

Abstract
Infection with HIV usually results in chronic activation of the immune system, with profound quantitative and qualitative changes in the T cell compartment. To better understand the mechanistic basis for T cell dysfunction and to discern whether such mechanisms are reversed after effective antiviral treatment, we analyzed changes in signaling pathways of human CD4+ and CD8+ T cells from 57 HIV-infected subjects in varying stages of disease progression and treatment, including long-term nonprogressors, progressors, and chronically infected subjects provided effective antiretroviral therapy (responders). A previously described PhosFlow method was adapted and optimized so that protein phosphorylation could be visualized in phenotypically defined subpopulations of CD4+ and CD8+ T cells (naive, memory, and effector) by flow cytometry. T cell signaling induced by TCR cross-linking, IL-2, or PMA/ionomycin was found to be blunted within all T cell subpopulations in those with progressive HIV disease compared with long-term nonprogressors and responders. Although alterations in cellular signaling correlated with levels of basal phosphorylation, viral load, and/or expression of programmed death-1, it was the level of basal phosphorylation that appeared to be the factor most dominantly associated with impaired signaling. Notably, provision of effective antiretroviral therapy was associated with a normalization of both basal phosphorylation levels and T cell signaling. These data, in aggregate, suggest that generalized dysfunction of the T cell compartment during progressive HIV disease may be in part dependent upon an increased basal level of phosphorylation, which itself may be due to the heightened state of immune activation found in advanced disease.
The T cell plays a major role in the cellular response to the HIV infection, inhibiting virus replication either through secretion of IFNs and other suppressive factors or by direct cytotoxicity (1-9). Even in the face of a vigorous T cell response to HIV, however, viral replication usually proceeds un-checked and disease progression occurs.
CD4+ T cells play an important role in immune responses, both by providing help to B cells and by facilitating the generation and activity of CD8+ CTLs (10). A major characteristic of HIV disease is the decline of CD4+ T cell numbers due to a shortened survival time and a failure to increase T cell production (11-15). In addition to the loss of numbers, CD4+ T cells have functional defects, reduced proliferative capacity, abnormal cytokine profiles, and defective responses to and production of IL-2 (16-20).
Changes in the number and function of CD4+ T cells alone, however, would not fully explain HIV disease progression. CD4+ T cells are critical for effective CD8+ T cell responses (10) and loss of numbers and function within the CD4+ T cell compartment might, in turn, have detrimental effects on CD8+ T cell compartment. By example, HIV-specific CD8+ T cells have been reported to express only low levels of perforin, a key mediator of cytolytic activity, even though they can still produce antiviral cytokines and chemokines (21-23). Defective cytolytic activity has been associated with a specific CD8+ T cell subpopulation, the less differentiated CD8+CD27+ T cell subset (22), suggesting an association between impaired T cell function with skewed maturation and an immature phenotype (24, 25). Finally, CD8+ T cells have lower levels of CD3ζ and CD28, features associated with defects in stimulation via the TCR (26, 27).
Multiple mechanisms might underlie impaired T cell function, including defects in differentiation (28, 29), impaired signaling via the TCR (30, 31), and down-modulation and/or modification of molecules critical for T cell signaling (26, 27, 32, 33). Signaling defects have also been found in rhesus macaques after experimental SIV infection (34, 35). However, cellular function and signaling properties are in large part dictated by the stage of T cell activation and/or differentiation (36-38), making it difficult to discern cell-specific defects when heterogeneous populations of cells are analyzed, e.g., using Western blot analysis to examine phosphorylation events in PBMCs.
The aim of this study was to determine whether signaling alterations exist in specific CD4+ and CD8+ T cells subpopulations during the course of HIV disease progression and treatment. We adapted and optimized a previously described flow cytometric assay (39-41) to visualize phosphorylation of known intermediates of cellular signaling pathways in defined T cell subpopulations. We demonstrate that signaling mediated by TCR cross-linking, IL-2, and PMA/ionomycin is blunted in cells from untreated subjects with progressive disease (progressors, PROGs)4 compared with long-term nonprogressors (LTNP) and to those who have been successfully provided antiretroviral treatment (responders, RESPs). These signaling alterations are not restricted to a specific T cell subpopulation and some are correlated with levels of basal phosphorylation of proteins involved in various T cell signaling cascades, viral load (VL), and/or expression of programmed death-1 (PD-1), a CD28 family member that negatively regulates T cell function in the context of HIV disease (42-46). Of these influences, a high level of basal phosphorylation was found to have the greatest impact on the magnitude of signaling changes to specific stimuli. Finally, altered signaling found in PROGs was observed to be reversible with antiretroviral treatment.
Materials and Methods
Study participants
HIV-infected people were recruited from the San Francisco Bay Area into the Study of the Consequences of the Protease Inhibitor Era. Samples of PBMCs for the current study were taken from three distinct groups: (i) LTNP with a CD4 T cell count > 500 cells/μl despite at least 10 years of untreated HIV infection, and a plasma VL < 2,000 copies/ml; (ii) PROGs with a CD4 T cell count < 200 cells/μl, plasma VL > 10,000 copies/ml, and no antiretroviral therapy (ART) at the time of blood sampling; and (iii) RESPs who were on a stable antiretroviral regimen, had an undetectable VL, and had a previous CD4 T cell count nadir < 200/μl, but at the time of sampling had a CD4 count > 500/μl. Analyses were conducted on archived PBMCs that had been viably frozen and stored at the University of California-San Francisco AIDS Specimen Bank. PBMCs from HIV-uninfected individual controls were isolated from buffy coats from whole blood (Stanford Blood Center) and viably frozen for subsequent analyses.
Measurements
Plasma HIV RNA levels were determined by the branched DNA assay (Quantiplex HIV RNA, version 3.0; Chiron Corporation). CD4 cell counts were determined by flow cytometry.
Cell culture and flow cytometric analyses
Before analysis and stimulation, frozen PBMCs were thawed in 15 ml RPMI 1640 cell culture medium (Mediatech) containing 5% FBS (Hy-Clone; RPMI+), washed in PBS containing 2% FBS (PBS+), and then rested at 5 × 106 cells/ml in RPMI+ at 37°C, 5% CO2, overnight. The following day, cells were washed with ice-cold PBS+ and transferred to a 96-well V-bottom plate. Each sample was stained for expression of the cell surface markers CD3, CD4, CD8, CD27, CD45RA, and PD-1. An amine-reactive dye (violet Live/Dead; Invitrogen) was used to stain dead cells. Expression levels of cell surface markers were measured in terms of median fluorescence intensity (MFI). To account for interassay variability, MFI-values of HIV-infected patients were normalized by subtraction of MFI-values of an HIV-uninfected standard control (ΔMFI), which was included in all experiments.
For signaling analysis in T cell subpopulations, cells were initially stained on ice with Abs detecting CD3, CD8, CD27, and Live/Dead cell markers. The anti-CD45RA Ab was not included in the initial cell surface stain, as this Ab gives reasonable staining results only after fixation/permeabilization of the cells. To activate the TCR complex, cells were first preincubated with biotin-conjugated anti-CD3 (clone HIT3a) and anti-CD28 Abs, either alone or in combination with either biotin-conjugated anti-CD4 or anti-CD8 CD8 Ab for 20 min on ice. Subsequently, cells were transferred to streptavidin in PBS+ at 37°C, thereby cross-linking and activating TCR-mediated signaling. To activate cytokine- or mitogen-induced signaling, cells were transferred to PBS+ containing IL-2, IL-4, or PMA/ionomycin at 37°C. Signaling was arrested after 15 min by immediate fixation, adding 4% paraformaldehyde to a final concentration of 2%. After 20 min of fixation and subsequent wash, cells were permeabilized in 70% ice-cold methanol for 20 min on ice. Cells were washed and stained with an Ab mixture containing phospho- (p-) and CD45RA-specific Abs for 60 min on ice. Before analysis, cells were washed and resuspended in PBS+ containing 0.05% formaldehyde. Unstimulated control cells underwent the same manipulations. Cells were analyzed on a customized LSR II Flow Cytometer (BD Biosciences). Analysis of data was performed using FlowJo (Tree Star). Fold-changes in phosphorylation were calculated as the ratio of MFI of stimulated cells over unstimulated cells.
Calcium flux response after TCR cross-linking was assessed with the fluorescent calcium indicator, Indo-1 AM. Calcium release was measured by flow cytometry over time by the change in emission spectrum from blue to violet. Indo-1 is excited in the UV and fluoresces at different wavelengths depending on whether it is bound to calcium (∼420 nm) or free (∼510 nm). The ratio of these two wavelengths indicates changes in intracellular calcium concentration. TCR activation was induced by adding streptavidin during FACS acquisition to cross-link biotinylated Abs: anti-CD3 in combination with anti-CD4 or anti-CD8. The ionophore, ionomycin, was used as a positive control and levels before stimulation were used as negative control baseline level. An average of 2 × 106 total PBMCs were labeled for 30 min at 37°C with 2 μM Indo-1 AM, 0.02% pluronic F-127 in 2 ml of HBSS supplemented with 1% FBS, 1 mM CaCl2, and MgCl2 (HBSS Ca2+ buffer). Cells were then washed twice and resuspended in 500 μl of HBSS Ca2+ buffer containing biotinylated anti-CD3 and anti-CD8 or anti-CD4 Abs for 5 min at room temperature (RT). Cells were subsequently stained with a combination of anti-CD3, anti-CD8, and anti-CD4 Abs for 25 min at RT. Cells were washed twice and resuspended in 500 μl of HBSS Ca2+ buffer, then kept on ice until being warmed to RT before FACS acquisition on a FACSDiva flow cytometer. After 25 s of acquisition, 5 μl of 2 μM streptavidin solution or 5 μl of ionomycin at 0.1 mg/ml were added, and calcium release was measured during the remaining 3−5 min. FACS data were analyzed with the calcium flux platform from FlowJo software on live (Indo-1) CD3+CD8+CD4− T lymphocytes. No cross-blocking activity was observed between CD3-, CD4-, or CD8-biotinylated and -fluoresceinated murine mAbs with this combination.
