

Abstract
Vitronectin and plasminogen activator inhibitor-1 (PAI-1) are proteins that interact in the circulatory system and pericellular region to regulate fibrinolysis, cell adhesion, and migration. The interactions between the two proteins have been attributed primarily to binding of the somatomedin B (SMB) domain, which comprises the N-terminal 44 residues of vitronectin, to the flexible joint region of PAI-1, including residues Arg-103, Met-112, and Gln-125 of PAI-1. A strategy for deletion mutagenesis that removes the SMB domain demonstrates that this mutant form of vitronectin retains PAI-1 binding (Schar, C. R., Blouse, G. E., Minor, K. M., and Peterson, C. B. (2008) J. Biol. Chem. 283, 10297–10309). In the current study, the complementary binding site on PAI-1 was mapped by testing for the ability of a battery of PAI-1 mutants to bind to the engineered vitronectin lacking the SMB domain. This approach identified a second, separate site for interaction between vitronectin and PAI-1. The binding of PAI-1 to this site was defined by a set of mutations in PAI-1 distinct from the mutations that disrupt binding to the SMB domain. Using the mutations in PAI-1 to map the second site suggested interactions between α-helices D and E in PAI-1 and a site in vitronectin outside of the SMB domain. The affinity of this second interaction exhibited a KD value ∼100-fold higher than that of the PAI-1-somatomedin B interaction. In contrast to the PAI-1-somatomedin B binding, the second interaction had almost the same affinity for active and latent PAI-1. We hypothesize that, together, the two sites form an extended binding area that may promote assembly of higher order vitronectin-PAI-1 complexes.
Vitronectin is a circulatory protein that interacts with several other plasma components that regulate coagulation and fibrinolysis, cell lysis via the complement cascade, and cell binding and migration in an integrin-dependent and urokinase-type plasminogen activator receptor-dependent fashion (1, 2). An interaction with vitronectin that has been widely studied occurs with the serine protease inhibitor (or serpin), PAI-1,2 the primary inhibitor of urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA). Most circulating PAI-1 is found in a complex with vitronectin (3). The formation of the complex between vitronectin and PAI-1 is presumed to be physiologically significant in several respects. Binding of vitronectin to PAI-1 stabilizes the active conformation of the serpin, increasing the half-life for its conversion to the inactive, so-called latent state, and maintains its protease inhibitory properties for a prolonged period (4, 5). PAI-1 binds to the N-terminal ∼50-amino acid somatomedin B (SMB) domain of vitronectin. The binding of PAI-1 competes with the binding of the urokinase-type plasminogen activator receptor to this domain and with the binding of integrins to the adjacent RGD sequence (for a review, see Ref. 6). Moreover, PAI-1 has been reported to induce oligomerization of vitronectin (7–10).
The structural basis for the transition of PAI-1 from an active to inactive or latent serpin is understood in exquisite detail thanks to several x-ray crystallographic studies on the two forms of the inhibitor (11–15). PAI-1 exhibits the canonical serpin fold (reviewed in Ref. 16), with a central five-strand β-sheet from which protrudes the reactive center loop (RCL), the peptide sequence that houses the specific target sequence recognized by proteases, tPA and uPA. The conversion of PAI-1 to its inactive, latent form results from repositioning of the RCL, whereby it becomes incorporated as a sixth strand in the central β-sheet, and the overall fold of the protein adopts a structure that is more thermodynamically favored (17). The identification of the primary binding site for vitronectin on PAI-1 as the flexible joint region, localized in an area defined by s2A, hE, and hF and comprising, among others, residues Arg-103, Met-112, and Gln-125 (18), was instructive for understanding the mechanism by which vitronectin stabilizes PAI-1 (16, 18–23). By binding to this flexible joint region at the “base” of central β-sheet A, vitronectin opposes expansion of the β-sheet, which is required to accommodate the RCL as a sixth strand. In this way, the active conformation of PAI-1 is stabilized by vitronectin. Besides this purely steric effect, other evidence suggests that the SMB domain induces extensive conformational changes in PAI-1 (24, 25).
Although there is ample information on the three-dimensional structure of PAI-1, much less is known about vitronectin. The only domain for which there is high-resolution information is the SMB domain (15, 26–31), which is the region of vitronectin that binds to the flexible joint region in PAI-1 and stabilizes the serpin. A co-crystal of a recombinant SMB domain and PAI-1 has provided useful information about the intermolecular contacts made in this complex (15). The PAI-1-binding site within the SMB domain is mostly limited to a single-turn α-helix, the only recognizable secondary structural element in this small disulfide-rich domain (15, 26–28).
In a recent study (32), we demonstrated that a deletion mutant of vitronectin lacking the SMB domain retains PAI-1 binding activity, although with a reduced affinity. These data suggest that PAI-1 should contain a second vitronectin-binding site to provide the needed complementary interactions required for binding to vitronectin outside of the SMB domain. To test this idea, the deletion mutant of vitronectin lacking the SMB domain (rΔsBVN (32)) was used to screen for binding to a battery of PAI-1 mutants. The results define a second vitronectin-binding site on PAI-1 that is distinct from the SMB-binding site, both in location and binding properties, although it is localized close to the SMB-binding site. Key questions to be addressed regarding the significance of this other binding site include the following. Where is the second site located relative to the flexible joint region of PAI-1? Does binding of vitronectin to this site stabilize the active conformation of PAI-1? How do the kinetics of binding to the second site compare with the binding to the SMB domain?
EXPERIMENTAL PROCEDURES
Materials—Native vitronectin was purified from human plasma using a modified protocol of the method developed by Dahlbäck and Podack (33, 34). A recombinant form of vitronectin lacking the somatomedin B domain (rΔsBVN) was expressed in the baculovirus FastBac system and purified as described (32). PAI-1 was produced according to the method outlined by Jensen et al. (21). The numbering used to denote the amino acid positions in PAI-1 throughout the present work is Ser-1—Ala-2—Val-3—His-4—His-5, according to Andreasen et al. (35). Latent PAI-1 was generated by incubating PAI-1 in running buffer for 7 days at 25 °C at a concentration of 5 μm. Incubation with uPA followed by SDS-PAGE to test for covalent, inactive serpin-uPA complexes was used to assay for PAI-1 activity and conversion to the latent form over time. Two versions of the SMB domain were used. One form (residues 1–51) was purified from a cyanogen bromide digest of human vitronectin as described (26). Another form (residues 1–47) was produced as a recombinant protein in Pichia pastoris and was a kind gift from Michael Ploug (Finsen Laboratory, Copenhagen, Denmark). The two forms are functionally and structurally indistinguishable (29). All other reagents were of analytical reagent grade or better.
