

Abstract
Background and Purpose
Protein unfolding and aggregation are dominant early pathogenic events in neurons after brain ischemia. This study used a transient cerebral ischemia model to investigate whether overproduction of unfolded proteins after brain ischemia is a consequence of proteasome dysfunction.
Methods
Proteasome peptidase activity and proteasome subcellular redistribution and assembly were studied by peptidase activity assay, Western blot analysis, and size-exclusion chromatography.
Results
Proteasome peptidase activity, as determined with the peptide substrate succinyl-LLVY-7-amino-4-methylcoumarin, was moderately decreased, and the 26S proteasome was disassembled during the early period of reperfusion after transient brain ischemia. Furthermore, the proteasome subunits, particularly the 19S components, were deposited into the protein aggregate-containing fraction after an episode of transient cerebral ischemia.
Conclusions
These results clearly demonstrate that after an episode of brain ischemia, proteasomes are disassembled and aggregated and thus fail to function normally. Deposition of proteasomes into protein aggregates may also indicate that proteasomes attempt to degrade ubiquitin-conjugated proteins (ubiproteins) overproduced after brain ischemia. However, ubiproteins are too numerous to be degraded and trap some of the proteasomes into their aggregates after brain ischemia.
Keywords: brain ischemia/reperfusion, delayed neuronal death, protein aggregation, protein misfolding, proteosomes, ubiquitin, size-exclusion chromatography
The ubiquitin-proteasome pathway represents the chief cellular proteolytic machinery responsible for the degradation of abnormal proteins. In this pathway, abnormal proteins to be degraded are first conjugated by polyubiquitin chains and then degraded by the proteasomes. Protein ubiquitination involves several enzymatic reactions that ligate ubiquitin to its substrate proteins via isopeptide bonds. The 26S proteasome is the active form of a large, multisubunit protease complex consisting of a 20S proteolytic core and 2 19S regulatory caps on each end.1 An insufficient proteasome degradation capability to cope with overproduced abnormal proteins has been implicated in numerous neuropathologic conditions.2
Neuronal death after a brief episode of brain ischemia does not occur immediately but takes place after 2 to 3 days of reperfusion, the so-called delayed neuronal death.3 Delayed neuronal death has been intensively studied because this type of neuronal death provides a window of opportunity for studying ongoing molecular processes before neuronal death after brain ischemia. During the delayed period, cerebral blood flow, cellular ATP, and ionic homeostasis gradually recover, and most neurons destined to die appear normal under light microscopy (LM).4 However, under electron microscopy (EM), a dominant pathologic event is the continuous accumulation of ubiquitin-conjugated protein (ubiprotein) aggregates, which takes place mainly in vulnerable ischemic neurons from the onset of reperfusion onward until delayed neuronal death occurs after 2 to 3 days of reperfusion.5–9
To study whether overproduction of ubiproteins after brain ischemia is due to proteasome dysfunction, we investigated proteasome activity, disassembly, and deposition into protein aggregates after brain ischemia. This study suggests that unfolded proteins are too numerous to be degraded by proteasomes, but they trap the “escorting” proteasomes into their aggregates after brain ischemia.
Materials and Methods
Ischemia Model
The 2-vessel occlusion (2VO) ischemia model in rats was used in this study, according to the method of Smith et al.10 All experimental procedures were approved by the Animal Care and Use Committee at the University of Miami and were performed in compliance with National Institutes of Health guidelines on the ethical use of animals. In brief, male Wistar rats (250 to 300 g) were fasted overnight. Anesthesia was induced with halothane. Catheters were inserted into a femoral and a tail artery to allow blood sampling, arterial blood pressure recording, and drug infusion. Blood gases were measured and adjusted to PaO2 >90 mm Hg, PaCO2 35 to 45 mm Hg, and pH 7.35 to 7.45 before induction of ischemia and 15 minutes after ischemia. Brain ischemia was induced by withdrawing blood from the femoral artery catheter to produce a mean arterial blood pressure of 50 mm Hg, followed by clamping of both carotid arteries for 20 minutes. Mean arterial blood pressure was maintained at 50 mm Hg during the ischemic period by withdrawing or infusing blood through the femoral artery catheter. At the end of ischemia, the clamps were removed, halothane was discontinued, and all wounds were sutured. Brain temperature was maintained at 37°C before, during, and after ischemia. Brains were collected at the end of 0.5, 4, 24, and 72 hours of reperfusion. Each experimental group consisted of 4 rats. For biochemical studies, brains were frozen in situ with LN2.11 For EM, rats were perfusion-fixed with 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 mol/L cacodylate buffer.
