

Abstract
Notch signaling plays a key role in various cell differentiation processes including bone homeostasis. However, the specific involvement of Notch in regulating osteoclastogenesis is still controversial. In the present study, we show that RANKL induces expression of Jagged1 and Notch2 in bone marrow macrophages during osteoclast differentiation. Suppression of Notch signaling by a selective γ-secretase inhibitor or Notch2 short hairpin RNA suppresses RANKL-induced osteoclastogenesis. In contrast, induction of Notch signaling by Jagged1 or by ectopic expression of intracellular Notch2 enhances NFATc1 promoter activity and expression and promotes osteoclastogenesis. Finally, we found that Notch2 and p65 interact in the nuclei of RANKL-stimulated cells and that both proteins are recruited to the NFATc1 promoter, driving its expression. Taken together, our results show a new molecular cross talk between Notch and NF-κB pathways that is relevant in osteoclastogenesis.
Osteoclasts are bone-resorbing multinucleated cells derived from the monocyte-macrophage lineage (5, 8, 56). The differentiation and activation of osteoclasts are closely regulated by osteoblasts or bone marrow-derived stromal cells (8, 56). Receptor activator of NF-κB ligand (RANKL) and macrophage colony-stimulating factor (M-CSF) are both crucial for osteoclast development (8, 56). RANKL is a member of the tumor necrosis factor (TNF) family of cytokines, and its expression is regulated by a number of bone-resorbing factors including 1α,25-dihydroxyvitamin D3 [1α,25(OH)2D3], interleukin-1 (IL-1), IL-6, and TNF-α. M-CSF induces the proliferation and survival of monocyte/macrophage osteoclast precursor cells, whereas RANKL mainly regulates differentiation and activation of these precursors (5).
RANK is expressed as a transmenbrane heterotrimer on the surfaces of hematopoietic osteoclast progenitors and mature osteoclasts. The in vivo significance of the RANKL-RANK signaling pathway has been verified in mice by targeted disruption of either of the associated genes, which results in severe osteopetrosis and a total lack of osteoclasts (14, 29). Although the RANK cytoplasmic domain interacts with various TNF receptor-associated factors (TRAFs) (TRAF1 to -6, except for TRAF4) only TRAF6 is essential for RANK-dependent osteoclast differentiation, as evidenced by the osteopetrosis developed in mice lacking TRAF6 (41). The RANKL-RANK interaction promotes osteoclast differentiation through activation of several intracellular pathways including nuclear factor of activated T cells (NFAT) (55), NF-κB (18, 25), Fos (19), and mitogen-activated protein kinase (24) pathways. In agreement with this, c-Fos-deficient mice and NF-κB p50 and p52 double-knockout mice developed typical osteopetrosis, accompanied by growth retardation, excessive calcification in the long bones, and impaired tooth eruption due to lack of osteoclasts. Recently, Asagiri et al. demonstrated the requirement for NFATc1 in the generation of osteoclasts in vivo by transferring NFATc1-deficient hematopoietic stem cells into c-Fos-deficient mice (4). Recent studies showed that, in addition to RANKL-RANK signaling, other costimulatory signals such as those involving multiple immunoglobulin-like receptors associated with immunoreceptor tyrosine-based activation motif-harboring adapters, Fc receptor common γ chain subunit (FcRγ), and DNAX-activating protein 12 are important in regulating osteoclast differentiation (28). Furthermore, calcineurin-NFATc1 calcium signaling activates the calcium/calmodulin-activated kinase-CREB (cyclic AMP response element binding protein) pathway, which also plays a critical role in osteoclastogenesis (46).
Notch signaling is a highly conserved signaling pathway that plays a critical role in a variety of cellular functions including cell proliferation, differentiation, and apoptosis (3). In vertebrates, four Notch receptors (Notch1 to -4) and five ligands (Delta1, -3, and -4 and Jagged1 and -2) have been identified; all are single-span transmembrane polypeptides that respond to cell-cell interactions (9). Notch receptors contain multiple epidermal growth factor-like repeats and three cysteine-rich Notch/LIN-12 repeats in the extracellular domain, whereas the intracellular domain contains seven cdc10/ankyrin repeats, a glutamine-rich domain, and a PEST sequence. Notch ligands also have epidermal growth factor repeats in the extracellular domain in addition to a unique cysteine-rich N-terminal region, referred to as the Delta:Serrate:LAG2 domain, and a small intracellular domain. Notch is activated through binding to the appropriate ligand present on neighboring cells, which results in the proteolytic cleavage of Notch receptor by presenilin/γ-secretase and the nuclear translocation of the Notch intracellular domain (NICD) (3, 9). Once in the nucleus, NICD interacts with the DNA-binding protein CSL [named for CBF1/RBPJκ, Su (H), and LAG1] and activates transcription of its target genes such as the hairy enhancer of split 1 (HES-1) and HES-5 genes (20). Alternatively, it has been reported to transmit signals through CSL-independent pathways by interacting with other signaling molecules, such as phosphatidylinositol 3-kinase, Src, and NF-κB (40, 50, 51, 52, 58).
Mice deficient in several Notch family members such as Notch1, Notch2, Jagged1, and Delta1 die during embryogenesis before bone formation (23, 36, 53, 61); thus, it is impossible to use these models for the study of osteoclastogenesis. However, multiple pieces of evidence indicate that Notch signaling dysfunction results in bone disease. For example, presenilin 1-deficient mice and Delta3 mutant mice (Pudgy) exhibit axial skeletal defects (10, 30, 35, 48) and Alagille syndrome (OMIM no. 118450 and 610205), which is caused by mutation in the Jagged1 or Notch2 gene and is characterized by the presence of “butterfly” vertebrae (36, 37, 38). Moreover, parathyroid hormone/parathyroid hormone-related peptide receptor transgenic mice that show increased Jagged1 expression in the osteoblastic stromal cells and contain a higher number of NICD-positive hematopoietic stem cells in bone marrow than wild-type mice display increased trabecular bone volume and decreased cortical bone thickness of the long bones (11, 12). In addition, in vitro studies also support a role for the Notch pathway in osteoblastogenesis and bone formation (13, 43, 47, 57, 63). Recently two different groups demonstrated that specific deletion of presenilin 1 and 2 in the skeletogenic mesenchyme in the limb and the calvaria leads to increased trabecular bone mass. At the biochemical level, they showed that Notch signaling inhibits osteoblast differentiation by repressing the Runx2 transactivation function. Furthermore, knockout mice developed an age-related osteoporosis resulting from increased osteoclastic activity, likely due to decreased osteoprogerin mRNA expression (15, 21). These results are in agreement with previous in vitro data showing that Notch activation by Delta1 reduces the surface levels of the M-CSF receptor, c-Fms, in osteoclast precursor cells and enhances the expression of osteoprogerin in stromal cells, resulting in the downregulation of osteoclastogenesis (62).
To further study the functional involvement of Notch in RANKL-dependent osteoclastogenesis, we first analyzed the expression patterns and the activation status of several Notch family members and determined the effects of inhibiting or activating the Notch pathway at different stages of osteoclast differentiation. In addition, we addressed the molecular mechanisms by which Notch modulates RANKL-induced osteoclastogenesis at the chromatin level.
MATERIALS AND METHODS
Animals.
This study was approved by the Council on Animal Care and Gene Transfer at Fukuoka Dental College. Male ddY mice (3 to 5 weeks old) were purchased from the Kyudo (Tosu, Saga, Japan). Mice were housed at the Animal Center of Fukuoka Dental College.
Reagents.
Recombinant human RANKL and M-CSF were purchased from PeproTech Inc. (Rocky Hill, NJ). γ-Secretase inhibitor X (L685,458) (GSI), a selective inhibitor of Notch signaling, was purchased from Calbiochem (La, Jolla, CA). Cleaved Notch1 (Val1744) and anti-Notch1 antibodies (C-20) were purchased from Cell Signaling (Beverly, MA) and Santa Cruz Biotechnology (Santa Cruz, CA), respectively. The anti-Notch2 antibody (C651.6DbHN) was purchased from Developmental Studies Hybridoma Bank (University of Iowa, Iowa City). The anti-RBPJκ antibody was purchased from the Institute of Immunology (Tokyo, Japan). Anti-NFATc1, -p65, -p50, -RANK, -IκBα, -histone deacetylase 1 (HDAC1), and -c-Fos antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). FLAG-fused human Jagged1 (Jagged-1 FL) was cloned and then purified as described previously (43, 62).