Assay for Rate of Conversion of PAI-1 to a Latent Form—The assay for measuring the rate at which PAI-1 converts to the latent form was performed as described (18). PAI-1 (10 μg/ml final concentration) and vitronectin (2-fold molar excess over PAI-1) were incubated at 37 °C in HEPES-buffered saline with 0.25% gelatin. At various time points over a period of 2.5 h, samples were withdrawn for determination of the specific inhibitory activity of PAI-1 by titration against a fixed concentration of uPA. The PAI-1 activity was plotted as a function of incubation time, and the functional half-life of PAI-1 was calculated by fitting the data to a simple exponential decay.
Analysis of PAI-1 Binding to rΔsBVN and Full-length Vitronectin by Surface Plasmon Resonance (SPR)—For matters of convenience with multiple instruments in the various laboratories, SPR experiments for analysis of binding of PAI-1 variants to rΔsBVN or full-length vitronectin were performed on a Biacore X™, Biacore 3000™, or Biacore T100™ instrument equipped with CM5™ chips. There were no discernable differences between the results obtained by the three instruments. The running buffer used contained 10 mm HEPES, pH 7.4, 140 mm NaCl, 3 mm EDTA, 0.005% Tween-20, and 0.1% polyethylene glycol 8000. All solutions were filtered and degassed prior to use in the SPR experiments. Full-length vitronectin or rΔsBVN was coupled directly to the surface of a CM5™ chip with the standard amine coupling protocol. A solution of 30 μg/ml rΔsBVN or vitronectin in 10 mm sodium acetate, pH 4.5, was injected for coupling until the desired density was achieved. (Note that higher or lower densities of protein were coupled strategically to measure equilibrium saturation binding or determine kinetics for on- and off-rates.) Ethanolamine-blocked empty flow cells were used as reference. When necessary, regeneration of the flow cells between runs was achieved by injecting pulses of 10 mm glycine/HCl, pH 2.0, and/or 0.05% SDS in running buffer until base-line levels were acquired.
To determine the affinity of PAI-1 binding to full-length vitronectin or rΔsBVN, samples of PAI-1 diluted in running buffer were injected into an array of concentrations ranging from 0.25 to 1500 nm at a flow rate of 30 μl/min. After discontinuation of injection, the dissociation reaction was followed for a period of time. Rate constants for association (kon) and dissociation (koff) and equilibrium dissociation constants (KD) were determined in one of three ways. In most cases, the constants were determined by fitting the data to a 1:1 binding model using of the Biacore software. In other cases, koff values were first determined manually from the time courses of dissociation using the Biacore software. The calculated koff values, evaluated over a concentration range of 10 to 250 nm PAI-1, were shown to be concentration-independent, as expected for dissociation of the complex. The kon values were determined by following the association phase at different PAI-1 concentrations. In either case, the apparent association rate constants (kon,app) were determined by a fit (global with the Biacore software package or individual curve first for the manual data treatment) to Equation 1,
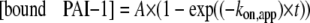 |
(Eq. 1) |
where A is the steady state level.
The true kon values were then determined from Equation 2.
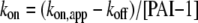 |
(Eq. 2) |
The equilibrium dissociation constant, KD, was then calculated as koff/kon.
Finally, in some cases the association and dissociation rates were too fast to be determined. In these cases, the KD values were calculated according to the method outlined by Rich and Myszka (36) for use of SPR in an equilibrium binding approach. The steady state binding values were plotted against the PAI-1 concentration and fit by nonlinear least squares to the equation,
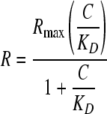 |
(Eq. 3) |
where R is the observed response, Rmax is the maximal response, C is the analyte concentration, and KD is the equilibrium dissociation constant.
To form the SMB domain-PAI-1 complexes, varying molar ratios of native SMB to a fixed concentration of active or latent PAI-1 (0.7 μm) were incubated for 30 min at room temperature before injection at a flow rate of 20 μl/min over either monomeric vitronectin or rΔsBVN.
For the monoclonal antibody blocking experiments, the antibodies were incubated with PAI-1 at 1:1 and 1:3 (PAI-1:antibody) molar ratios for 10 min at room temperature in running buffer. After this incubation period, the sample was injected at a flow rate of 20 μl/min over immobilized rΔsBVN.
RESULTS
The Deletion Mutant of Vitronectin Lacking the SMB Domain Does Not Stabilize the Active Conformation of PAI-1—One of the key consequences of vitronectin binding to PAI-1 is stabilization of the active serpin, with its RCL exposed and poised to act as bait for target proteases. The structural details of this stabilizing interaction have been attributed to binding of PAI-1 to the SMB domain of vitronectin (15, 37, 38). The intrinsic rate of conversion of wild-type PAI-1 in the absence and presence of added full-length vitronectin or rΔsBVN was measured by monitoring inhibition of uPA over time. In isolation, PAI-1 exhibits a half-life of 67 ± 3 min (n = 15) for conversion to the latent, inactive form. Full-length vitronectin stabilizes the serpin, increasing the half-life of the active form to 121 ± 17 min (n = 12, p = 0.0001), whereas the deletion mutant appears to have no effect on PAI-1, which exhibits a half-life of 75 ± 30 min (n = 8) in the presence of added rΔsBVN (p = 0.013).
Comparison of Binding of PAI-1 to rΔsBVN and Full-length Vitronectin—The binding of PAI-1 to either full-length vitronectin or rΔsBVN was followed using SPR. Initial experiments designed to detect weaker PAI-1-vitronectin interactions were performed with high PAI-1 concentrations (400 nm) and a high density of vitronectin or the rΔsBVN variant on the chip. The sensorgrams of active and latent PAI-1 binding to either vitronectin or rΔsBVN are shown in Fig. 1. The data in Fig. 1, top panel, with immobilized full-length vitronectin confirm the substantially weaker binding that latent PAI-1 exhibits for full-length vitronectin and clarify kinetic contributions to the overall low affinity of the interaction. Although the association phase with latent PAI-1 shows interaction with vitronectin, dissociation occurs at a high rate (t½ ∼ 10 s), yielding an overall weak affinity (Fig. 1, top panel, dashed line). The binding behavior is clearly different with active PAI-1, which exhibits a slow off-rate in the dissociation phase of the sensorgram (Fig. 1, top panel, solid line).
FIGURE 1.

Characterization of the binding of active and latent PAI-1 to full-length vitronectin and rΔsBVN by SPR. Active (solid lines) or latent (dotted lines) wild-type PAI-1 at a concentration of 400 nm was injected at a flow rate of 20 μl/min over a chip with 8000 RU of full-length vitronectin (top panel) or a chip with 6000 RU of rΔsBVN (bottom panel).
SPR experiments with active and latent PAI-1 confirmed that both forms of PAI-1 bind to rΔsBVN (Fig. 1, bottom panel); latent wild-type PAI-1 binds rΔsBVN slightly more weakly, with an approximate 20% reduction in the steady state level compared with active PAI-1. Nevertheless, the differences between association and dissociation kinetics with rΔsBVN immobilized on the chip are not nearly as marked as those observed in the case of full-length vitronectin. Notably, the dissociation phases are quite rapid for both active and latent forms of PAI-1, with half-lives around 5 s, akin to the dissociation kinetics observed with latent PAI-1 binding to full-length vitronectin and clearly different from the very slow dissociation of active PAI-1 from full-length vitronectin.