EM and LM
Tissue sections were stained by the conventional osmium-uranyl-lead method for EM and with celestine blue and acid fuchsin for LM.8 In brief, coronal brain sections were cut consecutively with a vibratome at a thickness of 150 μm for EM and at 30 μm for LM. The EM brain sections were postfixed with 4% glutaraldehyde for 1 hour and then with 1% OsO4 for 2 hours in 0.1 mol/L cacodylate buffer (pH 7.4). EM brain sections were stained with 1% aqueous uranyl acetate overnight, dehydrated in an ascending series of ethanols followed by dry acetone, and then embedded in Durcopan ACM. Ultrathin sections (0.1 μm) were cut, stained with 3% lead citrate, and examined with a Zeiss EM (Germany). The vibratome brain sections (30 μm) were stained with celestine blue and acid fuchsin and examined by LM. Neuronal histopathology was determined according to the method of Smith et al.10
Isolation of Proteasomes by Size-Exclusion Chromatography
The dorsal neocortical tissues from frozen coronal brain sections were dissected and then weighted in a −12°C glove box according to a method described previously.5 The neocortical tissues were homogenized with a Dounce homogenizer (50 strokes) in 10 volumes of ice-cold extraction buffer containing 20% glycerol, 10 mmol/L Tris-base/HCl, pH 7.5, 1 mmol/L EDTA, 4 mmol/L dithiothreitol, and 2 mmol/L ATP. Homogenates were centrifuged at 17 000g at 4°C for 10 minutes to obtain pellet (often referred to as P1+P2) and supernatant (S2) fractions.7 The pellet fractions were suspended in extraction buffer. The protein contents in the S2 fractions were measured and adjusted to 4 mg/mL. Size-exclusion chromatographic separation of proteasome subcomplexes was carried out with a fast protein liquid chromatography (FPLC) system on a Superose 6 column (Pharmacia, Uppsala, Sweden). The column was equilibrated with the same extraction buffer at a flow rate of 0.2 mL/min. The supernatant (S2) fraction was passed through a 0.2-μm filter and loaded into the FPLC sample loop in a volume of 400 μL. The proteasomal fractions were eluted with extraction buffer at a flow rate of 0.2 mL/min. Absorbance at the UV wavelength of 280 nm in each fraction was monitored. Forty 0.6-mL fractions were collected. In separate experiments, the molecular size markers albumin (66 kDa, 25 mg/mL), alcohol dehydrogenase (150 kDa, 12.5 mg/mL), apoferritin (443 kDa, 25 mg/mL), thyroglobulin (670 kDa, 20 mg/mL), and dextran blue (2000 kDa, 5 mg/mL) (Sigma-Aldrich, St. Louis, Mo) were loaded onto the FPLC column for calculation of the molecular sizes of the proteasome complexes. Aliquots of all fractions (20 μL each) were assayed for proteasome peptidase activity with the succinyl-LLVY-7-amino-4-methylcoumarin (AMC) substrate. Aliquots of 60 μL of each fraction were also analyzed by Western blotting.
Proteasome Peptidase Activity
Ten microliters (1 μg/μL) of each freshly made supernatant (S2) or pellet (P1+P2) fraction was incubated in a 96-well plate at 37°C for 30 minutes with 10 μL of 300 μmol/L of succinyl-LLVY-AMC substrate and 85 μL of assay buffer (20 mmol/L Tris-HCl, pH 7.5, and 20% glycerol, with or without 30 μmol/L lactacystin). For proteasome peptidase activity assay of the FPLC-eluted fractions (see previous sections), 20-μL aliquots of all chromatographic fractions were incubated in a 96-well plate at 37°C for 30 minutes with 10 μL (300 μmol/L) of succinyl-LLVY-AMC and 75 μL of assay buffer containing 50 mmol/L Tris-HCl, pH 7.5, 1 mmol/L dithiothreitol, 5 mmol/L MgCl2, 10 μg/mL ovalbumin, and a freshly added ATP-regenerating system (2 mmol/L ATP, 20 mmol/L phosphocreatine, and 200 μg/mL creatine phosphokinase). Release of fluorescent AMC was measured with a spectrofluorometer (Perkin-Elmer Life and Analytical Sciences, Inc, Wellesley, Mass) at 440 nm with an excitation wavelength of 380 nm.
