

Abstract
The advent of high-throughput sequencing (HTS) methods has enabled direct approaches to quantitatively profile small RNA populations. However, these methods have been limited by several factors, including representational artifacts and lack of established statistical methods of analysis. Furthermore, massive HTS data sets present new problems related to data processing and mapping to a reference genome. Here, we show that cluster-based sequencing-by-synthesis technology is highly reproducible as a quantitative profiling tool for several classes of small RNA from Arabidopsis thaliana. We introduce the use of synthetic RNA oligoribonucleotide standards to facilitate objective normalization between HTS data sets, and adapt microarray-type methods for statistical analysis of multiple samples. These methods were tested successfully using mutants with small RNA biogenesis (miRNA-defective dcl1 mutant and siRNA-defective dcl2 dcl3 dcl4 triple mutant) or effector protein (ago1 mutant) deficiencies. Computational methods were also developed to rapidly and accurately parse, quantify, and map small RNA data.
Keywords: small RNA, sequencing-by-synthesis, CASHX, SAM-seq, oligoribonucleotide standards, statistical methods
INTRODUCTION
Most eukaryotic organisms contain one or more classes of small RNA that function as guides in association with ARGONAUTE (AGO) proteins for regulation at the post-transcriptional or transcriptional level. Diverse small RNA can derive through distinct biogenesis routes and function through specialized classes of AGO proteins (Chapman and Carrington 2007; Faehnle and Joshua-Tor 2007; Peters and Meister 2007; Farazi et al. 2008). microRNA (miRNA) form through multistep processing of self-complementary foldback structures through the activities of DICER (or DICER-LIKE) and other RNaseIII-type nucleases, resulting in products with a 5′ monophosphate and 3′ hydroxyl. Endogenous classes of 5′ monophosphate-containing short interfering RNA (siRNA) form by DICER-mediated processing of long dsRNA, which can arise from bidirectional transcription, self-complementary foldback structures within transcripts, or the activity of RNA-dependent RNA polymerases (RdRPs) (for review, see Voinnet 2008). Several DICER-dependent classes of siRNA have been characterized in plants (Chapman and Carrington 2007), and were recently discovered in flies and mice (Czech et al. 2008; Ghildiyal et al. 2008; Kawamura et al. 2008; Okamura et al. 2008; Tam et al. 2008; Watanabe et al. 2008). In Caenorhabditis elegans, secondary siRNA that contain 5′ triphosphate arise through an RdRP-dependent, but DICER-independent, route (Pak and Fire 2007; Sijen et al. 2007). miRNA and siRNA associate with members of the AGO subclass of ARGONAUTE proteins. In animal lineages, several types of small RNA associate with members of the PIWI subclass of ARGONAUTE proteins. These are generally referred to as Piwi-interacting RNA (piRNA), but subtypes include repeat-associated siRNA (flies) and 21U-RNA (C. elegans) (Vagin et al. 2006; Brennecke et al. 2007; Batista et al. 2008; Wang and Reinke 2008). piRNA form through AGO/PIWI-dependent, but DICER-independent, mechanisms, and function to promote germline development and suppress transposons (for review, see Klattenhoff and Theurkauf 2008). In part, discovery of the existence or expanse of these and other distinct small RNA classes involved high-throughput sequencing, as originally shown by Lu et al. 2005.
High-throughput sequencing has also emerged as a direct small RNA profiling method (for examples, see Lu et al. 2006; Ruby et al. 2006; Kasschau et al. 2007; Lister et al. 2008). Compared to finite-sample platforms, such as microarray or PCR-based assays, HTS profiling permits semi-open-ended analysis of both known and unknown small RNAs. In principle, because HTS-based profiling is essentially a random-sampling method, the effective linear range should be broad. Picoliter-scale pyrosequencing (Margulies et al. 2005) has been useful for quantitative analysis of small RNA populations in silencing mutants and differential tissues, and in association with specific AGO proteins (Girard et al. 2006; Henderson et al. 2006; Lu et al. 2006; Qi et al. 2006; Rajagopalan et al. 2006; Ruby et al. 2006; Brennecke et al. 2007; Kasschau et al. 2007; Kawamura et al. 2008). This method can be done in a single-sample or multiplexed format. Recently, short-read sequencing-by-synthesis (SBS) of amplified DNA colonies (Bentley 2006) has emerged as a small RNA profiling method (Czech et al. 2008; Gregory et al. 2008; Lister et al. 2008; Mi et al. 2008; Montgomery et al. 2008a; Okamura et al. 2008; Tam et al. 2008). SBS methods facilitate far greater sequencing depth per sample relative to previous methods. These have been combined with relatively simple analytical methods, usually with relatively low statistical power, for over- or underrepresentation analyses between samples (for examples, see Henderson et al. 2006; Kasschau et al. 2007; Czech et al. 2008; Montgomery et al. 2008a). A consensus regarding rigorous, standardized experimental designs and statistical methods based on replicate samples to analyze individual small RNA or small RNA classes has not yet emerged.
As HTS small RNA data sets increase in size, computational problems intensify. For example, rapid and accurate mapping of small RNA sequences from 107 or more reads to a reference genome is a significant computational challenge. Analysis of small RNA data sets presents another set of issues, such as how to normalize data and quantitatively assess differences between multiple samples. Representational artifacts can occur, particularly when the abundance of whole small RNA classes differs significantly between samples. We developed and tested new computational approaches to rapidly parse, quantify, map, and analyze small RNA reads from SBS data sets. We also developed a method using synthetic standards to objectively and quantitatively compare small RNA populations between samples. In addition, we adapted and tested microarray-based statistical methods to identify differentially expressed small RNA between sample sets.
RESULTS AND DISCUSSION
Mapping of SBS reads using CASHX
The SBS platform (Illumina 1G Genome Analyzer) uses bridge-PCR with primers fixed to a silicon slide to amplify DNA clones from single initial molecules (Bentley 2006). For all small RNA populations analyzed here, an adaptor was ligated to the 3′ end in an ATP-independent manner (Pfeffer et al. 2005), and then joined to 5′ adaptors by a second enzymatic ligation (Supplemental Fig. 1A). cDNA was generated and amplified by 14 PCR cycles, and the resulting population of DNA molecules was subjected to SBS using single-plex loading of individual flow-cell lanes. This generally yielded 5,000,000–9,000,000 raw reads/lane. Raw reads were processed through a pipeline to parse small RNA sequences from the 3′ adapter, collapse the data to a uniread set, count the number of reads per unique sequence, map sequences to the reference genome, and annotate sequences with basic information (Supplemental Fig. 1B).
