

Abstract
Functional proteomics approaches that comprehensively evaluate the biological activities of human cDNAs may provide novel insights into disease pathogenesis. To systematically investigate the functional activity of cDNAs that have been implicated in breast carcinogenesis, we generated a collection of cDNAs relevant to breast cancer, the Breast Cancer 1000 (BC1000), and conducted screens to identify proteins that induce phenotypic changes that resemble events that occur during tumor initiation and progression. Genes were selected for this set using bioinformatics and data mining tools that identify genes associated with breast cancer. Greater than 1000 cDNAs were assembled and sequence verified with high-throughput recombination-based cloning. To our knowledge, the BC1000 represents the first publicly available sequence-validated human disease gene collection. The functional activity of a subset of the BC1000 collection was evaluated in cell-based assays that monitor changes in cell proliferation, migration and morphogenesis in MCF10A mammary epithelial cells expressing a variant of ErbB2 that can be inducibly activated through dimerization. Using this approach, we identified many cDNAs, encoding diverse classes of cellular proteins, that displayed activity in one or more of the assays, thus providing insights into a large set of cellular proteins capable of inducing functional alterations associated with breast cancer development.
Introduction
The human adult breast is an organized, branch-like structure comprising ducts, lobes and intervening stromal tissue. Lobes are composed of multiple individual units termed acini, which have hollow lumen encased by a single layer of polarized epithelial cells that are surrounded by myoepithelial cells and a basement membrane. Disruption of this well-ordered structure is a prerequisite for the initiation of carcinoma. Increased proliferation of epithelial cells, loss of acinar structure and filling of luminal space are common characteristics of the premalignant stages of breast cancer, such as ductal carcinoma in situ (DCIS). The transformation from premalignant stages to invasive disease is commonly associated with loss of anchorage dependence, invasion through the basement membrane and metastasis. The genetic events that trigger the conversion of normal breast acini to premalignant stages of breast cancer and the subsequent progression to malignancy are only partially understood. The loss of tumor suppressor function or the gain of oncoprotein activity can induce some of these changes. Since these proteins can act as master switches and regulate many downstream pathways, their dysfunction can lead to broad reaching consequences.
Although many tumor suppressors and oncoproteins have been characterized with respect to their biological activities, numerous genomic and proteomic studies have revealed hundreds of alterations in uncharacterized proteins in breast tumor cells1–7.. One limitation of such analyses is the inability to distinguish whether the candidate cDNAs or proteins are the cause or the effect of these oncogenic transitions.
One approach to identify cDNAs that control tumorigenic transitions is to use phenotypic assays to screen for cDNAs that can alter the behavior of cells in a defined manner. In this context, a causal relationship can be established between protein and phenotype, and with further testing, a role in disease development can be further examined. The value of phenotypic assays can be enhanced using cell culture systems in which cells are allowed to organize into structures that resemble the natural architecture of breast acini, such as three-dimensional (3D) basement membrane cultures of MCF-10A cells. MCF-10A cells are immortalized, non-transformed breast epithelial cells that require epidermal growth factor (EGF) for proliferation8. In 3D basement membrane cultures, they form hollow, growth-arrested structures that resemble normal human mammary acini9–11. Using 3Dbasement membrane cultures of MCF-10A cells, we have demonstrated that 3D cultures provide a context to monitor a broad spectrum of biological activities. Indeed, each of the cDNAs that we have found to induce hyperproliferation provoke distinct changes in the architecture of acinar structures12, 13. For example, hyperproliferative structures induced by cyclin D are hollow due to clearance of the proliferating, centrally-localized cells through programmed cell death; whereas the luminal space of structures expressing activated ErbB2 homodimers or CSF-1 receptors are filled with cells as a consequence of the anti-apoptotic activity of these activated receptor tyrosine kinases10, 14, 15. In addition, structures expressing activated CSF-1R show distinct alterations in cell-cell adhesion, leading to dissociation of cells within the structures.
The progression from premalignancy to malignancy is in part marked by the appearance of cell migratory behavior. We previously conducted a pilot screen of 30 candidate cDNAs to identify proteins that cooperate with ErbB2 activation to induce cell migration in MCF-10A cells in the absence of motogenic factors such as EGF16. Two members of the Transforming Growth Factor β (TGFβ) family, TGFβ1 and TGFβ3, were identified as pro-migratory. Furthermore, co-activation of ErbB2 and TGFβ signaling pathways appeared to promote invasive behavior in MCF-10A cells. Several reports have provided supporting evidence that TGFβ can collaborate with ErbB2 in mice to promote metastasis17–19. These data suggest that the TGF signaling pathway operates synergistically with ErB2 signaling to promote both metastatic and invasive behavior in premalignant cells.