Abs and reagents
The following Abs were used for detection of cell surface markers: CD3 (clone SP34−2, Alexa700-conjugated at a dilution 1/100, purchased from Invitrogen or clone UCHT1 from eBiosciences), CD4 (clone S3.5, PE-Cy7, 1/100; Invitrogen), CD8 (clone 3B5, PE-Cy5.5, 1/1000; Invitrogen), CD27 (clone 0323, 1/100, allophycocyanin-Alexa750; eBioscience), CD45RA (clone 2H4, ECD, 1/100; Beckman Coulter), and PD-1 (clone MIH4, PE). Dead cells were stained with a violet-fluorescent fixable Live/Dead amine-reactive dye (1/1000; Invitrogen). The following Abs (all BD Biosciences), either alone or in combination, were used at a dilution of 1/25 for TCR cross-linking: CD3-biotin (clone HIT3), CD4-biotin (clone RPA-T4), CD8-biotin (clone SK1), and CD28-biotin (clone CD28.2). Streptavidin (Sigma-Aldrich) was used at a final concentration of 80 μg/ml. The following p-specific Abs (all BD Biosciences) were used at a dilution of 1/20: Zap70 (phosho-tyrosine (pY)319/Syk pY352, clone 4, Alexa647-conjugated), lymphocyte specific kinase (Lck) (pY505, PE), linker for activation of T cells (Lat) (pY226; clone J96 −1238−58.93, Alexa488), ERK1/2 (pthreonine (pT)202/pY204, Alexa488), p38 (pT180/pY182, Alexa647), Akt (pT308, PE), Stat5 (pY694, PE), and Stat6 (pY641, Alexa647).
For stimulation, IL-2 (Sigma-Aldrich) was used at 100 ng/ml, IL-4 (R&D Systems) at 100 ng/ml, PMA (Sigma-Aldrich) at 100 ng/ml, and ionomycin calcium salt (Sigma-Aldrich) at 1 μg/ml. For fixation of cells, we used final concentration of 2% paraformaldehyde (Electron Microscopy Sciences; 15710). Cells were permeabilized with 70% methanol (Fisher Scientific). The fluorescent calcium indicator Indo-1 AM (Molecular Probes) was used at a final concentration of 2 μM.
Statistical analyses and heatmaps
To compare expression of cell surface markers, signaling, and levels of basal phosphorylation between groups, a nonparametric two-tailed Mann-Whitney U test for unpaired data sets was used. Two-tailed Spearman's rank correlation was used to analyze relationship between signaling and levels of VL, levels of basal phosphorylation, or expression of PD-1. Differences were statistically significant with a value of p < 0.05 (*, p < 0.05; **, p < 0.01; ***, p < 0.001). Statistical analyses were performed using Prism 4 (GraphPad). TIGR MeV v3.1 was used to create heatmaps (www.tm4.org/index.html).
Results
HIV-infected subjects and study design
This study included 57 HIV-infected individuals at distinct stages of disease progression, including chronically HIV-infected untreated LTNP (n = 20), untreated PROGs (n = 18), and RESPs (n = 19) with treatment-mediated viral suppression (Table I). The LTNP were selected on the basis of being infected for at least 10 years, having a CD4 T cell count > 500 cells/μl (median 770), having a plasma VL < 2000 copies/ml (median 114), and not having received ART. PROGs had a low CD4 count (<200 cells/μl; median 55), a high plasma VL (>10,000 copies/ml; median 65,864), and were not on treatment at the time of blood sampling. Antiretroviral “RESPs” to treatment were selected on the basis of having had a CD4 nadir of <200/μl, which had increased to over 500 cells/μl (median 703) at the time of sampling, and at the same time having a VL < 75 copies/ml. The median number of CD8+ T cells was slightly reduced (median 770) in HIV PROGs compared with LTNP (1146) and RESPs (1088). The median age and year of having been tested HIV positive were comparable in all groups. A majority of all participants were men.
Table I.
Characteristics of participantsa
| Characteristic | LTNP CD4 > 500/μl VL Typical < 2000 Copies/ml > 10 Years No Therapy n = 20 | PROG CD4 < 200/μl VL > 10,000 Copies/ml No Therapy n = 18 | RESP CD4 > 500/μl, Previously < 200/μl VL Undetectable on Therapy n = 19 |
|---|---|---|---|
| Median CD4 count, cells/μl (IQR) | 770 (685 to 955) | 55 (22 to 124) | 703 (596 to 836) |
| Median CD8 count, cells/μl (IQR) | 1146 (935 to 1693) | 770 (424 to 973) | 1088 (920 to 1476) |
| Median Plasma HIV RNA level, copies/ml (IQR) | 114 (<75 to 1438) | 65864 (36702 to 154765) | <75 |
| Median age, years (IQR) | 48 (44 to 52) | 45 (42 to 51) | 49 (42 to 60) |
| Median year 1st HIV+ test (IQR) | 1988 (1986 to 1990) | 1990 (1986 to 1993) | 1989 (1987 to 1992) |
| Ethnicity percentage B/AA, W/C, A, M, H/L, PI | 50, 35, 5, 10, 0, 0 | 17, 44, 0, 6, 28, 6 | 21, 58, 5, 0, 11, 5 |
| Female gender | 25% | 11.1% | 21.1% |
IQR, Interquartile range; B/AA, Black/African-American; W/C, White/Caucasian; A, Asian; M, Mixed; H/L, Hispanic/Latino; PI, Pacific Islander.
Phenotypic analysis of PBMCs
PBMCs from all participants were analyzed for expression levels of the cell surface markers CD3, CD4, and CD8, and the maturation markers CD45RA and CD27 were used to subdivide CD3+CD4+ and CD3+CD8+ T cells into subpopulations of naive (CD45RA+CD27+), memory (CD45RA−CD27+), memory-effector (CD45RA−CD27−), and effector (CD45RA+CD27−) T cells (see Fig. 1A for gating strategy). As expected, and when compared with LTNP and RESPs, those with progressive disease had an inverted CD4:CD8 ratio with a very low percentage of CD4+ T cells (mean 7%, 12.9% SD) within their CD3+ T cell pool (Fig. 1B, left), a higher frequency of circulating memory/memory-effector T cells, and a lower frequency of naive cells (Fig. 1B, middle for CD4 and right for CD8). Interestingly, LTNP had a significantly higher representation of effector CD8+ T cells than the other two groups, whereas RESPs had more naive CD8+ T cells than the other groups ( p < 0.05 for each pairwise comparison) (Fig. 1B, right). Under the conditions used for staining in these experiments, the cell surface expression levels of CD3, CD4, and CD8 (measured in terms of MFI) were found to be comparable in all T cell subpopulations in each of the three groups studied (Fig. 1C).
FIGURE 1.

A, Representative flow cytometric plots of staining and gating strategy of fixed/permeabilized cells used in signaling analyses by PhosFlow. Lymphocytes and singlets were identified in forward and sideward scatter dot plots. Dead cells were excluded by amine stain, and live cells were gated for expression of CD3. The CD3+CD4+ and CD3+CD8+ T cells were further separated by expression of CD45RA and CD27 to identify naive (CD45RA+CD27+), memory (CD45RA−CD27+), memory-effector (CD45RA−CD27−), and effector (CD45RA+CD27−) T cells as indicated. B, Mean frequencies of CD4+ and CD8+ T cells within the CD3+ compartment (left) and of naive, memory, memory-effector, and effector T cells within CD4+ (middle) and CD8+ T cells (right) in the three HIV-infected groups of LTNP, PROGs, and RESPs. Error bars indicate SD. C, Cell surface expression levels of CD3 on CD3+ T cells (left) and CD4 or CD8 on naive, memory, memory-effector, and effector CD4+ or CD8+ subpopulations (middle and left, respectively) from three HIV-infected groups as indicated. Expression levels of cell surface markers were measured as MFI and normalized (ΔMFI) to an HIV-uninfected standard control, which was included in all experiments. For the box-and-whisker graph, the lines in the boxes represent median values, the boxes range from the 25th to 75th percentiles, and the error bars indicate the lowest and highest values. Groups in B and C were compared using the nonparametric two-tailed Mann-Whitney U test; statistically significant differences are indicated by the lines below the plots. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Simultaneous analysis of cellular phenotype and intracellular signaling events within heterogeneous cell subpopulations by flow cytometry (PhosFlow)
To study cell signaling in defined subpopulations of T cells, the PhosFlow assay (39-41) was adapted so that it could be used reliably to analyze human PBMC. We systematically tested a series of different protocols and conditions for fixation, permeabilization, activation, and cell staining so that the phenotypic markers used to demarcate specific T cell subpopulations could be visualized at the same time as intracellular p-proteins (data not shown).
Different cell culture conditions, e.g., resting time, cell density, serum concentration, and origin (e.g., human vs bovine), did not greatly affect levels of basal phosphorylation or of stimulation. However, resting cells overnight, as compared with resting cells for 90 min, resulted in stronger fold-changes in phosphorylation after stimulation. To establish optimal conditions for the analysis of TCR-mediated signaling, a protocol for cross-linking components of the TCR complex on the cell surface was developed, using combinations of biotinylated anti-CD3, -CD28, -CD4, and -CD8 Abs together with streptavidin (see below and Ref. 47). Cross-linking did not alter cell surface expression or detection of CD3, CD4, or CD8 (data not shown).