Among the steps in the pipeline, accurate mapping of unireads to the reference genome was the most computationally intensive. The traditional DNA search algorithms BLAST and BLAT (Altschul et al. 1990; Kent 2002) were used in initial tests of randomly sampled populations of 10–107 small RNA reads (50% perfect Arabidopsis genome match, 50% mismatch). At 104 queries and higher, BLAT performed faster than BLAST (Fig. 1). For example, BLAT mapped 106 reads 3.2-fold faster than BLAST (Fig. 1). The faster speed of BLAT with larger read sets is due to the database indexing method (Kent 2002). However, at 107 reads, BLAT required ∼78.8 h, which was judged to be unacceptably slow for SBS data sets.
FIGURE 1.

Processing speed to query 10–108 small RNA sequences (50% Arabidopsis genome perfect match, 50% mismatch) using BLAT, BLAST, and CASHX. Each data point represents the average of five independent runs. CASHX was run with and without precaching. Due to the extensive time requirement, a maximum of 106 and 107 queries were done by BLAST and BLAT, respectively.
An alternative mapping program, cache-assisted hash search with XOR digital logic (CASHX), was developed to map small RNA reads efficiently to a reference genome. This program utilizes a 2 bit-per-base binary format of query and reference genome sequences to reduce computational weight. The reference genome is divided into all possible 30 nucleotide (nt) sequences, each of which is linked to data for chromosome, strand, and start/end coordinates. Each 30-mer is indexed by a preamble string of 4 nt at the 5′ end within a HASH database. The initial HASH database, therefore, has 256 (44) containers of 30-mer sequences, where each sequence within a container has the same first four nucleotides. The CASHX algorithm searches the HASH index in 0(1) constant time (fast) and the containers in 0(1) linear time (slow). Therefore, the amount of data within a container impacts processing speed disproportionately compared to the number of indexed containers. To increase processing speed, the HASH database, indexed to a 4 nt preamble, is easily transformed to a user-defined preamble string of 8–12 nt to optimize the number of containers with the number of sequences in each container. In the case of a 12 nt preamble, the CASHX database built from the Arabidopsis genome was created in less than 8 min, used 7.2G of memory, and generated 16,777,216 containers of 30-mer sequences.
Next, the genome HASH database is searched with each small RNA-derived query sequence. First, the query preamble sequence is identified within the HASH database using key value pairs, thereby locating a container. This search can be done after preloading the HASH database into cache memory, or by searching directly from file space. If the HASH database is not precached, a key value pair hit loads the container contents into memory. Second, each sequence within a hit container is searched using an XOR digital logic string. Sequences that pass through the XOR gate with an outcome of zero correspond to a perfect match. Default CASHX output files contain sequence information, number of reads/sequence in the library, and a list of perfect genome hits, including strand and start/stop coordinates. The output can also be formatted for compatibility with BLAT PSL/PSLX formats (Kent 2002). The minimum searchable sequence length is 15 nt. Sequences over 30 nt in length are divided into 30-mers and aligned to the CASHX HASH database. Consecutive hits on the genome are identified to reconstruct the full sequence match. CASHX was tested successfully using sequences up to 10,000 nt in length.
CASHX was tested using 10–108 sequences (50% Arabidopsis genome matched, 50% mismatched), with and without precaching of the HASH database. Without precaching, processing time for 103 queries was comparable to BLAT and BLAST (Fig. 1). However, CASHX processing speed accelerated as numbers of queries increased above 103. This was due to the impact of on-the-fly data caching of recurring searches within a given container, and because searching in cache memory space is significantly faster than searching in file space. For example, 103 CASHX searches done after precaching finished ∼500-fold faster than the same number of CASHX searches done using file space (Fig. 1). Compared to BLAT, CASHX run with precaching was ∼500–900-fold faster for 103 or more queries (Fig. 1). Only CASHX performed at speeds deemed practical under normal circumstances with 107 queries or greater.
Other programs, such as ELAND (Illumina, http://www.illumina.com) and SOAP (Li et al. 2008), can be used to map HTS reads to a reference genome. Using a 5′ ligation-dependent SBS data set of Arabidopsis small RNA (6,668,228 parsed reads of 18–29 nt), ELAND and SOAP both identified reads with genomic hits with speed comparable to, or slightly slower than, CASHX (Table 1). All reads and unique sequences returned using CASHX were returned with ELAND, and these were confirmed to be bona fide hits to the Arabidopsis genome by using a direct string comparison between the query sequence and the sequence retrieved by FASTACMD (Johnson et al. 2008) using the coordinates supplied by CASHX. SOAP, which was run using an 8 nt seed size and searching for reads with zero mismatches, returned fewer total reads and unique sequences. CASHX identified more genomic loci with perfect hits than did ELAND or SOAP (Table 1). ELAND returns fewer genomic hits because repetitive hits are reported only once. The basis for fewer hits identified by SOAP is not clear, but may relate to the method of reference genome indexing used prior to search.
TABLE 1.
Perfect Arabidopsis genomic hits identified by three programs

Reproducibility of SBS small RNA data sets
To assess the SBS method as a quantitative profiling tool, biological and technical replicates of Arabidopsis small RNA SBS runs were compared for reproducibility. Two small RNA classes—miRNA families, and 24 nt siRNA within genomic windows or bins (50,000 nt, 10,000 nt scroll)—were quantified separately. Read counts were normalized to adjust for differences in library size, or sequencing depth, to reads per million (RPM). Data were also repeat normalized to distribute read counts evenly among all loci with a perfect match, and small RNA corresponding to rRNA, tRNA, snRNA, and snoRNA were removed.
The correlation of normalized miRNA family and 24 nt siRNA bin reads between biological (two distinct samples processed independently and run on two lanes) replicates was very high (Spearman's ρ for miRNA and 24 nt siRNA bins was 0.947261 and 0.9453777, respectively) (Fig. 2A, upper panel). For technical replicates (one amplicon preparation divided among two lanes in one flow cell), the correlation for normalized miRNA family and 24 nt siRNA bin reads was even higher (Spearman's ρ = 0.9818703 and 0.9569397, respectively) (Fig. 2B, upper panel).
FIGURE 2.

Reproducibility of SBS data sets. Comparison of (A) biological and (B) technical replicates of Arabidopsis small RNA samples prepared using the 5′ ligation-dependent amplicon preparation method. Upper graphs show normalized small RNA reads in one replicate versus another. Lower graphs show absolute residual error versus fitted read values. In all graphs, miRNA are plotted as black dots and 24 nt siRNA bins (50,000 nt windows, 10,000 nt scroll) are plotted as gray dots. Data were normalized to reads/million.
