

Abstract
To gain insight into the molecular mechanism(s) whereby macrophages produce large amounts of IL-10, we analyzed IL-10 gene expression and temporally correlated it with modifications to chromatin associated with the IL-10 promoter. In resting cells, which make essentially no cytokines, the IL-10 promoter is associated with histones containing little or no detectable modifications. Macrophages stimulated in the presence of immune complexes begin to produce high levels of IL-10 pre-mRNA transcripts within minutes of stimulation. Coincident with this transcription was a rapid and dynamic phosphorylation of histone H3 at specific sites in the IL-10 promoter. Histone phosphorylation was closely followed by the binding of transcription factors to the IL-10 promoter. Blocking the activation of ERK prevented histone phosphorylation and transcription factor binding to the IL-10 promoter. In contrast to histone phosphorylation, the peak of histone acetylation at this promoter did not occur until after transcription had peaked. Inhibition of histone deactylase did not alter IL-10 gene expression, suggesting that phosphorylation but not acetylation was the proximal event responsible for IL-10 transcription. Our findings reveal a rapid and well-orchestrated series of events in which ERK activation causes a rapid and transient phosphorylation of histone H3 at specific regions of the IL-10 promoter, resulting in a transient exposure of the IL-10 promoter to the transcription factors that bind there. This exposure is essential for the efficient induction of IL-10 gene expression in macrophages. To our knowledge, this represents a unique way in which the expression of a cytokine gene is regulated in macrophages.
Macrophages are important mediators of host defense. They can function as major immune effector cells and are potent secretory cells. The cytokines they secrete can influence the magnitude and the character of an adaptive immune response. The typical response to bacterial products is the production of a variety of inflammatory cytokines from macrophages, including TNF-α, IL-1, IL-6, and IL-12 (1). These cytokines are an important component of an innate immune response, but their production must be tightly regulated to prevent autoimmune pathologies. TNF-α is one of the first cytokines to be produced after stimulation (2) and is followed closely by IL-1 and IL-6. The synthesis of IL-10 under these conditions usually comes after the induction of TNF-α and IL-1 and is thought to prevent the overproduction of the inflammatory cytokines. For most of the inflammatory cytokines, the transcription factors that induce cytokine gene expression and the signal transduction pathways leading to cytokine production have been identified (3). The regulation of IL-10 remains less well understood.
IL-10 is a class II α-helical cytokine and the founding member of a growing family of structurally related cytokines, which includes IL-19, IL-20, IL-22, IL-24, and IL-26. The binding of IL-10 to IL-10R1 induces a conformational change in the receptor, allowing it to dimerize with IL-10R2 leading to signal transduction in target cells (4). IL-10 is one of a small number of cytokines that have been well documented to have potent immune inhibitory capacity. IL-10 was originally called “cytokine synthesis inhibiting factor,” and it can inhibit the transcription and translation of a variety of inflammatory cytokines (5). IL-10 exerts potent inhibitory effects on accessory cells, resulting in a reduction in Ag presentation and either an inhibition (6) or biasing of (7) T cell activation. The administration of exogenous IL-10 to macrophages can render them refractory to IFN-γ (8) and diminish their responses to LPS (9).
We have previously shown that stimulation of macrophages in the presence of immune complexes (IC)3 induces the production of high amounts of IL-10 (8), and we demonstrated that activation of ERK was required but not sufficient for such event (10). In the present work, we examined the kinetics and the molecular mechanisms of IL-10 production. We show that the superinduction of IL-10, which occurs following stimulation of macrophages in the presence of IC, happens remarkably rapidly. Within minutes of stimulation, IL-10 pre-mRNA transcripts are detectable, and within an hour cytokine production can be measured in the supernatants. These kinetics are even more rapid than the production of TNF-α in response to endotoxin (2). As we examined the chromatin modifications across the IL-10 promoter in resting and stimulated macrophages, we found that the rapid induction of cytokine synthesis depended not only on the activation of transcription factors, but also on a transient remodeling of the IL-10 promoter that occurs as a result of ERK activation. We herein describe a specific, rapid, and evanescent phosphorylation of histone H3 along the IL-10 promoter that correlates temporally with transcription factor binding and with IL-10 gene expression. These data reveal a critical role for chromatin phosphorylation in regulating IL-10 gene expression in macrophages.
Materials and Methods
Reagents
The MEK/ERK inhibitor, PD98059, and the histone deacetylase (HDAC) inhibitor, trichostatin A (TSA), were obtained from Calbiochem (EMD Biosciences). Ultrapure LPS from Escherichia coli K12 strain was purchased from InvivoGen. Anti-phosphorylated histone H3 (Ser10) Ab, anti-acetylated histone H3 (Lys14), and anti-Sp1 Ab were obtained from Upstate. TRIzol reagent was purchased from Invitrogen Life Technologies. RNase-free DNase I and micrococcal nuclease (MNase) were obtained from Roche Diagnostics.
Mice
Six- to 8-wk-old BALB/c mice were purchased from Taconic Farms. All mice were maintained in HEPA (High Efficiency Particulate Air)-filtered Thoren units (Thoren Caging Systems) at the University of Maryland (College Park, MD). Mice were used at 6−10 wk of age as a source of bone marrow-derived macrophages (BMMϕ). All procedures were reviewed and approved by the University of Maryland Institutional Animal Care and Use Committee.
BMMϕ and IC
BMMϕ were prepared as described previously (11). Briefly, BM was flushed from the femurs and tibiae of mice, and cells were plated in petri dishes in complete medium, which consists of DMEM/F12 supplemented with 10% FBS, penicillin/streptomycin, glutamine, and 20% L cell conditioned medium. On days 2 and 5, cells were fed with complete medium. On day 7, cells were removed from petri dishes and cultured on tissue culture dishes in complete medium without L cell conditioned medium. On the next day, cells were subject to experimentation.
Rabbit polyclonal IgG to chicken egg albumin (anti-OVA IgG) was supplied by Cappel (MP Biomedicals). After the lyophilized powder was reconstituted, the contaminating endotoxin was removed by using EndoClean from BioVitage. Chicken egg albumin (OVA) was purchased from Worthington Biochemical Corporation and polymyxin B-agarose (Sigma-Aldrich) was used to remove LPS. IC consisting of IgG-OVA were prepared by mixing 181 μl of RPMI 1640 with 14 μl of chicken OVA (1 mg/ml) and 117 μl of anti-OVA IgG (4 mg/ml) for 30 min on a rotary platform at room temperature, as described (8).
Quantitative real-time PCR (QRT-PCR)
QRT-PCR was performed on an ABI Prism 7700 Sequence Detection System using SYBR Green PCR reagents purchased from Bio-Rad. Melting curve analyses were performed after PCR amplification to ensure that a single product with the expected melting curve characteristics was obtained, as described previously (10).
Cytokine measurement
Cytokines were measured by sandwich ELISA using Ab pairs provided by BD Pharmingen (IL-12p70, 9A5, and C17.8; IL-10, JES-2A5, and JES-16E3) according to the manufacturer's instructions.