The optimized PhosFlow protocol for TCR-mediated signaling was validated by incubating heterogeneous cell subpopulations of PBMCs with or without a combination of biotinylated Abs binding CD3, CD28, and CD4 (CD3 + 28 + 4) or CD3, CD28, and CD8 (CD3 + 28 + 8) for 20 min on ice, followed by a cross-linking step with streptavidin at 37°C to activate the TCR. After 15 min, cells were fixed with paraformaldehyde, permeabilized with methanol, and stained with a fluorochrome-conjugated p-Zap70-specific Ab. Using multicolor flow cytometry, phosphorylation levels of Zap70 (p-Zap70) were then analyzed in CD4+ and CD8+ T cells. As expected, p-Zap70 was most evident in CD4+ T cells after cross-linking of CD3 + 28 + 4 (Fig. 2A, left). Incubation with anti-CD3 + 28 + 8 Abs still cross-links CD3 + 28 on CD4+ T cells and therefore induced a small change of p-Zap70 when compared with unstimulated control. Reciprocal results were obtained in CD8+ T cells, in which cross-linking of CD3 + 28 + 8 resulted in the highest levels of Zap70 phosphorylation (Fig. 2A, right). To better quantify and to visualize changes in levels of phosphorylation, the MFI of the p-specific signal was measured and fold-changes in phosphorylation were calculated as a ratio of MFI in stimulated vs unstimulated cells (Fig. 2B). To show that the TCR cross-linking protocol provides a functional response, Ca2+ influx was analyzed after TCR-linking and in comparison with ionomycin, a potent stimulator of calcium influx (Fig. 2C). Indeed, TCR cross-linking induced cellular Ca2+-influx, although not as strong as ionomycin, possibly reflecting a more physiological TCR-mediated stimulation. When used in the absence of cross-linking, the biotinylated or fluoresceinated Abs did not induce any detectable calcium release when added to unstained cells (data not shown). Moreover, cross-linking was specific for either CD4 or CD8 T cell populations, depending on whether anti-CD4 or anti-CD8 Abs, respectively, were used.
FIGURE 2.
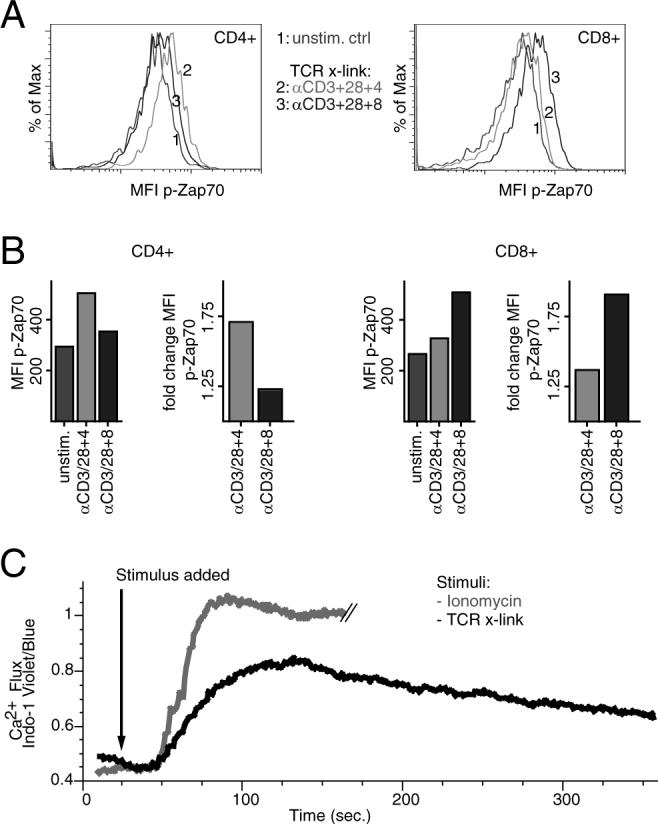
T cell-specific stimulation by TCR cross-linking induces specific signaling analysis, as analyzed by PhosFlow, and Ca2+-influx. A, Histograms of MFI of p-Zap70 in CD4+ (left) and CD8+ T cells (right). PMBCs were stained for expression of CD3, CD4, and CD8 and subsequently incubated with biotinylated Abs binding either CD3 + 28 + 4 or anti-CD3 + 28 + 8 or without Abs. TCR-mediated signaling was activated by cross-linking biotinylated Abs with streptavidin for 15 min. After fixation and permeabilization, cells were stained with an Ab detecting p-Zap70. Levels of p-Zap70 within CD4+ and CD8+ T cells were analyzed by flow cytometry. B, Bar graphs represent levels of p-Zap70 as quantified by MFI and fold-changes (stimulated over unstimulated) of MFI after specific TCR cross-linking, as indicated below, in CD4+ (left) and CD8+ T cells (right). C, TCR cross-linking induces cellular Ca2+-influx. PBMCs were incubated with biotinylated Abs binding CD3 + 8 before staining with a combination of anti-CD3, anti-CD8, and anti-CD4 Abs. The TCR signaling was activated by adding streptavidin, and Ca2+ influx was measured with the fluorescent calcium indicator, Indo-1. Calcium release was measured by flow cytometry over time by the change in emission spectrum from blue to violet (Indo-1 AM). Stimulation with the calcium ionophore, ionomycin, served as a positive control.
PhosFlow analysis of T cells from HIV-infected patients at different stages of disease progression
Using the PhosFlow protocol as described above, PBMCs from HIV-infected LTNP, PROGs, and RESPs were stained for cell surface phenotype, stimulated for 15 min, and tested with a panel of different p-specific Abs. Changes in signaling were detected as a fold-change in phosphorylation, comparing the MFI of selected phosphoproteins (including the TCR proximal kinases Lck, Zap70, and Lat, the downstream kinases ERK1/2 and p38, as well as Akt, a kinase linked to the costimulatory molecule CD28) in stimulated and unstimulated cells within different subpopulations of CD4+ and CD8+ T cells (except for CD4+ effector T cells, of which there were too few cells to analyze in most subjects). In response to TCR-mediated signaling, significant responses to stimulation and differences between HIV-infected groups were found in phosphorylation of Lck and Zap70 (Fig. 3A). Changes in phosphorylation were blunted in PROGs when compared with LTNP and RESPs. The blunted signaling responses did not seem to be restricted to or especially pronounced in specific CD4+ or CD8+ T cell subpopulations. Overall, changes in p-Lck tended to be more evident in the naive compartment (Fig. 3A, top), whereas those in p-Zap70 resided preferentially in more differentiated T cell sub-populations (Fig. 3A, bottom).
FIGURE 3.

Signaling is comparable between T cells from HIV-uninfected individuals and HIV-infected LTNP, but blunted in T cells from HIV PROGs. Comparing stimulated over unstimulated cells, fold-changes in phosphorylation were analyzed in naive, memory, memory-effector, and effector subpopulations of CD4+ (no effector T cells analyzed) and CD8+ T cells. Protein phosphorylation after specific stimulation was analyzed between (A–C) HIV-infected individuals and (D–F) LTNP and HIV-uninfected individuals for (A and D) Lck and Zap70 after TCR stimulation, (B and E) Stat5 after stimulation with IL-2, and (C and F) ERK1/2 and p38 after stimulation with PMA/ionomycin. The groups were compared using the nonparametric two-tailed Mann-Whitney U test; statistically significant differences are indicated by the lines below the plots. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Signaling and functional responses to stimulation with IL-2 have previously been described as defective in HIV-infected subjects (16 -20). We accordingly tested signaling responses to cytokine signaling in subpopulations by PhosFlow, monitoring p-Stat5 after IL-2 stimulation and p-Stat6 and p-p38 after IL-4 stimulation. When compared with LTNP and RESPs, cells (particularly CD8+ T cells) from PROGs again had blunted responses to IL-2 (Fig. 3B), and these altered responses were not restricted or specific to a given T cell subpopulation. We did not see significant differences in CD25 expression in different CD4+ or CD8+ T cell subpopulations between the three groups (data not shown). Therefore, it is unlikely that expression levels of CD25, the α-chain of the tripartite high affinity IL-2 receptor, account for differences in signaling after IL-2 stimulation. In contrast to p-Stat5 after stimulation with IL-2, fold-changes of p-Stat6 and p-p38 in response to IL-4 were similar in all T cell subpopulations in each of the three HIV-infected groups (data not shown).
To test whether T cell subpopulations from HIV-infected PROGs have altered signaling responses to even more general stimuli, PhosFlow analyses were conducted after treatment with PMA/ionomycin. Even under these conditions, CD4+ and CD8+ T cells from PROGs had a significantly blunted ability to phosphorylate the MAPK ERK1/2 when compared with LTNP and RESPs (Fig. 3C, top). These signaling defects were not restricted to any of the T cell subpopulations analyzed. Although fold-changes in phosphorylation of the MAPK p38 tended to be lower in PROGs, no statistically significant differences were found between them and LTNP or RESPs (Fig. 3C, bottom).
To compare “normal” T cell signaling with that found in the context of HIV infection, we compared a group of six HIV-uninfected controls with the group of LTNP. Signaling in both groups was comparable (Fig. 3, D–F), most likely reflecting the low VLs and high numbers of CD4+ T cells associated with the group of LTNP.
With the stimulations analyzed here, phosphorylation levels were found to increase in T cell subpopulations overall, rather than within a subset of each subpopulation (data not shown). However, and as can also be seen in Fig. 3, CD4+ and CD8+ T cells as well as subpopulations thereof respond with a different magnitude of changes in phosphorylation after stimulation.