Despite the high degree of correlation between replicates, variability of individual miRNA family or 24 nt siRNA bins was not constant. Residual error after fitting a linear model was low for highly abundant small RNA, while residual error for less abundant small RNA was high (Fig. 2A,B, lower panels). This nonuniform variance, or heteroscedasticity, was significant (P ≤ 4.9 × 10−4, Goldfeld–Quandt and Breusch–Pagan tests). Heteroscedasticity is also a common feature of microarray data where probes associated with higher intensity signal are more reproducibly measured than probes with lower signal (Fan et al. 2004). Significant heteroscedasticity will cause standard error estimates to be inflated, resulting in a decrease in statistical power and an increase in Type I error (Allison 2006; Montgomery et al. 2006). Various techniques, such as those described by Tusher et al. 2001, attempt to correct for this expression level-dependent variance.
Spike-in standards to compare small RNA SBS data sets
Although the trends shown in Figure 2 are clear, the profiling methods presented above suffer from a lack of objective standards against which to compare small RNA data. Thus, given the semi-open-ended nature of the SBS platform, comparisons between different small RNA depend largely on relative, not absolute, abundance. To overcome this limitation, three unique oligoribonucleotides (Std2, Std3, Std6) were designed to mimic canonical 21 nt small RNA (5′ monophosphate, 3′ hydroxyl) and tested as spike-in standards with small RNA SBS libraries (Fig. 3A). In initial tests, the three oligoribonucleotides were each added to four Arabidopsis total RNA samples (100 μg) in four amounts (0.01, 0.1, 1.0, and 10.0 pmol), and preparations were subjected to SBS sequencing. Note that the standards were included in all preparatory steps, including the initial purification of small RNA by gel elution. In each library, the normalized reads for each of Std2, Std3, and Std6 were similar and read counts increased in a linear progression in samples containing between 0.01 and 1.0 pmol initial spike-in amounts (Fig. 3A). At 10 pmol, however, standard reads approached saturation on the flow cell (Fig. 3A). Additionally, five other oligoribonucleotide standards were tested in the same concentration range, each yielding linear increases between 0.01 and 1.0 pmol initial amounts, although the efficiency of sequencing individual RNA varied among different standards (data not shown).
FIGURE 3.

Use of synthetic, 21 nt oligoribonucleotide standard spike-in controls for SBS sequencing. (A) Comparison of normalized reads for Std2, Std3, and Std6 added to Arabidopsis total RNA samples at four different amounts. Data show reads per million (RPM). (B) Comparison of standards, miRNA, miRNA*, tasiRNA, and 24 nt siRNA bins in samples prepared from wild-type (Col-0) and dcl2-1 dcl3-1 dcl4-2 (left) or dcl1-7 (right) mutant plants. Scatter plots show log2-scale RPM for each small RNA class or standard. Standards are shown with a fitted linear regression model.
Given the similar efficiencies of sequencing Std2, Std3, and Std6, a spike-in cocktail containing standards in three different amounts (Std 2, 0.01; Std 3, 0.1; Std 6, 1.0 pmol) per 100 μg total sample RNA was tested in two small RNA profiling experiments. In one, small RNA were compared between wild-type Arabidopsis (Col-0) and the dcl1-7 mutant, which accumulates lower levels of most miRNA (Park et al. 2002; Reinhart et al. 2002). In the other, wild type was compared to the dcl2-1 dcl3-1 dcl4-2 triple mutant, which is deficient in all known classes of siRNA (Zhang 2008). The standards formed an objective reference curve against which experimental samples, including canonical miRNA, canonical miRNA*, 21 nt tasiRNA, and 24 nt siRNA bins (1000 nt windows, 200 nt scroll), were normalized and converted to picomoles.
In the dcl2-1 dcl3-1 dcl4-2 triple mutant, tasiRNA and 24 nt siRNA populations were depressed relative to the standard curve, while miRNA and miRNA* were generally unaffected (Fig. 3B, left). Conversely, in the dcl1-7 sample, miRNA, miRNA*, and tasiRNA reads were generally low relative to the standard curve, whereas 24 nt siRNA were largely unaffected (Fig. 3B, right). Loss of tasiRNA in the dcl1-7 mutant was expected, as tasiRNA biogenesis requires transcript cleavage at a miRNA target site. These differences were analyzed statistically using a nonparametric t-test (Mann–Whitney–U/Wilcoxon rank sum test) to compare the quantities of miRNA and miRNA*, tasiRNA, and 24 nt siRNA bins between wild type and each mutant sample. As expected, miRNA, miRNA*, and tasiRNA groups were significantly underrepresented in dcl1-7 versus wild type (miRNA and miRNA* were 5.5-fold underrepresented, P = 4.04 × 10−8; tasiRNA were ∼32-fold underrepresented, P < 2.2 × 10−16). The 24 nt siRNA class was not significantly affected in the dcl1-7 mutant (P = 0.1943). In contrast, miRNA and miRNA* were not significantly affected in the dcl2-1 dcl3-1 dcl4-2 triple mutant (P = 0.311), but tasiRNA and 24 nt siRNA were significantly underrepresented (tasiRNA were ∼33-fold underrepresented, P < 2.2 × 10−16; 24 nt siRNA were ∼18-fold underrepresented, P < 2.2 × 10−16).
These data indicate that synthetic oligoribonucleotide standards can be used effectively in profiling experiments to objectively compare and normalize small RNA populations between independent samples.
Statistical analysis of small RNA profiles
Although comparisons between entire small RNA populations are often useful, identification of differentially expressed individual small RNA is often desired. Meaningful analysis of small RNA differences between samples requires statistical power at rigorous significance levels, and this is obtained through increased sample size (replicate data sets). We reasoned that abundance of a given sequence in an SBS data set is roughly analogous to signal intensity in a single channel microarray experiment. The commonly used significance analysis of microarrays (SAM) method (Tusher et al. 2001) was adapted. Library size-normalized small RNA from immature flowers of wild-type Col-0 and ago1-25 mutant plants, each of which was represented by three biological replicates (Montgomery et al. 2008b), were compared. miRNA and tasiRNA, but not 24 nt siRNA, are known to be moderately affected in the hypomorphic ago1-25 mutant (Morel et al. 2002). Therefore, we considered these data sets to be well suited for testing the statistical power and sensitivity of SAM as applied to sequencing-by-synthesis data (SAM-seq). The SAM procedure uses a relative difference score d(i) as a statistic to test for significant differential expression (Tusher et al. 2001). The d(i) are compared to a null distribution of scores, determined using a permutation-based resampling method, to determine the significance of individual scores. In this case, the relative difference of a small RNA between ago1-25 and wild-type Col-0 is
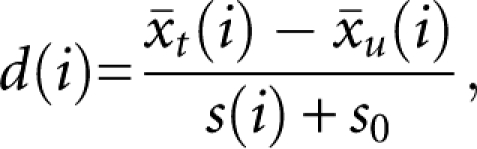 |
where 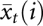 and
and 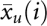 are the average RPM for the ith small RNA in ago1-25 and wild-type Col-0, respectively. The value s(i) is the combined standard deviation of replicate measurements of small RNA i in both ago1-25 and wild-type Col-0 and is given by
are the average RPM for the ith small RNA in ago1-25 and wild-type Col-0, respectively. The value s(i) is the combined standard deviation of replicate measurements of small RNA i in both ago1-25 and wild-type Col-0 and is given by
 |
where n and m are the number of replicates in ago1-25 and Col-0, respectively. The value s0 is a small, positive constant that is used to minimize the coefficient of variation for the data set (Tusher et al. 2001). Therefore, d(i) is essentially a t-statistic that is modified to use a variance shrinkage procedure that increases inferential power (for review, see Allison et al. 2006). Also, rather than using a P-value cutoff, SAM allows for control of the false discovery rate (FDR)—the percentage of false positive tests expected out of all tests called significant—using a Q-value measure (Tusher et al. 2001).
