

Abstract
Interventions that enhance plasminogen activation within the lung consistently limit the fibrosis that follows alveolar injury. However, this protective effect cannot be attributed solely to accelerated clearance of fibrin that forms as a provisional matrix after lung injury. To explore other mechanisms, we considered interactions between the plasminogen activation system and hepatocyte growth factor (HGF). HGF is known to have antifibrotic activity, but to do so, it must be both released from its sites of sequestration within extracellular matrix (ECM) and activated by proteolytic cleavage. A recent study using bleomycin-exposed mice showed that manipulations of the plasminogen activation system influenced the amount of free HGF within bronchoalveolar lavage fluid without affecting total lung HGF mRNA or protein. To elucidate the mechanisms, we studied the role of plasminogen activation in fibroblast-mediated HGF release and activation. We found that NIH3T3 and mouse lung fibroblasts release ECM-bound HGF in a plasminogen-dependent fashion. The plasminogen effect was lost when lung fibroblasts from urokinase-type plasminogen activator (uPA)–deficient mice were used, and was increased by fibroblasts from plasminogen activator inhibitor (PAI)-1–deficient mice. Plasminogen addition to NIH3T3 or mouse lung fibroblasts increased conversion of pro-HGF to its active form. The plasminogen effect on activation was lost when uPA-deficient fibroblasts were used and accentuated by PAI-1–deficient fibroblasts. In conjunction with the previous in vivo study, these results suggest that plasminogen activation can protect the lung against fibrosis by increasing the availability of active HGF.
Keywords: fibroblasts, heparan sulfate proteoglycans, plasminogen activators, plasminogen activator inhibitors, pulmonary fibrosis
CLINICAL RELEVANCE
This work demonstrates how the plasminogen activation system can influence the fibrosis that follows lung injury by increasing the activation and release of hepatocyte growth factor from the extracellular matrix.
The lung has a considerable capacity to repair itself after many types of alveolar injury. The repair process involves a well-orchestrated series of events, in which alveolar epithelium and endothelium are restored, debris is cleared, and excessive mesenchymal cells and provisional matrix are removed. Unfortunately, this repair process may become dysfunctional and ineffective, resulting in a fibroproliferative reaction that obliterates lung architecture and replaces it with collagenous scars. The determinants of whether the repair process reconstitutes normal lung tissue or results in pulmonary fibrosis are complex, and multiple inter- and intracellular signaling systems appear to be involved (1). In an effort to understand these processes for the purpose of developing treatments, we have been investigating two interacting systems that appear to have pivotal roles in lung repair; namely, the plasminogen activation system and hepatocyte growth factor (HGF).
Abundant evidence from human and animal studies demonstrates that the plasminogen activation system is centrally involved in lung repair (reviewed in Ref. 2). The fibrinolytic activity that is normally present in bronchoalveolar lavage (BAL) fluid is depressed in patients with many types of lung injury due to increased levels of plasminogen activator inhibitors, particularly plasminogen activator inhibitor (PAI)-1 (3–7). The importance of this perturbation has been demonstrated in animal models where suppression of plasminogen activator activity worsens fibrosis, whereas enhanced plasminogen activator activity limits scarring (8–11). An obvious explanation for the link between suppressed fibrinolysis and fibrosis is that successful repair requires the timely removal of the fibrin-rich provision matrix that accumulates after tissue injury. However, the finding that pulmonary fibrosis occurred after bleomycin administration in mice genetically deficient in fibrinogen indicates that fibrin deposition is not required (12, 13). Although a number of mechanisms may be involved in the plasminogen activation system's effect on lung repair, recent experimental results have led us to consider how this system affects HGF within the lung (14).
HGF is a polypeptide growth factor that is produced by mesenchymal cells, and acts upon target tissues via the c-met receptor (15). HGF has particular importance in lung repair due to its ability to stimulate proliferation and migration of alveolar epithelial cells, which are required to resurface denuded alveolar surfaces (reviewed in Ref. 16). HGF also counteracts some of the profibrotic effects of transforming growth factor-β, which, in excess, leads to pulmonary fibrosis (17, 18). In human diseases and experimental animals, HGF levels are found to be increased in blood and BAL fluid during lung injury/repair (19–21). These increases are significant because inhibition of HGF activity in the lung in animal models worsens lung fibrosis (14). Conversely, interventions designed to increase HGF reduce scarring (22–24). For HGF to have its effect on target cells, it must undergo several extracellular processing events (15, 16). HGF is synthesized and secreted as a latent, single-chain protein. Internal proteolytic cleavage is required to generate the active disulfide-linked dimer. Furthermore, secreted HGF becomes tightly bound to the heparan sulfate moiety of proteoglycans (25) that are abundant in extracellular matrices within the lung (26). Bound HGF must be released from these sites of sequestration so that it can access the c-met receptor on neighboring target cells (27).
Direct links between the plasminogen activation system and HGF have become evident through a variety of investigative approaches. In vitro, high concentration of urokinase-type plasminogen activator (uPA) can activate latent, single-chain HGF by cleaving at the appropriate peptide bond in a stoichiometric reaction (28, 29). Plasmin has been shown to release another growth factor, basic fibroblast growth factor, from the extracellular matrix (ECM), where it binds to heparan sulfate proteoglycans (30). Plasmin does so by cleaving the core protein of the proteoglycan, thereby releasing fragments containing the glycosaminoglycan to which basic fibroblast growth factor remains noncovalently bound. It is unknown whether plasmin can do the same for HGF.
The likelihood that the plasminogen activation system could influence lung fibrosis through an HGF-related mechanism was further suggested by experiments using bleomycin-induced lung injury in mice. In particular, mice genetically deficient in PAI-1 (PAI-1−/−) released more HGF into BAL fluid after bleomycin exposure than did wild-type (WT) mice (14). Total lung HGF mRNA and protein, on the other hand, were not different in the two groups. An inhibitor of plasminogen activation and plasmin activity, tranexamic acid, blocked HGF release (14) and worsened lung fibrosis (12). Administration of uPA to WT mice after bleomycin also increased HGF in BAL fluid and reduced the development of fibrosis (14).
Having demonstrated through in vivo experiments the tight association between plasminogen activator activity, HGF BAL fluid levels, and protection from fibrosis (14), we now report the results of in vitro studies that were designed to elucidate the mechanisms by which these in vivo events might occur. We found that fibroblasts activate and release HGF from ECM by a mechanism that is heavily influenced by the plasminogen activation system.
MATERIALS AND METHODS
Materials
L-ascorbic acid, heparin, heparin-agarose, guanidine-HCl, leupeptin, ε-aminocaproic acid, PMSF, and aprotinin were purchased from Sigma Chemical Company (St. Louis, MO). Human plasmin and α2-antiplasmin were obtained from Calbiochem (La Jolla, CA). Human Glu-plasminogen was purchased from American Diagnostica Inc. (Greenwich, CT). Dulbecco's modified Eagle's media (DMEM) and FBS were purchased from Invitrogen (Carlsbad, CA). Penicillin-streptomycin-amphotericin B mixture was purchased from Cambrex (East Rutherford, NJ). Carrier-free human HGF, neutralizing anti-human HGF antibody, and normal goat IgG were purchased from R&D Systems (Minneapolis, MN). Goat anti-human HGFα antibody that was raised against a peptide mapping to the N terminus of human HGFα was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). This antibody cross-reacts with murine HGF. Rabbit anti-goat horseradish peroxidase–conjugated antibody was purchased from Bio-Rad (Hercules, CA).