Correlation of cellular signaling with levels of HIV VL
To ascertain whether any of these alterations in T cell signaling might be associated with VL, the fold-changes in response to each of the above stimuli (TCR cross-linking, IL-2, and PMA/ionomycin) were plotted against the log10VL (Fig. 4). In general, greater fold-changes in phosphorylation after stimulation were associated with lower VLs. This inverse correlation was statistically significant for changes in p-Lck after TCR cross-linking and p-ERK1/2 after PMA/ionomyconin in all CD4+ T cell subpopulations analyzed. Moreover, the change in p-p38 after stimulation with PMA/ionomycin was inversely correlated with VL in memory-effector CD4+ T cells. Other statistically significant inverse correlations of VL and signaling were found within the CD8+ T cell compartments: p-ERK1/2 after PMA/ionomycin in naive, memory, and memory-effector T cells; and p-Lck after TCR cross-linking in naive and effector T cells.
FIGURE 4.
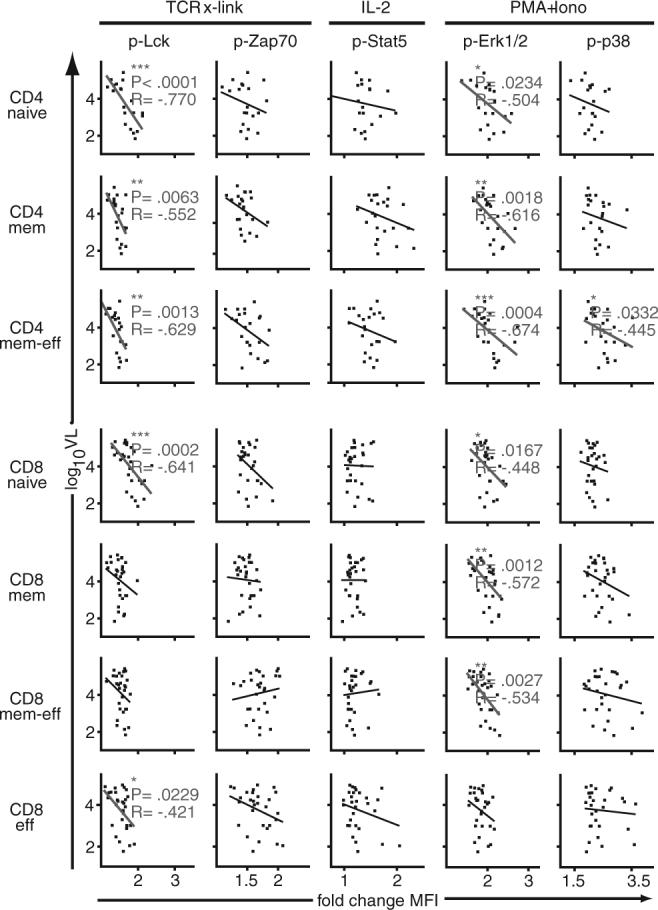
Higher levels of HIV VL are associated with blunted cellular signaling. Fold-changes in phosphorylation (x-axis) after stimulation (as indicated on top) were correlated with levels of HIV VL (y-axis) in CD4+ and CD8+ T cell subpopulations (as indicated on left). These analyses were performed for a subset of LTNPs and PROGs, those that had detectable VL. Two-tailed Spearman's rank correlation was used to analyze the relationship between signaling and levels of VL; statistically significant correlations are indicated. *, p < 0.05; **, p < 0.01; ***, p < 0.001; R, correlation coefficient.
PD-1 expression and correlation with cellular signaling
The CD28 family member PD-1 has been recently shown to be highly expressed on viral-specific T cells during chronic viral infections and to negatively regulate T cell function (42, 43, 45, 46). To determine whether levels of PD-1 expression correlate with cellular signaling, the MFI of PD-1 expression was analyzed in each of the CD4+ and CD8+ subpopulations from the three patient groups described above. As expected, higher levels of PD-1 expression were observed on most if not all of the different T cell subpopulations from PROGs compared with LTNP and RESPs (Fig. 5A). In addition, expression of PD-1 tended to be higher on CD8+ T cells (right) when compared with CD4+ T cells (left).
FIGURE 5.
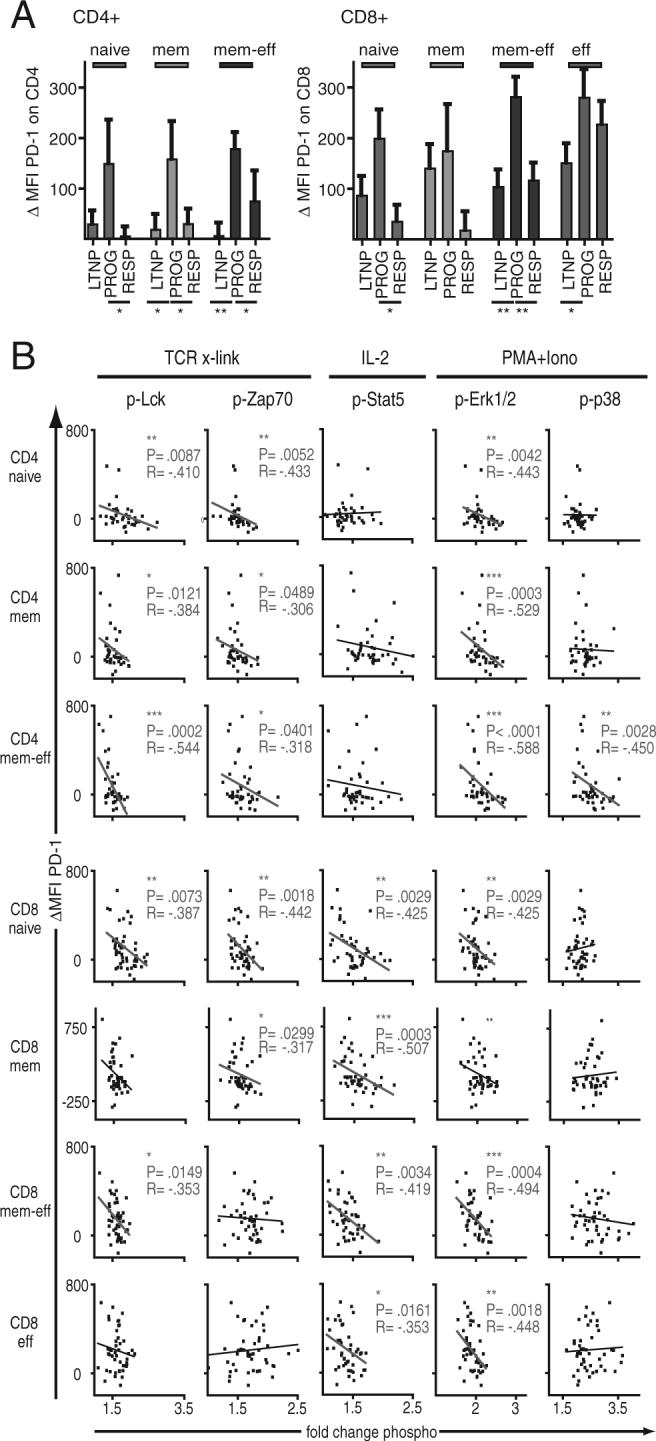
Levels of PD-1 expression are correlated with cellular signaling in HIV infection. A, Mean cell surface expression levels of PD-1 on naive, memory, memory-effector, and effector subpopulations of CD4+ and CD8+ T cells (left and right, respectively) from three HIV-infected groups. Expression levels of PD-1 were measured as MFI and normalized (ΔMFI) to an HIV-uninfected standard control, which was included in all experiments. Error bars indicate SE. HIV-infected groups were compared using the nonparametric two-tailed Mann-Whitney U test, statistically significant differences are indicated by the lines below the plots. B, Fold-changes in phosphorylation (x-axis) after stimulation (as indicated on top) were correlated with expression levels of PD-1 (y-axis) in CD4+ and CD8+ T cell subpopulations (as indicated on left). Two-tailed Spearman's rank correlation was used to analyze the relationship between signaling and levels of VL; statistically significant correlations are indicated. *, p < 0.05; **, p < 0.01; ***, p < 0.001; R, correlation coefficient.
We correlated expression levels of PD-1 (Fig. 5B, y-axis) with changes in cellular signaling (Fig. 5B, x-axis) after stimulation with TCR cross-linking, IL-2, or PMA/ionomycin. Overall, higher levels of PD-1 expression were associated with lower fold-changes in protein phosphorylation after stimulation. The strongest correlations between the levels of PD-1 expression and blunted signaling were found in CD4+ T cell subpopulations for p-Lck and p-Zap70 after TCR-stimulation and p-ERK1/2 after stimulation with PMA/Iono. In CD8+ T cell subpopulations, higher levels of PD-1 expression were associated with blunted phosphorylation of Stat5 and of ERK1/2 after stimulation with IL-2 and PMA/ionomycin, respectively.
Analysis of basal phosphorylation levels in T cells from HIV-infected patients at varying stages of disease progression
Because the observed alterations in signaling were measured as a fold-change over baseline (unstimulated) levels of phosphorylation, an observed “decrease” in signaling could be due either to decreased levels of induced phosphorylation and/or to increased levels of basal phosphorylation. We accordingly analyzed basal phosphorylation levels of selected proteins in CD4+ and CD8+ T cell subpopulations from LTNP, PROGs, and RESPs. Overall, basal levels of phosphorylation (particularly those associated with p-Lck and p-ERK1/2) were higher in T cells from patients with progressive disease when compared with LTNP and RESPs (Fig. 6). Many of these differences reached high levels of significance and were observed in all of the CD4+ and CD8+ T cell subpopulations analyzed (Fig. 6, A and C, respectively). In the case of p-Stat5, the differences between groups were more pronounced in CD8+ than in CD4+ T cells (Fig. 6B). By contrast, the basal levels of p-Zap70 (Fig. 6A) and p-p38 (Fig. 6C) were more uniform between the three patient groups, with only isolated differences in patterns found in PROGs compared with LTNP and RESPs.