In an initial series of tests, a post hoc power analysis, with varying sample size (two, three, four, or five replicates), was done using the SAM package for R (Tibshirani 2006; R Development Core Team 2008). The power of a test (power = 1—false negative rate [FNR]) is affected by both sample size and effect size (Tibshirani 2006). For instance, with duplicate samples for wild-type Col-0 and ago1-25 and a small effect size (100 small RNA) the FNR was estimated to be over 90% (Fig. 4A). Although the FNR decreases with increasing effect size, addition of replicates also decreased the FNR (Fig. 4A). For instance, with three replicates compared to two replicates, the FNR was decreased by almost 40%; however, even with five replicates, the FNR is still predicted to range from ∼5%–25%, depending on effect size (Fig. 4A). In contrast, the FDR, which is controlled for using SAM-seq, was predicted to be relatively low (generally under 5%) for all replicated sample sizes and effect sizes (Fig. 4A). Therefore, increased numbers of replicate data sets are needed to increase statistical sensitivity (decrease the FNR).
FIGURE 4.

SAM-seq analysis of differentially expressed small RNA. (A) Statistical power assessment for the Col-0 versus ago1-25 analysis with 2, 3, 4, or 5 replicates modeled per sample. (B) Scatter plot of the mean (n = 3) RPM (log2-scale) in Col-0 versus ago1-25. Black data points and line show the mean oligoribonucleotide standards fit with a linear model. (C) Volcano plot of fold change (mean [n = 3] RPM in ago1-25 divided by mean [n = 3] RPM in wild type, log2-scale) versus absolute SAM score, d(i). (D) Scatter plot of mean (n = 3) RPM in Col-0 (log2-scale) versus absolute SAM score, d(i). In B–D, SAM-seq significant data points are color-coded red (overrepresented) and green (underrepresented).
Next, unique Arabidopsis small RNA sequences (n = 359,447) that were identified in at least three samples and did not originate from rRNA, tRNA, snRNA, or snoRNA were analyzed. SAM-seq yielded 1161 differentially expressed small RNA (321 over- and 840 underrepresented in ago1-25) that were each >twofold affected, with a FDR < 0.041 (Figs. 4B–D, 5; Supplemental Table 1). Of course, not all small RNA that were twofold-affected, particularly those with low read numbers, were called significant (Figs. 4C,D, 5). SAM-seq predicted that the FNR for underrepresented small RNA with d(i) between −0.13 and −0.75 was greater than 7%, while the FNR for overrepresented small RNA was 0% (Supplemental Table 2). This indicates that many more small RNA may be underrepresented in the ago1-25 mutant than can be identified with the statistical power available with three replicates.
FIGURE 5.

Genome-wide view of differentially expressed small RNA identified by SAM-seq. Fold change (mean [n = 3] RPM in ago1-25 divided by mean [n = 3] RPM in wild type, log2-scale) plotted for each unique small RNA analyzed by SAM-seq (n = 359,447) by position across each Arabidopsis chromosome. Small RNAs are color-coded gray (nonsignificant), red (significantly overrepresented), and green (significantly underrepresented). Chromosomes are illustrated in blue at the bottom of each graph with centromeres marked.
To assess the results from SAM-seq, significantly over- and underrepresented small RNA were categorized and analyzed for 5′ nucleotide content. Previous analyses revealed the preference of AGO1 for miRNA and tasiRNA with a 5′ uracil (5′U) (Mi et al. 2008; Montgomery et al. 2008a); therefore, 5′U-containing miRNA and tasiRNA were predicted to be disproportionately affected in ago1-25 plants. Based on annotated features, 58% and 38% of over- and underrepresented small RNA, respectively, were categorized as miRNA, miRNA*, tasiRNA, known phased siRNA or inverted repeat-derived siRNA (Fig. 6). Most overrepresented small RNA were derived from two large inverted repeats on chromosome 3, or corresponded to inaccurately processed or nonannotated species from MIRNA foldbacks (Fig. 6). Underrepresented small RNA were fairly evenly distributed among miRNA, tasiRNA, a collection of 21 nt siRNA from mRNA encoding pentatricopeptide repeat (PPR) proteins (Howell et al. 2007), and the inverted repeats from chromosome 3 (Fig. 6). However, underrepresented small RNA overwhelmingly possessed a 5′U, while overrepresented small RNA most often possessed a 5′ base other than a 5′U (P < 2.2 × 10−16 and P = 4.4 × 10−4, respectively, Fisher's exact test) (Fig. 6).
FIGURE 6.

Profile of over- and underrepresented small RNA in ago1-25 identified by SAM-seq. Small pie charts (left) represent the percentage of total over- or underrepresented small RNA in ago1-25 that were (light red or light green) or were not (dark red or dark green) categorized as previously annotated miRNA, annotated miRNA*, other MIRNA foldback-derived small RNA, 21 nt tasiRNA, other TAS locus-derived small RNA, RDR6/DCL4-dependent PPR-derived siRNA (Howell et al. 2007), RDR6/DCL4-dependent siRNA derived from non-PPR genes and inverted repeat-derived siRNA. Breakdowns of categorized small RNA are expanded in the larger pie charts. The larger pie charts (right) show the percentage of small RNA in each category listed in the key (top right). Outer rings around each pie chart show percentage of small RNA from each category that possess a 5′U.