Analysis of the Binding of Active and Latent PAI-1 to rΔsBVN—To compare the binding of active and latent PAI-1 to the two different forms of vitronectin in detail, we performed a series of experiments that would accomplish a more quantitative characterization of binding by SPR. The association and dissociation time courses and steady state binding were followed at different PAI-1 concentrations ranging from 0.4 to 1000 nm, using a chip with ∼600 RU of rΔsBVN. Binding isotherms were generated by measuring the level of steady state binding at varying concentrations of active and latent PAI-1 to rΔsBVN on the chip, as described previously (32). Consistent with our previous report, the equilibrium binding experiments with active PAI-1 gave a KD value of 29 ± 3 nm. Evaluation of the rates of association and dissociation under these conditions gave koff = 0.049 ± 0.010 s-1 and kon = 1.17 ± 0.17 × 106 m-1s-1 (n = 7). KD calculated from the kinetics in SPR is 42 nm. Latent PAI-1 binds to rΔsBVN with a KD value of ∼120 nm (Fig. 2). Thus, latent PAI-1 binds only somewhat more weakly to the second site on vitronectin, which lies external to the SMB domain.
FIGURE 2.

Determination of the KD value for binding of PAI-1 to rΔsBVN. SPR was used as a steady state method for determining the KD value for binding of PAI-1 forms to the mutant vitronectin lacking the SMB domain, as described under “Experimental Procedures.” Varying concentrations of active wild-type PAI-1 (•), latent PAI-1 (▪), or active PAI-1 R117E/R120E (▾) were injected across a chip with 600 RU of immobilized rΔsBVN. The steady state binding levels obtained from this concentration series were normalized and plotted versus the concentration of PAI-1. The points were fit to the 1:1 interaction model (Equation 3) to yield a KD of 29 ± 3 nm for the binding of active wild-type PAI-1, a KD of 120 nm for the binding of latent PAI-1, and a KD of 480 ± 70 nm for the binding of the active form of R117E/R120E to rΔsBVN.
Mapping the Binding Site in PAI-1 That Recognizes Residues Outside of the SMB Domain in rΔsBVN—To probe for an additional vitronectin-binding site within PAI-1 that is complementary to the site on vitronectin that lies external to the SMB domain (32), monoclonal antibodies specific for regions of PAI-1 were tested for their ability to block binding to rΔsBVN. The monoclonal antibodies were preincubated with both active and latent PAI-1 before being injected over an rΔsBVN chip. The monoclonal antibodies mAb-1 (39) and mAb-6 (40), with respective binding epitopes localized to α-helix C and α-helix F, did not inhibit the binding of PAI-1 to rΔsBVN. However, two monoclonal antibodies, 33B8 (41) and mAb-7 (42), blocked binding of PAI-1 to rΔsBVN, with 33B8 and mAb-7 reducing the binding of latent PAI-1 to 37 or 22% of its original value, respectively, and reducing the binding of active PAI-1 to less than 50% of its original value. The epitopes for these antibodies are composed of residues from the loop connecting α-helix D and β-strand 2A, from β-strand 3A, and from β-strand 2B; the epitope for mAb-7 was mapped to Trp-88, Lys-90, Asp-91, Lys-178, and His-231 (42, 43).
The general localization of the binding site, apparent from the experiments using monoclonal antibodies, was refined by testing a battery of PAI-1 mutants for binding to rΔsBVN. To screen a larger number of mutants, we measured the steady state level of their binding to rΔsBVN immobilized on a Biacore chip, passing latent PAI-1 over the chip in concentrations ranging from 1 to 2000 nm. The data are summarized in Table 1, reporting the steady state binding of the mutants relative to that of wild-type PAI-1 at a concentration of 400 nm, a value slightly above the KD value described above for binding of latent PAI-1 to rΔsBVN. Under these conditions, binding is subsaturating, and the KD is a major determinant of the total binding. We found that mutant forms of PAI-1 with a number of charge reversal mutations or multiple alanine substitutions reduced the binding substantially. The implicated residues are localized in α-helix E (Arg-117 and Arg-120), α-helix F (Lys-71, Arg-78, Tyr-81, Lys-82) or β-strand 3A (Trp-177). It appears that multiple mutations or charge reversal mutations are needed to observe a significant reduction in binding, as single alanine substitutions had less effect (Table 1). Remarkably, we also found a significant reduction in the binding of a PAI-1 variant with mutations in the flexible hinge region (R103A/M112A/Q125A), which we reported previously as having a strongly reduced binding via the SMB domain (18).
TABLE 1.
Effect of mutations on PAI-1 binding to rΔsBVN as analyzed by SPR PAI-1 (400 nm) was injected at a flow rate of 20 μl/min over a chip coated with 8000 RU of rΔsBVN. The steady state binding levels were normalized to the average active wild-type PAI-1 steady state binding level in the same experiment. Average values are given ± S.D. with the number of replicates in parentheses. Additional PAI-1 mutants with single-point mutations that were tested but for which binding was not significantly different (reduced by more than 50%) compared with the wild type were E55A, Q57A, Q58A, Q59A, Q61A, K71A, K71A/Y81A, R78A, K82A, M85A, P87A, W88A, K90A, D91A, E92A, S94A, T96A, F116A, S121A, T122A, K178A, K231A, and D233A. All of the mutants shown in this table had a significantly reduced binding as compared with wild-type PAI-1 (p < 0.01).
| PAI-1 variant | Latent | Active |
|---|---|---|
| Wild type | 79 ± 3 (6) | 100 |
| K82E | 48 ± 8 (2) | 28 ± 1 (2) |
| R117E/R120E | 8 ± 1 (6) | 11 ± 21 (6) |
| R103A/M112A/Q125A | 32 ± 10 (5) | 60 ± 21 (6) |
| K71A/R78A/Y81A/K82A | 12 ± 1 (2) | 27 ± 1 (4) |
| W177F | 8 ± 6 (2) | 37 ± 18 (5) |
Because the R117E/R120E mutant form of PAI-1 was severely impaired with regard to binding to the secondary site in vitronectin, we determined the KD value for its binding to rΔsBVN by SPR using steady state measurements (Fig. 2). The calculated dissociation constant from these data is ∼480 nm, compared with 25–50 nm for active, wild-type PAI-1.
Evaluation of the Binding of PAI-1 Mutants to Full-length Vitronectin—As described above, mapping of the second site for binding of PAI-1 to the deletion mutant of vitronectin revealed that there were relatively small differences in affinities for the active and latent forms of PAI-1. Furthermore, there was no stabilization of the active conformation by rΔsBVN, although full-length vitronectin gave the well characterized prolonged half-life. These observations indicated that it was important to reevaluate the binding of PAI-1 to full-length vitronectin in light of our new data indicating dual binding sites on both proteins. For this reason, we pursued studies with wild-type PAI-1 and the mutants identified in the mapping experiments that defined the second binding site using SPR measurements with full-length vitronectin immobilized on the chip surface.