These pilot studies have illustrated the value of expanding such phenotypic screens to much larger gene sets. Historically, large-scale screening experiments in mammalian cells have relied upon pooled cDNA libraries as the cDNA sources, carrying with them a number of limitations20–22. The methods used to produce these libraries result in many incomplete or out of frame cDNA products, missing cDNAs (not expressed in the mRNA source tissues), and, most importantly, a vast overrepresentation of proteins produced from abundant mRNAs. Consequently, millions of clones (a large fraction repetitive or aberrant) must be examined in order to ensure broader coverage of the genome, ultimately limiting the types of screens that can be implemented using pools of cDNAs, thus requiring deconvolution and/or rescue of responsible cDNA.
Ideally, a functional screen of the proteome would entail testing every distinct protein independently, requiring an indexed collection of full length sequence verified clones encompassing the entire proteome. The availability of such a cataloged clone set would enable efficient proteomic screens in which hits can be easily identified, information regarding all clones (even those with no response) extracted and the significance of each clone assimilated. With the caveat that not all cDNAs identified by these screens would prove to be directly related to the underlying biology, it is likely that their identification may contribute toward understanding the molecular pathways involved.
To examine the feasibility and validity of this approach, we sought to build and screen a moderately sized collection of breast cancer-related cDNAs. Our screening strategy involved assembling a ready-to-use clone collection of candidate cDNAs and screening the first several hundred genes in this collection in three well- characterized cell-based phenotypic assays to identify cDNAs that induce cancer-like phenotypes. We have assembled the Breast Cancer 1000 (BC1000), a sequence-validated cDNA collection of 1000 breast cancer-related cDNAs ranging from well-studied oncogenes to less conspicuous breast cancer-associated cDNAs. A subset of cDNAs in this collection was transferred to retroviral vectors, transduced into MCF-10A cells, and assayed for their ability to induce phenotypic characteristics associated with premalignancy and malignancy. Specifically we tested for EGF-independent proliferation, disruption of acinar morphogenesis, and cell migratory behavior independently or cooperatively with activated ErbB2. Through this comprehensive approach, we identified potentially novel functional activities for known breast cancer-associated cDNAs.
Materials and Methods
Bioinformatics
MedGene, an advanced literature-mining software developed in-house, was utilized to systematically identify all breast cancer-related cDNAs (http://hipseq.med.harvard.edu/MEDGENE/)23. Various statistical methods were used to estimate the strength of gene-disease relationships using MedGene, including chi-square analysis, Fisher’s exact probabilities, relative risk of gene, relative risk of disease and the product of frequency. All statistical scores were log-transformed to obtain a normal distribution. The validation of MedGene was done using 3 different approaches: 1) a comparison of MedGene results with manually annotated disease databases, 2) a detailed analysis on the 1467 genes related to prostate cancer, and 3) a global analysis of 1000 diseases23. As previously described, the results demonstrated that MedGene was more comprehensive than manually annotated disease databases, with an estimated 70–80% of identified gene-disease relationships representing true relationships. All aspects of clone production were tracked and managed by our internally-designed laboratory information management system (LIMS), termed the FLEX database. This relational database has a three-tier architecture: data storage (Oracle 8i), business logic (Java, Java Servlets, Java Server Pages, the Struts framework, Tomcat) and presentation layers (HTML). Oligonucleotides incorporating the appropriate cloning recombination sites and clone format were designed based on the nearest-neighbor algorithm using our oligonucleotide generator software. The FLEXGene database and the oligonucleotide generator have web-based interfaces that users can query (www.hip.harvard.edu>Informatics>Programs).