Interestingly, three new miRNA were identified in the significantly affected sets (Supplemental Table 3). Two miRNA (miR1886.2 and miR2111a/b) possessed a 5′U and were in the underrepresented set. The third miRNA (miR2112), which derives from an intron at the At1g01650 locus, possessed a cytosine base at the 5′ end of the major small RNA from both the 5′ and 3′ arms (Supplemental Table 3). Each of these was represented by miRNA and miRNA* sequences in these or previously published libraries (Rajagopalan et al. 2006) and originated from foldbacks that fulfill consensus MIRNA requirements (Supplemental Fig. 2; Ambros et al. 2003; Meyers et al. 2008), but none were conserved in poplar, cassava or rice. miR1886.2 was previously identified as a candidate miRNA by Rajagopalan et al. 2006 and is derived from the recently identified MIR1886 foldback (German et al. 2008). miR1886.2 is the most abundant small RNA from MIR1886, according to small RNA libraries available at the Arabidopsis SBS database (http://mpss.udel.edu/at_sbs), and is offset from the annotated miR1886.1 sequence by 9 nt (Supplemental Fig. 2). Both miR1886.1 and miR1886.2 have miRNA* sequences represented in the public databases (ASRP, http://asrp.cgrb.oregonstate.edu/db and Arabidopsis SBS, http://mpss.udel.edu/at_sbs) and may represent abundant, offset variants like those seen from MIR161 (Allen et al. 2004).
In addition to analysis of individual sequences, SAM-seq was used to identify AGO1-dependent siRNA clusters. Such clusters may be composed of sets of low-abundance siRNA that, individually, may occur at levels insufficient for reliable SAM-seq analysis. Reads in each sample were library size-normalized, repeat-normalized, then masked for previously annotated miRNA, tasiRNA, and sequences from rRNA, tRNA, snRNA and snoRNA. siRNA of 21 or 24 nt were independently binned using the scrolling window method (1000 nt window, 200 nt scroll). Only bins with reads from at least three replicates were included in the analysis. SAM-seq identified 146 differentially represented 21 nt siRNA bins (16 over- and 130 underrepresented in ago1-25) that were all more than twofold affected, with a FDR < 0.046 (Fig. 7A, left; Supplemental Table 4). SAM-seq also identified 276 differentially expressed 24 nt siRNA bins (114 over- and 162 underrepresented in ago1-25) that were all more than twofold affected, with a FDR < 0.035 (Fig. 7B, left; Supplemental Table 5). The majority (68.5%) of down-affected 21 nt siRNA bins corresponded to RDR6/DCL4-dependent siRNA, such as those from the PPR gene family (Axtell et al. 2006; Lu et al. 2006; Rajagopalan et al. 2006; Howell et al. 2007), that were identified previously based on different criteria (Fig. 7A, right). In fact, 90.5% of all PPR loci that were identified previously as RDR6/DCL4-dependent siRNA-generating loci were recognized as significantly affected in this analysis (Fig. 7A, right). The other 21 nt siRNA-generating loci corresponded to 31% of all other (non-PPR) such loci identified previously (Fig. 7A, right). Additionally, two of the underrepresented 21 nt bins represented heterogeneous small RNA derived from the MIR839 locus, and three corresponded to two of the novel or recently identified miRNA families identified above (MIR1886 and MIR2111) (Supplemental Table 3). Among the remaining over- and underrepresented 21 and 24 nt bins, 124 corresponded to transposable element loci, and 104 did not overlap any currently annotated features (Fig. 7A,B). The far greater number of underrepresented, compared to overrepresented, 21 nt siRNA bins is likely a reflection of direct or indirect dependence of 21 nt siRNA on AGO1 for stability or biogenesis.
FIGURE 7.

SAM-seq analysis of differentially expressed small RNA-containing bins between wild-type Col-0 and ago1-25. Analysis of (A) 21 and (B) 24 nt siRNA bins (1000 nt windows, 200 nt scroll). Scatter plots (left) of the mean (n = 3) RPM for small RNA-containing bins in Col-0 versus ago1-25. Red and green data points show statistically significant over- and underrepresented bins, respectively. Black data points and lines show the oligoribonucleotide standards fit with a linear model. Gray data points show nonsignificant bins. Pie charts (right) show the percentage of bins identified in each category. The outer arcs represent the percentage of PPR and non-PPR genes that were identified as significant 21 nt siRNA bins by SAM-seq, and that were identified in previous analyses of RDR6/DCL4-dependent siRNA clusters (Axtell et al. 2006; Howell et al. 2007).
CONCLUSIONS
In this study, we presented methods to generate, parse, map, quantify, standardize, and analyze large SBS-derived data sets, and demonstrated that SBS profiling of diverse small RNA populations can be done quantitatively and reproducibly. Although in this study we generated SBS data for Arabidopsis small RNA populations, these methods are not limited to plants. We introduce CASHX, a new mapping program developed to identify and quantify perfect genome hits for HTS data sets to a reference genome. Along with some other search programs (for example, Ning et al. 2001; Kahveci and Singh 2003), CASHX takes advantage of HASH database structure and cache memory to rapidly identify genome loci with matches to small RNA. Additionally, CASHX is not limited to small RNA data sets. It also works well with other types of SBS data, such as those resulting from mRNA transcript profiling or genomic resequencing (data not shown). Additionally, the CASHX pipeline is suitable for processing SAGE-like reads containing an adaptor linked to a cDNA sequence tag, as the CASHX parsing tool effectively separates the adaptor from tag sequence before alignment to the reference genome. CASHX can also work with longer reads, such as those produced by 454 pyrosequencing (Margulies et al. 2005) or traditional Sanger sequencing.
The application of SBS as a small RNA profiling tool is enabled by the high quantitative reproducibility between like samples using the Illumina platform. Even with the consistency between replicates, we show that the use of synthetic oligoribonucleotides as spike-in standards can facilitate more objective, quantitative comparisons of small RNA data sets from different samples, and should reduce problems associated with interpreting proportional representation differences. Although we did not multiplex samples in this analysis, variant (barcoded) synthetic standards should work equally well with mixed samples.
We also demonstrate the usefulness of adapting the microarray-based method SAM (Tusher et al. 2001) as a statistical method for analyzing replicate SBS data sets. By using SAM-seq, we were able to detect individual, differentially expressed small RNA or differentially expressed small RNA clusters with a low false discovery rate. The false negative rate, or sensitivity, of SAM-seq is limited by number of replicate samples. The applicability of SAM-seq to SBS data sets was shown through quantitative discrimination of known small RNA classes and subclasses in wild-type and small RNA-defective mutants. The utility was also demonstrated through discovery of new miRNA based on a quantitative threshold.
The profiling methods presented here are not without shortcomings. Most notably, low-abundance small RNA are difficult to analyze with high statistical confidence. Improvements in SBS or other ultra-high-throughput sequencing technology will undoubtedly lower the abundance threshold at which significant calls can be made. Additionally, the use of spike-in standards as objective references is limited by the relatively few data points compared to the number of experimental data points. Inclusion of additional standards will reduce this limitation.