The SPR analysis of binding of latent PAI-1 to full-length vitronectin (Fig. 3) indicated that the binding kinetics fit well to a simple 1:1 model. This is reasonable because the predominant binding that occurs with the SMB domain is not observed with latent forms of PAI-1, so that the sole binding interaction is that mediated via the second site. Fitting the data to this 1:1 model showed that latent PAI-1 bound to full-length vitronectin with an association rate constant of (3.86 ± 2.56) × 105 m-1s-1, a dissociation rate constant of (3.39 ± 2.11) × 10-2 s-1, and an equilibrium dissociation constant of 94 ± 27 nm (n = 9) (Table 2). Using an alternative approach, determining the KD value from the concentration dependence of the steady state binding yielded a value of 177 ± 98 nm. By both measures, the KD value is thus ∼2 orders of magnitude weaker than observed for the binding of active PAI-1 (18). Significantly, these rate constants, as well as the equilibrium dissociation constant, were in good agreement with those for the binding of active and latent PAI-1 to rΔsBVN (Fig. 2).
FIGURE 3.

Analysis of the binding of latent PAI-1 to full-length vitronectin by SPR. The indicated PAI-1 variants (latent form), in concentrations of 250 (black lines), 125 (gray lines), 63 (red lines), 32 (green lines), 16 (blue lines), 8 (orange lines), and 4 nm (magenta lines), were injected over a chip with ∼3000 RU of full-length vitronectin at a flow rate 30 μl/min. After discontinuation of the injection, dissociation was followed for an additional 200 s.
TABLE 2.
Effect of mutations on the binding of latent PAI-1 to full-length vitronectin as analyzed by SPR Latent PAI-1, in concentrations of 500, 250, 125, 63, 32, 16, 8, 4, and 0 nm, were passed over a chip with approximately 3000 RU of full-length vitronectin at a flow rate of 30 ml/min. After 120 s, the injection was discontinued, and the dissociation phase was followed. Means ± S.D. and the number of experiments (in parentheses) for the estimated KD values are indicated in the table. Other PAI-1 mutants with single-point mutations that were tested but for which binding was not significantly different (reduced by more than 50%) from the wild type were K71A, R78A, Y81A, K82A, M85A, P87A, W88A, R117A, R120A, N174A, K178A, H231A, D233A, K90A, S94A, T96A, E92A, and D91A.
| PAI-1 variant | KD |
|---|---|
| nm | |
| Wild type | 94 ± 27 (8)a |
| 177 ± 98 (8)b | |
| K82E | 538 ± 208 (3)b |
| R117E/R120E | 5282 ± 4487 (7)b |
| R103A/M112A/Q125A | 811 ± 359 (5)b |
| K71A/R78A/Y81A/K82A | 769 ± 299 (5)b |
| W177F | >10,000 (5)b |
The data were fit to a 1:1 binding model using Biacore software. The KD value for wild-type PAI-1 was calculated as the ratio of the on- and off-rates: kon = 3.86 ± 2.56 × 105 m–1s–1 and koff = 3.39 ± 2.11 × 10–2s–1 (8 measurements each)
KD values were calculated manually from the PAI-1 concentration dependence of the steady state binding values according to Equation 3 using the equilibrium binding approach of Rich and Myzska (36) (see “Experimental Procedures”)
We also tested latent forms of PAI-1 from the battery of mutants used for the mapping studies for binding to full-length vitronectin (Fig. 3). Among this large series of PAI-1 mutants, K82E, R117E/R120E, R103A/M112A/Q125A, K71A/R78A/Y81A/K82A, and W177F exhibited an increased KD value as compared with wild-type PAI-1 (Table 2). Indeed, these are the same mutant forms of PAI-1 that exhibit reduced binding for rΔsBVN. Thus, latent PAI-1 binds to full-length vitronectin with kon, koff, and KD values similar to those found for the binding of active and latent PAI-1 to rΔsBVN. Moreover, the effects of the individual mutations in PAI-1 were similar in the two cases.
Nonetheless, a close inspection of the effects of some mutations revealed small but significant differences in the binding of latent PAI-1 to rΔsBVN and full-length VN. With rΔsBVN on the chip, the R103A/M112A/Q125A mutant showed reduced binding, but the K71A/R78A/Y81A/K82A form of PAI-1 showed more severely impaired binding (Table 1). In contrast, in experiments using full-length vitronectin on the chip, the R103A/M112A/Q125A mutation reduced the binding 10-fold, whereas K71A/R78A/Y81A/K82A had a much weaker effect (Table 2, Fig. 3).
Comparison of the Binding of Active and Latent Forms of PAI-1 Mutants to Full-length Vitronectin—The binding site for the SMB domain on PAI-1 has been well defined by x-ray crystallography (15, 31) and site-directed mutagenesis (19, 21). Because the mutants identified in this study (K82E, R117E/R120E, R103A/M112A/Q125A, K71A/R78A, Y81A/K82A, and W177F) interact in the latent form at a site that is separate from the recognition sequences for the SMB domain, it was important to complete our analysis and test for any effects of these mutagenized forms of PAI-1 in their active conformation on binding to full-length vitronectin, which contains the SMB domain. In other words, the effects on affinity of these mutants in their latent form, where binding is mediated solely through the second site, would be expected to be negligible for the active forms of these mutants, in which binding to full-length vitronectin is dominated by the high affinity interaction at the SMB domain.
We tested for these effects using SPR, with full-length vitronectin immobilized on the chip and injection of the PAI-1 variants in their active form, in concentrations ranging from 0.4 to 50 nm (Fig. 4). The binding kinetics were characterized by a fast association and a very slow dissociation. Because of the near immeasurable off-rates, we relied on our calculations for the initial association rates to permit a meaningful comparison of the relative binding of the mutants in a quantitative fashion. The rates were linearly related to the PAI-1 concentration (Table 3). It is apparent from this analysis that the mutant forms of PAI-1 with substitutions of K82E, R117E/R120E, and W177F differed little if at all from the wild-type protein in binding rates. However, PAI-1 with K71A/R78A/Y81A/K82A had a somewhat reduced binding, and the R103A/M112A/Q125A mutant exhibited no measurable binding.
FIGURE 4.

SPR analysis of the binding of active PAI-1 to full-length vitronectin. The indicated PAI-1 variants (active form), in concentrations of 50 (black lines), 25 (gray lines), 12.5 (red lines), 6.25 (green lines), 3.13 (blue lines), 1.56 (orange lines), and 0.78 nm (magenta lines), were injected over a chip with ∼500 RU of full-length vitronectin at a flow rate 30 μl/min. After discontinuation of the injection, dissociation was followed for an additional 200 s.