Cell culture.
Bone marrow-derived macrophages (BMMs) were prepared as osteoclast precursors from 3- to 5-week-old male ddY mice. Bone marrow cells obtained from the mouse tibia were suspended in 60-mm-diameter dishes for 16 h in the presence of M-CSF (50 ng/ml) in α-minimal essential medium containing 10% fetal bovine serum. Then, nonadherent cells were harvested and further cultured for 2 days with M-CSF (50 ng/ml). The adherent cells, most of which expressed macrophage-specific antigens such as Mac-1, Moma-2, and F4/80, were used as BMMs. BMMs were cultured for 3 days with RANKL (50 ng/ml). The macrophage cell line RAW 264.7 (RAW cells) (ATCC, Manassas, VA) was cultured for 3 days with RANKL (20 ng/ml). Cultures were fixed with 3.7% formaldehyde, and osteoclasts were detected by staining for tartrate-resistant acid phosphatase (TRAP). TRAP-positive multinucleated cells (MNCs) containing more than three nuclei were observed under a microscope and counted as osteoclasts. Soluble or immobilized Jagged1 was used as described elsewhere (43, 62).
Gene chip analysis.
RAW cells were stimulated for 3 days with or without RANKL (20 ng/ml). Total RNA from untreated or RANKL-treated RAW cells was extracted from three independent experiments using Trizol (Invitrogen, Carlsbad, CA). Total RNA (15 μg) was used for cDNA synthesis by reverse transcription followed by synthesis of biotinylated cRNA via in vitro transcription. After cRNA fragmentation, hybridization with a mouse U74Av2 GeneChip (Affymetrix, Santa Clara, CA) displaying probes for 12,000 mouse genes/expressed sequence tags was performed according to the manufacturer's protocol. Chips were washed, stained with streptavidin-phycoerythrin, and analyzed using a scanner and accompanying gene expression software GeneSpring (Agilent Technologies, Santa Clara, CA).
PCR analysis.
For reverse transcription-PCR (RT-PCR), total RNA prepared using Trizol was amplified using Superscript II and Taq polymerase (Invitrogen). Primer sequences were as described by Yamada et al. (62). The PCR conditions were as follows: 94°C (3 min), 60°C (2 min), and 72°C (3 min) for the primary cycle and 94°C (45 s), 60°C (1 min), and 72°C (1.5 min) for the following 30 cycles. The fluorescence of each PCR product was detected using an image analyzer (fluoro image analyzer FLA-2000F; Fuji Film, Tokyo, Japan).
Coprecipitation and immunoblotting.
Cells were lysed in TNT buffer (20 mM Tris-HCl, pH 7.5, 200 mM NaCl, 1% Triton X-100, 1 mM dithiothreitol) containing protease inhibitors (Roche, Basel, Switzerland). Cytosolic, membrane, and nuclear fraction proteins were extracted from cells using a Calbiochem Proteo Extract subcellular proteosome extraction kit (Merck KGaA, Darmstadt, Germany) by following the manufacturer's protocol. For coprecipitation experiments, nuclear extracts were incubated for 6 h at 4°C with anti-Notch2 antibody coupled to protein A/G-Sepharose beads. The immune complex was extensively washed with TNT buffer, and samples were boiled and analyzed by immunoblotting.
Protein content was measured with Pierce reagent by following the manufacturer's protocol. Twenty micrograms of protein was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane using 100 V for 1 h at 4°C. These membranes were then incubated with antibodies at 1:500 to 1:1,000 dilutions in 5% dry milk solution plus 0.01% azide overnight at 4°C. Subsequently, blots were washed in TTBS (10 mM Tris-HCl, 50 mM NaCl, 0.25% Tween 20) and incubated with a horseradish peroxidase-conjugated secondary antibody. The immunoreactive proteins were visualized using enhanced chemiluminescence (Amersham).
Immunofluorescence staining.
Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline and incubated with 0.3% H2O2 in methanol for 30 min followed by 10% horse serum for 1 h. The slides were then incubated with primary antibodies for Notch1 and Notch2 overnight at 4°C. These primary antibodies were detected by goat anti-rabbit Alexa 488 and goat anti-rat Alexa 568 (Molecular Probes) and visualized by fluorescence microscopy (TMD 300; Nikon, Tokyo, Japan). After fluorescence analysis, cells were stained with TRAP and nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI).
shRNA cell lines.
For tetracycline-regulated expression of short hairpin RNAs (shRNAs) (Tet-on) that target Notch1 and Notch2 constructs, we designed four variant shRNAs to target the mRNA on a Block-iT RNA interference (RNAi) designer (https://rnaidesigner.invitrogen.com/rnaiexpress) (Invitrogen). First, double-stranded oligonucleotides that encoded the shRNAs were cloned into the pENTR/H1/TO vector using a Block-iT inducible H1 RNAi entry vector kit by following the manufacturer's protocol (Invitrogen). We screened the pENTR/H1/TO Notch1 and Notch2 shRNA clones by transfecting them into RAW cells stably expressing pcDNA6/TR. Transfection was performed using Lipofectamine 2000 (Invitrogen), and cells were maintained in 50 μg/ml blasticidin or 100 μg/ml zeocin (Invitrogen) for selection. Resistant cell lines were evaluated for their ability to attenuate Notch1 and Notch2 expression by RT-PCR and immunoblotting. Four of the tested constructs were then used to attenuate Notch1 and Notch2 shRNA-expressing cell lines (RAW-teton-shNotch1 and RAW-teton-shNotch2). Tetracycline-resistant pcDNA6/TR-positive clones were used as parent RAW cells.
RNAi assay.
The RNAi assay was described previously (32). Three small interference RNA (siRNA) molecules targeting RBPJκ (siRBPJκ) (Rbpsuh Stealth Select 3 RNAi [MSS208567, MSS208565, and MSS208566]) were purchased from Invitrogen. We used scrambled sequences as a negative control. We used immunoblotting to validate the silencing effect on RBPJκ.
DNA constructs.
Constructs that activate Notch1 (N1ICΔOP) and Notch2 (N2ICΔOP) with a myc tag in pCS2+6MT have been previously described (1, 16). The retroviral vector pMX-IRES-EGFP and Plat-E cells were kindly provided by T. Kitamura (Tokyo University, Japan) (27, 44). The constructs were subcloned from the EcoRI/XhoI site of N1ICΔOP/pCS2+6MT and N2ICΔOP/pCS2+6MT into the EcoRI/XhoI sites of pMX-IRES-EGFP, generating pMX-N1ICΔOP-IRES-EGFP and pMX-N2ICΔOP-IRES-EGFP, respectively. Retrovirus packaging was performed by transfecting the plasmids into Plat-E cells using Lipofectamine 2000 (Invitrogen). Supernatants of pMX-N1ICΔOP-IRES-EGFP- and pMX-N2ICΔOP-IRES-EGFP-transfected Plat-E cell culture medium were used to infect primary BMMs in the presence of 8 μg/ml Polybrene. The pCMX-N/RBP-Jκ construct was described elsewhere (20).
ChIP.
Chromatin immunoprecipitation (ChIP) was performed with a ChIP assay kit (Upstate Biotechnology) according to the manufacturer's instructions using antibodies against NFATc1, p50, p65, c-Fos, Notch2, RBPJκ, and normal immunoglobulin G. The purified DNA was analyzed by PCR using primers that detect sequences containing the NFATc1-P1 promoter, 5′-CCGGGACGCCCATGCAATCTGTTAGTAATT-3′ (sense) and 5′-GCGGGTGCCCTGAGAAAGCTACTCTCCCTT-3′ (antisense), and the Hes-1 promoter, 5′-CACATCCTCTTTACCTTGTTCCCTC-3′ (sense) and 5′-CCCTTTGCTAACTCTTTCCTCTGG-3′ (antisense).