FIGURE 6.
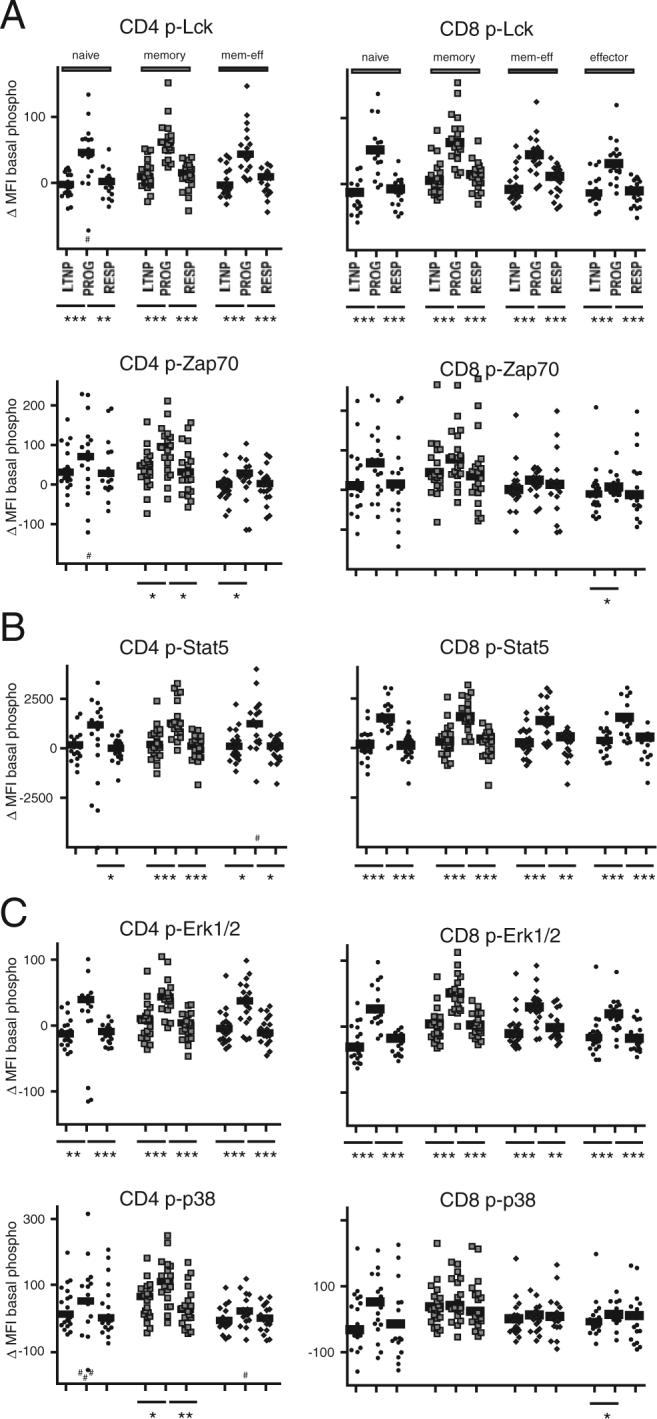
Basal phosphorylation levels are elevated in T cells from HIV PROGs. Levels of basal phosphorylation were analyzed in naive, memory, memory-effector, and effector subpopulations of CD4+ (left; no effector T cells analyzed) and CD8+ T cells (right). Protein phosphorylations were analyzed for (A) Lck and Zap70, (B) Stat5, and (C) ERK1/2 and p38. The HIV-infected groups were compared using the nonparametric two-tailed Mann-Whitney U test; statistically significant differences are indicated by the lines below the plots. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Higher basal phosphorylation levels, in turn, might contribute to the dysregulated and blunted cellular signaling responses seen in progressive stages of HIV infection. To obtain a more global view of this possibility, basal levels of p-Lck, p-Zap70, p-Stat5, p-ERK1/2, and p-p38 (Fig. 7, y-axis) were correlated with respective fold-changes in phosphorylation after stimulation (Fig. 7, x-axis). In CD4+ and CD8+ T cell subpopulations, high basal phosphorylation levels were associated with lower changes of phosphorylation for p-Lck after TCR-stimulation, p-ERK1/2 after PMA/Iono, and p-Stat5 after stimulation with IL-2. No correlations between basal and fold-change in phosphorylation were apparent for Zap70 and p-38.
FIGURE 7.

High levels of basal phosphorylation are associated with blunted signaling in HIV infection. Fold-changes in phosphorylation (x-axis) after stimulation (as indicated on top) were correlated with basal levels of phosphorylation (y-axis) in CD4+ and CD8+ T cell subpopulations (as indicated on left). Two-tailed Spearman's rank correlation was used to analyze the relationship between signaling and levels of VL; statistically significant correlations are indicated. *, p < 0.05; **, p < 0.01; ***, p < 0.001; R, correlation coefficient.
Discussion
HIV disease is associated with chronic immune activation and multiple tiers of T cell dysfunction (16, 20, 48, 49). In this study, we have adapted a multiparameter flow cytometric technique (PhosFlow) (39-41) to interrogate T cell signaling pathways in discrete subpopulations of CD4+ and CD8+ T cells obtained from subjects in varying stages of HIV disease and treatment. We show that PhosFlow enables simultaneous analysis of specific signaling pathways within discrete and multiple subpopulations of CD4+ and CD8+ T cells. Using this approach, we wished to know: (a) whether and which defects in T cell signaling might be associated with advanced disease; (b) whether such defects were generalized across the CD4+ and/or CD8+ T cell lineages; and (c) whether any observed defects were reversed by effective antiviral treatment. Our results indicate that, in subjects with advanced disease, T cell signaling responses to TCR cross-linking, IL-2, and PMA/ionomycin were blunted within many if not all CD4+ and CD8+ T cell subpopulations. Interestingly, changes in phosphorylation of key signaling intermediates were not so much associated with defects in induced phosphorylation per se but with an increased level of basal phosphorylation. When effective antiviral treatment was initiated, basal phosphorylation levels returned to normal, as did signaling responses to multiple T cell stimuli. These observations highlight the indirect effects that HIV infection has on the T cell compartment and may in part provide a mechanistic basis to observed features of T cell dysfunction found in late-stage disease.
PhosFlow analysis showed that, in response to TCR cross-linking, phosphorylation of Lck and Zap70 was most significantly reduced in PROGs compared with LTNP and RESPs, especially in naive T cells for p-Lck and in more differentiated T cell subpopulations for p-Zap70. After stimulation with IL-2, the extent of phosphorylation of Stat5 in PROGs was also diminished in most T cell subpopulations, and especially in CD8+ T cells. Finally, blunted phosphorylation of ERK1/2 was observed in most T cell subpopulations after stimulation of cells from PROGs with PMA/ionomycin. These results are consistent with previous reports in the literature. For example, Cayota et al. (1994) (32) and Stefanova et al. (1996) (33) showed that HIV disease progression is associated with defective tyrosine phosphorylation and altered levels of or post-translational modifications of T cell signaling molecules. Likewise, differential display of protein tyrosine kinases in CD4+ T cells revealed dysregulation of multiple protein kinases in the setting of pathogenic SIV infection (34, 35). Down-regulation of CD3ζ and CD28 on CD8+ T cells has been associated with defects in TCR stimulation (26, 27), whereas defects in IL-2 receptor expression in HIV disease have been linked to impaired activation of Stat5 and upstream kinases (19). Of note, all of these findings were made in the context of heterogeneous populations of CD4+ and CD8+ T within PBMCs, making it difficult to determine whether they might simply reflect changes in the relative frequencies of individual T cell subpopulations. In this study, using the single cell analytical platform provided by Phosflow, we show that abnormalities in protein phosphorylation and signaling are found in multiple discrete subpopulations of both CD4+ and CD8+ T cells, suggesting a generalized impact of progressive HIV infection on all. Technical limitations, i.e., primary cells available from HIV+ individuals, did not allow us to directly include more detailed analyses of protein expression levels here.
Potential drivers of such generalized dysregulation of T cell signaling might include the chronically activated state that attends progressive HIV disease (50) and/or circulating virus (or viral proteins). For instance, cross-linking of CD4 by HIV envelope glycoprotein gp120 and/or gp120-specific Abs has been shown to inhibit CD4+ T cell function and activation (51, 52). Moreover, gp120 has been found to induce TCR desensitization and to alter signal transduction through Lck, possibly by affecting its association with CD4 (53, 54). Interestingly, in the present study, only changes in p-Lck, but not p-Zap70, after TCR-stimulation were significantly affected by VL. This suggests that T cells in the presence of high VL and higher levels of circulating gp120 are more prone to lose activation of the immediate-early Lck-mediated TCR signaling and that this altered activation of the CD4-associated Lck kinase does not fully translate to the downstream Zap70 kinase. However, signaling through Lck is also altered in CD8+ T cells, suggesting that gp120 alone does not account for blunted signaling. The more pronounced impairment of p-ERK1/2 after stimulation with PMA/Iono, when compared with p-p38, possibly reflects the fact that blunted changes in p-ERK1/2 are associated with higher VLs, whereas p-p38 is less affected. High VLs do not seem to be the causative agent of blunted IL-2 signaling, as analyzed by change in p-Stat5 after stimulation. To more directly analyze the effect of cellular activation on signaling in HIV infection, we correlated basal phosphorylation and signaling with cellular activation, i.e., as measured by expression of the activation markers CD38 and/or HLA-DR, for a subset of patients (9 LTNP, 3 PROG, and 10 RESP). Data from these patients had all been gathered within 12 mo of the PhosFlow analysis, and the patients had shown no change in clinical status during the intervening time frame. There was a clear trend of higher activation correlated with higher basal phosphorylation and blunted signaling (data not shown). However, with the limited data set available, the correlations were not statistically significant and we are, at this point, not able to directly show a correlation between cellular activation and basal levels of phosphorylation or signaling. However, studies addressing this important question are underway.