TABLE 3.
Concentration dependence of initial association rate of active PAI-1 binding to full-length vitronectin in SPR experiments The indicated PAI-1 variants (active form), in concentrations of 50, 25, 12.5, 6.25, 3.13, and 1.56 nm, were injected over a chip with approximately 500 RU of full-length vitronectin. The observed binding curves are shown in Fig. 4. For each PAI-1 variant, the slopes of the initial linear portions of the association curves were calculated by linear regression analysis. The rates vary linearly with the PAI-1 concentration. The table shows means ± S.D. for the indicated number of independent experiments (in parentheses).
| PAI-1 variant | Rate |
|---|---|
| Rμs–1 nm–1 | |
| Wild type | 0.178 ± 0.027 (6) |
| K82E | 0.156 ± 0.022 (3) |
| R117E/R120E | 0.155 ± 0.031 (5) |
| R103A/M112A/Q125A | 0.000 ± 0.000 (5) |
| K71A/R78A/Y81A/K82A | 0.059 ± 0.023 (5) |
| W177F | 0.182 ± 0.04 (5) |
Thus, the effects of specific mutations on the binding of active PAI-1 to full-length vitronectin are unrelated to their effects on the binding of latent PAI-1 to full-length vitronectin. Likewise, they are not directly correlated to the binding behavior observed with the rΔsBVN deletion mutant.
Tests for Competition of the Isolated SMB Domain for Binding to Full-length Vitronectin and rΔsBVN—The residues in PAI-1 implicated for binding to the second site on vitronectin are found outside of the previously identified SMB binding area. Thus, the two vitronectin-binding sites on PAI-1 appear to be separate and distinct, and thus binding of the isolated SMB domain to PAI-1 should not prevent binding of PAI-1 to the second site on vitronectin. This possibility was explored using SPR. Complexes of PAI-1 and the isolated SMB domain were formed and then injected over immobilized rΔsBVN. The isolated SMB domain, in slight molar excess, causes a partial competition of the binding of 700 nm active PAI-1 to rΔsBVN (see representative data in Fig. 5A); in four independent experiments, isolated SMB caused a reduction in binding of active PAI-1 to (0.45 ± 0.09 times the control value). In contrast, and as predicted, equimolar amounts of isolated SMB strongly competed the binding of active PAI-1 to full-length vitronectin (Fig. 5B). In either case, with rΔsBVN or full-length vitronectin on the chip, the isolated SMB domain competed poorly for the binding of latent PAI-1 to either form of vitronectin; this result is as expected because latent PAI-1 has a very low affinity for the SMB domain.
FIGURE 5.

Effect of isolated SMB domain on the binding of PAI-1 to rΔsBVN or full-length vitronectin. A, binding to rΔsBVN. PAI-1 (0.7 μm) was incubated for 30 min at room temperature in the absence or presence of a 2-fold molar excess of isolated SMB domain before injection at a flow rate of 20 μl/min over a chip coated with 6000 RU of rΔsBVN. B, binding to full-length vitronectin. PAI-1 (100 nm) was passed over a Biacore chip coated with 3000 RU of monomeric vitronectin in the absence or presence of a 1.5-fold molar excess of isolated SMB domain. The results in both panels for binding of PAI-1 are shown in the absence (solid lines) or presence (dashed lines) of the SMB domain. In both panels, binding in the presence of the SMB domain is normalized relative to the of binding observed for PAI-1 alone.
In both cases, there is little or no competition from the isolated SMB domain for binding to the second site on PAI-1, either in binding to rΔsBVN or in binding of latent forms of PAI-1 to full-length vitronectin. These results substantiate a second, weaker site for interaction between PAI-1 and vitronectin, which determines the interaction between both active and latent PAI-1 with rΔsBVN, as well as the interaction of latent PAI-1 with full-length vitronectin.
Characterization of PAI-1 Mutants with Defective Binding to the Second Site on Vitronectin—All of the mutant forms of PAI-1 that were tested were fully active inhibitors of uPA as measured by the formation of stable, covalent serpin-uPA complexes observed in SDS-PAGE. To further characterize the mutants that had a reduced binding to the second site, we measured the half-life for their conversion to the latent state upon incubation at 37 °C (Table 4). In agreement with previous findings, wild-type PAI-1 had a half-life of a little more than 1 h. The R103A/M112A/Q125A and K71A/R78A/Y81A/K82A forms of PAI-1 did not differ from wild-type PAI-1 with respect to the half-life for transition to the latent form. The R117E/R120E mutant had a somewhat prolonged half-life, and the K82E form of PAI-1 exhibited a 3-fold shorter half-life. All variants but R103A/M112A/Q125A PAI-1 exhibited the expected prolonged half-life in the presence of vitronectin. This was as expected because the R103A/M112A/Q125A form of PAI-1 has mutations in the flexible hinge region to disrupt binding to the SMB domain. Because the W177F mutant was previously reported to have a strongly prolonged half-life of more than 400 min (44), the effects of vitronectin on the rate of conversation of this variant to a latent form were not tested.
TABLE 4.
Half-lives for conversion of PAI-1 variants to the latent form For each PAI-1 variant, the specific inhibitory activity toward uPA was monitored over time at 37 °C; the rate of conversion to the latent form is expressed as the functional half-life, t1/2. The averages ± S.D. for the indicated number of independent experiments (in parentheses) are given. ND, not determined.
| PAI-1 variant | Vitronectin | t1/2 |
|---|---|---|
| min | ||
| Wild type | – | 67 ± 3 (15) |
| + | 121 ± 17 (12)a | |
| R117E/R120E | – | 109 ± 8 (3)b |
| + | 158 ± 22 (3)a | |
| R103A/M112A/Q125A | – | 59 ± 3 (3) |
| + | 45 ± 4 (3) | |
| K71A/R78A/Y81A/K82A | – | 59 ± 4 (3) |
| + | 127 ± 4 (3)a | |
| W177F | – | 432 ± 18 (3)b |
| + | ND | |
| K82E | – | 19 ± 4 (3)b |
| + | 138 ± 4 (3)a |
Significantly larger than the corresponding value in the absence of vitronectin (p ≤ 0.01)
Significantly different from the corresponding value for wild-type PAI-1 (p ≤ 0.01)
DISCUSSION
Structural Determinants of the Second Binding Site on PAI-1 Are Localized in the Distal Ends of Helices D and E and Are Separate from the SMB-binding Site—The results presented here are compatible with the hypothesis of a second, weaker interaction between PAI-1 and vitronectin, different from the SMB domain-PAI-1 binding. This conclusion is based on three types of evidence.