Luciferase reporter assay.
pNFATc1P1 0.8 kb-Luc (pNFATc1-pr-luc) was constructed by inserting the pGL3 basic promoter vector according to the method of Asagiri et al. (4). The RBPJκ site mutation in the pNFATc1P1 promoter (pNFATc1-RBPJκ mut-pr-luc) was constructed by PCR amplification using the following primers: 5′-ATTTAGCGGGAGGGGAATTTCC-3′ (sense) and 5′-GGAAATTCCCCTCCCGCTAAAT-3′ (antisense); the following primers were used to generate a mutation of the NF-κΒ binding site in the pNFATc1P1 promoter (pNFATc1-RBPJκ mut-pr-luc): 5′-CGGGATGGGAATTTCGTTACTCCC-3′ (sense) and 5′-GGGAGTAACGAAATTCCCATCCCG-3′ (antisense). The plasmids were transfected into RAW cells using Lipofectamine 2000 (Invitrogen), and luciferase activity was measured with the dual-luciferase reporter assay system (Promega, Madison, WI).
Data analysis.
Data are shown as mean values ± standard errors of means (SEM; n = number of culture wells). All experiments were performed at least three times, and similar results were obtained. Statistical differences were analyzed using one-way analysis of variance, and P values less than 0.05 were considered significant.
RESULTS
Notch is expressed and activated during osteoclast differentiation.
By microarray screening to identify mRNAs that were differentially expressed in untreated versus RANKL-treated RAW cells, a macrophage cell line that differentiates into osteoclasts in response to RANKL, we found that several Notch family members and Notch targets were upregulated following RANKL stimulation, concomitant with the expression of osteoclast differentiation markers (Table 1). We confirmed these results for RANKL-stimulated BMMs using semiquantitative RT-PCR and found that expression of Notch2 mRNA was increased within 24 h after RANKL stimulation (Fig. 1A) together with a weak induction of Notch1. The expression of the Hes-1 gene, a Notch target gene, was similar to that of Notch2, suggesting that Notch signaling was activated during osteoclast differentiation induced by RANKL. On the other hand, other Notch family members such as Notch3 and Notch4 were undetectable in these cells (Fig. 1A). We next examined the expression and cellular localization of Notch1 and Notch2 proteins in RANKL-treated BMMs undergoing terminal differentiation of multinucleated osteoclasts. By Western blotting and immunofluorescence, we detected the presence of the intracellular active Notch2 form, N2ICD, at 24 h, and N2ICD was still present after 3 days of RANKL treatment (Fig. 1B and C). To further demonstrate that Notch2 was translocated into the nucleus following RANKL stimulation, we obtained membrane, cytoplasmic, and nuclear fractions from BMM cells and performed Western blot analysis with anti-Notch2 antibody. N2ICD was observed only in the nuclear fractions after RANKL stimulation (Fig. 1D). RANKL, IκBα, and HDAC1 were used to determine the purities of the different cellular fractions. In addition, total levels of Notch2 protein in both membrane and nuclear fractions were significantly increased after the RANKL stimulation compared to those in untreated cells. These results indicated that RANKL induced Notch2 protein expression and activated Notch signaling during osteoclastogenesis.
TABLE 1.
Genes preferentially expressed in RAW264.7 cell-derived osteoclasts
| Group | Entrez gene symbol | GenBank accession no. | Fold increasea |
|---|---|---|---|
| Notch receptor | Notchl | NM_008714 | 1.1 |
| Notch2 | NM_010928 | 3.8 | |
| Notch3 | NM_008716 | 0.9 | |
| Notch4 | NM_010929 | NDb | |
| Notch ligand | Delta1 | NM_007865 | 1.2 |
| Delta3 | NM_007866 | 0.4 | |
| Delta4 | NM_019454 | ND | |
| Jagged1 | NM_013822 | 3.6 | |
| Jagged2 | NM_010588 | 1.1 | |
| Notch-dependent | Hes-1 | NM_008235 | 1.7 |
| genes | Hey-1 | NM_010423 | 1.4 |
| CSL DNA binding protein | RBPJκ | NM_009035 | 1.0 |
| Osteoclast | Calcitonin receptor | NM_007588 | 6.8 |
| markers | TRAP | NM_021330 | 7.1 |
| Cathepsin K | NM_007588 | 5.7 | |
| RANK | NM_011613 | 3.9 | |
| NFATc1 | NM_016791 | 4.4 |
Increases in the mRNA expression level in RANKL-stimulated RAW cell-derived osteoclasts compared with that in unstimulated RAW cells. GeneChip analysis was repeated several times and yielded similar results; a representative set of data is shown. The increases were calculated with GeneSpring.
ND, not determined.
FIG. 1.

Notch is expressed and activated during osteoclast differentiation. (A) BMM cells were treated with 50 ng/ml of RANKL for the indicated times. Total RNA was isolated from BMMs, and expression levels of Notch1 to -4, Hes-1, RANK, cathepsin K, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA were measured by RT-PCR. (B) RANKL activates Notch signaling in BMMs. Total cell lysates from RANKL-treated BMMs (50 ng/ml) were immunoblotted with anti-Notch1 or anti-Notch1 ICD (left) or anti-Notch2 (right) antibodies. The images of anti-Notch1 and anti-Notch1 ICD were taken from the same membrane by reblotting. (C) Immunostaining of Notch2 protein in BMMs stimulated with RANKL (50 ng/ml) (top). The middle panels show nuclei stained with DAPI. The lower panels show merged images of Notch2 and DAPI staining. Scale bar = 50 μm. (D) Active form of Notch2 translocates to nuclei. BMMs were treated with or without RANKL (50 ng/ml) for 30 min, and then cytoplasm (C), membrane (M), and nuclear (N) extracts were prepared and immunoblotted with anti-Notch2 antibodies. As fractionation controls we used anti-IκBα antibodies for cytosol, anti-RANK antibodies for the membrane, and anti-HDAC1 antibodies for the nuclear fraction. Similar results were obtained in three independent experiments.
GSI suppresses RANKL-induced osteoclastogenesis.
To further examine the role of Notch2 in osteoclast differentiation, we added GSI, which blocks Notch signaling, to bone marrow cell cultures together with RANKL and M-CSF. GSI inhibited RANKL-induced osteoclastogenesis from bone marrow cells and the expression of Hes-1 in a dose-dependent manner (Fig. 2A). It was previously shown that osteoclast differentiation consists of at least two steps: (i) the proliferation of osteoclast progenitors and their differentiation into osteoclast precursors (BMMs) induced by M-SCF and (ii) the subsequent differentiation of osteoclast precursors into osteoclasts induced by RANKL (54). We examined which step was affected by GSI treatment. Incubation with GSI together with M-CSF for 3 days and then further culture with RANKL gave rise to the generation of numerous osteoclasts (Fig. 2B), whereas expression of Hes-1 did not change (Fig. 2B). However, when added to the culture together with RANKL, GSI strongly inhibited osteoclast formation together with the expression of Hes-1 (Fig. 2C). These findings suggest that Notch signaling is essential during the second wave of osteoclastogenesis, which is mediated by RANKL.
FIG. 2.

GSI suppresses RANKL-induced osteoclastogenesis. (A) Mouse bone marrow cells were treated with M-CSF (50 ng/ml) for 3 days in the presence of GSI and further cultured in the presence of 50 ng/ml RANKL together with GSI for 3 more days. (B) Mouse bone marrow cells were treated with M-CSF for 3 days in the presence of GSI. The culture medium was removed, and cells were washed with phosphate-buffered saline twice and were further cultured in the presence of 50 ng/ml RANKL for 3 days. (C) Mouse bone marrow cells were treated with M-CSF (50 ng/ml) for 3 days and further cultured in the presence of RANKL (50 ng/ml) together with GSI for 3 days. Cells were fixed and stained for TRAP. TRAP+ MNCs were counted as osteoclasts. Scale bar = 100 μm. Data shown are the numbers of TRAP+ MNCs per culture well (values are means ± SEM, n = 3). Hes-1 and GAPDH mRNA expression levels were determined by RT-PCR.
Inhibition of RANKL-induced osteoclastogenesis by silencing Notch2 mRNA.