RNA isolation and RT-PCR
BMMϕ (3−5 × 106 cells per reaction) were subjected to RNA extraction using TRIzol reagent. The contaminating DNA was then removed by RNase-free DNase I treatment. ThermoScript RT-PCR system (Invitrogen Life Technologies) was used to generate cDNA from RNA by using random hexamers or oligo(dT)20. QRT-PCR was used to measure both mature and premature IL-10 mRNA levels. Premature IL-10 mRNA was analyzed by using random hexamers-generated cDNA and the following primer pairs: sense, 5′-CATTCCAGTAAGTCACACCCA-3′ (intronic primer), and antisense, 5′-TCTCACCCAGGGAATTCAAA-3′, and GAPDH primer pairs: 5′-TGTTCCTACCCCCAATGTGT-3′, and antisense, 5′-TCCCAAGTCACTGTCACACC-3′ (intronic primer). Mature IL-10 mRNA was amplified by using oligo(dT)20-generated cDNA and the following primer pairs: sense, 5′-AAGGACCAGCTGGACAACAT-3′, and antisense, 5′-TCTCACCCAGGGAATTCAAA-3′, and GAPDH primer pairs: sense, 5′-TGTTCCTACCCCCAATGTGT-3′, and antisense, 5′-GGTCCTCAGTGTAGCCCAAG-3′.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed following ultrasonic shearing conditions that result in relatively uniform DNA fragment size of ∼300 bp. The remaining procedures were conducted as previously described (10). The primers used to amplify specific regions of the IL-10 promoter are listed in the table of Fig. 1. TNF-α primers used for QRT-PCR were as follows: 5′-CCACAT GAGATCATGGTTTTCTC-3′ and 5′-CTGGCTAGTCCCTTGCTGTC-3′. IL-12p40 primers used for QRT-PCR were as follows: 5′-TTTC GACGTCTATATTCCCTCTG-3′ and 5′-AGCTGCCTGGTCTGATGTG-3′. IL-12p35 primers used for QRT-PCR were as follows: 5′-CGACG CACTTGTCCTTGAGAT-3′ and 5′-ACTGAGAGGAGCTGCTGGAT-3′. TdT primers used for QRT-PCR were as follows: 5′-ACCAAGACT GACAACCCACGTT-3′ and 5′-GTGGCAGTCAGAGGCATCTTT-3′.
FIGURE 1.
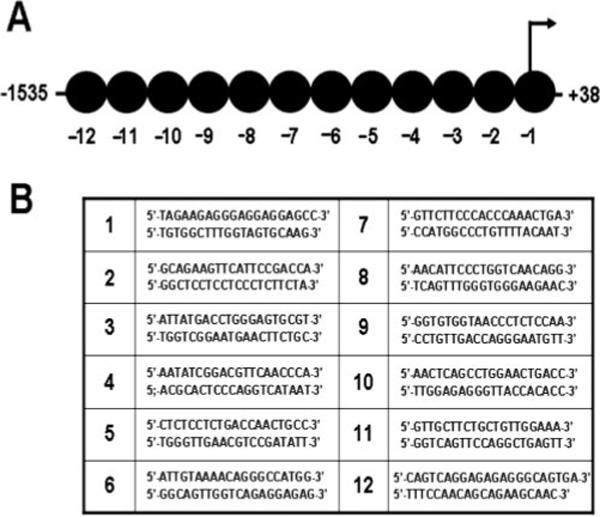
DNA fragmentation and ChIP. A, Schematic illustration representing the nucleosomes along the promoter region of the IL-10 gene and the primer pairs used to amplify each segment of the IL-10 promoter. B, The primers used to amplify specific regions of the IL-10 promoter are listed.
MNase and DNase I accessibility assay
MNase accessibility assay was performed on cells grown in 100-mm tissue culture dishes that were stimulated with LPS/IC. At different time intervals, formaldehyde was added for 15 min at room temperature at a final concentration of 1%, followed by glycine (0.125 M) to neutralize formaldehyde. Cells were washed and lysed in 4 ml of ice-cold Nuclei EZ lysis buffer (Sigma-Aldrich), centrifuged at 500 × g for 4 min, and the nuclei were resuspended with an additional 4 ml of ice-cold Nuclei EZ lysis buffer. Washed nuclei were pooled and resuspended in MNase digestion buffer (10 mM Tris-HCl (pH 7.4), 15 mM NaCl, 60 mM KCl, 0.15 mM spermine, and 0.5 mM spermidine). After centrifugation at 120 × g for 10 min at 4°C, the nuclei were resuspended in MNase digestion buffer containing 1 mM CaCl2. MNase was then added and incubated at room temperature for 5 min. The reaction was terminated by adding MNase stop buffer (100 mM EDTA and 10 mM EGTA in 10 mM Tris-HCl (pH 7.4)). Proteinase K (100 μg) and RNase A (10 μg) were then added and incubated at 37°C overnight. DNA was purified with phenol/chloroform extraction and ethanol precipitation. The purified DNA fragments were subjected to QRT-PCR analysis. DNase I accessibility was determined as described previously (10).
Western blotting analysis of histone H3 acetylation
BMMϕ (3−5 × 106 cells per reaction) were treated with TSA for 1 h. The cells were lysed by whole-cell lysis buffer (20 mM Tris-HCl (pH 7.5), 0.15 M NaCl, 1% Triton X-100, and protease inhibitor and phosphatase inhibitor mixtures). After sonication, equal amounts of whole-cell lysate were subjected to SDS-PAGE analysis. Western blotting assay was then performed to detect histone H3 acetylation using rabbit polyclonal anti-acetylyl-histone H3 Ab (06−599) and mouse monoclonal anti-histone H3 Ab (05−499) (Upstate Biotechnology).
Data analysis
The relative differences among samples were determined using the ΔΔCT methods as described in the Applied Biosystems protocol. A ΔCT value was determined for each sample using the CT value from input DNA to normalize ChIP assay results. The CT value for GAPDH was used to normalize loading in the RT-PCRs. For the MNase or DNase I accessibility assays, ΔCT values were determined by subtracting the CT value from each MNase or DNase I concentration from the zero enzyme control CT value. A ΔΔCT value was then obtained by subtracting control ΔCT values from the corresponding experimental ΔCT. The ΔΔCT values were converted to fold difference compared with the control by raising 2 to the ΔΔCT power. Student's t test was used for statistical analysis. Values of p < 0.05 were considered to be statistically significant.
Results
Phosphorylation at Ser10 of histone H3 across the IL-10 promoter region
We previously demonstrated that the stimulation of macrophages with TLR agonists (LPS) combined with IC resulted in the production of large amounts of IL-10 (11). This superinduction of IL-10 was due, in part, to the activation of the MAPK pathway (10). In the present work, we quantitatively and spatiotemporally analyzed changes in histone H3 phosphorylation across the promoter region of the IL-10 gene following stimulation. We generated chromatin fragments with an average size of 300 bp (Fig. 1), which in combination with QRT-PCR allowed a high resolution profiling of histone modifications across the first 1600 bases of the IL-10 promoter.