Cells
NIH3T3 cells and Madin Darby canine kidney (MDCK) cells were obtained from the American Type Culture Collection (Rockville, MD) and grown to subconfluence in DMEM supplemented with 10% FBS at 37°C in a 5% CO2, humidified environment. Subclones of MDCK cells were selected that form tightly aggregated colonies when grown in the absence of HGF. Primary cultures of mouse lung fibroblasts were prepared from three strains of mice. WT C57BL/6 mice were purchased from Charles River Laboratories, Inc. (Portage, MI). PAI-1−/− mice were originally obtained from Dr. Peter Carmeliet (University of Leuven, Leuven, Belgium) and have been back-crossed with C57BL/6 mice for at least eight generations. C57BL/6 mice genetically deficient in uPA (uPA−/−) were purchased from Jackson Laboratories (Bar Harbor, ME). Lung fibroblasts were isolated and cultured by a previously described technique (31). Briefly, mice were killed by sodium pentobarbital overdose, exsanguinated, and the lungs lavaged with PBS. After removing major airways and pulmonary blood vessels, the lungs were minced with scissors and the pieces placed into 100-cm2 dishes in DMEM containing 15% FBS and penicillin-streptomycin-amphotericin B. The cells that grew from the tissue fragments were maintained in media at 37°C in a 5% CO2 incubator and were serially passed a total of five times to yield pure populations of lung fibroblasts, as previously described (31). The fibroblasts were used after the fifth passage.
125I Labeling of HGF
Carrier-free HGF was radioiodinated by modifications of the procedure of Lyon and colleagues (25). Briefly, 5 μg of recombinant human HGF was adsorbed to a 150-μl suspension of heparin-agarose in 0.5 M NaCl, 20 mM sodium phosphate, pH 7.0. Nal25I (0.25 mCi) was added, followed by 50 μl of 0.1% (wt/vol) chloramine-T in 0.5 M NaCl, 20 mM sodium phosphate, pH 7.0. After 1 min, an additional 50 μl of chloramine-T solution was added. One minute later, the gel suspension was transferred to a small disposable column and washed extensively with 0.5 M NaCl, 20 mM sodium phosphate, pH 7.0, to remove all free l25I. Bound l25I-HGF (1,200–3,000 dpm/fmol) was then step-eluted with a small volume of 1.5 M NaCl, 20 mM sodium phosphate, 2 mg/ml BSA, pH 7.0, and stored at −20°C until used. The integrity of the l25I-HGF was confirmed by SDS-PAGE and autoradiography.
Preparation of ECM-Coated Culture Plates
ECMs were generated by NIH3T3 cells using the method of Cukierman (32). Briefly, NIH3T3 cells were added to 24-well plates and incubated for 5–7 d past confluence in DMEM, 10% FCS, antibiotic/antimycotic, and 50 μg/ml L-ascorbic acid. Cells and soluble proteins were removed by incubation for 5 min in 0.5% Triton X-100 plus 20 mM NH4OH at room temperature, followed by four washes in PBS. For the experiments testing the effects of added plasminogen, matrices were also incubated for 2 h at 37°C with 5 mM PMSF to minimize residual serine protease activity within the matrices.
Binding and Release of 125I-HGF from NIH3T3-Derived ECM
ECMs were incubated with 10 or 50 ng/ml 125I-HGF for 2 h at 4°C in PBS. After extensive washing, the labeled matrices were immediately used for experiments. To detect inhibition of l25I-HGF binding by soluble heparin, ECMs were incubated for 2 h at 4°C with 10 ng/ml of 125I-HGF and various concentrations of heparin. Unbound 125I-HGF was removed by repeated washing, after which the ECMs were solubilized with 5 M guanidine HCl and radioactivity was measured using a γ counter (PerkinElmer, Boston, MA). To measure release of HGF from ECM, serum-free DMEM containing various reagents or cell types were added to wells containing l25I-HGF–labeled matrices. After incubation for various times at 37°C, media were collected and their radioactivity measured in a γ counter. The amount of radioactivity released from ECM when incubated with DMEM alone was subtracted as background.
Analysis of Released HGF
The molecular weights of the radiolabeled HGF that was released from ECM were analyzed by SDS-PAGE run under reducing conditions followed by autoradiography. To measure HGF bioactivity, an MDCK scatter assay was used (33). One hundred MDCK cells were added to each well of a 96-well plate and incubated for 4 d in DMEM with 10% FBS. The attached colonies were washed, and media from the HGF-release experiments were added. DMEM alone or containing 50 ng/ml recombinant HGF were added to a set of wells as negative and positive control conditions, respectively. To verify that the scatter activity observed in conditioned media was caused by HGF, 10 μg/ml of neutralizing anti-human HGF antibody was used. As an isotype-matched control antibody, normal goat IgG was used. After overnight incubation at 37°C, the cells were stained with 0.5% crystal violet in 50% ethanol for 10 min. Photographic images were digitally captured (Spot RT Slider camera and software; Diagnostic Instruments, Sterling Heights, MI) and the percentage of scattered colonies to total colonies in each well was determined. A colony was counted as “scattered” if the colony contained more than ten cells and half of them had lost contact with their neighbors and exhibited a fibroblastic phenotype (33).
Activation of Endogenously Produced HGF by NIH3T3 Cells or Mouse Lung Fibroblasts
NIH3T3 cells (1 × 105 cells/well) were plated on 24-well tissue culture plates and incubated in DMEM, 10% FBS, 50 μg/ml ascorbic acid, with or without 20 μg/ml leupeptin, for 7 d. Media were changed every 2–3 d. To evaluate the activation state of HGF, conditioned media were collected and analyzed by SDS-PAGE followed by Western blot. To test the effects of plasminogen and plasmin on HGF activation, cells were cultured for 7 d with leupeptin, after which the cells were washed with PBS. Serum-free DMEM was added containing various concentrations of plasminogen or plasmin and with or without 20 μg/ml α2-antiplasmin or 10 μg/ml aprotinin. After 24 h, media were collected and 20 μl of 50% slurry of heparin-agarose was added to concentrate the HGF.