Subcellular Fractionation
The dorsal neocortical tissue (≈50 mg) was homogenized with a Dounce homogenizer (50 strokes) in 8 volumes of ice-cold homogenization buffer containing 50 mmol/L Tris-HCl, pH7.5, 20 μmol/L ATP, 5 mmol/L MgCl2, 1 mmol/L dithiothreitol, and 20% glycerol. The homogenate was centrifuged at 17 000g for 10 minutes at 4°C to obtain pellet (P1+P2) and supernatant (S2) fractions. The supernatant fraction (S2) was further centrifuged at 165 000g for 60 minutes at 4°C to obtain cytosolic (S3) and microsomal (P3) fractions. The pellet (P1+P2) fractions were suspended in homogenization buffer containing 1% Triton-X100 (TX) and 400 mmol/L KCl, sonicated 3 times each for 5 seconds, washed on a shaker for 1 hour at 4°C, and then centrifuged at 20 000g at 4°C for 10 minutes to obtain TX-soluble and -insoluble fractions. The protein concentration in subcellular fractions was determined by the microbicinchoninic acid method (Pierce, Rockford, Ill).
Western Blot Analysis
Western blot analysis was carried out by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) according to a method described previously.7 Samples for Western blotting contained 20 μg protein in the TX-insoluble and -soluble fractions, as well as in the cytosolic (S3) and microsomal (P3) fractions. The blot membranes were probed with antibodies against ubiquitin (Cell Signaling, Beverly, Mass), the proteasomal 20Sα-1 and 19S-S10B subunits (Affiniti Research Products Inc, Plymouth Meeting, Pa), and actin-β (Sigma, St. Louis, Mo). The blots were developed with an ECL detection system (Cell Signaling, Beverly, Mass) and then exposed to Kodak film to so that the protein bands on the films were not saturated. The optical densities of protein bands on the films were quantified by Kodak 1D gel analysis software.
Results
Protein Aggregation and Histopathology
We first evaluated protein aggregation and neuronal injury in the neocortical neurons after 20 minutes of ischemia. Under EM, neocortical neurons from control rats contained rosette-shaped polyribosomes (arrows) and a normal nucleus, endoplasmic reticulum, and mitochondria (Figure 1A, sham). After ischemia, the dominant ultrastructural changes were a progressive accumulation of large quantities of abnormal aggregates (Figure 1A, 24 hours, arrows) and intracellular vacuoles (Figure 1A, 24 hours, arrowheads). In contrast, under LM, no obvious morphological changes were seen before 24 hours of reperfusion, suggesting that ischemia-induced accumulation of protein aggregates and intracellular vacuoles is invisible by LM with acid fuchsin and celestine blue staining (Figure 1B). Twenty minutes of cerebral ischemia eventually led to delayed neuronal death in ≈20% to 40% of dorsolateral neocortical neurons after 72 hours of reperfusion, as demonstrated by EM (Figure 1A, 72 hours) and LM (Figure 1B, 72 hours). Delayed neuronal death also took place in almost all CA1 neurons and a few CA3 and DG neurons after 20 minutes of ischemia in this 2VO ischemia model (data not shown). Under EM, ischemic dead neocortical neurons at 72 hours of reperfusion showed clumped chromatin in the nucleus and amorphous organelles in the cytoplasm (Figure 1A, 72 hours, arrows). Under LM, ischemic dead neurons revealed a shrunken, acidophilic cytoplasm, as well as dark and shrunken or polygonal nuclei (Figure 1B, 72 hours, arrows). All of these results are consistent with previous studies.7,10
Figure 1.