MATERIALS AND METHODS
Plant materials and small RNA libraries
Mutant lines (Col-0 background) included dcl1-7 (Xie et al. 2005), dcl2-1 dcl3-1 dcl4-2 (Deleris et al. 2006), and ago1-25 (Morel et al. 2002). Small RNA libraries were constructed as in Kasschau et al. (2007), but with the following modifications. Spike-in control oligoribonucleotides were added to 100 μg of total RNA before amplicon preparation was started (see above). The 3′ adaptor was replaced with the miRNA cloning linker-1 (Integrated DNA Technologies, www.idtdna.com), which is 5′ adenylated to allow for ATP-independent ligation, and has a 3′ dideoxycytosine to prevent adaptor self ligation. The 5′ adaptor was replaced with an RNA oligonucleotide (5′-GUUCAGAGUUCUACAGUCCGACGAUC-3′). cDNA was amplified by PCR using Phusion High-Fidelity DNA Polymerase (New England Biolabs, www.neb.com), 5′ PCR primer (5′-AATGATACGGCGACCACCGACAGGTTCAGAGTTCTACAGTCCGA-3′), and 3′ PCR primer (5′-CAAGCAGAAGACGGCATACGAATTGATGGTGCCTACAG-3′). PCR primers contained sequences required for cluster generation on the Illumina Genome Analyzer system (Illumina, http://www.illumina.com). DNA amplicons were recovered from preparative 6% polyacrylamide gels by electro-transfer to DE81 paper, high-salt elution, and ethanol precipitation. DNA amplicons were then sequenced (36 cycles) using an Illumina 1G (Illumina, http://www.illumina.com). Amplicons (2.5 pmol) were added to each flow-cell lane. Sequencing primer (5′-GTTCAGAGTTCTACAGTCCGA-3′; 200 pmol/μL working stock) was prepared according to the Illumina protocol. A current, full protocol is available at http://jcclab.science.oregonstate.edu/?q=node/view/54596.
CASHX programs and scripts
The CASHX package is available at http://jcclab.science.oregonstate.edu/?q=node/view/54596. The suite is composed of Perl scripts and C++ programs that parse, quantify, and map reads and populate the resulting data into a MySQL database (Supplemental Fig. 1B). The mapping component of the CASHX package contains several programs and scripts for CASHX database formatting, database searching, and result processing. The program cashx_formatDB is used to convert the reference sequence from FASTA format to a HASH-indexed, 2-bit-per-base binary format. cashx_formatDB uses file space for work to minimize memory requirements, but as a result, is relatively slow. CASHX databases can also be created with the much faster program cashx_formatDBmem, which uses memory instead of file space (Supplemental Fig. 1B). Additional details about CASHX are provided as Supplementary Information.
Statistical analyses
All statistical analyses were done using R v2.7.0 (R Development Core Team 2008). For comparisons shown in Figure 2, an ordinary least squares (OLS) linear model (R function “lm,” “stats” package [R Development Core Team 2008]) was fitted to the miRNA families and 24 nt siRNA bins. The residual error analysis was done by plotting the absolute value of the residual error for each miRNA family or bin from the linear model versus the fitted y-values. The Goldfeld–Quandt and Breusch–Pagan tests (R functions “gqtest” and “bptest,” “lmtest” package (Zeileis and Hothorn 2002]) were used to check for significant heteroscedasticity.
For comparisons involving the spike-in standards shown in Figure 3, a standard curve was generated for each sample (wild-type Col-0, dcl1-7 mutant and dcl2-1 dcl3-1 dcl4-2 triple mutant) by fitting an OLS linear model to the log-transformed oligoribonucleotide standard reads versus the log-transformed pmol of standard. The standard curves were of the general form p = mr+b, where p is pmol of small RNA, r is reads and m and b are the slope and intercept of the model, respectively. The standard curves were p = 1.105r − 16.54 for wild-type Col-0, p = 1.049r − 15.85 for dcl1-7 mutant and p = 1.008r − 15.29 for dcl2-1 dcl3-1 dcl4-2 triple mutant. Plugging in log-transformed observed reads for r returns log-transformed pmol, and after back transformation returns an estimate of pmol of a particular small RNA in the original 100 μg of total RNA. Small RNA quantities were calculated for each canonical (previously annotated) miRNA and miRNA*, for each possible 21 nt tasiRNA from any of the eight TAS loci, and for 24 nt siRNA bins (1000 nt windows, 200 nt scroll). A Mann–Whitney–U/Wilcoxon rank sum test (R function “wilcox.test,” “stats” package [R Development Core Team 2008]) was used to evaluate whether there were small RNA population level differences between wild-type and either dcl1-7 mutant or dcl2-1 dcl3-1 dcl4-2 triple mutant samples.
All SAM-seq analyses were done by adapting the samr package for R (Tusher et al. 2001). Statistical power analysis was done using the samr “samr.assess.samplesize” function for two, three, four, or five replicates per sample and an expected difference of twofold. The SAM-seq analysis was done using the “samr” function with two class unpaired data, a standard test statistic, noncentering, and 1000 permutations. A delta value was chosen that kept the FDR less than 0.05 for each SAM-seq run. Delta tables for all SAM-seq analyses are available in supplemental information (Supplemental Tables 6–8). Miss rate tables for all SAM-seq analyses are also available in supplemental information (Supplemental Tables 2, 9, 10).
Small RNA data sets
SBS small RNA data sets used in this paper are available from Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo). GEO accessions are as follows: biological and technical replicates of wild-type Col-0 (GSE14694) and wild-type Col-0, dcl1-7 mutant and dcl2-1 dcl3-1 dcl4-2 triple mutant plants (GSE14695). Data for wild-type Col-0 and ago1-25 mutant flower tissue were previously described by Montgomery et al. 2008b and are available from GEO (GSE13605).
SUPPLEMENTAL MATERIAL
Supplemental material can be found at http://www.rnajournal.org.
ACKNOWLEDGMENTS
We thank Goretti Nguyen for assistance with small RNA library preparations, and Amy Shatswell for technical assistance. We also thank Detlef Weigel, Stefan Henz, Korbinian Schneeberger, and Stephan Ossowski for helpful discussions. This work was supported by grants from NSF (MCB-0618433), NIH (AI43288), and USDA-NRI (2006-35301-17420) to J.C.C., and a postdoctoral fellowship from the Damon Runyun Cancer Fund to S.D.G.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.1473809.