The immunoreceptor PD-1 suppresses TCR signaling, likely via a recruitment of SHP phosphatase activity, resulting in decreased phosphorylation of the CD3ζ activation motifs, attenuated Zap70 activation, and inhibition of downstream signal transduction (55). Recently, up-regulation and expression of PD-1 has been associated with T cell dysfunction, and cellular exhaustion in chronic lymphocytic choriomeningitis virus and HIV infections (42-46). Therefore, expression of PD-1 might well account for some of the decreased signaling seen here. PD-1 expression was highest on cells from patients with progressive disease and elevated levels were associated with blunted changes in p-Lck and p-Zap70 in most T cell subpopulations after TCR stimulation. Interestingly, high levels of PD-1 expression were also associated with blunted IL-2 signaling in CD8+, but not CD4+, T cells, possibly reflecting different IL-2 signaling networks or requirements in these cell types.
Progressive HIV disease is associated with a chronic inflammatory state that induces T cell activation (48, 50) and the secretion of multiple proinflammatory cytokines and chemokines. These mediators, in turn, can have profound effects on the expression of cellular proteins involved in cell-cell interactions. For instance, HIV-induced stimulation of IFNα from plasmacytoid dendritic cells results in up-regulation of MHC class I proteins in vivo, which in turn can interact with TCR of circulating cells (56, 57). These more global and indirect factors may affect the basal phosphorylation state of key signaling intermediates within CD4+ and CD8+ T cells. Thus, a constitutive activation of Stat1 and Stat5 in PBMCs has previously been reported in the setting of HIV infection (58). Our results show that levels of Stat5 phosphorylation are highest in CD4+ and CD8+ T cells from patients with progressive disease and that these cells are precisely those with impaired signaling. Similar results were found for Lck and ERK1/2 (but not Zap70 or p38) and, again, high levels of basal phosphorylation of these kinases were associated with lower changes in induced phosphorylation after TCR, IL-2, and PMA/ionomycin stimulation.
To determine whether blunted signaling is the consequence of elevated basal phosphorylation and/or down-regulated signaling pathways, we also compared the MFI of the p-signals after stimulation (data not shown). The results indicate that the “end point levels” of phosphorylation are similar in the three groups. Although in some instances (most evident for p-Stat5 after stimulation with IL-2) PROG had slightly higher end levels, these differences were not as striking and significant as those found in the case of basal phosphorylation (shown in Fig. 6). These results suggest that blunted responses can, at least in part, be explained by high levels of basal phosphorylation in T cells from HIV PROGs and are consistent with a model in which higher basal phosphorylation levels only allow for blunted changes in phosphorylation of “pre-activated” signaling proteins. Such higher basal phosphorylation might reflect activation and/or perturbed regulation of cellular signaling in the setting of HIV infection. However, other mechanisms, such as differential expression of signaling receptors or proteins, e.g., down-regulated IL-2R, might also be contributory.
Elevated levels of basal phosphorylation and dysregulated signaling might very well be related to or caused by cellular alterations in the cellular redox balance. Thus, HIV disease progression has been shown to be associated with decreasing levels of glutathione (GSH), the major redox buffer in almost all cells (59-62). Alterations in GSH levels affect the activity of redox-sensitive enzymes, including protein kinases and phosphatases. Such changes, in turn, appear to impact upon cellular signal transduction pathways (63). In the specific case of HIV disease, depletion of GSH was shown to result in elevated levels of basal phosphorylation and in cellular dysfunction, e.g., reduced calcium flux and proliferation in response to TCR stimulation (61). Importantly, GSH deficiency has also been associated with numerous other disease states (64). It is accordingly conceivable that, in the context of the chronic immune activation found in late-stage HIV disease, a dysregulated redox balance will result in increased levels of basal phosphorylation. If so, specific stimuli may not be able to generate sufficient levels of additional phosphorylation to transduce appropriate intracellular signals. Interestingly, very little is known about how basal phosphorylation levels affect signaling networks, their regulation, and cellular function. In a subset of patients with acute myeloid lymphoma, members of the Stat proteins have been reported to be constitutively activated, and up-regulated basal state of phosphorylation has been connected to the disability to activate further signaling past basal phosphorylation levels (65). These and our results underscore the need to better understand the role of basal levels of phosphorylation in regulating or perturbing cell signaling in health and in disease.
Finally, we found that the blunted signaling responses in progressive disease resolve upon ART. Although we cannot discriminate between normalization due to cell replacement (e.g., cells produced de novo from progenitor pools) or to reversion (e.g., of previously dysfunctional cells), this observation indicates that the lesion in signaling is reversible, as long as HIV VLs are brought under control.
In summary, we have demonstrated that, in the setting of HIV infection, CD4+ and CD8+ T cell signaling is blunted in cells from untreated subjects with progressive disease compared with LTNP and RESPs. The observed signaling alterations are not restricted to or manifest within a specific T cell subpopulation, suggesting a generalized state of unresponsiveness. Some alterations in cellular signaling correlated with levels of basal phosphorylation, VL, and/or expression of signaling-regulatory protein PD-1. Of these influences, it was the level of basal phosphorylation that appeared to be the most dominant (Fig. 8). Altered signaling found in PROGs was reversible with antiretroviral treatment, indicating that signaling dysfunctions can be restored. More detailed analyses of specific signaling pathways and of levels of basal phosphorylation might suggest ways to correct these T cell signaling dysfunctions and to help restore function of CD4+ and CD8+ T cells.
FIGURE 8.
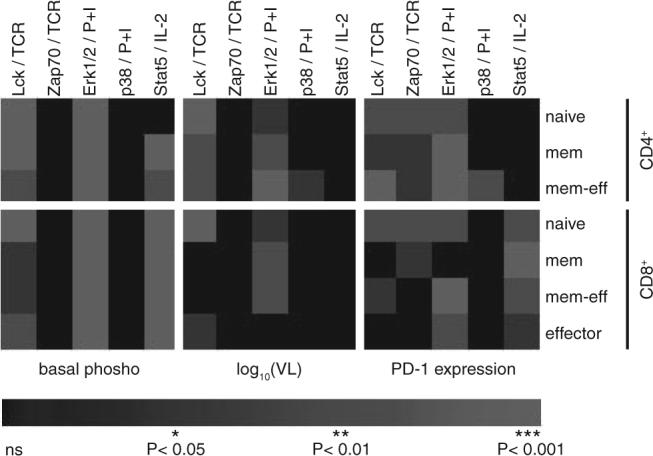
Overview of significances of correlations analyzed using a heat map representation. Values of p from previous correlation analyses are here color-coded (*, p < 0.05 in dark gray; **, p < 0.01 in gray; ***, p < 0.001 in light gray; not significant (ns) in black). Columns show p values for correlation of signaling (Lck and Zap70 after TCR stimulation; ERK1/2 and p38 after stimulation with PMA/ionomycin; and Stat5 after IL-2 stimulation) with (i) basal phosphorylation (left; compare Fig. 8), (ii) VL (middle; compare Fig. 4), and (iii) cell surface expression of PD-1 (right; compare Fig. 5). Corresponding CD4+ and CD8+ T cell subpopulations analyzed are ordered in rows as indicated on the right.
Acknowledgments
We thank the study volunteers Garry P. Nolan, Andrew W. Lee, and Omar D. Perez for their initial input on setting up the PhosFlow assay and for reviewing the manuscript, and Brinda Emu, Kristin Ladell, Peter Hunt, and Jason D. Barbour for insightful discussions.
Footnotes
This work was supported in part by grants from the University-wide AIDS Research Program (F05-GI-219), the National Institutes of Health (R01 AI40312, AI47062, AI52745, K24 AI69994, and M01 RR00083), American Foundation for AIDS Research (106710−40-RGRL), the University of California Center for AIDS Research (P30 AI27763, P30 MH59037, and CC99-SF-001), and the University of California Clinical and Translational Research Institute (UL1 RR024131), a component of the National Institutes of Health Roadmap for Medical Research. J.M.M. is a recipient of the Burroughs Wellcome Fund Clinical Scientist Award in Translational Research and the National Institutes of Health Director's Pioneer Award Program, part of the National Institutes of Health Roadmap for Medical Research, through Grant DPI OD00329.