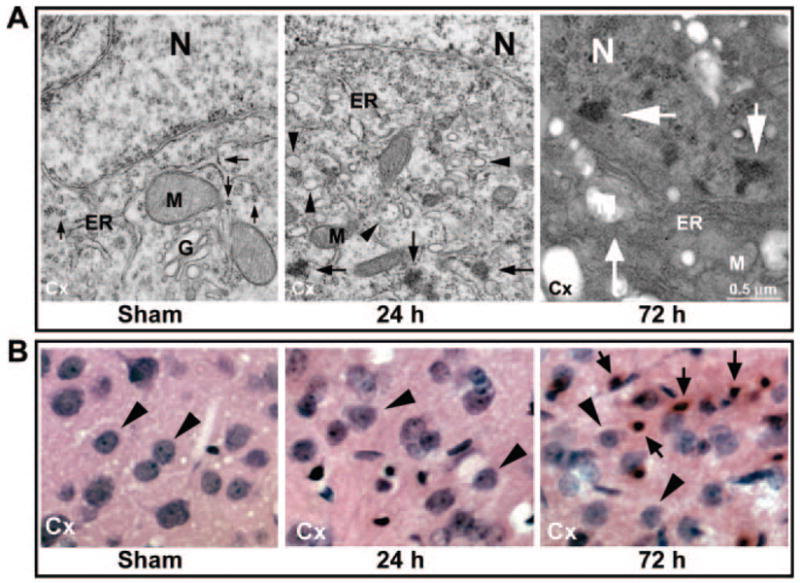
A, Electron photomicrographs of neocortical neurons from a sham-operated control rat and rats subjected to 20 minutes of ischemia followed by 24 and 72 hours of reperfusion. Sham control neurons contained normal polyribosomes (arrow) and cellular organelles. Large quantities of abnormal aggregates (arrows) and intracellular vacuoles (arrowheads) had accumulated in neurons at 24 hours of reperfusion. Ischemic dead neurons at 72 hours of reperfusion contained no viable organelles (small arrow) and clumped chromatin (large arrows). N indicates nucleus; ER, rough endoplasmic reticulum; M, mitochondria. B: Acid fuchsin– and celestine blue–stained neocortical neurons in brain sections adjacent to the brain sections used for the EM study (see Methods). Neuronal nuclei from controls and after 24 hours of reperfusion are round (arrowheads). Ischemic dead neurons at 72 hours of reperfusion have acidophilic cytoplasm, as well as dark and shrunken or polygonal nuclei (arrowheads).
Protein Ubiquitination After Brain Ischemia
When proteins become aggregated, their TX solubility decreases dramatically.12,13 Therefore, TX-insoluble pellet fractions were prepared for Western blot analysis of ubiprotein deposition into protein aggregate–containing fractions (Figure 2). The neocortical tissue samples were obtained from sham-operated rats and rats subjected to 20 minutes of 2VO cerebral ischemia followed by 0.5, 4, 24, and 72 hours of reperfusion. Ubiproteins were drastically and continuously accumulated in TX-insoluble pellet fractions from 0.5 to 24 hours of reperfusion (Figures 2A and 2B). This suggests that protein ubiquitination and aggregation were ongoing events during the reperfusion period before delayed neuronal death took place after brain ischemia. By comparison with the pellet fractions, ubiproteins were increased only slightly, if at all, in the tissue supernatant (S2) fractions after ischemia,14 consistent with the fact that ubiproteins were either aggregated or associated with subcellular structures.6 By 72 hours of reperfusion, ubiproteins were reduced drastically (Figures 2A and 2B), most probably because delayed neuronal death had occurred in some neocortical neurons. This agrees with the previous finding that ubiproteins are present only in living neurons after brain ischemia.5
Figure 2.

A and B, Immunoblots of ubi-proteins in TX-insoluble aggregates. Brain samples were obtained from neo-cortical tissues of sham-operated control rats and rats subjected to 20 minutes of cerebral ischemia followed by 0.5, 4, 24, and 72 hours of reperfusion. TX-insoluble fractions were immunoblotted with antibody to ubiproteins. A, Two separate samples derived from 2 different rats in each experimental group are shown. B, Changes in optical density of ubiproteins from 4 different individual rat samples were evaluated. Data are mean±SD (n=4). *P<0.05 between control and experimental conditions, 1-way ANOVA followed by Dunnett’s test.
Inhibition of Proteasome Peptidase Activity After Brain Ischemia
To study whether overproduction of ubiprotein aggregates in living neurons after brain ischemia is a consequence of proteasome inhibition, we analyzed proteasome peptidase activity after brain ischemia. Proteasome peptidase activity was determined with the succinyl-LLVY-AMC substrate in freshly made brain samples from the same rat tissues used for Western blotting of ubiproteins, as shown in Figure 2. Proteasome peptidase activity in the supernatant (S2 or tissue lysates; Figure 3, white bars) and pellets (Figure 3, black bars) was calculated by subtracting lactacystin-insensitive (or nonproteasome) peptidase activity from total peptidase activity. (Lactacystin is a specific inhibitor of the 26S proteasome.) The proteasome peptidase activity per microgram of sample protein was ≈60-fold more active in the tissue supernatant (Figure 3, white bars) than in the pellet fractions (Figure 3, black bars), suggesting that the proteasomes in tissue lysates were the major active forms. There was only an ≈20% to 30% reduction in proteasome activity from 30 minutes of reperfusion onward after transient cerebral ischemia (Figure 3).
Figure 3.