To evaluate the ability of primary lung fibroblasts from WT, uPA−/−, and PAI-1−/− mice to activate HGF, cells (5 × 105 cells/well) were plated onto 6-well tissue culture plates and incubated in DMEM, 15% FBS, 50 μg/ml ascorbic acid, with 20 μg/ml leupeptin, for 7 d. The cells were washed with PBS, and media were changed to serum-free DMEM containing 10 μg/ml plasminogen or 370 mU/ml plasmin (a concentration that is equivalent to 10 μg/ml plasminogen after full conversion). After 12 h, media were collected and added to heparin-agarose. The media-heparin-agarose mixtures were incubated at 4°C for 12 h with gentle rolling. After centrifugation and washing the heparin-agarose three times with PBS, SDS sample buffer (2% SDS, 50 mM dithiothreitol, 10% glycerol, and 0.1% bromphenol blue in 62.5 mM Tris HCl, pH 7.4) was added and the mixture placed in a 100°C water bath for 5 min. The samples were separated by SDS-PAGE, and the protein bands were transferred to a polyvinylidene fluoride membrane (Immobilon; Millipore, Bedford, MA). Blots were incubated with goat primary antibodies against HGFα diluted 1:500 in blocking solution for 1 h at room temperature, followed by incubation for 1 h at room temperature with rabbit anti-goat horseradish peroxidase–conjugated secondary antibody at a 1:5,000 dilution. Proteins were visualized after incubation of the blots in Western blot chemiluminescent substrate (Pierce Biotechnology, Rockford, IL), according to the manufacturer's protocol, and exposure to Hyperfilm (Amersham Bioscience, Buckinghamshire, UK). The densities of the pro-HGF and α-chain HGF were compared by densitometric analysis using Scion Image software (Scion Corporation, Frederick, MD).
Statistical Analysis
All values are shown as means ± SEM. The significance of differences between groups was assessed using ANOVA followed by Tukey's multiple comparison test for pairwise comparisons. Analyses were performed using Prism software (GraphPad Software, San Diego, CA), accepting a P < 0.05 as statistically significant.
RESULTS
125I-HGF Binding to ECM and Release by Plasmin
To study the binding and release of HGF, ECMs were generated by NIH3T3 cells, after which the cells and soluble proteins were removed by treatment with Triton X-100 and NH4OH. The cell-free ECMs were then labeled with 125I-HGF. To demonstrate that HGF was binding to ECM via heparan sulfate moieties, soluble heparin was added to the wells during the labeling period to compete with ECM binding sites. Figure 1A demonstrates that heparin was able to inhibit 125I-HGF binding to the ECM at concentrations known to block HGF binding to heparan sulfate (34). NaCl (2 M) was also able to elute 125I-HGF from the ECM (data not shown). To assess the release of radiolabel from the ECM, the amount of 125I-HGF in the media was measured at various times. Incubation with DMEM alone led to the release of a small amount of radioactivity (Figure 1B). This release was considered background and subtracted from the results of subsequent experiments. Treatment of the labeled matrices with 10 and 100 mU/ml plasmin resulted in an increased release of bound radiolabel into the media in a dose- and time-dependent fashion (Figure 1B).
Figure 1.


HGF binding to NIH3T3-derived ECM and release by plasmin. (A) Soluble heparin inhibits HGF binding to ECM. Fibroblast-derived ECMs were incubated with 10 ng/ml 125I-HGF together with various concentration of heparin for 2 h at 4°C. After extensive washing, ECMs were collected and their radioactivities in cpm were measured. Values are means ± SEM (n = 3). (B) Plasmin releases HGF from ECM in a dose- and time-dependent fashion. 125I-HGF–labeled ECMs were incubated without plasmin (open triangle), or with 10 mU/ml (open circle) or 100 mU/ml (closed circle) plasmin in PBS for the indicated times. The supernatants were collected and their radioactivities (cpm) were measured. Values are means ± SEM (n = 3). Statistically significant differences (P < 0.01) are present between all treatments at all time intervals.
Release of 125I-HGF from ECM by NIH3T3 Cells
To determine if cells were able to release HGF from ECM, fresh NIH3T3 cells were added to 125I-HGF–labeled cell-free matrices in the presence of various concentrations of plasminogen. The amount of radiolabel released into the media after 6 h of incubation was measured. The addition of plasminogen or cells alone released small amounts of radiolabel from the ECM (Figure 2A); however, when NIH3T3 cells (which are known to produce uPA [35–37]) and plasminogen were combined, the release of radioactivity from the matrices increased in a plasminogen dose–dependent fashion. Extending the incubation time of cells plus 10 μg/ml plasminogen to 24 h caused release of greater than 90% of the bound radioactivity (data not shown).
Figure 2.


HGF release from ECM by fibroblasts and/or plasminogen and inhibition by ε-aminocaproic acid. ECMs were produced by NIH3T3 cells in 24-well plates, the fibroblasts were removed, and the ECMs were labeled with 10 ng/ml 125I-HGF. (A) HGF is released from ECM synergistically by fibroblasts and plasminogen. NIH3T3 cells (3T3) (2 × 105) and various concentrations of plasminogen (Plg) were added to wells, and the plates were incubated for 6 h. Media were collected, and the amounts of radioactivity released into the media were measured. Values are means ± SEM (n = 3). Connecting lines between designated groups indicate statistically significant differences (*P < 0.001). (B) The plasmin inhibitor, ε-aminocaproic acid, inhibits fibroblast/plasminogen-induced release of HGF from ECM. NIH3T3 cells (3T3)(2 × 105) and various concentrations of plasminogen (Plg) and/or ε-aminocaproic acid (εACA) were added to wells, and the plates were incubated for 6 h. Media were collected and their radioactivities measured. Values are means ± SEM. Connecting lines between designated groups indicate statistically significant differences (*P < 0.01; **P < 0.001).
To further assess the role of plasminogen activation in the release of HGF, ε-aminocaproic acid was used to inhibit plasminogen activation and plasmin activity (38). Plasminogen and NIH3T3 cells were added to 125I-HGF–labeled matrices in the absence or presence of ε-aminocaproic acid, and the release of radiolabel was measured after a 6-h incubation (Figure 2B). As shown previously, plasminogen or cells alone caused a small release of radiolabel into the media. ε-Aminocaproic acid inhibited the plasminogen-mediated release, but did not affect the amount of growth factor released by cells alone. Importantly, ε-aminocaproic acid inhibited the synergistic effect of cells and plasminogen in a dose-dependent fashion.
Characterization of the HGF Released from ECM by NIH3T3 Cells
To determine if the radioactivity released into the media by the NIH3T3 cells represented intact HGF rather than degraded peptides, supernatants from matrices exposed for 5 h to DMEM with or without 10 μg/ml plasminogen and/or 2 × 105 NIH3T3 cells were separated by SDS-PAGE under reducing conditions. The resulting bands were visualized by autoradiography (Figure 3A). A control sample containing the stock 125I-HGF that was used to label the matrices was included (Figure 3A, leftmost lane). The radioactivity in the stock 125I-HGF migrated as bands of molecular weight appropriate for intact pro-HGF (∼ 92 kD) and the active, cleaved α-chain and β-chain forms (∼ 69 kD and 34 kD, respectively). The majority of this stock 125I-HGF was in the cleaved, active conformation that migrates as α- and β-chains. Exposure of 125I-HGF–labeled ECM to DMEM alone revealed no radioactive bands in the media after incubation for 5 h. ECM exposed to plasminogen or 3T3 cells alone released radioactive material that migrated in bands similar to the stock 125I-HGF. In accordance with our previous results, the intensities of the bands were greatest from ECM that was exposed to both cells and plasminogen.
Figure 3.