We next examined whether specific loss of function of Notch1 or Notch2 homologues affects osteoclast formation. Since both Notch1- and Notch2-deficient mice die at the early embryonic stage (37, 53), there is little information about the physiological role of these proteins in bone development. Thus, we generated RAW cell lines carrying tetracycline-inducible shRNA that specifically targets the expression of Notch1 or Notch2 (RAW-teton-shNotch1 or -shNotch2). We confirmed that endogenous Notch1 and Notch2 expression was suppressed in RAW-teton-shNotch1 and -shNotch2 cells in response to tetracycline (Fig. 3A and B, right). We found that RANKL-induced differentiation was not affected in RAW-shNotch1 cells compared to the control cells (Fig. 3A). In contrast, knocking down Notch2 expression significantly inhibited osteoclast formation in RAW-teton-shNotch2 cells (Fig. 3B).
FIG. 3.

Inhibition of RANKL-induced osteoclastogenesis by silencing Notch2 mRNA. Data for RAW cells with stable expression of tetracycline-regulated expression of shRNAs (Tet-on) that target Notch1 (RAW-teton-shNotch1) (A) and Notch2 (RAW-teton-shNotch2) (B) are shown. RAW-teton-shNotch1 and RAW-teton-shNotch2 cells were treated with RANKL (20 ng/ml) for 3 days. TRAP+ MNCs were counted as osteoclasts. Scale bar = 100 μm. The graph values are means ± SEM (n = 3). Both RAW-teton-shNotch1 and RAW-teton-shNotch2 cells were treated with or without tetracycline for 3 days. Total cell lysates were immunoblotted with anti-Notch1 or anti-Notch1 ICD (A, right) or anti-Notch2 (B, right) antibodies. β-Actin is shown as a loading control. The images of anti-Notch1 or anti-Notch1 ICD were taken from same membrane by reblotting.
The active form of Notch2 positively regulates RANKL-induced osteoclastogenesis.
To further investigate the role of Notch signaling, we examined the effects of expressing the active forms of Notch1 or Notch2 in BMMs. We constructed pMX-N1ICΔOP-IRES-GFP and pMX-N2ICΔOP-IRES-EGFP, which were engineered to express both active forms of Notch1 or Notch2 and green fluorescence protein (GFP) and infected BMMs with the different constructs. Infection efficiency was monitored by measuring GFP expression (Fig. 4A), and expression of ectopic Notch1 or Notch2 was detected by immunostaining with the anti-Notch1 and -Notch2 intracellular domains (ICDs) or the anti-myc tag antibodies (Fig. 4B). Ectopic expression of activated Notch2 but not Notch1 in BMMs resulted in efficient induction of osteoclast differentiation in a RANKL dose-dependent manner (Fig. 4C).
FIG. 4.

The active form of Notch2 positively regulates RANKL-induced osteoclastogenesis. BMMs were transduced with N1ICΔOP, N2ICΔOP, or empty pMX-IRES-EGFP retroviral vectors. (A) Images of GFP+ MNCs from BMM cells induced by RANKL stimulation. Scale bar = 100 μm. (B) Total cell lysates were immunoblotted with anti-Notch1, -Notch2, and -myc antibodies or anti-β-actin as a loading control. (C) Transduced BMMs were treated with RANKL (0, 25, or 50 ng/ml) for 3 days. TRAP+ MNCs were counted as osteoclasts. The values are means ± SEM (n = 3).
Expression of Notch ligands in osteoclast precursors.
Previous data showed RANKL induced not only Notch2 expression but also activation of Notch signaling (Fig. 1B to D), thus suggesting that RANKL regulates Notch ligand expression. Therefore, we examined the expression of Notch ligands in RANKL-treated BMMs and found that Jagged1 expression gradually increased from day 1 (1.23-fold) to day 3 (3.21-fold) of treatment (Fig. 5A). To determine whether Jagged1 was responsible for activating Notch2 during osteoclast differentiation, we incubated BMMs or RAW-teton-shNotch2 cells with either soluble recombinant FLAG-Jagged1 or Jagged1 immobilized on the culture plate. We found that Jagged1 enhanced RANKL-induced osteoclastogenesis in both culture conditions (Fig. 5B). As a control, addition of tetracycline inhibited RANKL-dependent osteoclast formation from RAW-teton-shNotch2 (Fig. 5C), but not RAW-teton-shNotch1, cells (data not shown) incubated with FLAG-Jagged1. Even though we cannot exclude the possibility that other RANKL-induced effects also affect Notch activation, these results strongly suggest that Jagged1 activates Notch signaling via Notch2 and that this activation enhanced the osteoclast formation induced by RANKL.
FIG. 5.
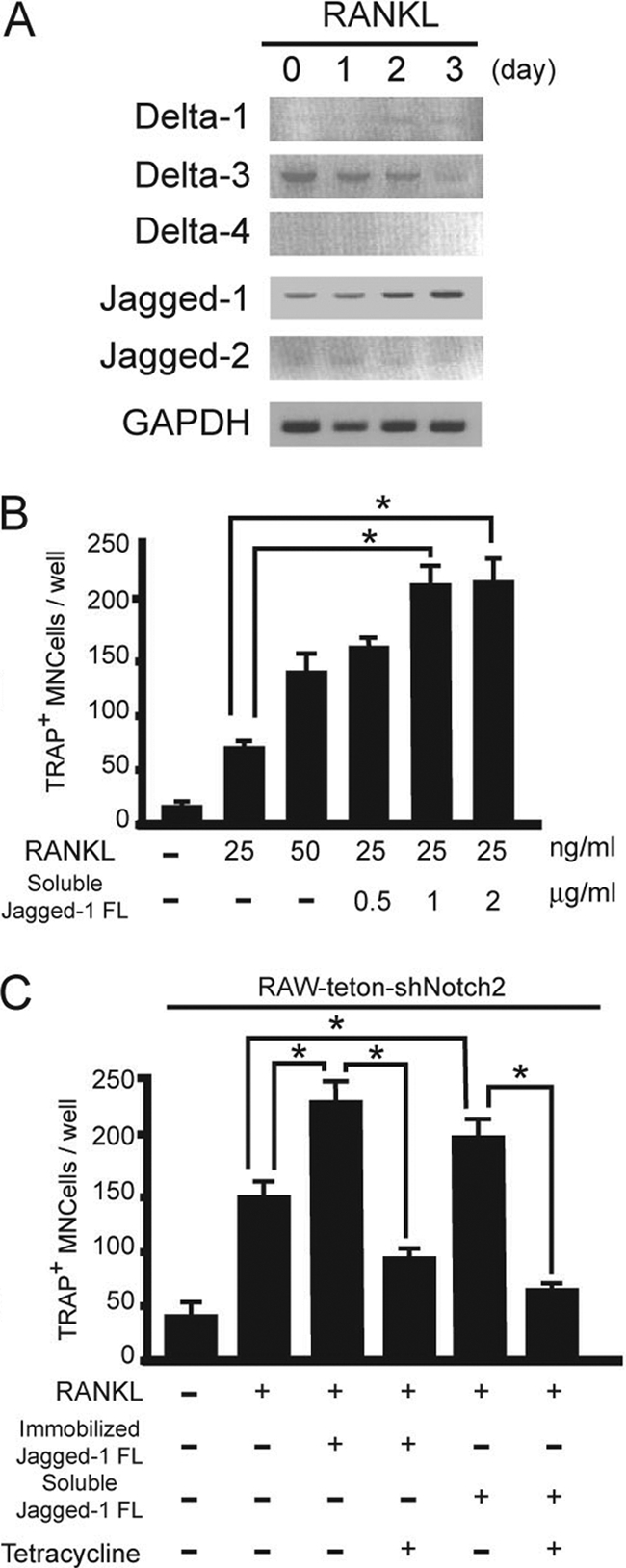
Expression of Notch ligands in osteoblasts and osteoclast precursors. (A) Expression of Notch ligand-related genes in mice treated with RANKL (50 ng/ml). Total RNA was isolated from osteoblasts or BMMs, and expression levels of indicated genes were measured by RT-PCR. (B) BMMs were treated with RANKL (50 ng/ml) in the presence of soluble FLAG-fused Jagged1 (Jagged-1 FL) (0.5, 1, or 2 μg/ml) for 3 days. (C) RAW-teton-shNotch2 cells were treated with RANKL (20 ng/ml) in the presence of either the soluble form or immobilized Jagged1 for 3 days with or without tetracycline. TRAP+ MNCs were counted as osteoclasts. Values are means ± SEM (n = 3).