Proteasome peptidase activity in supernatant and pellet fractions after brain ischemia. Brain samples were obtained from the same sets of samples as those described in Figure 2. Four different individual rat samples in each group were assayed with the succinyl-LLVY-AMC substrate in the presence or absence of 30 μmol/L lactacystin. Proteasome activity was calculated by subtracting the lactacystin-insensitive (or nonproteasome) peptidase activity from total peptidase activity. White bars indicate the supernatant. Black bars indicate the pellet. Data are mean±SD (n=4). *P<0.05 between control and experimental conditions, 1-way ANOVA followed by Dunnett’s test.
Deposition of Proteasomal Subunits Into Protein Aggregates After Brain Ischemia
We next investigated whether accumulated ubiproteins trapped proteasomal components into protein aggregates by using the same tissue samples as those for Western blotting of ubiproteins shown in Figure 2. TX-insoluble and -soluble fractions, as well as cytosolic (S3) and microsomal (P3) fractions, were immunoblotted with antibodies against 20Sα-1 and 19S-S10B. Proteasomal subunit 20Sα-1 was highly accumulated in TX-insoluble fractions at 30 minutes of reperfusion but gradually declined thereafter (Figures 4A and 4B). In comparison, 19S-S10B 40/42-kDa subunits were continuously accumulated in TX-insoluble fractions, particularly in the later periods of reperfusion (Figures 4C and 4D). Correspondingly, these proteasomal subunits were decreased at 0.5 to 4 hours of reperfusion in the cytosolic (S3) fraction. The proteasome 20Sα-1 subunit rebounded to above control levels in the cytosolic fraction after 4 hours of reperfusion (Figures 4C and 4D). These proteasomal subunits were virtually unchanged in the microsomal fractions (P3) and TX-soluble fractions after brain ischemia (data not shown). Equal amounts of protein content among the samples were loaded onto the SDS-PAGE system for Western blot analysis. In addition, actin-β, used as an endogenous protein loading control, was not significantly altered among samples after brain ischemia (Figure 4). These results suggested that proteasome subunits, particularly the 19S components, were deposited into the protein aggregate–containing fractions as a result of transient ischemia.
Figure 4.

Immunoblots (A and C) and quantitative analysis (B and D) of preoteasome subunits and actin in TX-insoluble and cytosolic fractions after brain ischemia. Brain samples were obtained from the same sets of rats as those described in Figure 2. Proteasome subunits were immunoblotted with antibodies against 20Sα-1 (25 kDa) and 19S-S10B (40/42 kDa). Actin-β was used as an endogenous protein loading control. Molecular sizes are indicated by arrows. Data are mean±SD (n=4). *P<0.05 between control and experimental conditions, 1-way ANOVA followed by Dunnett’s test.
Disassembly of Proteasomes After Brain Ischemia
We next analyzed whether inhibition of proteasomal peptidase activity was due to proteasomal disassembly. The 26S, 20S, and 19S proteasomal subcomplex–containing fractions were separated by size-exclusion chromatography on a Superose 6 FPLC column. These fractions were then identified by proteasome peptidase activity (Figure 5A) and immunoblotting of proteasomal subunits (Figure 5B). Proteasome succinyl-LLVY-AMC–hydrolyzing peptidase activity was measured in chromatographic fractions, and the graphs were smoothed with the Microsoft Excel program to reflect the continuous nature of chromatography (Figure 5A). Proteasomal peptidase activity in sham-operated control brains showed the typical peaks: a main peak located at the ≈1500-kDa fractions (fractions 18 to 22), representing 26S proteasome activity, and a shoulder peak (Figure 5A, solid line).15 Proteasome peptidase activity declined in brains subjected to 20 minutes of brain ischemia followed by 30 minutes and 24 hours of reperfusion (Figure 5A, dashed lines). Immunoblotting of proteasomal subunits in the chromatographic fractions indicated that the main peak activity was situated in the 26S proteasome fractions because they contained both proteasomal 20S and 19S subunits, followed by 20S and 19S proteasomal peaks, respectively (Figure 5B, immunoblot panels). The antibody against 20Sα-1 also recognized an unknown protein band lower than the 20Sα-1 band in fractions 38 to 40 (Figure 5B, immunoblot panels, arrows).
Figure 5.