Macrophages were stimulated with LPS plus IC and the phosphorylation of histone H3 at Ser10 along the promoter region of the IL-10 gene was analyzed by ChIP. Initially, we examined the second nucleosome from the transcription start site, because this is the location of the binding site to the transcription factor Sp1 (12). Phosphorylation of histone H3 at this site is rapid and peaks within 30 min of stimulation (Fig. 2A). This peak of histone phosphorylation is remarkably transient and is reduced to baseline levels by 60 min poststimulation (Fig. 2A). We performed a similar analysis to quantify phosphorylation over the entire proximal 1600 bp of the IL-10 promoter. Although there was a substantial increase in histone phosphorylation throughout much of this region following stimulation, there were clear differences in the degree to which individual nucleosomes were modified. The highest degree of phosphorylation was observed at regions corresponding to the first two nucleosomes, but regions corresponding to nucleosomes −4 and −6 were also phosphorylated with similar kinetics (Fig. 2A and unpublished data). These regions correspond to segments of the IL-10 promoter that are binding sites to the transcription factors implicated in IL-10 gene expression (13). The distal-most segments analyzed, which correspond to nucleosomes −12 and −11, were not phosphorylayted to any substantial degree, nor were some of the intermediate nucleosomes, e.g., −3, −7 (Fig. 2A and unpublished data). Thus, there appears to be a rapid and transient phosphorylation of nucleosomes across the IL-10 promoter in stimulated cells. These phosphorylation events do not appear to be random, but rather generally correspond to, or are adjacent to, segments of the promoter that bind to transcription factors.
FIGURE 2.
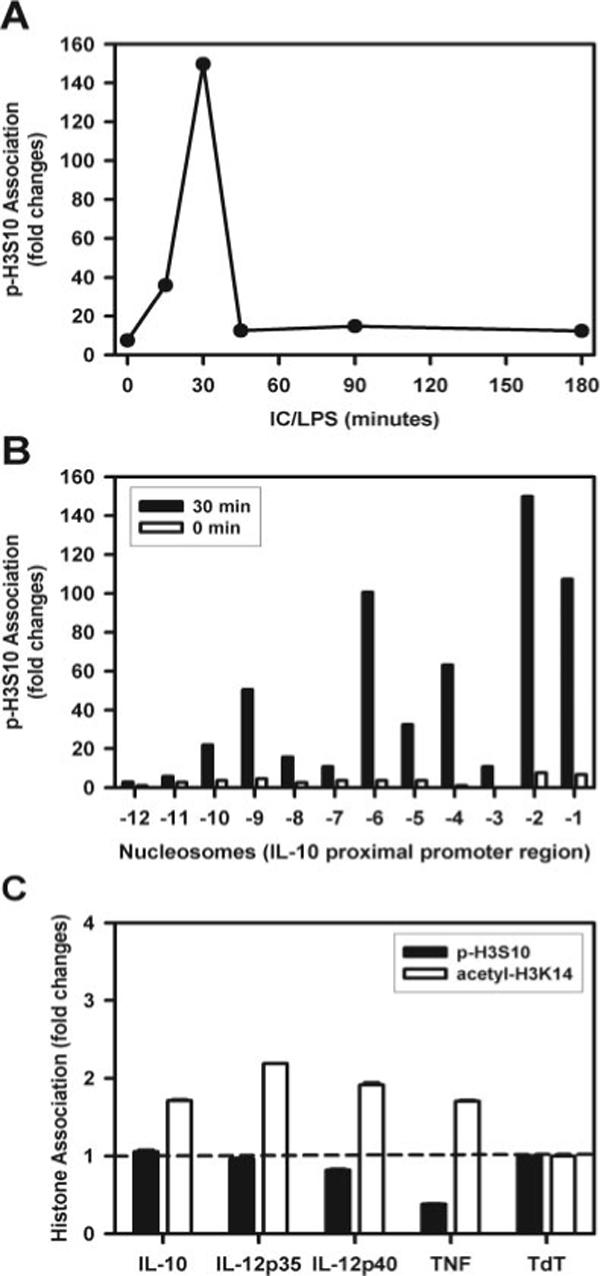
Histone H3 phosphorylation of Ser10 in nucleosomes associated with the promoter region of IL-10. A, BMMϕ were stimulated with IC plus 10 ng/ml LPS (IC/LPS) for 0, 15, 30, 45, 90, and 180 min. Cross-linked chromatin fragments were immunoprecipitated with anti-phosphorylated histone H3 at Ser10 Ab. The DNA was purified and examined for the presence of IL-10 promoter sequences corresponding to nucleosome −2 by QRT-PCR. The data were normalized to inputs at each time point and plotted graphically as fold changes relative to the data at 0 min. B, Immunoprecipitated DNA from the 30-min ChIP assay described in A was amplified with primers specific to each of the 12 nucleosomes by QRT-PCR. One representative experiment from three independent experiments is presented. C, ChIP assay to amplify segments of the IL-10, IL-12 (p35 and 40), and TNF-α promoters following immunoprecipitations with Ab to phosphorylated (■) or acetylated (□) histone H3. Levels were normalized to amplified TdT segment that was arbitrarily set as 1.
In unstimulated macrophages, there was little detectable phosphorylation at any of these locations (Fig. 2B, □). The negligible degree of histone H3 phosphorylation and acetylation in resting cells was similar for three different cytokines analyzed (IL-10, IL-12, and TNF-α) and comparable to the lymphocyte-specific TdT gene that is not expressed in macrophages (14) (Fig. 2C).
Acetylation of histone H3 at Lys14 on the IL-10 promoter
The acetylation of histones is an important modification that has been implicated in controlling gene expression (15). We examined the acetylation of histone H3 at Lys14 across the IL-10 promoter region over time. Although low levels of transient lysine acetylation of histone H3 at nucleosome 2 were detectable within the first 15 min of stimulation, the main peak of acetylation was not detected until 60 min after stimulation (Fig. 3A). By 120 min, this peak of acetylation had returned to baseline. The pattern and extent of acetylation was distinct from that observed with phosphorylation (Fig. 3B). The extent of acetylation at nucleosome 2 was modest relative to the extent of phosphorylation, and there were low levels of acetylation spanning a broad region corresponding to nucleosomes −3 to −8. There was no detectable acetylation at the distal regions of this promoter. Importantly, the peak of acetylation did not occur until 60 min after stimulation, a time by which the phosphorylation of histones was returning to baseline.
FIGURE 3.