Notch2 modulates RANKL-induced osteoclastogenesis through NFATc1.
To investigate the molecular mechanisms by which Notch signaling enhances osteoclast formation induced by RANKL, we examined the expression of NFATc1, a master regulator of osteoclastogenesis (4, 55). NFATc1 was induced in BMMs at 24 h after RANKL stimulation (Fig. 6A). Activation of Notch signaling by Jagged1 or by ectopic expression of N2ICD enhances NFATc1 expression (Fig. 6A). Conversely, inhibition of Notch signaling by GSI or shRNA for Notch2 strongly suppressed NFATc1 expression (Fig. 6A). These results suggest that Notch modulates RANKL-induced NFATc1 expression. Analysis of the NFATc1 promoter revealed the presence of a putative RBPJκ binding site overlapping an NF-κB site (Fig. 6B). ChIP analysis showed that Notch2 was recruited to the NFATc1 promoter after 30 min of RANKL stimulation, reached a maximum level at 1 h, and declined after 6 h of treatment (Fig. 6C). In contrast, we found that RBPJκ was in the NFATc1 promoter before RANKL stimulation and that RBPJκ was undetectable at 30 min and 3 h after RANKL stimulation, coinciding with Notch2 recruitment. It has been recently reported that RANKL-induced NF-κB activity is important for the initial induction of NFATc1 in the early stage of osteoclastogenesis and that c-Fos recruitment to the NFATc1 promoter is important in the autoamplification phase of NFATc1 induction (4). Consistent with this, we found that both p50 and p65 NF-κB subunits were recruited to the NFATc1 promoter within 30 min after RANKL stimulation while NFATc1 and c-Fos were recruited to the promoter after 6 h (Fig. 6C). These data suggest the possibility that NF-κB and Notch2 cooperate in the activation of NFATc1 expression. In agreement with this, coimmunoprecipitation experiments demonstrated that Notch2 physically interacts with p65 in the nucleus in a RANKL-dependent manner (Fig. 6D). Next, we generated luciferase reporter constructs containing specific mutations in the NF-κB (NF-κB-mut) or RBPJκ (RBPJκ-mut) binding sites of the NFATc1 promoter. Cotransfection of Notch2 with p65 enhanced NFATc1 promoter activity compared with transfection with Notch2 alone in both the wild-type and the RBPJκ-mut constructs (Fig. 6E). In contrast, Notch2 alone or in combination with p65 failed to activate the NF-κB-mut promoter (Fig. 6E), suggesting that p65 plays an essential role in Notch2-mediated activation of the NFATc1 gene. To further study the putative role of RBPJκ in the regulation of NFATc1 expression, we transfected RBPJκ together with Notch2 or Notch2/p65 and then measured luciferase activity. In both conditions RBPJκ strongly suppressed NFATc1 promoter activity (Fig. 6F), suggesting that RBPJκ may compete with p65 for DNA binding. We next examined the binding of endogenous p65 and RBPJκ to the NFATc1 promoter in the absence of Notch2 using RAW-teton-shNotch2 cells. Interestingly, we found that RBPJκ constitutively bound the NFATc1 promoter in these conditions (Fig. 6G). In a reciprocal experiment, knocking down RBPJκ expression using specific siRNA slightly enhanced binding of p65 and Notch2 to the NFATc1 promoter (Fig. 6H). These results indicate that NFATc1 is downstream of both Notch2 and NF-κB during RANKL-mediated osteoclast differentiation.
FIG. 6.

Molecular mechanisms by which Notch modulates RANKL-induced osteoclastogenesis. (A) Expression of NFATc1 induced by RANKL in mouse BMMs in the presence of soluble Jagged1, transduced Notch2, shRNA for Notch2, or GSI. Cells were treated with RANKL (50 ng/ml) for the indicated times. Total cell lysates were immunoblotted with anti-NFATc1 antibody. IB, immunoblotting. (B) Schematic representation of the NFATc1 P1 promoter. The sequences of the NF-κB binding site and the overlapping putative RBPJκ binding site are shown. (C) Chromatin from RANKL-treated BMMs was precipitated using the indicated antibodies. The NFATc1 promoter was amplified by PCR from precipitated DNA and from 1% of the input to monitor the amount of chromatin used. (D) Interaction of Notch2 and p65 in BMMs upon RANKL stimulation. Nuclear extracts from RANKL-treated BMMs were immunoprecipitated (IP) with anti-Notch2 antibodies and processed for immunoblotting with anti-p65 antibodies. The membrane was stripped and reblotted with anti-Notch2 antibodies to determine the amount of precipitated proteins. (E) RAW cells were transfected with the NFATc1 p1 promoter and the indicated plasmids and assayed for luciferase activity after 24 h. (F) Luciferase activity of NFATc1 p1 when cotransfected with the indicated plasmids in RAW cells. (G) ChIP assay with anti-p65, anti-Notch2, anti-RBPJκ, or control immunoglobulin G from RAW-teton-shNotch2 cells treated with RANKL with or without tetracycline. Analysis of the NFATc1 promoter was performed by PCR. (H) Chromatin treated with RANKL from RAW cells with knocked down RBPJκ expression was precipitated with the indicated antibodies. The NFATc1 promoter was amplified from precipitated DNA and 1% of the input by PCR. Similar results were obtained in three independent experiments.
Altogether, these results demonstrate an essential role for Notch2 in the induction of the terminal osteoclast differentiation.
DISCUSSION
Notch signaling has been implicated in various processes including cell fate decisions, tissue patterning, and morphogenesis (3, 9). Using genome-wide screening techniques and RT-PCR results, we found that Notch2 and Jagged1 are upregulated during RANKL-induced osteoclastogenesis, suggesting that Notch modulates downstream of RANK signaling. Consistently, inhibition of Notch signaling using GSI or shRNA for Notch2, but not Notch1, decreased NFATc1 expression, resulting in inhibition of osteoclastogenesis. We demonstrate that NFATc1 is a direct target of the Notch2-NF-κB complex in response to RANKL stimulation.
Our current results are in apparent contradiction with a previous study showing that activation of Notch by Delta1 negatively regulated osteoclastogenesis by reducing the surface levels of c-Fms in osteoclast precursor cells together with enhanced osteoprotegerin expression in stromal cells (62) and with the work from Bai et al. demonstrating that deletion of Notch1 to -3 enhances osteoclast commitment from bone marrow precursors (6). In fact, several reports support the idea that the requirement for Notch strongly depends on the stage of osteoclast differentiation and the type of the cells that are targeted. Recently, it was found that specific deletion of presenilin 1 and 2 in the skeletogenic mesenchyme led to increased trabecular bone mass. Furthermore, deletion of both Notch1 and -2 under the control of Prx-Cre produced a postnatal skeletal phenotype qualitatively identical to that of presenilin 1 and 2 double-knockout mice. However, deletion of Notch1 and -2 in committed osteoblasts using Col1-Cre did not result in any obvious skeletal phenotype (21). We believe that Notch2 is specifically required for the late stage of osteoclast differentiation, which is mediated by RANKL, but not for the initial phase, which is dependent on M-CSF, as was shown by deletion of Notch1 and -3 under the control of lyzM-Cre (6). Our results also show a very specific requirement for Notch2 in RANKL-induced osteoclast differentiation, which in collaboration with NF-κB regulates NFATc1 expression in wild-type cells; this is in apparent contradiction with the enhanced osteoclast differentiation that occurs in the triple-knockout cells (6). We speculate that deletion of all Notch receptors in early differentiation stages of the myeloid cell lineage may activate different transcriptional complexes and result in enhanced osteoclastogenesis in the triple-knockout mice.
