

Abstract
Sir2 deacetylases (sirtuins) couple the deacetylation of lysine residues with conversion of NAD+ to O-acetyl-ADP-ribose (OAADPr) and nicotinamide. Sirtuins are potential targets for the treatment of diabetes, ageing, cancer, and neurodegenerative diseases. The most debated portion of the chemical mechanism is the initial catalytic step that forms nicotinamide and the O-alkylamidate intermediate. Here, utilizing a series of acetyl-lysine analogs that differ greatly in the electron-withdrawing nature of the substituents, we present evidence that the nucleophilicity of the acetyl-oxygen is directly tied to nicotinamide-ribosyl bond cleavage, consistent with an SN2-like mechanism for the initial step. The log of the rate of nicotinamide formation, which varied over 5-orders of magnitude, was plotted versus the inductive Taft constant, σ*, revealing a linear-free energy relationship with a steep negative slope (ρ* = -1.9). In addition, most acetyl-lysine analogs exhibited Kd values that were as low or lower than that of acetyl-lysine, indicating the diminished reactivity was due to chemistry and not substrate binding. Facile nicotinamide exchange was observed with the acetyl substrate (Vmax = 2.9 ± 0.2 s-1; Km = 406 ± 70 μM) but >400-fold slower exchange rates were observed with the fluorinated analogs. All analogs were converted to their corresponding O-acetyl-ADP-ribose analogs at a steady-state turnover rate similar to the rate of nicotinamide formation. An SN2-like mechanism in Sir2 deacetylases is unusual, as other examples of enzymatic nicotinamide-ribosyl bond cleavage proceed through an oxocarbenium intermediate in an SN1-like mechanism. These results have important implications on the selective inhibition of Sir2 over other NAD+-metabolizing enzymes.
Sir2 protein deacetylases (or sirtuins) play a critical role in a variety of biological processes including glucose homeostasis, lifespan extension, apoptosis, and neurodegeneration, suggesting that sirtuins may be targets for treatment of diabetes, ageing, cancer, and neurodegenerative diseases.1 Sirtuins couple the deacetylation of lysine residues with conversion of NAD+ to O-acetyl-ADP-ribose (OAADPr) and nicotinamide.
A major unresolved portion of the chemical mechanism is the initial catalytic step resulting in formation of the O-alkylamidate intermediate (Scheme 1). One possible mechanism is the concerted attack of acetyl-lysine at the 1’-carbon of the nicotinamide ribose, displacing nicotinamide in a direct-displacement SN2 (AN DN) reaction. Another possibility is that the acetyl group is not chemically involved in nicotinamide cleavage, but instead serves to position NAD+ in a destabilizing conformation that allows nicotinamide cleavage to occur in an SN1 (DN + AN) mechanism, forming a distinct oxocarbenium intermediate (Scheme 1). Subsequent attack of acetyl-lysine on the oxocarbenium would yield the O-alkylamidate intermediate.2 The actual mechanism of NAD+ cleavage lies on a continuum between these two possibilities. Subsequently, the 2’-hydroxyl is activated by an active-site histidine to attack the O-alkylamidate, forming a l’,2’-cyclic intermediate. Addition of water yields deacetylated peptide and OAADPr (Scheme 1).1 Here, we present evidence that the nucleophilicity of the acetyl-oxygen is directly tied to the rate of nicotinamide-ribosyl bond cleavage, consistent with an SN2-like mechanism.
Scheme 1.

Possible chemical mechanisms for initial acetyl-lysine analog attack and complete deacetylation mechanism.