Molecular size and biological activity of HGF released from ECM. (A) Fibroblasts/plasminogen release intact HGF from ECM. ECMs were labeled with 50 ng/ml 125I-HGF and, after washing, were incubated in media alone (media) or with 10 μg/ml plasminogen (Plg) and/or 2 × 105 NIH3T3 cells (3T3) per well. After 5 h, media were collected and separated by SDS-PAGE and were analyzed by autoradiography. The left lane contains a sample of the stock 125I-HGF used to label the matrices. The autoradiogram is representative of the pattern of results seen in three separate studies. (B) HGF released from ECM by fibroblasts/plasminogen is biologically active. ECMs were labeled with 50 ng/ml HGF and incubated with media alone (media) or 10 μg/ml plasminogen (Plg) and/or 2 × 105 NIH3T3 cells (3T3) for 5 h. Conditioned media (100 μl) were transferred to wells in a 96-well plate containing compact colonies of MDCK cells. Serum-free DMEM (Bkgd) or DMEM containing 50 ng/ml recombinant HGF (rHGF) were added to a set of wells as negative and positive control conditions, respectively. To verify that scatter activity depended on HGF, neutralizing anti-human HGF antibody (anti-HGF) or isotype-matched control antibody (iso) were used. After overnight incubation at 37°C, the percent of colonies that showed scattering were measured for each well. Values are means ± SEM (n = 3). The figure is representative of three independent experiments. Connecting lines between designated groups indicate statistically significant differences (*P < 0.01, **P < 0.001). Anti-HGF antibody blocked the scatter activity generated by fibroblasts plus plasminogen and by recombinant HGF (P < 0.001).
To determine if the released HGF retained biological activity, an MDCK scatter assay was used. Matrices were prepared, labeled with HGF, and then incubated with 10 μg/ml plasminogen and/or 2 × 105 NIH3T3 cells. After 5 h, the media were transferred to wells containing compact colonies of MDCK cells, and the amount of colony scattering measured after overnight incubation. DMEM and recombinant HGF (50 ng/ml) were used as negative and positive controls, respectively (Figure 3B). Media conditioned by exposure to HGF-labeled ECM for 5 h contained no detectable scatter activity. Plasminogen or cells alone released small amounts of scatter activity; however, the combination of cells and plasminogen had a synergistic effect on the release of scatter activity in condition media. The scatter activity generated by fibroblasts and plasminogen was inhibited by 10 μg/ml of an anti-HGF neutralizing antibody.
Release of 125I-HGF from ECM by Mouse Lung Fibroblasts
To further evaluate the role of plasminogen activation on the release of 125I-HGF from ECM, fibroblasts were isolated from the lungs of WT, uPA−/−, and PAI-1−/− mice, and 5 × 104 cells were plated onto radiolabeled matrices in the absence (control) or presence of 10 μg/ml plasminogen. After 10-h incubation, media were collected, and the amount of radioactivity was measured (Figure 4). Fibroblasts from WT mice released an increased amount of radiolabel above background (P = 0.03). The level was increased further when plasminogen was present in the culture environment (P < 0.001). Cells from uPA−/− mice also released a small amount of radiolabel, but the further addition of plasminogen had no incremental effect (P > 0.05). Finally, fibroblasts from PAI-1−/− mice caused a small release of radiolabel (P < 0.002) that was greatly accentuated with the addition of plasminogen (P < 0.001).
Figure 4.

HGF release from ECM by primary lung fibroblasts obtained from WT, uPA−/−, and PAI-1−/− mice. Lung fibroblasts (5 × 104 cells/well) from mice of each genotype were plated onto radiolabeled ECM (10 ng/ml 125I-HGF) in 24-well plates and cultured in serum-free DMEM with or without 10 μg/ml plasminogen. After 10-h incubation, media were collected and their radioactivities were measured. Values are means ± SEM (n = 3). The addition of plasminogen increased the release of radiolabel from ECM by WT and PAI-1−/− mice significantly (*P < 0.001). However the release of radioactivity by uPA−/− fibroblasts was unchanged with the addition of plasminogen (P > 0.05).
Activation of Endogenously Generated HGF
Commercially available sources of recombinant HGF are not well suited for studying processes involved in its activation because a high proportion of the HGF has already been cleaved to the active disulfide-linked heterodimer. An alternative approach was therefore taken, in which NIH3T3 cells or mouse lung fibroblasts were allowed to generate their own HGF in a manner that maintained the majority of HGF in the single-chain, pro-HGF conformation. This was accomplished by adding to the culture media the protease inhibitor leupeptin, which blocks an enzyme, HGF activator, which is present in bovine plasma and is activated by thrombin during FBS processing (39). The amount of HGF in the pro- and active form could then be assessed by Western analysis using an antibody that recognizes an epitope on the α-chain. Under reducing conditions, the larger pro-HGF can be easily distinguished from the smaller cleaved α-chain. If leupeptin is not added to the serum-containing media used during NIH3T3 monolayer formation, the majority of HGF is in the activated form, as indicated by the dominance of the α-chain band (Figure 5A). On the other hand, after 7 d in culture in the presence of leupeptin, the majority of the HGF is in the pro-HGF form.
Figure 5.
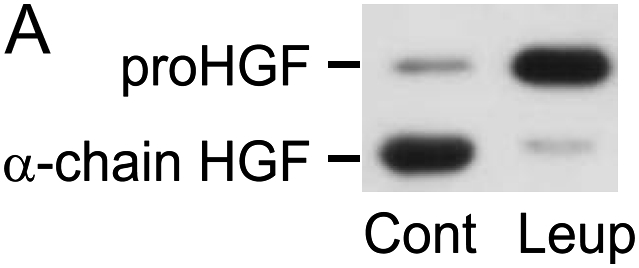


Activation of endogenously generated HGF by NIH3T3 cells. (A) Leupeptin inhibits the FBS-induced activation pro-HGF. NIH3T3 cells (1 × 105 cells/well) were plated into 24-well tissue culture plates and incubated in 10% FBS/DMEM containing 50 μg/ml of ascorbic acid, with or without 20 μg/ml leupeptin, for 7 d. Equal volumes of media were analyzed by SDS-PAGE run under reducing conditions, followed by immunoblotting for HGF. Results of a representative experiment (n > 10) are displayed for cells grown in the absence (Cont) or presence (Leup) of leupeptin. The locations of migration of pro-HGF and active, α-chain HGF are displayed. (B and C) NIH3T3 fibroblasts activate HGF in a plasminogen-dependent process that is inhibitable by α2-antiplasmin and aprotinin. NIH3T3 cells (1 × 105 cells/well) were plated into 24-well tissue culture plates and incubated in 10% FBS/DMEM containing 50 μg/ml of ascorbic acid with 20 μg/ml leupeptin. After 7 d in culture, cells were washed with PBS and media were changed to serum-free, leupeptin-free DMEM containing various concentrations of plasminogen (B) or plasmin (C) ± 20 μg/ml α2-antiplasmin or 10 μg/ml aprotinin. After 24 h, conditioned media were collected and subjected to SDS-PAGE, followed by immunoblotting for HGF. The data displayed are representative of results from three independent experiments.
To determine if NIH3T3 cells could activate pro-HGF using plasminogen, 7-d monolayers were washed and incubated in serum-free, leupeptin-free media containing various concentrations of reagent plasminogen or plasmin. After 24 h, conditioned media were collected and equal volumes were analyzed by SDS PAGE and immunoblotting (Figure 5B). In the absence of added plasminogen or plasmin, the majority of the HGF released into the media was in the pro-HGF form. Addition of plasminogen or plasmin caused activation of pro-HGF in a dose-dependent manner, as indicated by the accumulation of α-chain. Inclusion of aprotinin (inhibitor of serine proteases) or α2-antiplasmin (inhibitor of plasmin, but not plasminogen activators at the concentration used [40–42]) blocked the effect of plasminogen or plasmin on HGF activation.