Abbreviations used in this paper: PROG, progressor; LTNP, long-term nonprogressor; ART, antiretroviral therapy; RESP, chronically infected subjects responding to ART; VL, viral load (plasma HIV RNA copies/ml); PD-1, programmed death-1; p-, phospho-; MFI, median fluorescence intensity; Lck, lymphocyte specific kinase; Lat, linker for activation of T cell; RT, room temperature; GSH, glutathione.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Baier M, Werner A, Bannert N, Metzner K, Kurth R. HIV suppression by interleukin-16. Nature. 1995;378:563. doi: 10.1038/378563a0. [DOI] [PubMed] [Google Scholar]
- 2.Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1 α, and MIP-1 β as the major HIV-suppressive factors produced by CD8+ T cells. Science. 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 3.Gulzar N, Copeland KF. CD8+ T-cells: function and response to HIV infection. Curr. HIV Res. 2004;2:23–37. doi: 10.2174/1570162043485077. [DOI] [PubMed] [Google Scholar]
- 4.Koup RA, Safrit JT, Cao Y, Andrews CA, McLeod G, Borkowsky W, Farthing C, Ho DD. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 1994;68:4650–4655. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mackewicz CE, Blackbourn DJ, Levy JA. CD8+ T cells suppress human immunodeficiency virus replication by inhibiting viral transcription. Proc. Natl. Acad. Sci. USA. 1995;92:2308–2312. doi: 10.1073/pnas.92.6.2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ogg GS, Jin X, Bonhoeffer S, Dunbar PR, Nowak MA, Monard S, Segal JP, Cao Y, Rowland-Jones SL, Cerundolo V, et al. Quantitation of HIV-1-specific cytotoxic T lymphocytes and plasma load of viral RNA. Science. 1998;279:2103–2106. doi: 10.1126/science.279.5359.2103. [DOI] [PubMed] [Google Scholar]
- 7.Plata F, Autran B, Martins LP, Wain-Hobson S, Raphael M, Mayaud C, Denis M, Guillon JM, Debre P. AIDS virus-specific cytotoxic T lymphocytes in lung disorders. Nature. 1987;328:348–351. doi: 10.1038/328348a0. [DOI] [PubMed] [Google Scholar]
- 8.Walker BD, Chakrabarti S, Moss B, Paradis TJ, Flynn T, Durno AG, Blumberg RS, Kaplan JC, Hirsch MS, Schooley RT. HIV-specific cytotoxic T lymphocytes in seropositive individuals. Nature. 1987;328:345–348. doi: 10.1038/328345a0. [DOI] [PubMed] [Google Scholar]
- 9.Walker CM, Moody DJ, Stites DP, Levy JA. CD8+ lymphocytes can control HIV infection in vitro by suppressing virus replication. Science. 1986;234:1563–1566. doi: 10.1126/science.2431484. [DOI] [PubMed] [Google Scholar]
- 10.Kalams SA, Walker BD. The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J. Exp. Med. 1998;188:2199–2204. doi: 10.1084/jem.188.12.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hellerstein M, Hanley MB, Cesar D, Siler S, Papageorgopoulos C, Wieder E, Schmidt D, Hoh R, Neese R, Macallan D, et al. Directly measured kinetics of circulating T lymphocytes in normal and HIV-1-infected humans. Nat. Med. 1999;5:83–89. doi: 10.1038/4772. [DOI] [PubMed] [Google Scholar]
- 12.McCune JM. The dynamics of CD4+ T-cell depletion in HIV disease. Nature. 2001;410:974–979. doi: 10.1038/35073648. [DOI] [PubMed] [Google Scholar]
- 13.Sachsenberg N, Perelson AS, Yerly S, Schockmel GA, Leduc D, Hirschel B, Perrin L. Turnover of CD4+ and CD8+ T lymphocytes in HIV-1 infection as measured by Ki-67 antigen. J. Exp. Med. 1998;187:1295–1303. doi: 10.1084/jem.187.8.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolthers KC, Bea G, Wisman A, Otto SA, de Roda Husman AM, Schaft N, de Wolf F, Goudsmit J, Coutinho RA, van der Zee AG, et al. T cell telomere length in HIV-1 infection: no evidence for increased CD4+ T cell turnover. Science. 1996;274:1543–1547. doi: 10.1126/science.274.5292.1543. [DOI] [PubMed] [Google Scholar]
- 15.Zhang ZQ, Notermans DW, Sedgewick G, Cavert W, Wietgrefe S, Zupancic M, Gebhard K, Henry K, Boies L, Chen Z, et al. Kinetics of CD4+ T cell repopulation of lymphoid tissues after treatment of HIV-1 infection. Proc. Natl. Acad. Sci. USA. 1998;95:1154–1159. doi: 10.1073/pnas.95.3.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clerici M, Stocks NI, Zajac RA, Boswell RN, Lucey DR, Via CS, Shearer GM. Detection of three distinct patterns of T helper cell dysfunction in asymptomatic, human immunodeficiency virus-seropositive patients: independence of CD4+ cell numbers and clinical staging. J. Clin. Invest. 1989;84:1892–1899. doi: 10.1172/JCI114376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gruters RA, Terpstra FG, De Jong R, Van Noesel CJ, Van Lier RA, Miedema F. Selective loss of T cell functions in different stages of HIV infection. Early loss of anti-CD3-induced T cell proliferation followed by decreased anti-CD3-induced cytotoxic T lymphocyte generation in AIDS-related complex and AIDS. Eur. J. Immunol. 1990;20:1039–1044. doi: 10.1002/eji.1830200514. [DOI] [PubMed] [Google Scholar]
- 18.Imami N, Pires A, Hardy G, Wilson J, Gazzard B, Gotch F. A balanced type 1/type 2 response is associated with long-term nonprogressive human immunodeficiency virus type 1 infection. J. Virol. 2002;76:9011–9023. doi: 10.1128/JVI.76.18.9011-9023.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kryworuchko M, Pasquier V, Keller H, David D, Goujard C, Gilquin J, Viard JP, Joussemet M, Delfraissy JF, Theze J. Defective interleukin-2-dependent STAT5 signalling in CD8 T lymphocytes from HIV-positive patients: restoration by antiretroviral therapy. AIDS. 2004;18:421–426. doi: 10.1097/00002030-200402200-00007. [DOI] [PubMed] [Google Scholar]
- 20.Musey LK, Krieger JN, Hughes JP, Schacker TW, Corey L, McElrath MJ. Early and persistent human immunodeficiency virus type 1 (HIV-1)-specific T helper dysfunction in blood and lymph nodes following acute HIV-1 infection. J. Infect. Dis. 1999;180:278–284. doi: 10.1086/314868. [DOI] [PubMed] [Google Scholar]
- 21.Andersson J, Kinloch S, Sonnerborg A, Nilsson J, Fehniger TE, Spetz AL, Behbahani H, Goh LE, McDade H, Gazzard B, et al. Low levels of perforin expression in CD8+ T lymphocyte granules in lymphoid tissue during acute human immunodeficiency virus type 1 infection. J. Infect. Dis. 2002;185:1355–1358. doi: 10.1086/340124. [DOI] [PubMed] [Google Scholar]
- 22.Appay V, Nixon DF, Donahoe SM, Gillespie GM, Dong T, King A, Ogg GS, Spiegel HM, Conlon C, Spina CA, et al. HIV-specific CD8+ T cells produce antiviral cytokines but are impaired in cytolytic function. J. Exp. Med. 2000;192:63–75. doi: 10.1084/jem.192.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamin-Lewis R, Abdelwahab SF, Trang C, Baker A, DeVico AL, Gallo RC, Lewis GK. Perforin-low memory CD8+ cells are the predominant T cells in normal humans that synthesize the β-chemokine macrophage inflammatory protein-1β. Proc. Natl. Acad. Sci. USA. 2001;98:9283–9288. doi: 10.1073/pnas.161298998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, Ogg GS, King A, Lechner F, Spina CA, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 2002;8:379–385. doi: 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- 25.Champagne P, Ogg GS, King AS, Knabenhans C, Ellefsen K, Nobile M, Appay V, Rizzardi GP, Fleury S, Lipp M, et al. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature. 2001;410:106–111. doi: 10.1038/35065118. [DOI] [PubMed] [Google Scholar]
- 26.Trimble LA, Kam LW, Friedman RS, Xu Z, Lieberman J. CD3ζ and CD28 down-modulation on CD8 T cells during viral infection. Blood. 2000;96:1021–1029. [PubMed] [Google Scholar]
- 27.Trimble LA, Lieberman J. Circulating CD8 T lymphocytes in human immunodeficiency virus-infected individuals have impaired function and downmodulate CD3 ζ, the signaling chain of the T-cell receptor complex. Blood. 1998;91:585–594. [PubMed] [Google Scholar]
- 28.Lieberman J, Shankar P, Manjunath N, Andersson J. Dressed to kill? A review of why antiviral CD8 T lymphocytes fail to prevent progressive immunodeficiency in HIV-1 infection. Blood. 2001;98:1667–1677. doi: 10.1182/blood.v98.6.1667. [DOI] [PubMed] [Google Scholar]
- 29.van Baarle D, Kostense S, van Oers MH, Hamann D, Miedema F. Failing immune control as a result of impaired CD8+ T-cell maturation: CD27 might provide a clue. Trends Immunol. 2002;23:586–591. doi: 10.1016/s1471-4906(02)02326-8. [DOI] [PubMed] [Google Scholar]
- 30.Roos MT, Prins M, Koot M, de Wolf F, Bakker M, Coutinho RA, Miedema F, Schellekens PT. Low T-cell responses to CD3 plus CD28 monoclonal antibodies are predictive of development of AIDS. AIDS. 1998;12:1745–1751. doi: 10.1097/00002030-199814000-00005. [DOI] [PubMed] [Google Scholar]
