emsley, J., Critchley, D. R., Roberts, G. C., and Barsukov, I. L. (2006) Biochemistry 45 1805-1817 [DOI] [PubMed] [Google Scholar]
- 20.Gingras, A. R., Bate, N., Goult, B. T., Hazelwood, L., Canestrelli, I., Grossmann, J. G., Liu, H., Putz, N. S., Roberts, G. C., Volkmann, N., Hanein, D., Barsukov, I. L., and Critchley, D. R. (2008) EMBO J. 27 458-469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gingras, A. R., Ziegler, W. H., Bobkov, A. A., Joyce, M. G., Fasci, D., Himmel, M., Rothemund, S., Ritter, A., Grossmann, J. G., Patel, B., Bate, N., Goult, B. T., Emsley, J., Barsukov, I. L., Roberts, G. C., Liddington, R. C., Ginsberg, M. H., and Critchley, D. R. (2009) J. Biol. Chem. 284 8866-8876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gingras, A. R., Ziegler, W. H., Frank, R., Barsukov, I. L., Roberts, G. C., Critchley, D. R., and Emsley, J. (2005) J. Biol. Chem. 280 37217-37224 [DOI] [PubMed] [Google Scholar]
- 23.Hemmings, L., Rees, D. J., Ohanian, V., Bolton, S. J., Gilmore, A. P., Patel, B., Priddle, H., Trevithick, J. E., Hynes, R. O., and Critchley, D. R. (1996) J. Cell Sci. 109 2715-2726 [DOI] [PubMed] [Google Scholar]
- 24.Smith, S. J., and McCann, R. O. (2007) Biochemistry 46 10886-10898 [DOI] [PubMed] [Google Scholar]
- 25.Martel, V., Racaud-Sultan, C., Dupe, S., Marie, C., Paulhe, F., Galmiche, A., Block, M. R., and Albiges-Rizo, C. (2001) J. Biol. Chem. 276 21217-21227 [DOI] [PubMed] [Google Scholar]
- 26.Bretscher, A., Chambers, D., Nguyen, R., and Reczek, D. (2000) Annu. Rev. Cell Dev. Biol. 16 113-143 [DOI] [PubMed] [Google Scholar]
- 27.Molony, L., McCaslin, D., Abernethy, J., Paschal, B., and Burridge, K. (1987) J. Biol. Chem. 262 7790-7795 [PubMed] [Google Scholar]
- 28.Goksoy, E., Ma, Y. Q., Wang, X., Kong, X., Perera, D., Plow, E. F., and Qin, J. (2008) Mol Cell 31 124-133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oxley, C. L., Anthis, N. J., Lowe, E. D., Vakonakis, I., Campbell, I. D., and Wegener, K. L. (2008) J. Biol. Chem. 283 5420-5426 [DOI] [PubMed] [Google Scholar]
- 30.Wegener, K. L., Partridge, A. W., Han, J., Pickford, A. R., Liddington, R. C., Ginsberg, M. H., and Campbell, I. D. (2007) Cell 128 171-182 [DOI] [PubMed] [Google Scholar]
- 31.Wishart, D. S., Bigam, C. G., Yao, J., Abildgaard, F., Dyson, H. J., Oldfield, E., Markley, J. L., and Sykes, B. D. (1995) J. Biomol. NMR 6 135-140 [DOI] [PubMed] [Google Scholar]
- 32.Vranken, W. F., Boucher, W., Stevens, T. J., Fogh, R. H., Pajon, A., Llinas, M., Ulrich, E. L., Markley, J. L., Ionides, J., and Laue, E. D. (2005) Proteins 59 687-696 [DOI] [PubMed] [Google Scholar]
- 33.Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J., and Bax, A. (1995) J. Biomol. NMR 6 277-293 [DOI] [PubMed] [Google Scholar]
- 34.Cornilescu, G., Delaglio, F., and Bax, A. (1999) J. Biomol. NMR 13 289-302 [DOI] [PubMed] [Google Scholar]
- 35.Baxter, N. J., Hosszu, L. L., Waltho, J. P., and Williamson, M. P. (1998) J. Mol. Biol. 284 1625-1639 [DOI] [PubMed] [Google Scholar]
- 36.Herrmann, T., Guntert, P., and Wuthrich, K. (2002) J. Biomol. NMR 24 171-189 [DOI] [PubMed] [Google Scholar]
- 37.Linge, J. P., O'Donoghue, S. I., and Nilges, M. (2001) Methods Enzymol. 339 71-90 [DOI] [PubMed] [Google Scholar]
- 38.DeLano, W. L. (2004) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA
- 39.Ayed, A., Mulder, F. A., Yi, G. S., Lu, Y., Kay, L. E., and Arrowsmith, C. H. (2001) Nat. Struct. Biol. 8 756-760 [DOI] [PubMed] [Google Scholar]
- 40.Dominguez, C., Boelens, R., and Bonvin, A. M. (2003) J. Am. Chem. Soc. 125 1731-1737 [DOI] [PubMed] [Google Scholar]
- 41.de Vries, S. J., van Dijk, A. D., Krzeminski, M., van Dijk, M., Thureau, A., Hsu, V., Wassenaar, T., and Bonvin, A. M. (2007) Proteins 69 726-733 [DOI] [PubMed] [Google Scholar]
- 42.Blom, N., Gammeltoft, S., and Brunak, S. (1999) J. Mol. Biol. 294 1351-1362 [DOI] [PubMed] [Google Scholar]
- 43.Ratnikov, B., Ptak, C., Han, J., Shabanowitz, J., Hunt, D. F., and Ginsberg, M. H. (2005) J. Cell Sci. 118 4921-4923 [DOI] [PubMed] [Google Scholar]
- 44.Barsukov, I. L., Prescot, A., Bate, N., Patel, B., Floyd, D. N., Bhanji, N., Bagshaw, C. R., Letinic, K., Di Paolo, G., De Camilli, P., Roberts, G. C., and Critchley, D. R. (2003) J. Biol. Chem. 278 31202-31209 [DOI] [PubMed] [Google Scholar]
- 45.de Pereda, J. M., Wegener, K. L., Santelli, E., Bate, N., Ginsberg, M. H., Critchley, D. R., Campbell, I. D., and Liddington, R. C. (2005) J. Biol. Chem. 280 8381-8386 [DOI] [PubMed] [Google Scholar]
- 46.Ling, K., Doughman, R. L., Firestone, A. J., Bunce, M. W., and Anderson, R. A. (2002) Nature 420 89-93 [DOI] [PubMed] [Google Scholar]
- 47.Ling, K., Doughman, R. L., Iyer, V. V., Firestone, A. J., Bairstow, S. F., Mosher, D. F., Schaller, M. D., and Anderson, R. A. (2003) J. Cell Biol. 163 1339-1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garcia-Alvarez, B., de Pereda, J. M., Calderwood, D. A., Ulmer, T. S., Critchley, D., Campbell, I. D., Ginsberg, M. H., and Liddington, R. C. (2003) Mol Cell 11 49-58 [DOI] [PubMed] [Google Scholar]
- 49.Lee, H. S., Lim, C. J., Puzon-McLaughlin, W., Shattil, S. J., and Ginsberg, M. H. (2009) J. Biol. Chem. 284 5119-5127 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.