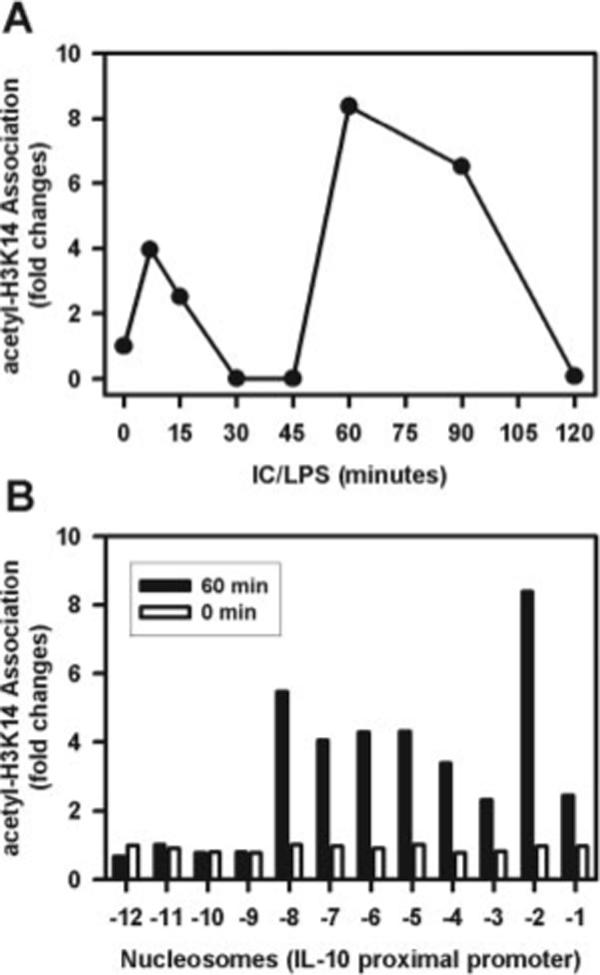
Histone H3 acetylation of Lys14 on nucleosomes associated with the promoter region of IL-10. A, BMMϕ were stimulated with IC plus LPS (10 ng/ml) for 0, 10, 15, 30, 45, 60, 90, and 120 min. Cross-linked chromatin fragments were immunoprecipitated with an Ab to acetylated histone H3 at Lys14. The DNA was purified and analyzed for the presence of IL-10 promoter sequences corresponding to nucleosomes −2 (●) by QRT-PCR. The data were normalized to inputs at each time point and plotted graphically as fold changes relative to the data at 0 min. B, The recovered DNA from the 60-min ChIP assay described in A was amplified with primers specific to each of the 12 nucleosomes by QRT-PCR. One representative from two independent experiments is presented.
Kinetics of Sp1 recruitment to the IL-10 promoter
Because Sp1 has been shown to play an important role in the regulation of IL-10 gene expression (12) and because this area of the promoter was rapidly phosphorylated, we assessed the kinetics of Sp1 recruitment to the IL-10 promoter. Macrophages were stimulated with LPS plus IC, and then Sp1 was immunoprecipitated for ChIP and analyzed over time. Sp1 could not be detected on the promoter for the first 30 min poststimulation. After this lag, however, Sp1 was rapidly recruited to the promoter, reaching a peak at 45 min posttreatment and gradually declining thereafter (Fig. 4A). The recruitment of Sp1 was highest in the area surrounding the Sp1 binding site (5′-GGAGGAGGAGCC-3′) in the nucleosome 2 (−196 to −76 bp); however, there was also a significant degree of Sp1 recruitment to the area corresponding to nucleosome 5 (−594 to −417 bp) that contained two potential Sp1 binding sites (5′-CTGCCCCACAGCAC-3′; 5′-AAAATCAGCCCTCT-3′) (Fig. 4B). Importantly, the kinetics of Sp1 recruitment to the area corresponding to nucleosome 2 temporally followed the phosphorylation of histone H3 at this site (see Fig. 2A).
FIGURE 4.
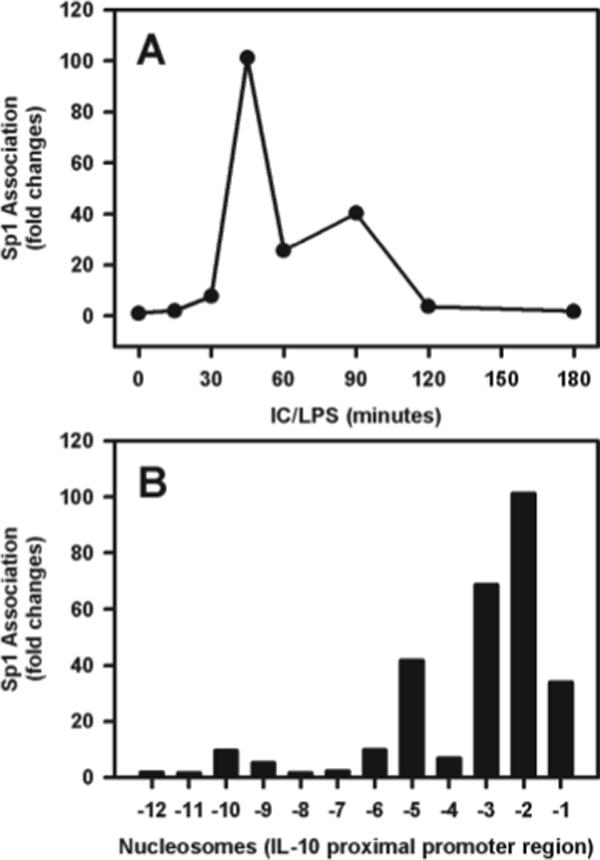
Recruitment of the Sp1 transcription factor to the IL-10 promoter. A, BMMϕ were stimulated with IC plus LPS (10 ng/ml) for 0, 15, 30, 45, 60, 90, 120, and 180 min. Cross-linked chromatin fragments were immunoprecipitated with anti-Sp1 Ab. The DNA was isolated and examined for the presence of IL-10 promoter sequences corresponding to nucleosome −2 by QRT-PCR. The data were normalized to inputs at each time point and plotted graphically as fold changes relative to the data at 0 min. B, The recovered DNA from the 45-min ChIP assay described in A was amplified with primers specific to each of the 12 nucleosomes by QRT-PCR. One representative from two independent experiments is presented.
Kinetic analysis of IL-10 gene transcription
We next compared the kinetics of histone modifications with the induction of IL-10 transcription. To do this we measured the levels of premature nuclear mRNA. This premature mRNA contains introns, and therefore the amplification of mRNA using an intron-specific primer can be used as an indicator of gene transcription (16). IL-10 pre-mRNA accumulation in macrophages treated with LPS plus IC was rapid and dynamic. It was detectable by as early as 15 min, and it peaked at 45 min poststimulation. By 120 min after stimulation, pre-mRNA levels had receded to background levels. As expected, mature IL-10 mRNA accumulation took slightly longer to be produced and it persisted longer (Fig. 5).
FIGURE 5.