Assessment of 26S proteasomes and 20S and 19S proteasomal subcomplexes by size-exclusion chromatography. Tissue samples from the same rats as those described in Figure 2 were used. A, Succinyl-LLVY-AMC–hydrolyzing proteasome activity was assayed as described in Methods. B, The chromatographic fractions 1 to 48 were subjected to SDS-PAGE and immunoblotted with antibodies to 20Sα-1 and 19S-S10B. The chromatographic fractions 17 to 40 containing 20Sα-1 and 19S-S10B protein bands are shown. C: Quantitative analysis of immunoblot intensity ratio between the 26S proteasomal peak and either the 20S or the 19S proteasomal peaks. The sums of immunoblot intensities of the 26S chromatographic peak (fractions 18 to 22), 20S peak (fractions 23 to 28), and 19S peak (fractions 29 to 35) were measured with Kodak 1D image software. Data are mean±SD (n=3). *P<0.05 between control and experimental conditions, 1-way ANOVA followed by Dunnett’s test.
Quantitative analysis of the ratio of 20S α-1 subunit immunoblot intensities to the 26S proteasome (fractions 18 to 22) and 20S proteasomal subcomplex (fractions 23 to 28) chromatographic peaks suggested that 20Sα-1 was significantly shifted from the 26S peak toward the 20S peak at 30 minutes of reperfusion, whereas the ratio change was insignificant at 24 hours of reperfusion (Figure 5C). The 19S-S10B peak was shifted insignificantly from the 26S proteasome peak (fractions 18 to 24) to the 19S peak (fractions 29 to 35) at 30 minutes of reperfusion (Figure 5C). This was probably because of either a low 19S-S10B content in the 19S subcomplex or 19S-S10B deposition into the pellet fraction during the early period of reperfusion (Figure 4). These results indicated that 26S proteasomal 20Sα-1 was significantly disassembled into its subunits during the early period of reperfusion after brain ischemia.
Discussion
Forebrain neurons after transient global ischemia do not die immediately but do so several days after ischemia. The present study has demonstrated that ubiproteins are continuously accumulated and aggregated in postischemic neurons from the onset of reperfusion until delayed neuronal death takes place after 2 to 3 days of reperfusion. Irreversible accumulation of ubiprotein aggregates in neurons after ischemia may be attributable, in part, to proteasomal inhibition.
Inhibition of Proteasomal Peptidase Activity
Succinyl-LLVY-AMC–hydrolyzing proteasomal peptidase activity was examined previously in tissue lysates from the gerbil cortex after 10 minutes of forebrain ischemia.16 Proteasome peptidase activity was transiently decreased by ≈50% and then returned to control levels after 1 hour of reperfusion. Subsequently, it was reported that inhibition of proteasome peptidase activity failed to recover in the vulnerable CA1 region after 30 minutes of forebrain ischemia in gerbils.17 Asai et al17 further suggested that the ATP-dependent formation of the active 26S proteasome was severely impaired in ischemic vulnerable neurons. These previous studies examined proteasomal peptidase activity in tissue lysates (S2 fraction) but discarded the pellet fractions from analysis. As discussed later, the pellet fractions contained proteasome aggregates. The present study is in agreement with previous studies showing that proteasomal peptidase activity is moderately reduced in the supernatant fractions after 20 minutes of ischemia in the rat 2VO ischemia model. This study further demonstrates proteasomal deposition into detergent-insoluble protein aggregate-containing fractions (Figure 4) and proteasomal disassembly into proteasomal subunits in the supernatants (Figure 5).
The present study demonstrated that reduction of proteosome succinyl-LLVY-AMC–hydrolyzing peptidase activity was moderate, ≈20% to 30%, in neocortical tissue lysates during reperfusion (Figure 3). This is inconsistent with the drastic accumulation of ubiproteins in vivo, which indicates that proteasomes in vivo are incapable of degradation of ubiproteins in postischemic neurons (Figure 2). This discrepancy may be due to the fact that peptidase activity measured in vitro with a small peptide substrate does not represent the total activity of whole proteasome degradation of polyubiquitin chain-conjugated proteins (polyubiproteins) in vivo. The molecular weights of polyubiproteins are mostly >50 kDa (Figure 2). To degrade polyubiproteins, the 20S proteasomes must cooperate with the 19S proteasomes that govern polyubiprotein movement into and out of the 20S catalytic chambers.18 To enter the 20S proteasome catalytic chambers, polyubiproteins must be unfolded and then de-ubiquitinated by the 19S proteasomal subunit in an ATP-dependent manner. The peptidase activity, measured by the current and previous studies with the tetrapeptide succinyl-LLVY-AMC substrate, requires little unfolding and de-ubiquitination effort from the 19S proteasome subcomplex and thus, reflects only the peptidase portion of the 26S proteasome function. Therefore, despite the fact that 19S subunits were aggregated and disassembled, proteoasome peptidase activity was reduced only moderately.