First, analyses of the binding kinetics led us to conclude that latent PAI-1 binds to full-length vitronectin and rΔsBVN with similar kon, koff, and KD values. Active PAI-1 binds to rΔsBVN with kinetics similar to that for the binding of latent PAI-1 to full-length vitronectin and rΔsBVN, but it binds much tighter to full-length vitronectin. Whereas SMB domain binding dominates the interaction of full-length vitronectin with active PAI-1, the absence of this interaction in the mutant form of vitronectin engineered without the SMB domain and in the binding of latent PAI-1 to full-length vitronectin makes it possible to map the contributions from the second, weaker site.
Second, from the hypothesis of two separate and distinct vitronectin-binding sites on PAI-1, one would predict that interactions of PAI-1 with the SMB domain should not prevent PAI-1 binding to the second site in vitronectin. In experiments designed to test this idea, complexes of active PAI-1 bound to the isolated SMB domain were shown to bind to rΔsBVN, albeit the binding was somewhat diminished compared with that for free PAI-1. In contrast, evaluation of the binding of active PAI-1 to full-length vitronectin by SPR, in the absence and presence of the SMB domain, showed that saturation of PAI-1 with the SMB domain virtually eliminates the high-affinity, slowly dissociating binding to native vitronectin. Steric effects may account for the small degree of inhibition observed with the SMB-PAI-1 complex binding to rΔsBVN because of the observed proximity of the SMB domain-binding site and the second binding site. The main PAI-1 binding determinants in the SMB domain are near a single-turn α-helix housing residues 26–30 and stabilized by an extensively disulfide-bonded domain structure (15, 26). Although the core of the SMB domain, including residues 18–41, is well structured, NMR measurements (26) and x-ray crystallography (15) indicate that the flanking regions extending beyond residue 42 are more disordered. It is therefore also possible that the slight SMB domain inhibition is due to the overlap of the SMB domain (residues 1–51 for the cyanogen bromide fragment (26) or residues 1–47 for the recombinant SMB (29)) and rΔsBVN (residues 41–458) and that this flexible region (amino acids 42–47) in the isolated SMB domain partially interferes with the binding of the PAI-1-SMB complex to full-length vitronectin and rΔsBVN.
Third, a panel of PAI-1 mutants was used to define the surface that corresponds to a secondary vitronectin-binding site on PAI-1. Several mutants with multiple alanine substitutions or charge reversal mutations had a strong impact on the binding. Mutants involving residues Lys-71, Arg-78, Tyr-81, Lys-82, Arg-117, Arg-120, and Trp-177 were implicated in the secondary site. These residues are clustered on the surface of α-helix D and α-helix E and at the C-terminal end of β-strand 3A (Fig. 6). Both ribbon and space-filling models, which highlight the location of the second binding site on PAI-1 concomitantly with the primary site in the flexible joint region, are shown in the figure. Likewise, monoclonal antibodies with epitopes in the α-helix D–β-strand 2A area were found to inhibit binding. Among the mutations tested, the most pronounced effects on binding were observed with PAI-1 mutants harboring cumulative mutations, whereas single amino acid substitutions had less striking effects. This fact, in conjunction with the relative large area over which the mutants are spread, suggests an extended interaction surface resulting from relatively weak contributions to the binding energy from individual residues.
FIGURE 6.

PAI-1 structures indicating the regions important in interacting with primary and secondary sites on vitronectin. A, a ribbon structure for active PAI-1 (Protein Data Bank accession code 1OC0), with the central β-sheet A shown in blue and the C-sheet in orange. The RCL, which is not observed in the crystal structure because of its mobility, is represented by the hashed line connecting the N- and C-terminal ends. B and C, space-filling models of active PAI-1 (Protein Data Bank accession code 1OC0) and latent PAI-1 (Protein Data Bank accession code 1LJ5), respectively. Amino acids identified as involved in binding to rΔsBVN in SPR experiments are color-coded, with residues associated with mutations that exhibit impaired binding to rΔsBVN shown in red (see Table 1). For reference, PAI-1 residues that are involved in binding to the SMB domain of vitronectin are colored green (15, 38), The mutations R103A/M112A/Q125A, which reduce binding to both the SMB domain and rΔsBVN, are shown in yellow, and the residues for which point mutations have no effect on binding to the to rΔsBVN chip are shown in cyan. The magenta ribbon structures in A and B represent the SMB domain complexed with PAI-1 (15).
Despite the fact that multiple mutations are most effective in disrupting binding to the second site, the effects of the mutations themselves do not appear to be the result of extensive structural changes or disordering. The mutant forms of PAI-1 typically exhibited normal function. For instance, the R103A/M112A/Q125A mutant has only a slightly increased rate at which it is converted to the latent form, and it exhibits normal binding to endocytosis receptors (18). Although two mutants (R117E/R120E and K82E) differed somewhat from wild-type PAI-1 with respect to the rates of transition to the latent form, they give the normal response upon addition of vitronectin, with prolonged half-lives for conversion to the latent conformation. Thus, most mutations resulting in a reduced binding to the second site did not affect the binding to full-length vitronectin via the SMB domain. Of course, an exception is PAI-1 with the R103A/M112A/Q125A mutations, which was engineered as a PAI-1 variant without measurable affinity to full-length vitronectin via the SMB domain (18). This mutant does not bind the SMB domain and thus did not respond to vitronectin in this fashion. Conversely, the W177F mutant identified in this study was previously shown to be “stable,” such that it converts to the latent form at an extremely slow rate (44) and does not require SMB binding to stabilize the active conformation. In summary, the remarkable clustering of the mutations identified to interact with the second binding site present on rΔsBVN indicates that these residues together define a more extended interaction surface for vitronectin than previously recognized.
The Second Binding Site Outside of the SMB Domain Only Weakly Discriminates between Active and Latent Forms of PAI-1—Latent PAI-1 is known to exhibit greatly diminished binding to vitronectin (45). This phenomenon is well understood, as there are significant structural differences between active and latent PAI-1 at the base of the A-sheet at the ends of helices D, E, and F that deform the primary binding surface in the region that interacts with the SMB domain (13, 15, 19, 21). In contrast, latent PAI-1 binds to the secondary site in vitronectin with similar kinetics and affinity compared with the binding observed with active PAI-1. Comparison of the structures of active and latent PAI-1 shows some changes in the positions of helices D and E, but the relatively contiguous nature of the secondary binding site surface on PAI-1 is maintained (Fig. 6).
Previous work, based on enzyme-linked immunosorbent assays to compare the various conformations of PAI-1 in terms of binding to vitronectin, suggests a large difference in binding affinity when comparing active and latent forms of PAI-1 (45). The current study supports these findings; however, detectable binding of latent PAI-1 to full-length and truncated vitronectin lacking the SMB domain was detected by SPR. Latent PAI-1 bound to vitronectin at this second site, and the SPR studies showed that the affinity was weaker than observed for the interaction within the SMB domain. The studies comparing binding of latent and active forms of PAI-1 to the second site revealed that the weak binding was due to appreciable off-rates (dissociation rate constants) that were nearly equivalent for latent and active forms of the serpin. Furthermore, binding the mutant form of vitronectin, which lacks the primary site for PAI-1 binding in the SMB domain, did not stabilize PAI-1 activity, so that it lost activity as it converted to the latent form at nearly the same rate as in the absence of vitronectin. Thus, this secondary binding site does not appear to play a role in one of the main known functions of vitronectin, that of stabilizing the active form of PAI-1.