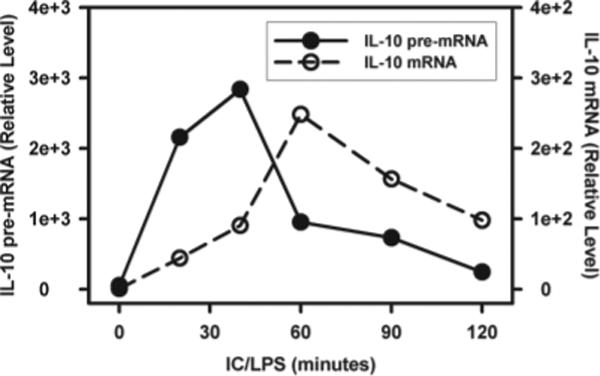
IL-10 gene transcription and mRNA accumulation in macrophages. BMMϕ were stimulated with IC plus LPS (10 ng/ml) for 0, 20, 40, 60, and 120 min. RNA isolated from the cytoplasm and nucleus was purified and treated with RNase-free DNase I. Cytoplasmic and nuclear RNA were reverse transcribed to cDNA using oligo(dT)20 primer and random hexamers, respectively. The generated cDNA was then subjected to QRT-PCR analysis. The IL-10 RNA levels were presented as arbitrary units that were derived from average normalization values of each sample at each time point by corresponding GAPDH. The IL-10 mRNA level at zero time point was arbitrarily set as 1. Results presented are one representative from two independent experiments run in triplicate.
There was a close temporal association between the production of IL-10 pre-mRNA, and both the phosphorylation of histones and the binding of Sp1 to the IL-10 promoter (Fig. 6). Phosphorylation at the Sp1 site peaked at 30 min. The binding of Sp1 and the production of IL-10 pre-mRNA transcripts peaked at 45 min (Fig. 6). The acetylation of histones, at this or other sites in the promoter, did not peak until 60 min, a time by which transcription was declining and Sp1 was no longer associated with the promoter. This later observation suggested that the acetylation of histones was not playing a primary role in IL-10 transcription.
FIGURE 6.
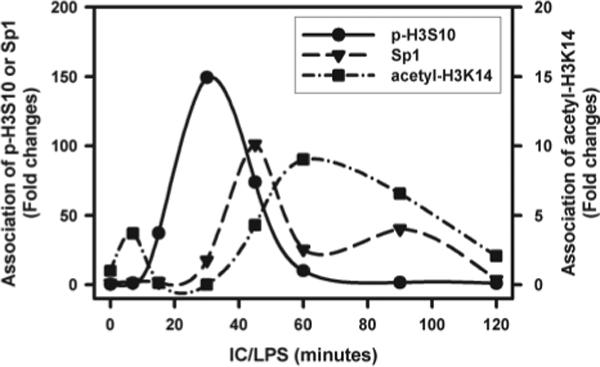
The kinetics of histone H3 phosphorylation, Sp1 recruitment, and histone H3 acetylation on nucleosome −2. BMMϕ were stimulated with IC plus LPS (10 ng/ml) for 0, 10, 15, 30, 45, 60, 90, and 120 min. Cross-linked chromatin fragments were immunoprecipitated with Ab to phosphorylated histone H3 at Ser10, Sp1 Ab, and Ab to acetylated histone H3 at Lys14, respectively. The DNA was purified and analyzed for the presence of IL-10 promoter sequences corresponding to nucleosome −2 by QRT-PCR. The data were normalized to inputs at each time point and plotted graphically as fold changes relative to the data at 0 min.
To further examine histone acetylation, macrophages were treated with increasing doses of the HDAC inhibitor, TSA, before stimulation. Both IL-10 mRNA and protein levels were then determined (Fig. 7). The inhibition of deacetylation failed to influence IL-10 protein production (Fig. 7A) and mRNA levels (B) when added to cells at concentrations that were sufficient to substantially inhibit IL-12 (A) and increase histone H3 acetylation (C). This observation is consistent with the hypothesis that an increase in histone H3 acetylation was not a prerequisite for IL-10 gene transcription following stimulation by LPS plus IC.
FIGURE 7.
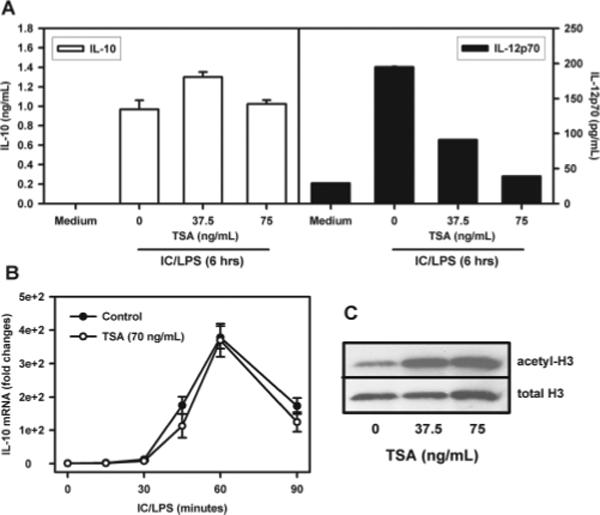
Cytokine changes following histone acetylation. TSA was added to BMMϕ for 1 h, and the cells were then washed with warmed medium before stimulation with 10 ng/ml LPS plus IC. A, After LPS plus IC stimulation for 6 h, the supernatants were collected and IL-10 and IL-12 were measured by ELISA. Results shown are one representative from two independent experiments conducted in triplicate (mean ± SD). B, BMMϕ were stimulated with IC plus LPS (10 ng/ml) for 0, 15, 30, 60, and 90 min. RNA isolation, cDNA synthesis, and QRT-PCR analysis were described above. The IL-10 mRNA levels were presented as arbitrary units that were derived from average normalization values of each sample at each time point by corresponding GAPDH. The IL-10 mRNA level at zero time point was arbitrarily set as 1. C, Different doses of TSA were added to BMMϕ for 1 h and whole-cell lysates were used for Western blotting analysis of histone H3 acetylation as detected by anti-acetyl histone H3 Ab.
The role of ERK in chromatin phosphorylation
We previously reported that maximal IL-10 production by macrophages depended on ERK activation (10), which has also been recently reported in RAW 264.7 cells (17). In the present work, we examined the levels of histone H3 phosphorylation and Sp1 recruitment to the IL-10 promoter following treatment of cells with PD98059, an inhibitor of ERK activation. Treatment of macrophages with PD98059 resulted in a substantial loss of histone H3 phosphorylation (Fig. 8). Sp1 recruitment was also dramatically reduced after treatment with PD98059 (Fig. 8). These data are consistent with the hypothesis that ERK activation leads to the phosphorylation of chromatin at the IL-10 promoter, allowing the recruitment of Sp1 to the promoter.
FIGURE 8.
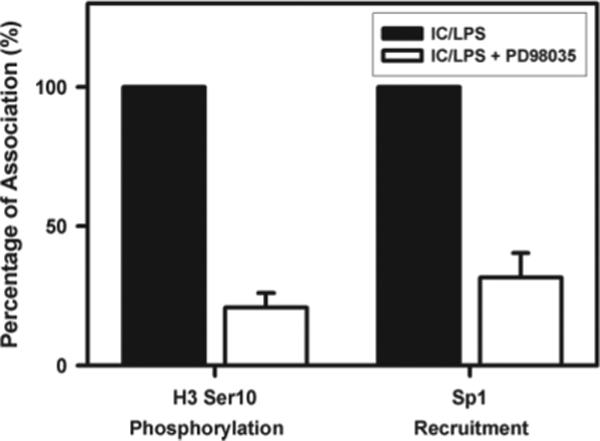
Inhibition of histone modifications by blocking ERK. BMMϕ were pretreated with PD98059 (15 μM) (□) or drug vehicle (■) for 1 h and then stimulated with IC plus LPS (10 ng/ml). Cross-linked chromatin fragments were immunoprecipitated with Ab to phosphorylated histone H3 at Ser10 or Ab to Sp1. The DNA was purified and analyzed for the presence of IL-10 promoter sequences corresponding to nucleosome −2 by QRT-PCR. The data were plotted graphically as the percentage change relative to drug vehicle (IC/LPS) at 30 min poststimulation for histone H3 phosphorylation, and at 45 min poststimulation for Sp1 recruitment. One representative from two independent experiments is presented.