To evaluate the role of uPA in HGF activation, 5 × 105 lung fibroblasts from WT, uPA−/−, and PAI-1−/− mice were added to wells and grown for 7 d in media containing serum and leupeptin. When cultured for another 24 h in serum-free and leupeptin-free media, the HGF generated by all 3 genotypes was found to be partially activated (Figure 6A, representative of duplicate experiments). The addition of plasmin caused the majority of the pro-HGF to be activated, regardless of genotype. When plasminogen was added to the fibroblast cultures, the increase in pro-HGF activation by PAI-1−/− cells was greater than the increase induced by plasminogen in the WT fibroblast cultures. In fact, the level of activation of HGF by PAI-1−/− fibroblasts rivaled that achieved by the addition of plasmin. In marked contrast, there was no plasminogen effect on uPA−/− fibroblasts. In particular, addition of plasminogen to uPA−/− fibroblasts caused no increase in pro-HGF activation above that which was induced by cells incubated in media alone. We note that the combined densities of the bands in the WT genotype lanes were less than those in the uPA−/− and PAI-1−/− lanes. This is because, in this experiment, the monolayer cell density of the WT fibroblasts was less than that of the other two genotypes, and equal volumes of conditioned media were analyzed. To semiquantify the results, the ratios of densities of the α-chain HGF to pro-HGF are shown in Figure 6B.
Figure 6.


Plasminogen-dependent pro-HGF activation by lung fibroblasts from WT, uPA−/−, and PAI-1−/− mice. (A) Primary lung fibroblasts (5 × 105 cells/well) obtained from WT, uPA−/−, and PAI-1−/− mice were placed into 6-well tissue culture plates and incubated for 7 d in 15% FBS/DMEM containing 50 μg/ml ascorbic acid with 20 μg/ml leupeptin. The monolayers were washed with PBS, and media were changed to serum-free DMEM (cont) containing 10 μg/ml plasminogen (Plg) or 370 mU/ml plasmin. After 24 h, equal volumes of conditioned media were collected and subjected to SDS-PAGE under reducing conditions, followed by immunoblotting for HGF. (B) Bar graph showing the density ratio of α-chain HGF to pro-HGF for the three genotypes, with and without plasminogen, as displayed in (A).
DISCUSSION
We and others have consistently observed that enhancement of plasminogen activator activity reduces the amount of fibrosis that follows lung injury. The experimental approaches used to increase plasminogen activation in the lung have included use of PAI-1−/− mice (8), delivery of uPA protein directly to the lung (14, 43), delivery of uPA genes using adenoviral vectors (9), and use of a transgenic mouse with doxycycline-inducible uPA activity expressed exclusively within the lung (11). In each situation, the lungs were relatively protected from bleomycin-induced fibrosis. We also reported that a polymorphism in the human PAI-1 promoter that is associated with higher PAI-1 expression and, by implication, less plasminogen activator activity, was more frequent in patients with nonspecific interstitial pneumonia than in control populations (44). Other groups have reported that BAL fluid from patients with interstitial lung diseases have suppressed plasminogen activator activity due to upregulation of PAI-1 (3).
Our initial hypothesis on the protective effects of plasminogen activator activity on pulmonary fibrosis had been that the efficient generation of plasmin allowed for accelerated clearance of accumulated extravascular fibrin in both acute and chronic lung injury (45). By speeding removal, fibroblasts would have less opportunity to invade the fibrin-rich provisional matrix and lay down collagenous scars (46). This hypothesis was severely challenged, however, when we and another research group found that mice genetically deficient in circulating fibrinogen still developed considerable lung scarring after bleomycin administration (12, 13). This caused us to consider a much broader range of possible mechanisms by which the plasminogen activation system reduces fibrosis.
Multiple connections between the plasminogen activation system and HGF motivated us to conduct the present studies. First, the HGF molecule is homologous to plasminogen in that both have an amino-terminal loop, several kringle domains (plasminogen 5, HGF 4), and a serine protease domain (15), the latter of which is proteolytically inactive in HGF due to changes in active site amino acids. Both plasminogen and HGF are synthesized and released extracellularly as single-chain, inactive proteins that require proteolytic cleavage for activation at a homologous Arg-Val peptide bond. For HGF, the proteases that have been shown to accomplish this are HGF activator (47), kallikrein (48), factor XIa (48), factor XIIa (47), matriptase (49), and the plasminogen activators, tPA and uPA (28, 39). Depending on the biological context, each of these activating proteases may have biologically relevant roles.
In the lung, HGF has been shown to increase alveolar epithelial cell proliferation and migration, and human studies have revealed that HGF expression is increased in response to a variety of lung injuries (16). Similar changes occur in animal models, including bleomycin-induced pulmonary fibrosis (23, 50). This increase in HGF expression is not an epiphenomenon, but is intimately linked to the repair process. Blocking HGF by inhibitory antibodies worsens lung fibrosis after bleomycin administration (14). Elevation of HGF levels by its exogenous delivery (22, 23) or by gene transfer (24) reduces the amount of fibrosis that occurs after lung injury. This antifibrotic effect is not unique to the lung, but occurs in other organs, including liver (51), kidney (52, 53), skin (54), and heart (55).
Once secreted, HGF binds avidly to heparan sulfate moieties of proteoglycans that are contained in the ECM (25). To activate the HGF receptor, c-met, on neighboring lung cells, HGF must be released from its sites of sequestration. Proteolytic digestion of proteoglycans has been shown to release other growth factors that bind to them (e.g., basic fibroblast growth factor [30]). Evidence suggesting that this phenomenon might be important for HGF comes from in vivo studies using the bleomycin mouse model (14). Increased plasminogen activator activity within the lung was found to be tightly associated with increased levels of free HGF within BAL fluid. In particular, after bleomycin administration, HGF protein levels were higher in BAL fluid from PAI-1−/− mice compared with that from WT mice, whereas total lung HGF protein and mRNA were unaffected by PAI-1 status. This increase in BAL HGF was suppressed by administering tranexamic acid, which inhibits plasminogen activation and plasmin activity, and increases collagen content in bleomycin-exposed PAI-1−/− mice (12). This increase in BAL HGF was likely important in promoting lung repair, because administration of an anti-HGF neutralizing antibody worsened fibrosis in the lungs of bleomycin-injured PAI-1−/− mice (14).
To elucidate the mechanism by which plasminogen activation can influence BAL HGF levels, we now present direct evidence that plasminogen activation releases HGF from the ECM. First, fibroblast-mediated release of HGF was greatly potentiated by the presence of plasminogen in the culture environment. Fibroblasts are known to produce plasminogen activators (35–37). Second, exposure of ECM to reagent plasmin leads to HGF release. Third, the addition of ε-aminocaproic acid, an inhibitor of plasminogen activation and plasmin activity, blocked HGF release. Fourth, fibroblasts from uPA−/− mice did not show a plasminogen-induced increase in HGF liberation. Conversely, fibroblasts from mice deficient in PAI-1, a major inhibitor of plasminogen activation, released more HGF from ECM than did fibroblasts from WT mice. This pattern of results is parallel to what was seen in vivo, where manipulations of the plasminogen activation system influenced the concentration of HGF found within BAL fluid in mice (see above and Ref. 14). In this respect, the previously published in vivo studies demonstrate the biological relevance of our in vitro studies.