Other NAD+-consuming enzymes such as ADP-ribosyltransferases and NAD+ hydrolases are postulated to proceed through oxocarbenium intermediates in which the incoming nucleophile is minimally involved in nicotinamide formation (bond order of the nucleophile is between 0 and 0.11 where kinetic isotope effects are known).3 With the Sir2 reaction, an acetylated lysine substrate is required to break the nicotinamide-ribosyl bond,4 suggesting that the acetyl-lysine residue might be chemically involved in nicotinamide-ribosyl bond cleavage. To determine how acetyl-lysine is involved, we measured the rate of nicotinamide formation under single-turnover conditions (Figure 1A) for six acetyl-lysine analogs that differ greatly in the electron-withdrawing nature of the substituents but are similar in steric size (Scheme 1). A rapid quenching approach was used so that the rate of chemical cleavage, independent of the physical dissociation of nicotinamide from the active site, could be determined. All acetyl-lysine analogs were incorporated into 11-mer peptides based on the N-terminal tail of histone H3 acetylated at lysine-14 (H2N-KSTGGK(Acetyl-analog)APRKQ-OH). If an SN2-like mechanism were operative, the rate of nicotinamide formation would be directly tied to the nucleophilicity of the acetyl group. With an SN1 mechanism, attack of acetyl-lysine occurs after nicotinamide formation and therefore the nucleophilicity of the amide oxygen would not greatly affect the rate of nicotinamide cleavage from NAD+.
Figure 1.
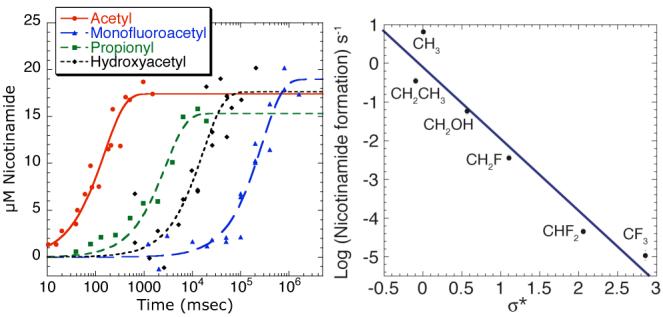
(Left) Single-turnover kinetics of acetyl-lysine analogs monitoring nicotinamide formation (Right) Plot of log rate of nicotinamide formation vs. the Taft σ* constant
Among the acetyl-lysine analogs, there was a dramatic dependence of rate (>5-orders of magnitude) on the electron-withdrawing potential of the substitution. The log of the rate of nicotinamide formation was plotted versus the inductive Taft constant, σ*,5 revealing a linear-free energy relationship with a steep negative slope of ρ* = -1.9 (Figure 1B). Notably, all analogs in this study were converted to the corresponding O-acetyl-ADP-ribose analogs, without significant formation of ADP-ribose6 (Supplemental Table 1). Where the steady-state turnover rate kcat could be measured, the first-order rate of nicotinamide formation was similar to that of the kcat values, indicating that the rate-limiting step was altered from product release2 to nucleophilic attack of the carbonyl oxygen on NAD+ (Table 1).
Table 1.
Physical and kinetic parameters of acetyl-lysine analogs
| Analog | Kd(μM) | Nicotinamide formation (s-1) |
kcat (s-1) |
|---|---|---|---|
| Acetyl | 21 ± 4 | 6.7 ± 0.9 × 100 | 2.0 ± 0.3 × 10-1 |
| Propionyl | 8.6 ± 0.2 | 3.6 ± 0.7 × 10-1 | 1.7 ± 0.3 × 10-1 |
| α-Hydroxyacetyl | 90 ± 50 | 6.0 ± 1.5 × 10-2 | 6.6 ± 0.6 × 10-2 |
| Monofluoroacetyl | 22 ± 5 | 3.7 ± 0.6 × 10-3 | 3.3 ± 0.5 × 10-3 |
| Difluoroacetyl | 20 ± 1 | 4.6 ± 0.8 × 10-5 | Not Determined |
| Trifluoroacetyl | 3.3 ± 0.7 | 1.1 ± 0.4 × 10-5 | Not Determined |
To ensure that the rate of nicotinamide formation was not reflective of diminished binding of the acetyl-lysine analogs in the ground-state, we measured the dissociation constant (Kd) of each analog to free enzyme, Hst2, by isothermal titration calorimetry (Table 1, Supplemental Figure 1). All acetyl-lysine analogs exhibited Kd values that were as low or lower than that of acetyllysine, with the exception of the α-hydroxyacetyl analog. Thus, the binding differences (<30-fold change among analogs) cannot account for the ∼600,000-fold range in nicotinamide formation rates observed. Instead, the negative slope likely reflects decreased nucleophilicity of the acetyl oxygen as the electron-withdrawing nature of the substituents were increased. This is best exemplified with the trifluoroacetyl analog that yielded the slowest rate of nicotinamide formation, but displayed the tightest binding to the enzyme.