Different expression patterns of Notch and Notch ligands together with the microarray data from RANKL-treated cells suggest that specific ligand-receptor pairs may play specific functions during osteoclast differentiation. Although there are no major functional disparities between Delta and Jagged or between the different Notch homologues in vitro, these proteins show distinct expression patterns (34, 60) and different, nonredundant in vivo functions (39). We here demonstrated that Jagged1 is specifically activated in response to RANKL treatment, thus inducing Notch2 activation in BMMs. As an additional mechanism, RANKL might directly induce Notch2. Alternatively, previous reports show increased Jagged1 expression in the osteoblastic stromal cells of PTH/PTHrP receptor transgenic mice (10, 11). We speculated that the source of Jagged1 during osteoclastogenesis is RANKL-stimulated BMMs as well as osteoblastic stromal cells. Moreover, Notch1 cannot compensate for Notch2 deficiency in RANKL-dependent osteoclast differentiation, in that the knockdown of Notch2 using shRNA absolutely eliminates the effects of Jagged1. These results are in agreement with the different roles for Notch1 and Notch2 in myeloid cell differentiation induced by granulocyte colony-stimulating factor or granulocyte-macrophage colony-stimulating factor (7). Another example showing that two different Notch ligands generate distinct responses in the same cell type has been recently published: Delta promotes Th1 responses, while Jagged instructs the Th2 lineage in naive CD4 T cells (2).
In this study, we demonstrated that, like the Notch2 ICD, Jagged1 enhanced RANKL-induced osteoclastogenesis. A previous report demonstrated that immobilized Notch ligand Delta1, but not soluble Delta1, induced Hes-1 gene expression or suppressed the differentiation from osteoclast precursors induced by M-CSF (62). We cannot explain the reasons underlying the reported controversial effects for the soluble and immobilized ligands; however, we speculate that experimental conditions and protein stability are crucial for success in the use of these molecules. In fact, soluble Jagged1 ligand or peptides have been extensively reported (26, 33, 42, 59). In this study, we found that both soluble and immobilized Jagged1 enhanced RANKL-induced osteoclastogenesis under the culture conditions of Yamada et al. (62) and that this effect was strictly dependent on Notch2.
Specific regulation of gene transcription is the combinatorial effect of multiple transcription factors. In this study, we identified a canonical RBPJκ site within the mouse NFATc1 P1 promoter that overlaps an NF-κB site. Based on the recognition sequence for RBPJκ (G/ATGGGAA), the occurrence of dual NF-κB/RBPJκ elements is predicted to occur at ∼15% of the NF-κB sites, which are the ones that contain the GGGAA NF-κB half-site (22, 31). This element, which is similar to those present in the IL-6, beta interferon, Hes-1, p52/NF-κB2, IκBα, and Bcl-3 genes, may define a distinct subclass of genes regulated by Notch and NF-κB (1, 16, 17, 45, 49, 58). We have found that RANKL induces association of NF-κB and Notch2 in the nucleus and that both NF-κB and Notch2 bind to the NFATc1 promoter in these conditions concomitant with reduced detection of RBPJκ. This result suggests that p65 functionally competes for RBPJκ binding to activate transcription; however, an alternative explanation could be that a ternary complex involving p65, Notch2, and RBPJκ may mask the epitope involved in antibody recognition. We demonstrated that the inhibition of Notch signaling by GSI leads to a dramatic decrease in RANKL-induced osteoclastogenesis by inhibiting NFATc1 expression. A comparable effect was observed by inhibiting endogenous expression of Notch2 with shRNA for Notch2 but not by inhibition of Notch1 by shRNA for Notch1. The fact that cross talk between the NF-κB and Notch pathways promotes synergistic signaling effects (16, 17, 45, 49, 58) suggests that Notch2 enhances NF-κB transcriptional activity. In addition, we have showed that RANKL induced Notch2 expression and activation. These data suggest that Notch2, like NFATc1, functions as an autoamplification factor in response to RANKL signaling. Further studies are required to determine the role of the molecular mechanism of Notch signaling in osteoclast differentiation.
In conclusion, we have shown in this study that RANKL induced Jagged1 and Notch2 expression during osteoclast differentiation. Moreover, inhibition of Notch2 signaling strongly suppressed, whereas ectopic expression of N2ICD enhanced, RANKL-induced osteoclastogenesis. Activated Notch2 cooperates with NF-κB to activate NFATc1 transcription, which is an important mediator of osteoclast maturation. These findings may provide a therapeutic opportunity to specifically target osteoclast activation in pathological situations.
Acknowledgments
Many thanks go to Lluís Espinosa for critical reading and helping in manuscript editing.
This work was supported by a grant-in-aid for scientific research from the Ministry of Education, Culture, Sports Science and Technology of Japan (no. 18791580) and a Frontier Research grant.
Footnotes
Published ahead of print on 18 August 2008.
REFERENCES
- 1.Aguilera, C., R. Hoya-Arias, G. Haegeman, L. Espinosa, and A. Bigas. 2004. Recruitment of IκBα to the hes1 promoter is associated with transcriptional repression. Proc. Natl. Acad. Sci. USA 10116537-16542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amsen, D., J. M. Blander, G. R. Lee, K. Tanigaki, T. Honjo, and R. A. Flavell. 2004. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell 117515-526. [DOI] [PubMed] [Google Scholar]
- 3.Artavanis-Tsakonas, S., M. D. Rand, and R. J. Lake. 1999. Notch signaling: cell fate control and signal integration in development. Science 284770-776. [DOI] [PubMed] [Google Scholar]
- 4.Asagiri, M., K. Sato, T. Usami, S. Ochi, H. Nishina, H. Yoshida, I. Morita, E. F. Wagner, T. W. Mak, E. Serfling, and H. Takayanagi. 2005. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 2021261-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asagiri, M., and H. Takayanagi. 2007. The molecular understanding of osteoclast differentiation. Bone 40:251-264. [DOI] [PubMed] [Google Scholar]
- 6.Bai, S., R. Kopan, W. Zou, M. J. Hilton, C. T. Ong, F. Long, F. P. Ross, and S. L. Teitelbaum. 2008. Notch1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via c-fos osteoblast lineage cells. J. Biol. Chem. 2836509-6518. [DOI] [PubMed] [Google Scholar]
- 7.Bigas, A., D. I. Martin, and L. A. Milner. 1998. Notch1 and Notch2 inhibit myeloid differentiation in response to different cytokines. Mol. Cell. Biol. 18:2324-2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boyle, W. J., W. S. Simonet, and D. L. Lacey. 2003. Osteoclast differentiation and activation. Nature 423337-342. [DOI] [PubMed] [Google Scholar]
- 9.Bray, S. J. 2006. Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 7678-689. [DOI] [PubMed] [Google Scholar]