REFERENCES
- Allen E., Xie Z., Gustafson A.M., Sung G.H., Spatafora J.W., Carrington J.C. Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat. Genet. 2004;36:1282–1290. doi: 10.1038/ng1478. [DOI] [PubMed] [Google Scholar]
- Allison D.B. DNA microarrays and related genomics techniques: Designs, analysis, and interpretation of experiments. Chapman and Hall/CRC; Boca Raton, FL: 2006. [Google Scholar]
- Allison D.B., Cui X., Page G.P., Sabripour M. Microarray data analysis: From disarray to consolidation and consensus. Nat. Rev. Genet. 2006;7:55–65. doi: 10.1038/nrg1749. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Ambros V., Bartel B., Bartel D.P., Burge C.B., Carrington J.C., Chen X., Dreyfuss G., Eddy S.R., Griffiths-Jones S., Marshall M., et al. A uniform system for microRNA annotation. RNA. 2003;9:277–279. doi: 10.1261/rna.2183803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axtell M.J., Jan C., Rajagopalan R., Bartel D.P. A two-hit trigger for siRNA biogenesis in plants. Cell. 2006;127:565–577. doi: 10.1016/j.cell.2006.09.032. [DOI] [PubMed] [Google Scholar]
- Batista P.J., Ruby J.G., Claycomb J.M., Chiang R., Fahlgren N., Kasschau K.D., Chaves D.A., Gu W., Vasale J.J., Duan S., et al. PRG-1 and 21U-RNAs interact to form the piRNA complex required for fertility in C. elegans. Mol. Cell. 2008;31:67–78. doi: 10.1016/j.molcel.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley D.R. Whole-genome re-sequencing. Curr. Opin. Genet. Dev. 2006;16:545–552. doi: 10.1016/j.gde.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Brennecke J., Aravin A.A., Stark A., Dus M., Kellis M., Sachidanandam R., Hannon G.J. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 2007;128:1089–1103. doi: 10.1016/j.cell.2007.01.043. [DOI] [PubMed] [Google Scholar]
- Chapman E.J., Carrington J.C. Specialization and evolution of endogenous small RNA pathways. Nat. Rev. Genet. 2007;8:884–896. doi: 10.1038/nrg2179. [DOI] [PubMed] [Google Scholar]
- Czech B., Malone C.D., Zhou R., Stark A., Schlingeheyde C., Dus M., Perrimon N., Kellis M., Wohlschlegel J.A., Sachidanandam R., et al. An endogenous small interfering RNA pathway in Drosophila. Nature. 2008;453:798–802. doi: 10.1038/nature07007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleris A., Gallego-Bartolome J., Bao J., Kasschau K.D., Carrington J.C., Voinnet O. Hierarchical action and inhibition of plant Dicer-like proteins in antiviral defense. Science. 2006;313:68–71. doi: 10.1126/science.1128214. [DOI] [PubMed] [Google Scholar]
- Faehnle C.R., Joshua-Tor L. Argonautes confront new small RNAs. Curr. Opin. Chem. Biol. 2007;11:569–577. doi: 10.1016/j.cbpa.2007.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J., Tam P., Woude G.V., Ren Y. Normalization and analysis of cDNA microarrays using within-array replications applied to neuroblastoma cell response to a cytokine. Proc. Natl. Acad. Sci. 2004;101:1135–1140. doi: 10.1073/pnas.0307557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farazi T.A., Juranek S.A., Tuschl T. The growing catalog of small RNAs and their association with distinct Argonaute/Piwi family members. Development. 2008;135:1201–1214. doi: 10.1242/dev.005629. [DOI] [PubMed] [Google Scholar]
- German M.A., Pillay M., Jeong D.H., Hetawal A., Luo S., Janardhanan P., Kannan V., Rymarquis L.A., Nobuta K., German R., et al. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat. Biotechnol. 2008;26:941–946. doi: 10.1038/nbt1417. [DOI] [PubMed] [Google Scholar]
- Ghildiyal M., Seitz H., Horwich M.D., Li C., Du T., Lee S., Xu J., Kittler E.L., Zapp M.L., Weng Z., et al. Endogenous siRNAs derived from transposons and mRNAs in Drosophila somatic cells. Science. 2008;320:1077–1081. doi: 10.1126/science.1157396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard A., Sachidanandam R., Hannon G.J., Carmell M.A. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006;442:199–202. doi: 10.1038/nature04917. [DOI] [PubMed] [Google Scholar]
- Gregory B.D., O'Malley R.C., Lister R., Urich M.A., Tonti-Filippini J., Chen H., Millar A.H., Ecker J.R. A link between RNA metabolism and silencing affecting Arabidopsis development. Dev. Cell. 2008;14:854–866. doi: 10.1016/j.devcel.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Henderson I.R., Zhang X., Lu C., Johnson L., Meyers B.C., Green P.J., Jacobsen S.E. Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nat. Genet. 2006;38:721–725. doi: 10.1038/ng1804. [DOI] [PubMed] [Google Scholar]
- Howell M.D., Fahlgren N., Chapman E.J., Cumbie J.S., Sullivan C.M., Givan S.A., Kasschau K.D., Carrington J.C. Genome-wide analysis of the RNA-DEPENDENT RNA POLYMERASE6/DICER-LIKE4 pathway in Arabidopsis reveals dependency on miRNA- and tasiRNA-directed targeting. Plant Cell. 2007;19:926–942. doi: 10.1105/tpc.107.050062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M., Zaretskaya I., Raytselis Y., Merezhuk Y., McGinnis S., Madden T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008;36:W5–W9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahveci T., Singh A. MAP: Searching large genome databases. Pac. Symp. Biocomput. 2003;8:303–314. [PubMed] [Google Scholar]
- Kasschau K.D., Fahlgren N., Chapman E.J., Sullivan C.M., Cumbie J.S., Givan S.A., Carrington J.C. Genome-wide profiling and analysis of Arabidopsis siRNAs. PLoS Biol. 2007;5:e57. doi: 10.1371/journal.pbio.0050057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura Y., Saito K., Kin T., Ono Y., Asai K., Sunohara T., Okada T.N., Siomi M.C., Siomi H. Drosophila endogenous small RNAs bind to Argonaute 2 in somatic cells. Nature. 2008;453:793–797. doi: 10.1038/nature06938. [DOI] [PubMed] [Google Scholar]
- Kent W.J. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klattenhoff C., Theurkauf W. Biogenesis and germline functions of piRNAs. Development. 2008;135:3–9. doi: 10.1242/dev.006486. [DOI] [PubMed] [Google Scholar]
- Li R., Li Y., Kristiansen K., Wang J. SOAP: Short oligonucleotide alignment program. Bioinformatics. 2008;24:713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- Lister R., O'Malley R.C., Tonti-Filippini J., Gregory B.D., Berry C.C., Millar A.H., Ecker J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C., Tej S.S., Luo S., Haudenschild C.D., Meyers B.C., Green P.J. Elucidation of the small RNA component of the transcriptome. Science. 2005;309:1567–1569. doi: 10.1126/science.1114112. [DOI] [PubMed] [Google Scholar]