Our studies also show that plasminogen promotes pro-HGF activation. To study this phenomenon, an experimental system was needed to generate HGF so that the majority of it was in the inactive, pro-HGF form. Because FBS contains a protease that artifactually activates pro-HGF during cell culture, the monolayers were grown with leupeptin in the media to inhibit the activating enzyme (39). The leupeptin was then removed and the monolayers studied in serum-free media. Using this system, our results show that NIH3T3 and mouse lung fibroblasts activate pro-HGF in a fashion that is significantly enhanced by plasminogen. Direct addition of plasmin accomplishes the same effect. α-2-Antiplasmin, which inhibits plasmin but not uPA (40–42), reduced pro-HGF activation. Plasminogen added to uPA−/− fibroblasts did not enhance pro-HGF activation, whereas the addition of plasminogen to PAI-1−/− cells greatly increased activation. It is notable that some background activation of pro-HGF occurred in the absence of plasminogen or uPA. It is likely that one or more of the previously mentioned HGF activating enzymes are responsible.
There has been uncertainty as to whether plasmin can directly activate pro-HGF. Three research groups have reported that the dominant effect of plasmin was to degrade HGF to inactive fragments (39, 47, 56). In contrast, another group reported that plasmin treatment of conditioned media from cells transfected with an HGF expression plasmid generated increased HGF-like activity in a bioassay (57). We also found that high concentrations of plasmin extensively degrade pro-HGF (data not shown). However, our data clearly demonstrate that addition of plasmin to cell cultures at lower concentrations cleaves pro-HGF to its dimeric form. The possibility still exits, however, that plasmin does not directly activate pro-HGF but, rather, activates another latent protease present in the cell culture, which in turn activates pro-HGF. At present, no such plasmin-activatable enzyme that activates pro-HGF has been described.
Although high concentrations of uPA can activate pro-HGF directly in a stoichiometric fashion (28, 29), it is unlikely that uPA is the direct pro-HGF–cleaving enzyme in our system. First, the amount of reagent two-chain (active) uPA that must be added to activate pro-HGF in our system (in the absence of plasminogen) is considerably greater than what is generated by the NIH3T3 or murine lung fibroblasts (data not shown). Second, addition of plasmin to uPA−/− fibroblasts caused pro-HGF activation, which would not have occurred if uPA was directly responsible for its activation. It is therefore likely that the role of uPA in our system is to activate plasminogen to plasmin, which then has downstream effects to promote HGF release and activation.
Our choice to use NIH3T3 for part of our study is appropriate because matrices formed by these cells contain many of the same macromolecular components found in lung interstitium (58) including types I, III, and IV collagens, fibronectin, laminin, and the heparan sulfate–containing proteoglycan, perlecan (32, 59–61). Importantly, the basic findings from our NIH3T3 cells were confirmed using primary cultures of mouse lung fibroblasts and were parallel to what was found in the in vivo mouse bleomycin studies (14).
In summary, we have demonstrated that fibroblasts activate pro-HGF and release it from sequestration sites in the ECM by converting plasminogen to plasmin (Figure 7). Because increased active HGF lessens fibrosis after lung injury, our results support the conclusion that one mechanism by which the plasminogen activation system can reduce pulmonary fibrosis is through its capacity to enhance the effects of HGF.
Figure 7.

Proposed mechanisms by which the plasminogen activation system modulates lung fibrosis. uPA produced by lung fibroblasts converts plasminogen to plasmin. The amount of plasmin generated depends upon the relative amounts of uPA and its inhibitor, PAI-1. The plasmin in turn, directly or indirectly, (1) releases HGF from its binding sites on heparan sulfate proteoglycans contained within the ECM, and (2) activates it. Although uPA can directly activate HGF, this process appears to be minor compared with the plasmin pathway. HGF once released and activated contributes to lung repair and prevention of fibrosis.
Acknowledgments
The authors thank Daniel S. McConnell, Ph.D., and Faith A. Bjork for performing the 125I labeling of HGF, and Daniel A. Lawrence, Ph.D., for valuable discussions and advice.
This work was supported by National Institutes of Health grants K08-HL-04434 and P50-HL-56402.
Originally Published in Press as DOI: 10.1165/rcmb.2006-0006OC on July 13, 2006
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med 2001;134:136–151. [DOI] [PubMed] [Google Scholar]
- 2.Sisson TH, Simon RH. Cell specific control of coagulation and fibrinolysis within the lung. Thromb Haemost 2004;92:435–437. [DOI] [PubMed] [Google Scholar]
- 3.Chapman HA, Allen CL, Stone OL. Abnormalities in pathways of alveolar fibrin turnover among patients with interstitial lung disease. Am Rev Respir Dis 1986;133:437–443. [DOI] [PubMed] [Google Scholar]
- 4.Idell S, James KK, Levin EG, Schwartz BS, Manchanda N, Maunder RJ, Martin TR, McLarty J, Fair DS. Local abnormalities in coagulation and fibrinolytic pathways predispose to alveolar fibrin deposition in the adult respiratory distress syndrome. J Clin Invest 1989;84:695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertozzi P, Astedt B, Zenzius L, Lynch K, LeMaire F, Zapol W, Chapman HA Jr. Depressed bronchoalveolar urokinase activity in patients with adult respiratory distress syndrome. N Engl J Med 1990;322:890–897. [DOI] [PubMed] [Google Scholar]
- 6.Idell S, Koenig KB, Fair DS, Martin TR, McLarty J, Maunder RJ. Serial abnormalities of fibrin turnover in evolving adult respiratory distress syndrome. Am J Physiol 1991;261:L240–L248. [DOI] [PubMed] [Google Scholar]
- 7.Kotani I, Sato A, Hayakawa H, Urano T, Takada Y, Takada A. Increased procoagulant and antifibrinolytic activities in the lungs with idiopathic pulmonary fibrosis. Thromb Res 1995;77:493–504. [DOI] [PubMed] [Google Scholar]