Few precedented examples exist where an amide oxygen acts as a nucleophile in an enzyme catalyzed reaction. In only one distantly related case has Taft free-energy analysis been performed. With β-N-acetylglucosamidases, which utilize anchimeric assistance of the 2’-acetamide for glycoside hydrolysis, ρ*-values ranging from -0.4 to -1.6 were determined for several homologs.7 These ρ*-values provided evidence for direct nucleophilic participation of the amide carbonyl oxygen in attack at the anomeric position. The large ρ*-value of -1.9 reported here suggests that Sir2 deacetylases utilize similar nucleophilic participation of the amide carbonyl in ribosylnicotinamide cleavage, consistent with an SN2-like mechanism.
We also examined the ability of the acetyl-lysine analogs to support Sir2-catalyzed transglycosidation (nicotinamide exchange) in which exogenously-added nicotinamide reacts with an intermediate to reform NAD+ in the presence of an acetyllysine substrate.4,8 With acetyl-lysine analogs that bind tightly but are weak nucleophiles (e.g. fluorinated analogs), efficient transglycosidation might be predicted in an SN1 mechanism as discrete oxocarbenium formation would be independent of the attacking nucleophile. Measurement of the transglycosidation rate with the acetyl, monofluoroacetyl, and trifluoroacetyl analogs revealed facile exchange with the acetyl substrate (Vmax = 2.9 ± 0.2 s-1; Km = 406 ± 70 μM) but >400-fold slower exchange rates with the fluorinated analogs (Supplemental Figure 2). These extremely low exchange rates argue against formation of a stable oxocarbenium but instead support an SN2-like mechanism where nicotinamide captures the O-alkylamidate to efficiently reverse the reaction. We cannot rule out the possibility that the steady-state oxocarbenium level in an SN1 mechanism is vanishingly low with the fluorinated analogs because of a fast subsequent catalytic step. However, this is unlikely as the next catalytic step would involve the attack of acetyl-analogs on the oxocarbenium, a competing reaction with respect to nicotinamide exchange. Thus, nicotinamide exchange would be expected to be faster with the less nucleophilic fluorinated analogs, opposite to what we observe.
An SN2-like mechanism for Sir2 deacetylases is unusual, as many studies of enzymatic nicotinamide-ribosyl bond cleavage postulate formation of an oxocarbenium intermediate.3,9 Our results suggest that Sir2 deacetylases use a NAD+-consuming reaction whereby nicotinamide-ribosyl bond cleavage involves considerable participation of the incoming nucleophile (the amide oxygen) to form an O-alkylamidate intermediate. These results have important implications on the selective inhibition of Sir2 over other NAD+-metabolizing enzymes. Design and development of compounds that mimic the attack of acetyl-lysine may be potent and selective inhibitors of Sir2 deacetylases.
Supplementary Material
Acknowledgment
This work was supported by NIH grant GM065386 (to J.M.D.) and by NIH Biotechnology Training Grant NIH 5 T32 GM08349 (to B.C.S.). We thank Dr. Alvan Hengge and Dr. Norman Oppenheimer for helpful discussions.