The ERK-dependent binding of Sp1 to the IL-10 promoter suggested that histone phosphorylation might increase chromatin accessibility, possibly due to nucleosome disassembly or sliding of nucleosomes away from the promoter (18). To address this, we examined the sensitivity of DNA to cleavage by nucleases in the presence or absence of ERK inhibition. Chromatin sensitivity to MNase was measured at nucleosome 2 and compared with nucleo-some 12 (Fig. 9A). In unstimulated cells (time 0), this region was relatively resistant to MNase cleavage. By 30 min poststimulation, there was a dramatic increase in MNase cleavage at nucleosome 2. Nucleosome 12 also exhibited a small increase in MNase sensitivity, but it was significantly lower than nucleosome 2. Cleavage accessibility at nucleosome 2 peaked at 60 min poststimulation and persisted for 120 min. Similar studies were performed using DNase (Fig. 9B). Similar to MNase treatment, nucleosome 2 was relatively resistant to cleavage in the absence of stimulation. The addition of LPS plus IC resulted in a dramatic increase in cleavage. Inhibiting ERK by the addition of PD98059 reduced sensitivity to DNase by greater than half (Fig. 9B). These data indicate that histone H3 phosphorylation, initiated by the activation of ERK, was a critical event in inducing chromatin accessibility to the transcription factors that induce IL-10 production in macrophages.
FIGURE 9.
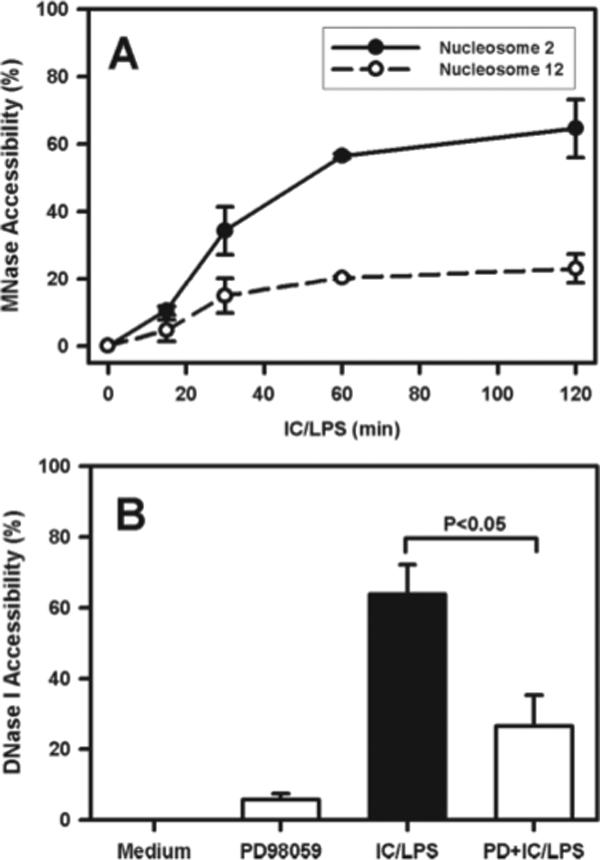
Enhanced chromatin accessibility following ERK activation. A, BMMϕ were stimulated with IC plus LPS (10 ng/ml) for 0, 15, 30, 60, and 120 min. Nuclei were isolated at each time point poststimulation and treated with or without MNase. DNA was purified and analyzed for the presence of sequences corresponding to nucleosomes 2 and 12 by QRT-PCR. The data were graphically plotted as percentage of MNase accessibility relative to the samples of stimulation at zero time point. B, BMMϕ were pretreated with PD98059 (15 μM) (□) or drug vehicle control (■) for 1 h and then stimulated with IC plus LPS (10 ng/ml) for 60 min. Nuclei were isolated from each group and treated with or without DNase I followed by extraction of genomic DNA. The presence of sequences corresponding to nucleosome −2 was examined by QRT-PCR. The data were expressed as percentage of DNase I accessibility relative to undigested genomic DNA sample and graphically plotted for each treated group. One representative from two independent experiments is presented. Each experiment was run in triplicate and shown as mean ± SD.
Discussion
We previously demonstrated that activating macrophages in the presence of IC leads to a specific hyperinduction of the cytokine, IL-10 (11). We recently made the observation that this hyperinduction requires activation of the MAPK pathway, leading to the phosphorylation of ERK (10). In the present work, we explore the underlying molecular mechanisms by systemically examining the spatiotemporal changes of chromatin remodeling along the macrophage IL-10 promoter. We show that the phosphorylation of histones occurs mainly at the IL-10 promoter, and specifically at transcription factor binding sites in this promoter. We also show that the kinetics of histone phosphorylation closely follow the kinetics of both transcription factor binding and transcriptional activation. These results strongly suggest that phosphorylation is causally related to transcription.
This work suggests that the transcription of an anti-inflammatory cytokine, such as IL-10, is regulated by a more complex mechanism than the inflammatory cytokines, such as TNF-α. In addition to transcription factor activation, a second level of regulation occurs at the level of chromatin. The speed and specificity of this second level of regulation are quite remarkable. The covalent modifications to histones occur within minutes of stimulation and they are reversed almost as quickly. The specificity of these modifications was also unexpected. Not only were these alterations specific to IL-10 and not to any of the other inflammatory cytokines examined, but also segments of the IL-10 promoter located only 500 bases away from highly modified regions underwent no detectable phosphorylation changes throughout the observation period. As far as we are aware, this type of regulation has not been reported for any other cytokine to date.
There are aspects of this regulation that closely resemble the “nucleosome response” previously reported by Mahadevan and colleagues (19). In a series of studies, this group demonstrated that the nucleosome response is initiated by the activation of the MAPK cascade in response to cellular stressors. This leads directly to immediate-early gene expression. This gene expression occurs in the absence of de novo protein synthesis (20), and in some cases the transcription factors are already associated with their respective DNA binding elements (19). We show that a similar cellular response occurs when macrophages encounter IC. The histones are similarly phosphorylated in response to MAPK activation. However, in the case of IL-10, this phosphorylation does not result in gene expression. Macrophages exposed to IC alone produce no IL-10, despite the activation of ERK and the phosphorylation of histones. Rather, transcription of the IL-10 gene requires a second set of signals provided by TLR stimulation. This stimulation activates the necessary transcription factors, allowing them to bind to the phosphorylated chromatin that is now accessible to them (Fig. 10). Thus, the induction of this anti-inflammatory cytokine is more complex than many of the inflammatory cytokines, whose synthesis mainly requires the recruitment of transcription factors and histone acetylation (21, 22).