- 8.Eitzman DT, McCoy RD, Zheng X, Fay WP, Shen T, Ginsburg D, Simon RH. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J Clin Invest 1996;97:232–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sisson TH, Hattori N, Xu Y, Simon RH. Treatment of bleomycin-induced pulmonary fibrosis by transfer of urokinase-type plasminogen activator genes. Hum Gene Ther 1999;10:2315–2323. [DOI] [PubMed] [Google Scholar]
- 10.Swaisgood CM, French EL, Noga C, Simon RH, Ploplis VA. The development of bleomycin-induced pulmonary fibrosis in mice deficient for components of the fibrinolytic system. Am J Pathol 2000;157:177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sisson TH, Hanson KE, Subbotina N, Patwardhan A, Hattori N, Simon RH. Inducible lung-specific urokinase expression reduces fibrosis and mortality after lung injury in mice. Am J Physiol Lung Cell Mol Physiol 2002;283:L1023–L1032. [DOI] [PubMed] [Google Scholar]
- 12.Hattori N, Degen JL, Sisson TH, Liu H, Moore BB, Pandrangi RG, Simon RH, Drew AF. Bleomycin-induced pulmonary fibrosis in fibrinogen-null mice. J Clin Invest 2000;106:1341–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilberding JA, Ploplis VA, McLennan L, Liang Z, Cornelissen I, Feldman M, Deford ME, Rosen ED, Castellino FJ. Development of pulmonary fibrosis in fibrinogen-deficient mice. Ann N Y Acad Sci 2001;936:542–548. [DOI] [PubMed] [Google Scholar]
- 14.Hattori N, Mizuno S, Yoshida Y, Chin K, Mishima M, Sisson TH, Simon RH, Nakamura T, Miyake M. The plasminogen activation system reduces fibrosis in the lung by a hepatocyte growth factor-dependent mechanism. Am J Pathol 2004;164:1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang WG, Martin TA, Parr C, Davies G, Matsumoto K, Nakamura T. Hepatocyte growth factor, its receptor, and their potential value in cancer therapies. Crit Rev Oncol Hematol 2005;53:35–69. [DOI] [PubMed] [Google Scholar]
- 16.Ware LB, Matthay MA. Keratinocyte and hepatocyte growth factors in the lung: roles in lung development, inflammation, and repair. Am J Physiol Lung Cell Mol Physiol 2002;282:L924–L940. [DOI] [PubMed] [Google Scholar]
- 17.Taipale J, Keski-Oja J. Hepatocyte growth factor releases epithelial and endothelial cells from growth arrest induced by transforming growth factor-β1. J Biol Chem 1996;271:4342–4348. [DOI] [PubMed] [Google Scholar]
- 18.Sakurai H, Nigam SK. Transforming growth factor-β selectively inhibits branching morphogenesis but not tubulogenesis. Am J Physiol 1997; 272:F139–F146. [DOI] [PubMed] [Google Scholar]
- 19.Sakai T, Satoh K, Matsushima K, Shindo S, Abe S, Abe T, Motomiya M, Kawamoto T, Kawabata Y, Nakamura T, et al. Hepatocyte growth factor in bronchoalveolar lavage fluids and cells in patients with inflammatory chest diseases of the lower respiratory tract: detection by RIA and in situ hybridization. Am J Respir Cell Mol Biol 1997;16:388–397. [DOI] [PubMed] [Google Scholar]
- 20.Verghese GM, McCormick-Shannon K, Mason RJ, Matthay MA. Hepatocyte growth factor and keratinocyte growth factor in the pulmonary edema fluid of patients with acute lung injury: biologic and clinical significance. Am J Respir Crit Care Med 1998;158:386–394. [DOI] [PubMed] [Google Scholar]
- 21.Yamanouchi H, Fujita J, Yoshinouchi T, Hojo S, Kamei T, Yamadori I, Ohtsuki Y, Ueda N, Takahara J. Measurement of hepatocyte growth factor in serum and bronchoalveolar lavage fluid in patients with pulmonary fibrosis. Respir Med 1998;92:273–278. [DOI] [PubMed] [Google Scholar]
- 22.Yaekashiwa M, Nakayama S, Ohnuma K, Sakai T, Abe T, Satoh K, Matsumoto K, Nakamura T, Takahashi T, Nukiwa T. Simultaneous or delayed administration of hepatocyte growth factor equally represses the fibrotic changes in murine lung injury induced by bleomycin: a morphologic study. Am J Respir Crit Care Med 1997;156:1937–1944. [DOI] [PubMed] [Google Scholar]
- 23.Dohi M, Hasegawa T, Yamamoto K, Marshall BC. Hepatocyte growth factor attenuates collagen accumulation in a murine model of pulmonary fibrosis. Am J Respir Crit Care Med 2000;162:2302–2307. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe M, Ebina M, Orson FM, Nakamura A, Kubota K, Koinuma D, Akiyama K, Maemondo M, Okouchi S, Tahara M, et al. Hepatocyte growth factor gene transfer to alveolar septa for effective suppression of lung fibrosis. Mol Ther 2005;12:58–67. [DOI] [PubMed] [Google Scholar]
- 25.Lyon M, Deakin JA, Mizuno K, Nakamura T, Gallagher JT. Interaction of hepatocyte growth factor with heparan sulfate: elucidation of the major heparan sulfate structural determinants. J Biol Chem 1994;269: 11216–11223. [PubMed] [Google Scholar]
- 26.Smits NC, Robbesom AA, Versteeg EM, van de Westerlo EM, Dekhuijzen PN, van Kuppevelt TH. Heterogeneity of heparan sulfates in human lung. Am J Respir Cell Mol Biol 2004;30:166–173. [DOI] [PubMed] [Google Scholar]
- 27.Birchmeier C, Gherardi E. Developmental roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends Cell Biol 1998;8:404–410. [DOI] [PubMed] [Google Scholar]
- 28.Mars WM, Zarnegar R, Michalopoulos GK. Activation of hepatocyte growth factor by the plasminogen activators uPA and tPA. Am J Pathol 1993;143:949–958. [PMC free article] [PubMed] [Google Scholar]
- 29.Naldini L, Vigna E, Bardelli A, Follenzi A, Galimi F, Comoglio PM. Biological activation of pro-HGF (hepatocyte growth factor) by urokinase is controlled by a stoichiometric reaction. J Biol Chem 1995; 270:603–611. [DOI] [PubMed] [Google Scholar]
- 30.Whitelock JM, Murdoch AD, Iozzo RV, Underwood PA. The degradation of human endothelial cell–derived perlecan and release of bound basic fibroblast growth factor by stromelysin, collagenase, plasmin, and heparanases. J Biol Chem 1996;271:10079–10086. [DOI] [PubMed] [Google Scholar]
- 31.Hogaboam CM, Lukacs NW, Chensue SW, Strieter RM, Kunkel SL. Monocyte chemoattractant protein-1 synthesis by murine lung fibroblasts modulates CD4+ T cell activation. J Immunol 1998;160:4606–4614. [PubMed] [Google Scholar]
- 32.Cukierman E. Preparation of extracellular matrices produced by cultured fibroblasts. In: Current protocols in cell biology. Vol. 10. Bonifacino J, Dasso M, Harford J, Lippincott-Schwartz J, Yamada K, editors. Somerset, NJ: John Wiley & Sons, Inc.; 2002. pp. 9.1–9.15.