What Is the Role of Two Complementary Binding Sites on Vitronectin and PAI-1?—The interaction between vitronectin via the SMB domain with PAI-1 has been well characterized. However, our mutagenesis approach and prior biophysical studies have pointed to a more complex binding scenario that involves regions outside of this primary binding site, between the flexible joint region and SMB domain. Early clues that suggested this model came from analytical ultracentrifugation on the PAI-1-vitronectin complex. That study demonstrated that two molecules of PAI-1 could bind to a single molecule of vitronectin, with assembly to a 4:2 (PAI-1:vitronectin) intermediate well populated at micromolar concentrations of the proteins (9). Further work using sedimentation velocity measurements in the ultracentrifuge clarified the concentration dependence of assembly of higher-order PAI-1-vitronectin complexes and suggested a step-wise mechanism for their assembly by which the primary, high-affinity site in the SMB domain is bound first followed by occupation of a second, lower affinity site at higher concentrations of PAI-1 (10). In the present study using SPR, any effects of PAI-1 that induce higher order complexes cannot occur and will not affect the results because vitronectin is covalently cross-linked to the chip.
The functional effects of these more extensive PAI-1-vitronectin interactions remain to be established. The KD for the binding of PAI-1 to the second site in isolation is high relative to circulating levels of PAI-1. With PAI-1 concentrations in vivo in the low nanomolar range, a KD value in the high nanomolar range would lead to very little binding. However, the concentration of PAI-1 in the extracellular environment in tissue has been reported to be much higher, approaching levels at which this binding event becomes significant, especially under pathological scenarios (46–50). Also, even at nanomolar concentrations, the presence of two binding sites will favor formation of a more extended binding surface comprised of both sites when the two proteins are juxtaposed by interaction at the primary, high-affinity site involving the SMB domain. A likely model for the PAI-1-vitronectin binding is that a single PAI-1 molecule binds one vitronectin molecule via an interaction involving both the first and the second site. The pathway for binding could consist of an initial docking of the SMB domain onto the flexible hinge region of PAI-1 followed by the establishment of an extended interaction that involves the secondary sites. An exciting possibility is that that the second site is necessary for inducing higher order structures of vitronectin oligomers, which might be deposited in the extracellular matrix and promote cell adhesion.
This work was supported, in whole or in part, by National Institutes of Health Grant HL50676 from NHLBI (to C. B. P.). This work was also supported by grants from the Danish Cancer Society, the Danish Research Agency, the University of Aarhus, and the European Commission (to P. A. A.) and by a grant from the Carlsberg Foundation (to G. E. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: PAI-1, plasminogen activator inhibitor type 1; RCL, reactive center loop; rΔsBVN, recombinant vitronectin lacking the SMB domain due to a deletion of residues 1–40; SMB, somatomedin B; SPR, surface plasmon resonance; uPA, urokinase-type plasminogen activator; tPA, tissue-type plasminogen activator; mAb, monoclonal antibody; RU, response unit(s).
References
- 1.Ekmekci, O. B., and Ekmekci, H. (2006) Clin. Chim. Acta 368 77-83 [DOI] [PubMed] [Google Scholar]
- 2.Preissner, K. T., and Seiffert, D. (1998) Thromb. Res. 89 1-21 [DOI] [PubMed] [Google Scholar]
- 3.Declerck, P. J., De Mol, M., Alessi, M. C., Baudner, S., Paques, E. P., Preissner, K. T., Muller-Berghaus, G., and Collen, D. (1988) J. Biol. Chem. 263 15454-15461 [PubMed] [Google Scholar]
- 4.Lindahl, T. L., Sigurdardottir, O., and Wiman, B. (1989) Thromb. Haemostasis 62 748-751 [PubMed] [Google Scholar]
- 5.Wun, T. C., Palmier, M. O., Siegel, N. R., and Smith, C. E. (1989) J. Biol. Chem. 264 7862-7868 [PubMed] [Google Scholar]
- 6.Durand, M. K., Bodker, J. S., Christensen, A., Dupont, D. M., Hansen, M., Jensen, J. K., Kjelgaard, S., Mathiasen, L., Pedersen, K. E., Skeldal, S., Wind, T., and Andreasen, P. A. (2004) Thromb Haemostasis 91 438-449 [DOI] [PubMed] [Google Scholar]
- 7.Seiffert, D., and Loskutoff, D. J. (1996) J. Biol. Chem. 271 29644-29651 [DOI] [PubMed] [Google Scholar]
- 8.Minor, K. H., and Peterson, C. B. (2002) J. Biol. Chem. 277 10337-10345 [DOI] [PubMed] [Google Scholar]
- 9.Podor, T. J., Shaughnessy, S. G., Blackburn, M. N., and Peterson, C. B. (2000) J. Biol. Chem. 275 25402-25410 [DOI] [PubMed] [Google Scholar]
- 10.Minor, K. H., Schar, C. R., Blouse, G. E., Shore, J. D., Lawrence, D. A., Schuck, P., and Peterson, C. B. (2005) J. Biol. Chem. 280 28711-28720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharp, A. M., Stein, P. E., Pannu, N. S., Carrell, R. W., Berkenpas, M. B., Ginsburg, D., Lawrence, D. A., and Read, R. J. (1999) Structure 7 111-118 [DOI] [PubMed] [Google Scholar]
- 12.Mottonen, J., Strand, A., Symersky, J., Sweet, R. M., Danley, D. E., Geoghegan, K. F., Gerard, R. D., and Goldsmith, E. J. (1992) Nature 355 270-273 [DOI] [PubMed] [Google Scholar]
- 13.Stout, T. J., Graham, H., Buckley, D. I., and Matthews, D. J. (2000) Biochemistry 39 8460-8469 [DOI] [PubMed] [Google Scholar]