- 31.Schellekens PT, Roos MT, De Wolf F, Lange JM, Miedema F. Low T-cell responsiveness to activation via CD3/TCR is a prognostic marker for acquired immunodeficiency syndrome (AIDS) in human immunodeficiency virus-1 (HIV-1)-infected men. J. Clin. Immunol. 1990;10:121–127. doi: 10.1007/BF00918194. [DOI] [PubMed] [Google Scholar]
- 32.Cayota A, Vuillier F, Siciliano J, Dighiero G. Defective protein tyrosine phosphorylation and altered levels of p59fyn and p56lck in CD4 T cells from HIV-1 infected patients. Int. Immunol. 1994;6:611–621. doi: 10.1093/intimm/6.4.611. [DOI] [PubMed] [Google Scholar]
- 33.Stefanova I, Saville MW, Peters C, Cleghorn FR, Schwartz D, Venzon DJ, Weinhold KJ, Jack N, Bartholomew C, Blattner WA, et al. HIV infection-induced posttranslational modification of T cell signaling molecules associated with disease progression. J. Clin. Invest. 1996;98:1290–1297. doi: 10.1172/JCI118915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bostik P, Wu P, Dodd GL, Villinger F, Mayne AE, Bostik V, Grimm BD, Robinson D, Kung HJ, Ansari AA. Identification of protein kinases dysregulated in CD4+ T cells in pathogenic versus apathogenic simian immunodeficiency virus infection. J. Virol. 2001;75:11298–11306. doi: 10.1128/JVI.75.23.11298-11306.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gale MJ, Jr., Ledbetter JA, Schieven GL, Jonker M, Morton WR, Benveniste RE, Clark EA. CD4 and CD8 T cells from SIV-infected macaques have defective signaling responses after perturbation of either CD3 or CD2 receptors. Int. Immunol. 1990;2:849–858. doi: 10.1093/intimm/2.9.849. [DOI] [PubMed] [Google Scholar]
- 36.Farber DL, Acuto O, Bottomly K. Differential T cell receptor-mediated signaling in naive and memory CD4 T cells. Eur. J. Immunol. 1997;27:2094–2101. doi: 10.1002/eji.1830270838. [DOI] [PubMed] [Google Scholar]
- 37.Meyaard L, Otto SA, Hooibrink B, Miedema F. Quantitative analysis of CD4+ T cell function in the course of human immunodeficiency virus infection: gradual decline of both naive and memory alloreactive T cells. J. Clin. Invest. 1994;94:1947–1952. doi: 10.1172/JCI117545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seder RA, Ahmed R. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat. Immunol. 2003;4:835–842. doi: 10.1038/ni969. [DOI] [PubMed] [Google Scholar]
- 39.Krutzik PO, Nolan GP. Intracellular phospho-protein staining techniques for flow cytometry: monitoring single cell signaling events. Cytometry A. 2003;55:61–70. doi: 10.1002/cyto.a.10072. [DOI] [PubMed] [Google Scholar]
- 40.Perez OD, Krutzik PO, Nolan GP. Flow cytometric analysis of kinase signaling cascades. Methods Mol. Biol. 2004;263:67–94. doi: 10.1385/1-59259-773-4:067. [DOI] [PubMed] [Google Scholar]
- 41.Perez OD, Nolan GP. Simultaneous measurement of multiple active kinase states using polychromatic flow cytometry. Nat. Biotechnol. 2002;20:155–162. doi: 10.1038/nbt0202-155. [DOI] [PubMed] [Google Scholar]
- 42.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 43.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 44.Freeman GJ, Wherry EJ, Ahmed R, Sharpe AH. Reinvigorating exhausted HIV-specific T cells via PD-1-PD-1 ligand blockade. J. Exp. Med. 2006;203:2223–2227. doi: 10.1084/jem.20061800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, Precopio ML, Schacker T, Roederer M, Douek DC, Koup RA. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J. Exp. Med. 2006;203:2281–2292. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, Boulassel MR, Delwart E, Sepulveda H, Balderas RS, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 47.Sommers CL, Park CS, Lee J, Feng C, Fuller CL, Grinberg A, Hildebrand JA, Lacana E, Menon RK, Shores EW, et al. A LAT mutation that inhibits T cell development yet induces lymphoproliferation. Science. 2002;296:2040–2043. doi: 10.1126/science.1069066. [DOI] [PubMed] [Google Scholar]
- 48.Deeks SG, Kitchen CM, Liu L, Guo H, Gascon R, Narvaez AB, Hunt P, Martin JN, Kahn JO, Levy J, et al. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood. 2004;104:942–947. doi: 10.1182/blood-2003-09-3333. [DOI] [PubMed] [Google Scholar]
- 49.Hazenberg MD, Otto SA, van Benthem BH, Roos MT, Coutinho RA, Lange JM, Hamann D, Prins M, Miedema F. Persistent immune activation in HIV-1 infection is associated with progression to AIDS. AIDS. 2003;17:1881–1888. doi: 10.1097/00002030-200309050-00006. [DOI] [PubMed] [Google Scholar]
- 50.Giorgi JV, Lyles RH, Matud JL, Yamashita TE, Mellors JW, Hultin LE, Jamieson BD, Margolick JB, Rinaldo CR, Jr., Phair JP, Detels R. Predictive value of immunologic and virologic markers after long or short duration of HIV-1 infection. J. Acquired Immune Defic. Syndr. 2002;29:346–355. doi: 10.1097/00126334-200204010-00004. [DOI] [PubMed] [Google Scholar]
- 51.Diamond DC, Sleckman BP, Gregory T, Lasky LA, Greenstein JL, Burakoff SJ. Inhibition of CD4+ T cell function by the HIV envelope protein, gp120. J. Immunol. 1988;141:3715–3717. [PubMed] [Google Scholar]
- 52.Mittler RS, Hoffmann MK. Synergism between HIV gp120 and gp120-specific antibody in blocking human T cell activation. Science. 1989;245:1380–1382. doi: 10.1126/science.2571187. [DOI] [PubMed] [Google Scholar]
- 53.Goldman F, Crabtree J, Hollenback C, Koretzky G. Sequestration of p56(lck) by gp120, a model for TCR desensitization. J. Immunol. 1997;158:2017–2024. [PubMed] [Google Scholar]
- 54.Juszczak RJ, Turchin H, Truneh A, Culp J, Kassis S. Effect of human immunodeficiency virus gp120 glycoprotein on the association of the protein tyrosine kinase p56lck with CD4 in human T lymphocytes. J. Biol. Chem. 1991;266:11176–11183. [PubMed] [Google Scholar]
- 55.Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, Qiu Y, Jussif JM, Carter LL, Wood CR, Chaudhary D. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3ζ signalosome and downstream signaling to PKCθ. FEBS Lett. 2004;574:37–41. doi: 10.1016/j.febslet.2004.07.083. [DOI] [PubMed] [Google Scholar]
- 56.Keir ME, Rosenberg MG, Sandberg JK, Jordan KA, Wiznia A, Nixon DF, Stoddart CA, McCune JM. Generation of CD3+CD8low thymocytes in the HIV type 1-infected thymus. J. Immunol. 2002;169:2788–2796. doi: 10.4049/jimmunol.169.5.2788. [DOI] [PubMed] [Google Scholar]
- 57.Keir ME, Stoddart CA, Linquist-Stepps V, Moreno ME, McCune JM. IFN-α secretion by type 2 predendritic cells up-regulates MHC class I in the HIV-1-infected thymus. J. Immunol. 2002;168:325–331. doi: 10.4049/jimmunol.168.1.325. [DOI] [PubMed] [Google Scholar]
- 58.Bovolenta C, Camorali L, Lorini AL, Ghezzi S, Vicenzi E, Lazzarin A, Poli G. Constitutive activation of STATs upon in vivo human immunodeficiency virus infection. Blood. 1999;94:4202–4209. [PubMed] [Google Scholar]
- 59.Droge W, Schulze-Osthoff K, Mihm S, Galter D, Schenk H, Eck HP, Roth S, Gmunder H. Functions of glutathione and glutathione disulfide in immunology and immunopathology. FASEB J. 1994;8:1131–1138. [PubMed] [Google Scholar]
- 60.Lim JS, Eck HP, Gmunder H, Droge W. Expression of increased immunogenicity by thiol-releasing tumor variants. Cell. Immunol. 1992;140:345–356. doi: 10.1016/0008-8749(92)90201-y. [DOI] [PubMed] [Google Scholar]
- 61.Staal FJ, Anderson MT, Staal GE, Herzenberg LA, Gitler C, Herzenberg LA. Redox regulation of signal transduction: tyrosine phosphorylation and calcium influx. Proc. Natl. Acad. Sci. USA. 1994;91:3619–3622. doi: 10.1073/pnas.91.9.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Staal FJ, Roederer M, Israelski DM, Bubp J, Mole LA, McShane D, Deresinski SC, Ross W, Sussman H, Raju PA, et al. Intracellular glutathione levels in T cell subsets decrease in HIV-infected individuals. AIDS Res. Hum. Retroviruses. 1992;8:305–311. doi: 10.1089/aid.1992.8.305. [DOI] [PubMed] [Google Scholar]
- 63.Kanner SB, Kavanagh TJ, Grossmann A, Hu SL, Bolen JB, Rabinovitch PS, Ledbetter JA. Sulfhydryl oxidation down-regulates T-cell signaling and inhibits tyrosine phosphorylation of phospholipase C γ 1. Proc. Natl. Acad. Sci. USA. 1992;89:300–304. doi: 10.1073/pnas.89.1.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Atkuri KR, Mantovani JJ, Herzenberg LA, Herzenberg LA. N-Acetylcysteine: a safe antidote for cysteine/glutathione deficiency. Curr. Opin. Pharmacol. 2007;7:355–359. doi: 10.1016/j.coph.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Irish JM, Hovland R, Krutzik PO, Perez OD, Bruserud O, Gjertsen BT, Nolan GP. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell. 2004;118:217–228. doi: 10.1016/j.cell.2004.06.028. [DOI] [PubMed] [Google Scholar]