FIGURE 10.
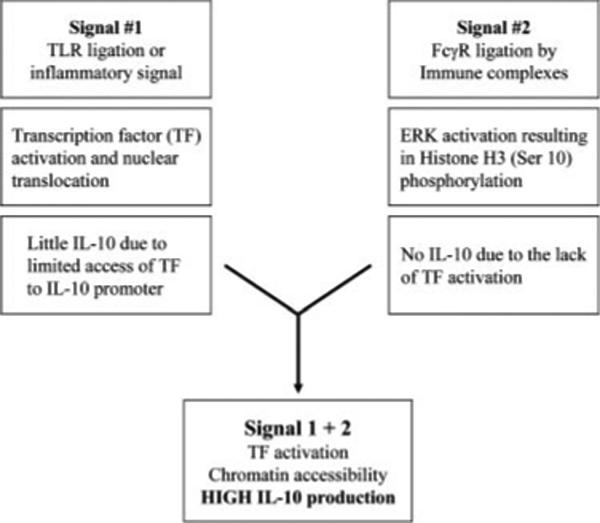
A schematic diagram of regulation of IL-10 gene expression in macrophages treated with IC in the presence of TLR ligands or other inflammatory signals.
The now well-described concept of the “histone code” predicts that patterns of histone modifications at specific genes can affect gene expression (23, 24). In the simplest terms, these modifications can influence the recruitment of transcription factors, polymerases, and coactivators to enhance or inhibit gene expression (25). In dividing cells, these modifications can be quite complex, including mono-, di-, and trimethylation, ubiquitination, or even sumoylation (26). These covalent modifications can “mark” chromatin for specific patterns of gene expression. In daughter cells, the gene expression profile is preserved over many generations, despite the condensation of chromosomes during mitosis. The marking of chromatin for cytokine gene expression has recently been reported in T cells. In the case of IL-4, Th2-specific enhancement of histone acetylation and DNA demethylation in the control locus allows Th2 cells to continue to produce IL-4 following daughter cell division (27, 28). A similar picture is emerging with regard to IL-10 production in T cells, where chromatin accessibility may predict stable IL-10 gene expression (13, 29). Unlike T cells, macrophages are end-stage cells that do not divide and therefore need no long-term marking of the IL-10 gene. In fact, the sustained production of IL-10 in Th2 cells and their progeny (30) is quite distinct from the transient expression of IL-10 observed in macrophages. Thus, it was likely that different mechanisms would exist for regulating IL-10 gene expressions in macrophages relative to T cells. In T cells, chromatin modifications accumulate slowly over generations and they are maintained in daughter cells. However, in macrophages, histone phosphorylation was dynamically regulated across the IL-10 proximal promoter. It peaks within 30 min of stimulation and then rapidly disappears. The time of histone H3 phosphorylation corresponds closely with the kinetics of the production of IL-10 pre-mRNA.
Similar to previous studies with other genes and other systems (31, 32), the acetylation of histone H3 at the IL-10 promoter follows histone H3 phosphorylation. Unlike many of these other systems, however, this modification does not appear to coincide with transcriptional activation. First, transcription factors bind to the promoter before acetylation occurs, and are in fact released from the promoter by the time that acetylation peaks. Second, the production of IL-10 pre-mRNA transcripts climaxes and declines before histone acetylation crests. Finally, treating macrophages with HDAC inhibitors does not affect IL-10 production. Thus, whereas histone H3 phosphorylation temporally correlates with IL-10 gene expression, acetylation does not.
Our results predict that the phosphorylation of histones is tightly associated with transcriptional activation of the IL-10 gene. In other cellular systems, the phosphorylation of histones is not always associated with gene activation. In fact, histone H3 is highly phosphorylated on serine 10 on condensed chromatin during mitosis (23). Unlike the phosphorylation events associated with mitosis, however, the IC-induced phosphorylation that we observe is specific to individual regions of the IL-10 promoter rather than uniform, and it is far more transient. Studies are underway to identify the kinase(s) responsible for histone phosphorylation, and the phosphatase(s) mediating the rapid dephosphorylation of histones following activation. We predict that mechanisms to prolong histone phosphorylation may be exploited to manipulate macrophage IL-10 production.
In summary, we provide a detailed analysis of the modifications of the nucleosomes associated with the promoter region of IL-10 gene. We show that the evanescent, ERK-dependent, phosphorylation of histone H3 at Ser10 is essential for IL-10 superinduction. These studies provide a molecular understanding of IL-10 gene regulation in macrophages, which may lead to novel ways to manipulate IL-10 levels. Because the production of high levels of IL-10 can either result in a beneficial anti-inflammatory response, or a pathological immunosuppressive response, new ways to manipulate the production of this cytokine can be used to enhance or inhibit immune responses, as necessary.
Acknowledgments
We are grateful to Ziyan Yang for her technical assistance, and to Drs. Shanjin Cao and Dalit Strauss-Ayali for their helpful suggestions.
Footnotes
This work was supported in part by National Institutes of Health Grant AI49383.
Abbreviations used in this paper: IC, immune complex; HDAC, histone deacetylase; TSA, trichostatin A; MNase, micrococcal nuclease; BMMϕ, bone marrow-derived macrophage; QRT-PCR, quantitative real-time PCR; ChIP, chromatin immunoprecipitation.
Disclosures
The authors have no financial conflict of interest.