- 10.Bulman, M. P., K. Kusumi, T. M. Frayling, C. McKeown, C. Garrett, E. S. Lander, R. Krumlauf, A. T. Hattersley, S. Ellard, and P. D. Turnpenny. 2000. Mutations in the human delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat. Genet. 24438-441. [DOI] [PubMed] [Google Scholar]
- 11.Calvi, L. M., N. A. Sims, J. L. Hunzelman, M. C. Knight, A. Giovannetti, J. M. Saxton, H. M. Kronenberg, R. Baron, and E. Schipani. 2001. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J. Clin. Investig. 107277-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calvi, L. M., G. B. Adams, K. W. Weibrecht, J. M. Weber, D. P. Olson, M. C. Knight, R. P. Martin, E. Schipani, P. Divieti, F. R. Bringhurst, L. A. Milner, H. M. Kronenberg, and D. T. Scadden. 2003. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 23841-846. [DOI] [PubMed] [Google Scholar]
- 13.Deregowski, V., E. Gazzerro, L. Priest, S. Rydziel, and E. Canalis. 2006. Notch 1 overexpression inhibits osteoblastogenesis by suppressing Wnt/β-catenin but not bone morphogenetic protein signaling. J. Biol. Chem. 2816203-6210. [DOI] [PubMed] [Google Scholar]
- 14.Dougall, W. C., M. Glaccum, K. Charrier, K. Rohrbach, K. Brasel, T. De Smedt, E. Daro, J. Smith, M. E. Tometsko, C. R. Maliszewski, A. Armstrong, V. Shen, S. Bain, D. Cosman, D. Anderson, P. J. Morrissey, J. J. Peschon, and J. Schuh. 1999. RANK is essential for osteoclast and lymph node development. Genes Dev. 132412-2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engin, F., Z. Yao, T. Yang, G. Zhou, T. Bertin, M. M. Jiang, Y. Chen, L. Wang, H. Zheng, R. E. Sutton, B. F. Boyce, and B. Lee. 2008. Dimorphic effects of Notch signaling in bone homeostasis. Nat. Med. 14299-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Espinosa, L., S. Santos, J. Inglés-Esteve, P. Muñoz-Canoves, and A. Bigas. 2002. p65-NF-κB synergizes with Notch to activate transcription by triggering cytoplasmic translocation of the nuclear receptor corepressor N-CoR. J. Cell Sci. 1151295-1303. [DOI] [PubMed] [Google Scholar]
- 17.Espinosa, L., J. Inglés-Esteve, A. Robert-Moreno, and A. Bigas. 2003. IκBα and p65 regulate the cytoplasmic shuttling of nuclear corepressors: cross-talk between Notch and NF-κB pathways. Mol. Biol. Cell 14491-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franzoso, G., L. Carlson, L. Xing, L. Poljak, E. W. Shores, K. D. Brown, A. Leonardi, T. Tran, B. F. Boyce, and U. Siebenlist. 1997. Requirement for NF-κB in osteoclast and B-cell development. Genes Dev. 113482-3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grigoriadis, A. E., Z. Q. Wang, M. G. Cecchini, W. Hofstetter, R. Felix, H. A. Fleisch, and E. F. Wagner. 1994. c-Fos: a key regulator of osteoclast-macrophage lineage determination and bone remodeling. Science 266443-448. [DOI] [PubMed] [Google Scholar]
- 20.Han, H., K. Tanigaki, N. Yamamoto, K. Kuroda, M. Yoshimoto, T. Nakahata, K. Ikuta, and T. Honjo. 2002. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int. Immunol. 14637-645. [DOI] [PubMed] [Google Scholar]
- 21.Hilton, M. J., X. Tu, X. Wu, S. Bai, H. Zhao, T. Kobayashi, H. M. Kronenberg, S. L. Teitelbaum, F. P. Ross, R. Kopan, and F. Long. 2008. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat. Med. 14306-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirano, F., H. Tanaka, Y. Hirano, M. Hiramoto, H. Handa, I. Makino, and C. Sheidereit. 1998. Functional interference of Sp1 and NF-κB through the same DNA binding site. Mol. Cell. Biol. 181266-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hrabe de Angelis, M., J. I. I. McIntyre, and A. Gossler. 1997. Maintenance of somite borders in mice requires the Delta homologue DII1. Nature 386717-721. [DOI] [PubMed] [Google Scholar]
- 24.Ikeda, F., R. Nishimura, T. Matsubara, S. Tanaka, J. Inoue, S. V. Reddy, K. Hata, K. Yamashita, T. Hiraga, T. Watanabe, T. Kukita, K. Yoshioka, A. Rao, and T. Yoneda. 2004. Critical roles of c-Jun signaling in regulation of NFAT family and RANKL-regulated osteoclast differentiation. J. Clin. Investig. 114475-484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iotsova, V., J. Caamaño, J. Loy, Y. Yang, A. Lewin, and R. Bravo. 1997. Osteopetrosis in mice lacking NF-κB1 and NF-κB2. Nat. Med. 31285-1289. [DOI] [PubMed] [Google Scholar]
- 26.Karanu, F. N., B. Murdoch, T. Miyabayashi, M. Ohno, M. Koremoto, L. Gallacher, D. Wu, A. Itoh, S. Sakano, and M. Bhatia. 2001. Human homologues of Delta-1 and Delta-4 function as mitogenic regulators of primitive human hematopoietic cells. Blood 971960-1967. [DOI] [PubMed] [Google Scholar]
- 27.Kitamura, T., Y. Koshino, F. Shibata, T. Oki, H. Nakajima, T. Nosaka, and H. Kumagai. 2003. Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp. Hematol. 311007-1014. [PubMed] [Google Scholar]
- 28.Koga, T., M. Inui, K. Inoue, S. Kim, A. Suematsu, E. Kobayashi, T. Iwata, H. Ohnishi, T. Matozaki, T. Kodama, T. Taniguchi, H. Takayanagi, and T. Takai. 2004. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature 428758-763. [DOI] [PubMed] [Google Scholar]
- 29.Kong, Y. Y., H. Yoshida, I. Sarosi, H. L. Tan, E. Timms, C. Capparelli, S. Morony, A. J. Oliveira-dos-Santos, G. Van, A. Itie, W. Khoo, A. Wakeham, C. R. Dunstan, D. L. Lacey, T. W. Mak, W. J. Boyle, and J. M. Penninger. 1999. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397315-323. [DOI] [PubMed] [Google Scholar]
- 30.Kusumi, K., E. S. Sun, A. W. Kerrebrock, R. T. Bronson, D. C. Chi, M. S. Bulotsky, J. B. Spencer, B. W. Birren, W. N. Frankel, and E. S. Lander. 1998. The mouse pudgy mutation disrupts Delta homologue Dll3 and initiation of early somite boundaries. Nat. Genet. 19274-278. [DOI] [PubMed] [Google Scholar]
- 31.Lee, S. H., X. Wang, and J. DeJong. 2000. Functional interactions between an atypical NF-κB site from the rat CYP2B1 promoter and the transcriptional repressor RBP-J.κ/CBF1. Nucleic Acids Res. 28:2091-2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li, J. P., H. Kajiya, F. Okamoto, A. Nakao, T. Iwamoto, and K. Okabe. 2007. Three Na+/Ca2+ exchanger (NCX) variants are expressed in mouse osteoclasts and mediate calcium transport during bone resorption. Endocrinology 1482116-2125. [DOI] [PubMed] [Google Scholar]
- 33.Li, L., L. A. Milner, Y. Deng, M. Iwata, A. Banta, L. Graf, S. Marcovina, C. Friedman, B. J. Trask, L. Hood, and B. Torok-Storb. 1998. The human homolog of rat Jagged1 expressed by marrow stroma inhibits differentiation of 32D cells through interaction with Notch1. Immunity 843-55. [DOI] [PubMed] [Google Scholar]
- 34.Lindsell, C. E., J. Boulter, G. diSibio, A. Gossler, and G. Weinmaster. 1996. Expression patterns of Jagged, Delta1, Notch1, Notch2, and Notch3 genes identify ligand-receptor pairs that may function in neural development. Mol. Cell. Neurosci. 814-27. [DOI] [PubMed] [Google Scholar]
- 35.Mastrangelo, P., P. M. Mathews, M. A. Chishti, S. D. Schmidt, Y. Gu, J. Yang, M. J. Mazzella, J. Coomaraswamy, P. Horne, B. Strome, H. Pelly, G. Levesque, C. Ebeling, Y. Jiang, R. A. Nixon, R. Rozmahel, P. E. Fraser, P. St. George-Hyslop, G. A. Carlson, and D. Westaway. 2005. Dissociated phenotypes in presenilin transgenic mice define functionally distinct γ-secretases. Proc. Natl. Acad. Sci. USA 102:8972-8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCright, B., X. Gao, L. Shen, J. Lozier, Y. Lan, M. Maguire, D. Herzlinger, G. Weinmaster, R. Jiang, and T. Gridley. 2001. Defects in development of the kidney, heart and eye vasculature in mice homozygous for a hypomorphic Notch2 mutation. Development 128491-502. [DOI] [PubMed] [Google Scholar]