The moderate reduction of peptidase activity after transient cerebral ischemia may also result from slight deposition of proteasome subunits into the TX-insoluble fraction, whereas the majority of proteasomes remain active. Therefore, a combination of accelerated production of ubiprotein aggregates and moderate reduction of proteasome activity may account for the drastic accumulation of ubiproteins in neocortical tissues in vivo after ischemia.
Proteasomal Disassembly and Aggregation
The 26S proteasome complex is formed by assembly of 20S and 19S subcomplexes in an ATP-dependent fashion.1 Neuronal ATP is completely depleted within a few minutes after the onset of brain ischemia and then gradually recovers to ≈80% of control levels at 30 minutes of reperfusion in the 2VO model.4 This may explain the results of the current study showing that 26S proteasomes are transiently disassembled during the early period of reperfusion. Because only the 26S proteasome is capable of degrading polyubiproteins in an ATP-dependent fashion,15 disassembly of the 26S proteasome may contribute to the accumulation of ubiproteins during the early postischemic phase.
Virtually all unfolded proteins in cells expose their toxic hydrophobic segments and must be escorted by molecular “chaperones.” We have previously demonstrated that molecular chaperones are irreversibly malfunctioning, leading to large amounts of unfolded proteins without chaperone protection in postischemic neurons.7–9 Without chaperone protection, unfolded polypeptides and their associated cellular machinery remain abnormal (toxic) and must be eliminated by the proteasome system after brain ischemia. However, ubiproteins after brain ischemia are too numerous to be degraded by the compromised proteasomes; they eventually aggregate after an episode of brain ischemia.
Proteasomal Dysfunction and Delayed Neuronal Death
The CA1 and DG tissue samples were not used in the present study because their amounts are insufficient for chromatographic preparation of proteasomal fractions. The microdissected CA1 or DG region from a frozen brain weighs ≈10 mg per rat (Hu et al, 2000, unpublished observations). To isolate proteasomes by chromatography, ≈30 mg is needed for each sample preparation. For this entire study, ≈3 sample preparations from each brain were prepared. The dorsal cortical tissue from each rat weighs ≈90 mg. For this reason, we used cortical tissue for sample preparation. To compensate for less damage in cortical neurons after transient cerebral ischemia, we used 20 minutes of ischemia, which leads to ≈20% to 40% of dorsal neocortical neurons that undergo delayed neuronal death. In our previous study, we have shown that EM-visible protein aggregates are present mostly in neurons that undergo delayed neuronal death after ischemia.6 Therefore, deposition of proteasome subunits into protein aggregates is likely to occur in neocortical neurons vulnerable to ischemia.
A key question is whether proteasome dysfunction and protein aggregation during the early period of reperfusion eventually lead to delayed neuronal death after brain ischemia. There is solid evidence demonstrating that defects in the ubiquitin-proteasome pathway induces protein aggregation and neuronal death virtually in all pathologic conditions.19,20 Conversely, enhancing proteasomal function by overexpression of proteasomal proteolytic subunits or ubiquitin ligases protects cells from lethal stress.21 We have previously proposed that protein unfolding and aggregation after brain ischemia reflect a quantitative imbalance between the amounts of toxic unfolded proteins and the capacity of protein quality control systems to eliminate them.7–9 Therefore, this study provides new evidence that ischemia-induced proteasome dysfunction contributes, at least in part, to delayed neuronal death after brain ischemia.
Acknowledgments
Sources of Funding
This work was supported by National Institutes of Health grants NS040407 and NS36810.
Footnotes
Disclosures
None.