- 14.Nar, H., Bauer, M., Stassen, J. M., Lang, D., Gils, A., and Declerck, P. J. (2000) J. Mol. Biol. 297 683-695 [DOI] [PubMed] [Google Scholar]
- 15.Zhou, A., Huntington, J. A., Pannu, N. S., Carrell, R. W., and Read, R. J. (2003) Nat. Struct. Biol. 10 541-544 [DOI] [PubMed] [Google Scholar]
- 16.Zhou, A., Carrell, R. W., and Huntington, J. A. (2001) J. Biol. Chem. 276 27541-27547 [DOI] [PubMed] [Google Scholar]
- 17.Baker, D., and Agard, D. A. (1994) Biochemistry 33 7505-7509 [DOI] [PubMed] [Google Scholar]
- 18.Jensen, J. K., Durand, M. K., Skeldal, S., Dupont, D. M., Bodker, J. S., Wind, T., and Andreasen, P. A. (2004) FEBS Lett. 556 175-179 [DOI] [PubMed] [Google Scholar]
- 19.Lawrence, D. A., Berkenpas, M. B., Palaniappan, S., and Ginsburg, D. (1994) J. Biol. Chem. 269 15223-15228 [PubMed] [Google Scholar]
- 20.Padmanabhan, J., and Sane, D. C. (1995) Thromb. Haemostasis 73 829-834 [PubMed] [Google Scholar]
- 21.Jensen, J. K., Wind, T., and Andreasen, P. A. (2002) FEBS Lett. 521 91-94 [DOI] [PubMed] [Google Scholar]
- 22.van Meijer, M., Gebbink, R. K., Preissner, K. T., and Pannekoek, H. (1994) FEBS Lett. 352 342-346 [DOI] [PubMed] [Google Scholar]
- 23.Arroyo De Prada, N., Schroeck, F., Sinner, E. K., Muehlenweg, B., Twellmeyer, J., Sperl, S., Wilhelm, O. G., Schmitt, M., and Magdolen, V. (2002) Eur. J. Biochem. 269 184-192 [DOI] [PubMed] [Google Scholar]
- 24.Gibson, A., Baburaj, K., Day, D. E., Verhamme, I., Shore, J. D., and Peterson, C. B. (1997) J. Biol. Chem. 272 5112-5121 [DOI] [PubMed] [Google Scholar]
- 25.Hansen, M., Busse, M. N., and Andreasen, P. A. (2001) Eur. J. Biochem. 268 6274-6283 [DOI] [PubMed] [Google Scholar]
- 26.Mayasundari, A., Whittemore, N. A., Serpersu, E. H., and Peterson, C. B. (2004) J. Biol. Chem. 279 29359-29366 [DOI] [PubMed] [Google Scholar]
- 27.Kamikubo, Y., De Guzman, R., Kroon, G., Curriden, S., Neels, J. G., Churchill, M. J., Dawson, P., Oldziej, S., Jagielska, A., Scheraga, H. A., Loskutoff, D. J., and Dyson, H. J. (2004) Biochemistry 43 6519-6534 [DOI] [PubMed] [Google Scholar]
- 28.Kamikubo, Y., Kroon, G., Curriden, S. A., Dyson, H. J., and Loskutoff, D. J. (2006) Biochemistry 45 3297-3306 [DOI] [PubMed] [Google Scholar]
- 29.Kjaergaard, M., Gardsvoll, H., Hirschberg, D., Nielbo, S., Mayasundari, A., Peterson, C. B., Jansson, A., Jørgensen, T. J., Poulsen, F., and Ploug, M. (2007) Protein Sci. 16 1934-1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li, X., Zou, G., Yuan, W., and Lu, W. (2007) J. Biol. Chem. 282 5318-5326 [DOI] [PubMed] [Google Scholar]
- 31.Zhou, A. (2007) Protein Sci 16 1502-1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schar, C. R., Blouse, G. E., Minor, K. H., and Peterson, C. B. (2008) J. Biol. Chem. 283 10297-10309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bittorf, S. V., Williams, E. C., and Mosher, D. F. (1993) J. Biol. Chem. 268 24838-24846 [PubMed] [Google Scholar]
- 34.Dahlbäck, B., and Podack, E. R. (1985) Biochemistry 24 2368-2374 [DOI] [PubMed] [Google Scholar]
- 35.Andreasen, P. A., Riccio, A., Welinder, K. G., Douglas, R., Sartorio, R., Nielsen, L. S., Oppenheimer, C., Blasi, F., and Dano, K. (1986) FEBS Lett. 209 213-218 [DOI] [PubMed] [Google Scholar]
- 36.Rich, R. L., and Myszka, D. G. (2001) J. Mol. Recognit. 14 223-228 [DOI] [PubMed] [Google Scholar]
- 37.Seiffert, D., Ciambrone, G., Wagner, N. V., Binder, B. R., and Loskutoff, D. J. (1994) J. Biol. Chem. 269 2659-2666 [PubMed] [Google Scholar]
- 38.Deng, G., Royle, G., Wang, S., Crain, K., and Loskutoff, D. J. (1996) J. Biol. Chem. 271 12716-12723 [DOI] [PubMed] [Google Scholar]
- 39.Bodker, J. S., Wind, T., Jensen, J. K., Hansen, M., Pedersen, K. E., and Andreasen, P. A. (2003) Eur. J. Biochem. 270 1672-1679 [DOI] [PubMed] [Google Scholar]
- 40.Wind, T., Jensen, M. A., and Andreasen, P. A. (2001) Eur. J. Biochem. 268 1095-1106 [DOI] [PubMed] [Google Scholar]
- 41.Gorlatova, N. V., Elokdah, H., Fan, K., Crandall, D. L., and Lawrence, D. A. (2003) J. Biol. Chem. 278 16329-16335 [DOI] [PubMed] [Google Scholar]
- 42.Mathiasen, L., Dupont, D. M., Chrsitsensen, A., Blouse, G. E., Jensen, J. K., Gils, A., Declerck, P. J., Wind, T., and Andreasen, P. A. (2008) Mol. Pharmacol. 74 641-653 [DOI] [PubMed] [Google Scholar]
- 43.Bijnens, A. P., Ngo, T. H., Gils, A., Dewaele, J., Knockaert, I., Stassen, J. M., and Declerck, P. J. (2001) Thromb. Haemostasis 85 866-874 [PubMed] [Google Scholar]
- 44.Blouse, G. E., Perron, M. J., Kvassman, J. O., Yunus, S., Thompson, J. H., Betts, R. L., Lutter, L. C., and Shore, J. D. (2003) Biochemistry 42 12260-12272 [DOI] [PubMed] [Google Scholar]
- 45.Lawrence, D. A., Palaniappan, S., Stefansson, S., Olson, S. T., Francis-Chmura, A. M., Shore, J. D., and Ginsburg, D. (1997) J. Biol. Chem. 272 7676-7680 [DOI] [PubMed] [Google Scholar]
- 46.Arteel, G. E. (2008) J. Gastroenterol. Hepatol. 23 Suppl. 1, S54-S59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ozel Demiralp, D., Aktas, H., and Akar, N. (2007) Clin. Appl. Thromb. Hemostasis, December 26, 2007 DOI: 10.1177/1076029607305081 [DOI] [PubMed]
- 48.Cale, J. M., and Lawrence, D. A. (2007) Curr. Drug Targets 8 971-981 [DOI] [PubMed] [Google Scholar]
- 49.Wanhainen, A., Nilsson, T. K., Bergqvist, D., Boman, K., and Bjorck, M. (2007) J. Vasc. Surg. 45 1109-1113 [DOI] [PubMed] [Google Scholar]
- 50.Aso, Y. (2007) Front. Biosci. 12 2957-2966 [DOI] [PubMed] [Google Scholar]