Footnotes
Supporting Information Available: Experimental procedures, complete Sir2 mechanism, representative isothermal titration calorimetry data, mass spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Sauve AA, Wolberger C, Schramm VL, Boeke JD. Ann. Rev. Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- (2).Smith BC, Denu JM. Biochemistry. 2006;45:272–282. doi: 10.1021/bi052014t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3)(a).Rising KA, Schramm VL. J. Am. Chem. Soc. 1997;119:27–37. [Google Scholar]; (b) Parikh SL, Schramm VL. Biochemistry. 2004;43:1204–1212. doi: 10.1021/bi035907z. [DOI] [PubMed] [Google Scholar]; (c) Scheuring J, Berti PJ, Schramm VL. Biochemistry. 1998;37:2748–2758. doi: 10.1021/bi972594x. [DOI] [PubMed] [Google Scholar]; (d) Berti PJ, Blanke SR, Schramm VL. J. Am. Chem. Soc. 1997;119:12079–12088. doi: 10.1021/ja971317a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Scheuring J, Schramm VL. Biochemistry. 1997;36:8215–8223. doi: 10.1021/bi970379a. [DOI] [PubMed] [Google Scholar]; (f) Scheuring J, Schramm VL. Biochemistry. 1997;36:4526–4534. doi: 10.1021/bi962841h. [DOI] [PubMed] [Google Scholar]
- (4)(a).Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5807–5811. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Borra MT, Langer MR, Slama JT, Denu JM. Biochemistry. 2004;43:9877–9887. doi: 10.1021/bi049592e. [DOI] [PubMed] [Google Scholar]
- (5).Hansch C, Leo A. Substituent Constants for Correlation Analysis in Chemistry and Biology. 1979. [DOI] [PubMed] [Google Scholar]
- (6).We have observed ADPr in previous mass spectrometry experiments2 with Hst2 H135A and comparison of mass spectra to HPLC absorbance traces indicates that ADPr ionizes with similar efficiency to corresponding OAADPr analogs. Therefore, we are confident that ADPr is not formed.
- (7)(a).Dennis RJ, Taylor EJ, Macauley MS, Stubbs KA, Turkenburg JP, Hart SJ, Black GN, Vocadlo DJ, Davies GJ. Nat. Struct. Mol. Biol. 2006;13:365–371. doi: 10.1038/nsmb1079. [DOI] [PubMed] [Google Scholar]; (b) Vocadlo DJ, Withers SG. Biochemistry. 2005;44:12809–12818. doi: 10.1021/bi051121k. [DOI] [PubMed] [Google Scholar]; (c) Macauley MS, Whitworth GE, Debowski AW, Chin D, Vocadlo DJ. J. Biol. Chem. 2005;280:25313–25322. doi: 10.1074/jbc.M413819200. [DOI] [PubMed] [Google Scholar]; (d) Macauley MS, Stubbs KA, Vocadlo DJ. J. Am. Chem. Soc. 2005;127:17202–17203. doi: 10.1021/ja0567687. [DOI] [PubMed] [Google Scholar]; (e) Jones CS, Kosman DJ. J. Biol. Chem. 1980;255:11861–11869. [PubMed] [Google Scholar]
- (8)(a).Denu JM. Trends Biochem. Sci. 2005;30:479–483. doi: 10.1016/j.tibs.2005.07.004. [DOI] [PubMed] [Google Scholar]; (b) Sauve AA, Schramm VL. Biochemistry. 2003;42:9249–9256. doi: 10.1021/bi034959l. [DOI] [PubMed] [Google Scholar]; (c) Jackson MD, Schmidt MT, Oppenheimer NJ, Denu JM. J. Biol. Chem. 2003;278:50985–50998. doi: 10.1074/jbc.M306552200. [DOI] [PubMed] [Google Scholar]
- (9)(a).Sauve AA, Munshi C, Lee HC, Schramm VL. Biochemistry. 1998;37:13239–13249. doi: 10.1021/bi981248s. [DOI] [PubMed] [Google Scholar]; (b) Oppenheimer NJ. Mol. Cell. Biochem. 1994;138:245–251. doi: 10.1007/BF00928468. [DOI] [PubMed] [Google Scholar]; (c) Cakir-Kiefer C, Muller-Steffner H, Schuber F. Biochem. J. 2000;349:203–210. doi: 10.1042/0264-6021:3490203. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Muller-Steffner HM, Augustin A, Schuber F. J. Biol. Chem. 1996;271:23967–23972. doi: 10.1074/jbc.271.39.23967. [DOI] [PubMed] [Google Scholar]; (e) Holbourn KP, Shone CC, Acharya KR. FEBS J. 2006;273:4579–4593. doi: 10.1111/j.1742-4658.2006.05442.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.