References
- 1.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 2.Meredith SC. Protein denaturation and aggregation: cellular responses to denatured and aggregated proteins. Ann N Y Acad Sci. 2005;1066:181–221. doi: 10.1196/annals.1363.030. [DOI] [PubMed] [Google Scholar]
- 3.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 4.Siesjo BK, Elmer E, Janelidze S, Keep M, Kristian T, Ouyang YB, Uchino H. Role and mechanisms of secondary mitochondrial failure. Acta Neurochir Suppl. 1999;73:7–13. doi: 10.1007/978-3-7091-6391-7_2. [DOI] [PubMed] [Google Scholar]
- 5.Hu BR, Janelidze S, Ginsberg MD, Busto R, Perez-Pinzon M, Sick TJ, Siesjo BK, Liu CL. Protein aggregation after focal brain ischemia and reperfusion. J Cereb Blood Flow Metab. 2001;21:865–875. doi: 10.1097/00004647-200107000-00012. [DOI] [PubMed] [Google Scholar]
- 6.Hu BR, Martone ME, Jones YZ, Liu CL. Protein aggregation after transient cerebral ischemia. J Neurosci. 2000;20:3191–3199. doi: 10.1523/JNEUROSCI.20-09-03191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu CL, Chen S, Kamme F, Hu BR. Ischemic preconditioning prevents protein aggregation after transient cerebral ischemia. Neuroscience. 2005;134:69–80. doi: 10.1016/j.neuroscience.2005.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu CL, Ge P, Zhang F, Hu BR. Co-translational protein aggregation after transient cerebral ischemia. Neuroscience. 2005;134:1273–1284. doi: 10.1016/j.neuroscience.2005.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang F, Liu CL, Hu BR. Irreversible aggregation of protein synthesis machinery after focal brain ischemia. J Neurochem. 2006;98:102–112. doi: 10.1111/j.1471-4159.2006.03838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith ML, Auer RN, Siesjo BK. The density and distribution of ischemic brain injury in the rat following 2–10 min of forebrain ischemia. Acta Neuropathol (Berl) 1984;64:319–332. doi: 10.1007/BF00690397. [DOI] [PubMed] [Google Scholar]
- 11.Ponten U, Ratcheson RA, Siesjo BK. Metabolic changes in the brains of mice frozen in liquid nitrogen. J Neurochem. 1973;21:1211–1216. [PubMed] [Google Scholar]
- 12.Kabakov AE, Gabai VL. Stress-induced insolubilization of certain proteins in ascites tumor cells. Arch Biochem Biophys. 1994;309:247–253. doi: 10.1006/abbi.1994.1109. [DOI] [PubMed] [Google Scholar]
- 13.Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman D. Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci U S A. 1999;96:11404–11409. doi: 10.1073/pnas.96.20.11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu CL, Martone ME, Hu BR. Protein ubiquitination in postsynaptic densities after transient cerebral ischemia. J Cereb Blood Flow Metab. 2004;24:1219–1225. doi: 10.1097/01.WCB.0000136706.77918.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sawada MT, Morinaga C, Izumi K, Sawada H. The 26S proteasome assembly is regulated by a maturation-inducing hormone in starfish oocytes. Biochem Biophys Res Commun. 1999;254:338–344. doi: 10.1006/bbrc.1998.9948. [DOI] [PubMed] [Google Scholar]
- 16.Kamikubo T, Hayashi T. Changes in proteasome activity following transient ischemia. Neurochem Int. 1996;28:209–212. doi: 10.1016/0197-0186(95)00071-2. [DOI] [PubMed] [Google Scholar]
- 17.Asai A, Tanahashi N, Qiu JH, Saito N, Chi S, Kawahara N, Tanaka K, Kirino T. Selective proteasomal dysfunction in the hippocampal CA1 region after transient forebrain ischemia. J Cereb Blood Flow Metab. 2002;22:705–710. doi: 10.1097/00004647-200206000-00009. [DOI] [PubMed] [Google Scholar]
- 18.Shibatani T, Carlson EJ, Larabee F, McCormack AL, Fruh K, Skach WR. Global organization and function of mammalian cytosolic proteasome pools: implications for PA28 and 19S regulatory complexes. Mol Biol Cell. 2006;17:4962–4971. doi: 10.1091/mbc.E06-04-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lang-Rollin I, Vekrellis K, Wang Q, Rideout HJ, Stefanis L. Application of proteasomal inhibitors to mouse sympathetic neurons activates the intrinsic apoptotic pathway. J Neurochem. 2004;90:1511–1520. doi: 10.1111/j.1471-4159.2004.02684.x. [DOI] [PubMed] [Google Scholar]
- 20.Mytilineou C, McNaught KS, Shashidharan P, Yabut J, Baptiste RJ, Parnandi A, Olanow CW. Inhibition of proteasome activity sensitizes dopamine neurons to protein alterations and oxidative stress. J Neural Transm. 2004;111:1237–1251. doi: 10.1007/s00702-004-0167-2. [DOI] [PubMed] [Google Scholar]
- 21.Chondrogianni N, Tzavelas C, Pemberton AJ, Nezis IP, Rivett AJ, Gonos ES. Overexpression of proteasome β5 subunit increases amount of assembled proteasome and confers ameliorated response to oxidative stress and higher survival rates. J Biol Chem. 2005;280:11840–11850. doi: 10.1074/jbc.M413007200. [DOI] [PubMed] [Google Scholar]