- 37.McCright, B., J. Lozier, and T. Gridley. 2002. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development 1291075-1082. [DOI] [PubMed] [Google Scholar]
- 38.McDaniell, R., D. M. Warthen, P. A. Sanchez-Lara, A. Pai, I. D. Krantz, D. A. Piccoli, and N. B. Spinner. 2006. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the Notch signaling pathway. Am. J. Hum. Genet. 79169-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moreno, A. R., J. Guiu, C. Ruiz-Herguido, M. E. López, J. Inglés-Esteve, L. Riera, A. Tipping, T. Enver, E. Dzierzak, T. Gridley, L. Espinosa, and A. Bigas. 5 June 2008. Impaired embryonic haematopoiesis yet normal arterial development in the absence of the Notch ligand Jagged1. EMBO J. doi: 10.1038/emboj. 2008.113. [DOI] [PMC free article] [PubMed]
- 40.Nair, P., K. Somasundaram, and S. Krishna. 2003. Activated Notch1 inhibits p53-induced apoptosis and sustains transformation by human papillomavirus type 16 E6 and E7 oncogenes through a PI3K-PKB/Akt-dependent pathway. J. Virol. 777106-7112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naito, A., S. Azuma, S. Tanaka, T. Miyazaki, S. Takaki, K. Takatsu, K. Nakao, K. Nakamura, M. Katsuki, T. Yamamoto, and J. Inoue. 1999. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells 4353-362. [DOI] [PubMed] [Google Scholar]
- 42.Nickoloff, B. J., J. Z. Qin, V. Chaturvedi, M. F. Denning, B. Bonish, and L. Miele. 2002. Jagged-1 mediated activation of notch signaling induces complete maturation of human keratinocytes through NF-κB and PPARγ. Cell Death Differ. 9842-855. [DOI] [PubMed] [Google Scholar]
- 43.Nobta, M., T. Tsukazaki, Y. Shibata, C. Xin, T. Moriishi, S. Sakano, H. Shindo, and A. Yamaguchi. 2005. Critical regulation of bone morphogenetic protein-induced osteoblastic differentiation by Delta1/Jagged1-activated Notch1 signaling. J. Biol. Chem. 28015842-15848. [DOI] [PubMed] [Google Scholar]
- 44.Nosaka, T., T. Kawashima, K. Misawa, K. Ikuta, A. L. Mui, and T. Kitamura. 1999. STAT5 as a molecular regulator of proliferation, differentiation and apoptosis in hematopoietic cells. EMBO J. 184754-4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oswald, F., S. Liptay, G. Adler, and R. M. Schmid. 1998. NF-κB2 is a putative target gene of activated Notch-1 via RBP-J.κ. Mol. Cell. Biol. 182077-2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sato, K., A. Suematsu, T. Nakashima, S. Takemoto-Kimura, K. Aoki, Y. Morishita, H. Asahara, K. Ohya, A. Yamaguchi, T. Takai, T. Kodama, T. A. Chatila, H. Bito, and H. Takayanagi. 2006. Regulation of osteoclast differentiation and function by the CaMK-CREB pathway. Nat. Med. 121410-1416. [DOI] [PubMed] [Google Scholar]
- 47.Sciaudone, M., E. Gazzerro, L. Priest, A. M. Delany, and E. Canalis. 2003. Notch 1 impairs osteoblastic cell differentiation. Endocrinology 1445631-5639. [DOI] [PubMed] [Google Scholar]
- 48.Shen, J., R. T. Bronson, D. F. Chen, W. Xia, D. J. Selkoe, and S. Tonegawa. 1997. Skeletal and CNS defects in presenilin-1-deficient mice. Cell 16629-639. [DOI] [PubMed] [Google Scholar]
- 49.Shin, H. M., L. M. Minter, O. H. Cho, S. Gottipati, A. H. Fauq, T. E. Golde, G. E. Sonenshein, and B. A. Osborne. 2006. Notch1 augments NF-κB activity by facilitating its nuclear retention. EMBO J. 25129-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Small, D., D. Kovalenko, D. Kacer, L. Liaw, M. Landriscina, C. Di Serio, I. Prudovsky, and T. Maciag. 2001. Soluble Jagged 1 represses the function of its transmembrane form to induce the formation of the Src-dependent chord-like phenotype. J. Biol. Chem. 27632022-32030. [DOI] [PubMed] [Google Scholar]
- 51.Small, D., D. Kovalenko, R. Soldi, A. Mandinova, V. Kolev, R. Trifonova, C. Bagala, D. Kacer, C. Battelli, L. Liaw, I. Prudovsky, and T. Maciag. 2003. Notch activation suppresses fibroblast growth factor-dependent cellular transformation. J. Biol. Chem. 27816405-16413. [DOI] [PubMed] [Google Scholar]
- 52.Song, L. L., Y. Peng, J. Yun, P. Rizzo, V. Chaturvedi, S. Weijzen, W. M. Kast, P. J. Stone, L. Santos, A. Loredo, U. Lendahl, G. Sonenshein, B. Osborne, J. Z. Qin, A. Pannuti, B. J. Nickoloff, and L. Miele. 16 June 2008. Notch-1 associates with IKKα and regulates IKK activity in cervical cancer cells. Oncogene doi: 10.1038/onc.2008.190. [DOI] [PubMed]
- 53.Swiatek, P. J., C. E. Lindsell, F. F. del Amo, G. Weinmaster, and T. Gridley. 1991. Notch1 is essential for postimplantation development in mice. Genes Dev. 113707-719. [DOI] [PubMed] [Google Scholar]
- 54.Takahashi, N., N. Udagawa, S. Tanaka, and T. Suda. 2003. Generating murine osteoclasts from bone marrow. Methods Mol. Med. 80129-144. [DOI] [PubMed] [Google Scholar]
- 55.Takayanagi, H., S. Kim, T. Koga, H. Nishina, M. Isshiki, H. Yoshida, A. Saiura, M. Isobe, T. Yokochi, J. Inoue, E. F. Wagner, T. W. Mak, T. Kodama, and T. Taniguchi. 2002. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 3889-901. [DOI] [PubMed] [Google Scholar]
- 56.Teitelbaum, S. L. 2000. Bone resorption by osteoclasts. Science 2891504-1508. [DOI] [PubMed] [Google Scholar]
- 57.Tezuka, K., M. Yasuda, N. Watanabe, N. Morimura, K. Kuroda, S. Miyatani, and N. Hozumi. 2002. Stimulation of osteoblastic cell differentiation by Notch. J. Bone Miner. Res. 17231-239. [DOI] [PubMed] [Google Scholar]
- 58.Vacca, A., M. P. Felli, R. Palermo, G. Di Mario, A. Calce, M. Di Giovine, L. Frati, A. Gulino, and I. Screpanti. 2006. Notch3 and pre-TCR interaction unveils distinct NF-κB pathways in T-cell development and leukemia. EMBO J. 251000-1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weijzen, S., M. P. Velders, A. G. Elmishad, P. E. Bacon, J. R. Panella, B. J. Nickoloff, L. Miele, and W. M. Kast. 2002. The Notch ligand Jagged-1 is able to induce maturation of monocyte-derived human dendritic cells. J. Immunol. 1694273-4278. [DOI] [PubMed] [Google Scholar]
- 60.Weinmaster, G., V. J. Roberts, and G. Lemke. 1991. A homolog of Drosophila Notch expressed during mammalian development. Development 113199-205. [DOI] [PubMed] [Google Scholar]
- 61.Xue, Y., X. Gao, C. E. Lindsell, C. R. Norton, B. Chang, C. Hicks, M. Gendron-Maguire, E. B. Rand, G. Weinmaster, and T. Gridley. 1999. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum. Mol. Genet. 8723-730. [DOI] [PubMed] [Google Scholar]
- 62.Yamada, T., H. Yamazaki, T. Yamane, M. Yoshino, H. Okuyama, M. Tsuneto, T. Kurino, S. Hayashi, and S. Sakano. 2003. Regulation of osteoclast development by Notch signaling directed to osteoclast precursors and through stromal cells. Blood 152227-2234. [DOI] [PubMed] [Google Scholar]
- 63.Zamurovic, N., D. Cappellen, D. Rohner, and M. Susa. 2004. Coordinated activation of notch, Wnt, and transforming growth factor-beta signaling pathways in bone morphogenic protein 2-induced osteogenesis. Notch target gene Hey1 inhibits mineralization and Runx2 transcriptional activity. J. Biol. Chem. 27937704-37715. [DOI] [PubMed] [Google Scholar]