- Lu C., Kulkarni K., Souret F.F., Muthuvalliappan R., Tej S.S., Poethig R.S., Henderson I.R., Jacobsen S.E., Wang W., Green P.J., et al. MicroRNAs and other small RNAs enriched in the Arabidopsis RNA-dependent RNA polymerase-2 mutant. Genome Res. 2006;16:1276–1288. doi: 10.1101/gr.5530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M., Egholm M., Altman W.E., Attiya S., Bader J.S., Bemben L.A., Berka J., Braverman M.S., Chen Y.J., Chen Z., et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers B.C., Axtell M.J., Bartel B., Bartel D.P., Baulcombe D., Bowman J.L., Cao X., Carrington J.C., Chen X., Green P.J., et al. Criteria for annotation of plant microRNAs. Plant Cell. 2008;20:3186–3190. doi: 10.1105/tpc.108.064311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi S., Cai T., Hu Y., Chen Y., Hodges E., Ni F., Wu L., Li S., Zhou H., Long C., et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell. 2008;133:116–127. doi: 10.1016/j.cell.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery D.C., Peck E.A., Vining G.G. Introduction to linear regression analysis. Wiley-Interscience; Hoboken, N.J: 2006. [Google Scholar]
- Montgomery T.A., Howell M.D., Cuperus J.T., Li D., Hansen J.E., Alexander A.L., Chapman E.J., Fahlgren N., Allen E., Carrington J.C. Specificity of ARGONAUTE7-miR390 interaction and dual functionality in TAS3 trans-acting siRNA formation. Cell. 2008a;133:128–141. doi: 10.1016/j.cell.2008.02.033. [DOI] [PubMed] [Google Scholar]
- Montgomery T.A., Yoo S.J., Fahlgren N., Gilbert S.D., Howell M.D., Sullivan C.M., Alexander A., Nguyen G., Allen E., Ahn J.H., et al. AGO1-miR173 complex initiates phased siRNA formation in plants. Proc. Natl. Acad. Sci. 2008b;105:20055–20062. doi: 10.1073/pnas.0810241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel J.B., Godon C., Mourrain P., Beclin C., Boutet S., Feuerbach F., Proux F., Vaucheret H. Fertile hypomorphic ARGONAUTE (ago1) mutants impaired in post-transcriptional gene silencing and virus resistance. Plant Cell. 2002;14:629–639. doi: 10.1105/tpc.010358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning Z., Cox A.J., Mullikin J.C. SSAHA: A fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K., Chung W.J., Ruby J.G., Guo H., Bartel D.P., Lai E.C. The Drosophila hairpin RNA pathway generates endogenous short interfering RNAs. Nature. 2008;453:803–806. doi: 10.1038/nature07015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak J., Fire A. Distinct populations of primary and secondary effectors during RNAi in C. elegans. Science. 2007;315:241–244. doi: 10.1126/science.1132839. [DOI] [PubMed] [Google Scholar]
- Park W., Li J., Song R., Messing J., Chen X. CARPEL FACTORY, a Dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr. Biol. 2002;12:1484–1495. doi: 10.1016/s0960-9822(02)01017-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters L., Meister G. Argonaute proteins: Mediators of RNA silencing. Mol. Cell. 2007;26:611–623. doi: 10.1016/j.molcel.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Pfeffer S., Sewer A., Lagos-Quintana M., Sheridan R., Sander C., Grasser F.A., van Dyk L.F., Ho C.K., Shuman S., Chien M., et al. Identification of microRNAs of the herpesvirus family. Nat. Methods. 2005;2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- Qi Y., He X., Wang X.J., Kohany O., Jurka J., Hannon G.J. Distinct catalytic and noncatalytic roles of ARGONAUTE4 in RNA-directed DNA methylation. Nature. 2006;443:1008–1012. doi: 10.1038/nature05198. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2008. [Google Scholar]
- Rajagopalan R., Vaucheret H., Trejo J., Bartel D.P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes & Dev. 2006;20:3407–3425. doi: 10.1101/gad.1476406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart B.J., Weinstein E.G., Rhoades M.W., Bartel B., Bartel D.P. MicroRNAs in plants. Genes & Dev. 2002;16:1616–1626. doi: 10.1101/gad.1004402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby J.G., Jan C., Player C., Axtell M.J., Lee W., Nusbaum C., Ge H., Bartel D.P. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell. 2006;127:1193–1207. doi: 10.1016/j.cell.2006.10.040. [DOI] [PubMed] [Google Scholar]
- Sijen T., Steiner F.A., Thijssen K.L., Plasterk R.H. Secondary siRNAs result from unprimed RNA synthesis and form a distinct class. Science. 2007;315:244–247. doi: 10.1126/science.1136699. [DOI] [PubMed] [Google Scholar]
- Tam O.H., Aravin A.A., Stein P., Girard A., Murchison E.P., Cheloufi S., Hodges E., Anger M., Sachidanandam R., Schultz R.M., et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature. 2008;453:534–538. doi: 10.1038/nature06904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R. A simple method for assessing sample sizes in microarray experiments. BMC Bioinformatics. 2006;7:106. doi: 10.1186/1471-2105-7-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher V.G., Tibshirani R., Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin V.V., Sigova A., Li C., Seitz H., Gvozdev V., Zamore P.D. A distinct small RNA pathway silences selfish genetic elements in the germline. Science. 2006;313:320–324. doi: 10.1126/science.1129333. [DOI] [PubMed] [Google Scholar]
- Voinnet O. Use, tolerance and avoidance of amplified RNA silencing by plants. Trends Plant Sci. 2008;13:317–328. doi: 10.1016/j.tplants.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Wang G., Reinke V. A C. elegans Piwi, PRG-1, regulates 21U-RNAs during spermatogenesis. Curr. Biol. 2008;18:861–867. doi: 10.1016/j.cub.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T., Totoki Y., Toyoda A., Kaneda M., Kuramochi-Miyagawa S., Obata Y., Chiba H., Kohara Y., Kono T., Nakano T., et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature. 2008;453:539–543. doi: 10.1038/nature06908. [DOI] [PubMed] [Google Scholar]
- Xie Z., Allen E., Fahlgren N., Calamar A., Givan S.A., Carrington J.C. Expression of Arabidopsis MIRNA genes. Plant Physiol. 2005;138:2145–2154. doi: 10.1104/pp.105.062943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeileis A., Hothorn T. Diagnostic checking in regression relationships. R News. 2002;2:7–10. [Google Scholar]
- Zhang X. The epigenetic landscape of plants. Science. 2008;320:489–492. doi: 10.1126/science.1153996. [DOI] [PubMed] [Google Scholar]