References
- 1.van der Poll T, van Deventer SJ. Cytokines and anticytokines in the pathogenesis of sepsis. Infect. Dis. Clin. North Am. 1999;13:413–426. doi: 10.1016/s0891-5520(05)70083-0. [DOI] [PubMed] [Google Scholar]
- 2.Waage A, Aasen AO. Different role of cytokine mediators in septic shock related to meningococcal disease and surgery/polytrauma. Immunol. Rev. 1992;127:221–230. doi: 10.1111/j.1600-065x.1992.tb01416.x. [DOI] [PubMed] [Google Scholar]
- 3.Kawai T, Akira S. Pathogen recognition with Toll-like receptors. Curr. Opin. Immunol. 2005;17:338–344. doi: 10.1016/j.coi.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 4.Zdanov A. Structural features of the interleukin-10 family of cytokines. Curr. Pharm. Des. 2004;10:3873–3884. doi: 10.2174/1381612043382602. [DOI] [PubMed] [Google Scholar]
- 5.Donnelly RP, Sheikh F, Kotenko SV, Dickensheets H. The expanded family of class II cytokines that share the IL-10 receptor-2 (IL-10R2) chain. J. Leukocyte Biol. 2004;76:314–321. doi: 10.1189/jlb.0204117. [DOI] [PubMed] [Google Scholar]
- 6.Conti P, Kempuraj D, Frydas S, Kandere K, Boucher W, Letourneau R, Madhappan B, Sagimoto K, Christodoulou S, Theoharides TC. IL-10 subfamily members: IL-19, IL-20, IL-22, IL-24 and IL-26. Immunol. Lett. 2003;88:171–174. doi: 10.1016/s0165-2478(03)00087-7. [DOI] [PubMed] [Google Scholar]
- 7.Grutz G. New insights into the molecular mechanism of interleukin-10-mediated immunosuppression. J. Leukocyte Biol. 2005;77:3–15. doi: 10.1189/jlb.0904484. [DOI] [PubMed] [Google Scholar]
- 8.Anderson CF, Mosser DM. Cutting edge: biasing immune responses by directing antigen to macrophage Fcγ receptors. J. Immunol. 2002;168:3697–3701. doi: 10.4049/jimmunol.168.8.3697. [DOI] [PubMed] [Google Scholar]
- 9.Kane MM, Mosser DM. The role of IL-10 in promoting disease progression in leishmaniasis. J. Immunol. 2001;166:1141–1147. doi: 10.4049/jimmunol.166.2.1141. [DOI] [PubMed] [Google Scholar]
- 10.Lucas M, Zhang X, Prasanna V, Mosser DM. ERK activation following macrophage Fcγ ligation leads to chromatin modifications at the IL-10 locus. J. Immunol. 2005;175:469–477. doi: 10.4049/jimmunol.175.1.469. [DOI] [PubMed] [Google Scholar]
- 11.Sutterwala FS, Noel GJ, Salgame P, Mosser DM. Reversal of proinflammatory responses by ligating the macrophage Fcγ receptor type I. J. Exp. Med. 1998;188:217–222. doi: 10.1084/jem.188.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brightbill HD, Plevy SE, Modlin RL, Smale ST. A prominent role for Sp1 during lipopolysaccharide-mediated induction of the IL-10 promoter in macrophages. J. Immunol. 2000;164:1940–1951. doi: 10.4049/jimmunol.164.4.1940. [DOI] [PubMed] [Google Scholar]
- 13.Im SH, Hueber A, Monticelli S, Kang KH, Rao A. Chromatin-level regulation of the IL10 gene in T cells. J. Biol. Chem. 2004;279:46818–46825. doi: 10.1074/jbc.M401722200. [DOI] [PubMed] [Google Scholar]
- 14.Zhou L, Nazarian AA, Smale ST. Interleukin-10 inhibits inter-leukin-12 p40 gene transcription by targeting a late event in the activation pathway. Mol. Cell. Biol. 2004;24:2385–2396. doi: 10.1128/MCB.24.6.2385-2396.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Struhl K. Transcriptional activation: mediator can act after preinitiation complex formation. Mol. Cell. 2005;17:752–754. doi: 10.1016/j.molcel.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 16.Goriely S, Van LC, Dadkhah R, Libin M, De WD, Demonte D, Willems F, Goldman M. A defect in nucleosome remodeling prevents IL-12(p35) gene transcription in neonatal dendritic cells. J. Exp. Med. 2004;199:1011–1016. doi: 10.1084/jem.20031272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu YW, Chen CC, Tseng HP, Chang WC. Lipopolysaccharide-induced transcriptional activation of interleukin-10 is mediated by MAPK-and NF-κB-induced CCAAT/enhancer-binding protein δ in mouse macrophages. Cell Signal. 2006 doi: 10.1016/j.cellsig.2005.12.001. In press. [DOI] [PubMed] [Google Scholar]
- 18.Boeger H, Griesenbeck J, Strattan JS, Kornberg RD. Removal of promoter nucleosomes by disassembly rather than sliding in vivo. Mol. Cell. 2004;14:667–673. doi: 10.1016/j.molcel.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 19.Thomson S, Clayton AL, Hazzalin CA, Rose S, Barratt MJ, Mahadevan LC. The nucleosomal response associated with immediateearly gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J. 1999;18:4779–4793. doi: 10.1093/emboj/18.17.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clayton AL, Mahadevan LC. MAP kinase-mediated phosphoacetylation of histone H3 and inducible gene regulation. FEBS Lett. 2003;546:51–58. doi: 10.1016/s0014-5793(03)00451-4. [DOI] [PubMed] [Google Scholar]
- 21.Saccani S, Pantano S, Natoli G. p38-dependent marking of inflammatory genes for increased NF-κB recruitment. Nat. Immunol. 2002;3:69–75. doi: 10.1038/ni748. [DOI] [PubMed] [Google Scholar]
- 22.Avni O, Lee D, Macian F, Szabo SJ, Glimcher LH, Rao A. TH cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nat. Immunol. 2002;3:643–651. doi: 10.1038/ni808. [DOI] [PubMed] [Google Scholar]
- 23.Cheung P, Allis CD, Sassone-Corsi P. Signaling to chromatin through histone modifications. Cell. 2000;103:263–271. doi: 10.1016/s0092-8674(00)00118-5. [DOI] [PubMed] [Google Scholar]
- 24.Wood A, Schneider J, Shilatifard A. Cross-talking histones: implications for the regulation of gene expression and DNA repair. Biochem. Cell Biol. 2005;83:460–467. doi: 10.1139/o05-116. [DOI] [PubMed] [Google Scholar]
- 25.Kornberg RD. Mediator and the mechanism of transcriptional activation. Trends Biochem. Sci. 2005;30:235–239. doi: 10.1016/j.tibs.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 26.Mellor J. The dynamics of chromatin remodeling at promoters. Mol. Cell. 2005;19:147–157. doi: 10.1016/j.molcel.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 27.Lee DU, Agarwal S, Rao A. Th2 lineage commitment and efficient IL-4 production involves extended demethylation of the IL-4 gene. Immunity. 2002;16:649–660. doi: 10.1016/s1074-7613(02)00314-x. [DOI] [PubMed] [Google Scholar]
- 28.Fields PE, Lee GR, Kim ST, Bartsevich VV, Flavell RA. Th2-specific chromatin remodeling and enhancer activity in the Th2 cytokine locus control region. Immunity. 2004;21:865–876. doi: 10.1016/j.immuni.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 29.Shoemaker J, Saraiva M, O'Garra A. GATA-3 directly remodels the IL-10 locus independently of IL-4 in CD4+ T cells. J. Immunol. 2006;176:3470–3479. doi: 10.4049/jimmunol.176.6.3470. [DOI] [PubMed] [Google Scholar]
- 30.O'Garra A, Vieira P. Regulatory T cells and mechanisms of immune system control. Nat. Med. 2004;10:801–805. doi: 10.1038/nm0804-801. [DOI] [PubMed] [Google Scholar]
- 31.Nowak SJ, Corces VG. Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet. 2004;20:214–220. doi: 10.1016/j.tig.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 32.He Z, Cho YY, Ma WY, Choi HS, Bode AM, Dong Z. Regulation of ultraviolet B-induced phosphorylation of histone H3 at serine 10 by Fyn kinase. J. Biol. Chem. 2005;280:2446–2454. doi: 10.1074/jbc.M402053200. [DOI] [PubMed] [Google Scholar]