- 33.Chen HC. Cell-scatter assay. Methods Mol Biol 2005;294:69–77. [DOI] [PubMed] [Google Scholar]
- 34.Mizuno K, Inoue H, Hagiya M, Shimizu S, Nose T, Shimohigashi Y, Nakamura T. Hairpin loop and second kringle domain are essential sites for heparin binding and biological activity of hepatocyte growth factor. J Biol Chem 1994;269:1131–1136. [PubMed] [Google Scholar]
- 35.Grimaldi G, Di Fiore P, Locatelli EK, Falco J, Blasi F. Modulation of urokinase plasminogen activator gene expression during the transition from quiescent to proliferative state in normal mouse cells. EMBO J 1986;5:855–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boxman IL, Quax PH, Lowik CW, Papapoulos SE, Verheijen J, Ponec M. Differential regulation of plasminogen activation in normal keratinocytes and SCC-4 cells by fibroblasts. J Invest Dermatol 1995;104: 374–378. [DOI] [PubMed] [Google Scholar]
- 37.Vayalil PK, Olman M, Murphy-Ullrich JE, Postlethwait EM, Liu RM. Glutathione restores collagen degradation in TGF-β–treated fibroblasts by blocking plasminogen activator inhibitor-1 expression and activating plasminogen. Am J Physiol Lung Cell Mol Physiol 2005; 289:L937–L945. [DOI] [PubMed] [Google Scholar]
- 38.Ellis V, Whawell SA, Werner F, Deadman JJ. Assembly of urokinase receptor-mediated plasminogen activation complexes involves direct, non-active-site interactions between urokinase and plasminogen. Biochemistry 1999;38:651–659. [DOI] [PubMed] [Google Scholar]
- 39.Naldini L, Tamagnone L, Vigna E, Sachs M, Hartmann G, Birchmeier W, Daikuhara Y, Tsubouchi H, Blasi F, Comoglio PM. Extracellular proteolytic cleavage by urokinase is required for activation of hepatocyte growth factor/scatter factor. EMBO J 1992;11:4825–4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walker JE, Ogston D. The inhibition of tissue activator and urokinase by human plasma. Thromb Haemost 1982;47:265–268. [PubMed] [Google Scholar]
- 41.Lijnen HR, Zamarron C, Blaber M, Winkler ME, Collen D. Activation of plasminogen by pro-urokinase: I. Mechanism. J Biol Chem 1986; 261:1253–1258. [PubMed] [Google Scholar]
- 42.Duval-Jobe C, Parmely MJ. Regulation of plasminogen activation by human U937 promonocytic cells. J Biol Chem 1994;269:21353–21357. [PubMed] [Google Scholar]
- 43.Gunther A, Lubke N, Ermert M, Schermuly RT, Weissmann N, Breithecker A, Markart P, Ruppert C, Quanz K, Ermert L, et al. Prevention of bleomycin-induced lung fibrosis by aerosolization of heparin or urokinase in rabbits. Am J Respir Crit Care Med 2003; 168:1358–1365. [DOI] [PubMed] [Google Scholar]
- 44.Kim KK, Flaherty KR, Long Q, Hattori N, Sisson TH, Colby TV, Travis WD, Martinez FJ, Murray S, Simon RH. A plasminogen activator inhibitor-1 promoter polymorphism and idiopathic interstitial pneumonia. Mol Med 2003;9:52–56. [PMC free article] [PubMed] [Google Scholar]
- 45.Kuhn C. Patterns of lung repair: a morphologist's view. Chest 1991; 99:11S–14S. [PubMed] [Google Scholar]
- 46.Basset F, Ferrans VJ, Soler P, Takemura T, Fukuda Y, Crystal RG. Intraluminal fibrosis in interstitial lung disorders. Am J Pathol 1986; 122:443–461. [PMC free article] [PubMed] [Google Scholar]
- 47.Shimomura T, Miyazawa K, Komiyama Y, Hiraoka H, Naka D, Morimoto Y, Kitamura N. Activation of hepatocyte growth factor by two homologous proteases, blood-coagulation factor XIIa and hepatocyte growth factor activator. Eur J Biochem 1995;229:257–261. [DOI] [PubMed] [Google Scholar]
- 48.Peek M, Moran P, Mendoza N, Wickramasinghe D, Kirchhofer D. Unusual proteolytic activation of pro-hepatocyte growth factor by plasma kallikrein and coagulation factor XIa. J Biol Chem 2002;277:47804–47809. [DOI] [PubMed] [Google Scholar]
- 49.Lee SL, Dickson RB, Lin CY. Activation of hepatocyte growth factor and urokinase/plasminogen activator by matriptase, an epithelial membrane serine protease. J Biol Chem 2000;275:36720–36725. [DOI] [PubMed] [Google Scholar]
- 50.Adamson IY, Bakowska J. Relationship of keratinocyte growth factor and hepatocyte growth factor levels in rat lung lavage fluid to epithelial cell regeneration after bleomycin. Am J Pathol 1999;155:949–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsuno Y, Iwata H, Umeda Y, Takagi H, Mori Y, Kosugi A, Matsumoto K, Nakamura T, Hirose H. Hepatocyte growth factor gene transfer into the liver via the portal vein using electroporation attenuates rat liver cirrhosis. Gene Ther 2003;10:1559–1566. [DOI] [PubMed] [Google Scholar]
- 52.Gao X, Mae H, Ayabe N, Takai T, Oshima K, Hattori M, Ueki T, Fujimoto J, Tanizawa T. Hepatocyte growth factor gene therapy retards the progression of chronic obstructive nephropathy. Kidney Int 2002;62:1238–1248. [DOI] [PubMed] [Google Scholar]
- 53.Yang J, Dai C, Liu Y. Hepatocyte growth factor gene therapy and angiotensin II blockade synergistically attenuate renal interstitial fibrosis in mice. J Am Soc Nephrol 2002;13:2464–2477. [DOI] [PubMed] [Google Scholar]
- 54.Wu MH, Yokozeki H, Takagawa S, Yamamoto T, Satoh T, Kaneda Y, Katayama I, Nishioka K. Hepatocyte growth factor both prevents and ameliorates the symptoms of dermal sclerosis in a mouse model of scleroderma. Gene Ther 2004;11:170–180. [DOI] [PubMed] [Google Scholar]
- 55.Li Y, Takemura G, Kosai K, Yuge K, Nagano S, Esaki M, Goto K, Takahashi T, Hayakawa K, Koda M, et al. Postinfarction treatment with an adenoviral vector expressing hepatocyte growth factor relieves chronic left ventricular remodeling and dysfunction in mice. Circulation 2003;107:2499–2506. [DOI] [PubMed] [Google Scholar]
- 56.Mizuno K, Takehara T, Nakamura T. Proteolytic activation of a single-chain precursor of hepatocyte growth factor by extracellular serine-protease. Biochem Biophys Res Commun 1992;189:1631–1638. [DOI] [PubMed] [Google Scholar]
- 57.Gak E, Taylor WG, Chan AM, Rubin JS. Processing of hepatocyte growth factor to the heterodimeric form is required for biological activity. FEBS Lett 1992;311:17–21. [DOI] [PubMed] [Google Scholar]
- 58.Dunsmore SE, Rannels DE. Extracellular matrix biology in the lung. Am J Physiol 1996;270:L3–L27. [DOI] [PubMed] [Google Scholar]
- 59.Alitalo K, Kuismanen E, Myllyla R, Kiistala U, Asko-Seljavaara S, Vaheri A. Extracellular matrix proteins of human epidermal keratinocytes and feeder 3T3 cells. J Cell Biol 1982;94:497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iozzo RV, Cohen IR, Grassel S, Murdoch AD. The biology of perlecan: the multifaceted heparan sulphate proteoglycan of basement membranes and pericellular matrices. Biochem J 1994;302:625–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science 2001;294:1708–1712. [DOI] [PubMed] [Google Scholar]