Cloning Strategy
Oligonucleotides specific for each target cDNA that incorporated the appropriate cloning recombination sites and clone format (closed or fusion) were generated based on the nearest-neighbor algorithm. PCR products were gel purified on 1% agarose gels in a size-based saw tooth pattern to avoid cloning aberrant products such as primer-dimer, alternative splice forms and cross-hybridizing sequences. Purified PCR products were captured into either pDONR201 (Gateway®) or pDNR-dual (Creator®) using the BP Clonase (Invitrogen) or In-Fusion™ enzyme (Clontech), repectively24. The resulting intermediate was then transformed into DH5α bacteria, robotically plated onto custom-designed bioassay dishes with 48 compartments and 4 independent colonies were picked automatically using the MegaPix™ (Genetix) colony-picking robot. Colonies were grown in liquid culture, which was then allocated to produce archived glycerol stocks and prepped to make DNA for sequencing and transfer to other vectors.
PCR amplification of BC1000 cDNAs
PCR reactions, done in 96-well format, consisted of 50uL reactions containing 0.1uM oligonucleotides, 1x Platinum® Pfx Amplification Buffer and Enhancer (Invitrogen), 0.3mM dNTPs, 1mM MgSO4, 1U Platinum® Pfx DNA Polymerase, and 100ng brain:placenta (1:4) first-strand cDNA (prepared in-house) or 200ng plasmid template. For Gateway® clones, a second PCR reaction with similar conditions was done to add recombination sites and to refresh enzyme. Annealing temperatures and times, as well as cycle numbers varied depending on cloning strategy, template, and gene size. The final PCR products were visualized on 1% agarose gels and correctly sized bands were excised. DNA was isolated through gel filtration.
Clone capture reactions
Capture of cDNAs into the entry vector pDONR201 for Gateway® or pDNR-dual for Creator® was performed by carrying out 10uL reactions in 96-well format. BD In-Fusion™ capture reactions contained 7uL of gel-purified and glass filtered PCR product, 1X In-Fusion™ reaction buffer, 0.1ng/ul pDNR-dual, 1X BSA, and 0.5 uL of In-Fusion™ enzyme concentrate diluted 1:10 in In-Fusion™ dilution buffer. Gateway® BP capture reactions contained 5ul of gel-purified and glass filtered PCR product, 1X BP Reaction Buffer, 1ng/uL pDONR201, and 2uL BP Clonase. Reagents were thawed and mixed on ice, and capture reactions were incubated at 25°C overnight.
Transformation of DH5α bacterial cells and isolation of clones
Transformations for Gateway® clones were performed with 5uL BP reaction, 45uL KCM (100mM KCL, 30mM CaCl2, and 50mM MgCl2), and 50uL DH5α cells. Reactions were incubated for 20 minutes on ice, followed by 10 minutes at room temperature. 90uL of TB was then added to each well and plates were shaken (370rpm) at 37°C for 1 hour. For In-Fusion™, 5uL of capture reaction diluted in water (1:5) was added to 45uL KCM (100mM KCL, 30mM CaCl2, and 50mM MgCl2), and 50uL DH5α cells. Reactions were incubated in ice water for 30 minutes, placed in 42°C for 45 seconds, and then transferred back to ice water for 2 minutes. 120uL SOC was then added to each well and plates were shaken (370rpm) at 37°C for 1 hour. Transformations were plated on 48 well grid LB-Agar plates containing either 50ug/mL Kanamycin (Gateway®) or 125ug/mL Ampicillin and 40ug/mL X-gal (BD In-Fusion™). Individual isolates were grown in 1mL LB containing the appropriate antibiotics. DNA preparations were done with Millipore Montage™ (Qiagen).
Clone validation
All clones were submitted for full-length DNA sequence analysis. We utilized a primer walking strategy, which encompassed end reads for all clones followed by internal sequencing for clones that required further sequencing. The sequencing reactions were done either in-house or by Agencourt™. We then assembled and aligned sequence reads for each clone to the expected target sequence using Sequencher™ software. Aligned sequences were manually scanned for potential mismatches. All discrepancies were confirmed by visual inspection of trace files.
Cell culture and materials
MCF-10A cells were cultured as previously described11 in Growth medium (DMEM/F12 (Invitrogen) supplemented with 5% horse serum, 20 ng/ml epidermal growth factor (EGF), 10 μg/ml insulin or 100ng/ml IGF-I (R&D Biosystems), 0.5 μg/ml hydrocortisone, 100 ng/ml cholera toxin, 50 U/ml penicillin, and 50 mg/ml streptomycin.). Matrigel was purchased from Collaborative Biosciences; the protein concentration of the lots used ranged between 9 and 11 mg ml−1. AP1510 was from ARIAD Pharmaceuticals.
Retroviral transduction and cell lines
The generation of MCF-10A cells expressing the chimeric receptor p75.B2 has been described10. cDNAs (with a STOP codon) captured in pDONR-201 were transferred into a modified version of pBABE-Puro, pBABE-puroATT, in which the Gateway™ cassette was inserted into the EcoR1 site according to the manufacturer’s protocol (Invitrogen). MCF-10As and MCF-10As with the inducibly activated ErbB2 receptor (10A.B2) were plated and infected with VSV-G pseudotyped retroviruses expressing cDNA from the BC1000 collection then selected for vector expression with Puromycin, as described16. Controls cells were generated by transfection of the empty pBabe-puroATT vector alone and simultaneous Puromycin selection11.
EGF-independent proliferation assay
As mammary epithelial cells require EGF to undergo normal cellular proliferation, 231 cDNAs were screened for the ability to confer EGF-independent proliferation. Cells were virally transduced with unique retroviral vectors encoding cDNAs from a subset of the BC1000 collection and selected for vector expression 48 hours after completion of antibiotic selection. EGF-independent proliferation in two-dimensional cultures was performed by plating 3 × 104 MCF-10A cells in 6.0cm-diameter dishes in Assay media (MCF-10A growth media containing 1% serum and no EGF). Cultures were maintained for 12–14 days. Proliferation was assessed after fixation and crystal violet staining. A cDNA was scored as a hit if the induced cellular proliferation was estimated to be more than three-fold above the vector-transfected MCF10A control.
Morphogenesis assays
Acinar structures were generated by plating individual 10A.B2 cells expressing each of 265 BC1000 cDNAs in the absence of dimerizer in 3D culture within a reconstituted basement membrane (Matrigel™). After 15–18 days in culture, acini were stained with 4’,6-diamidino-2-phenylindole (DAPI) to ascertain whether acinar units had basally localized nuclei and a hollow lumen. Three-dimensional assays were performed using the overlay method as described16. cDNAs were scored as a hit if any abnormalities were observed in acinar formation, morphology, or lumen formation or if invasion into Matrigel was observed. In addition to the initial screen, any validated migration hits were also examined with 10A.B2 cells in 3D basement membrane cultures in the presence AP1510. Cells that were examined under these conditions were initially plated in an identical manner, and then 4 days after seeding in 3D culture, EGF was withdrawn and Assay media with 500 nM AP1520AP1510 was added. Similar assays were performed with 10A.16 and 10A.21 cells without the addition of AP1510 and were monitored over 24 hours.
Transwell migration assay
MCF-10A cells as well as the 10A.B2 subtype (with or without dimerizer) will not migrate without additional stimuli. 10A.B2 cells were transduced with each of the 258 cDNAs from the BC1000 collection and were starved overnight in Assay media. Cells (1x105) were added to the top chambers of 24-well transwell plates (BD, 8μm pore size), and Assay media with 500 nM AP1510 was added to the bottom chambers. After overnight incubation, top (non-migrated) cells were removed with a cotton swab, and bottom (migrated) cells were fixed and stained with crystal violet to visualize cells. Addition of AP1510, the chimeric ErbB2 dimerizer, was utilized to activate ErbB2. cDNAs were scored as a hit if they initially induced an estimated 2-fold increase in migration over the vector-transduced control. Experiments were repeated a minimum of three times for cDNAs that scored as a hit, and considered a validated hit if a migratory phenotype was observed at least twice. Validated hits were also tested under similar conditions without the addition of 500 nM AP1510.
Results
Selection of BC1000 cDNAs
To provide a useful resource for studying the mechanisms involved in the development of breast cancer, we built a clone collection of more than 1000 cDNAs with particular relevance to this disease. Selection of the cDNAs for inclusion in the BC1000 was a collective effort: roughly 200 genes were suggested by local experts in breast cancer research, 50 genes were shown to be overexpressed in ductal carcinoma in situ (DCIS) by SAGE analysis4, and the remainder were identified by MedGene (http://hipseq.med.harvard.edu/MEDGENE/login.jsp). MedGene is a literature-mining software application that searches all titles and abstracts in the Medline database to identify cDNAs co-cited with a particular disease and utilizes statistical methods to rank the relative strengths of these gene-disease relationships based on the frequency of total citation and co-citation23. Based on our previous evaluation and validation of MedGene, we utilized the log of the product of frequency (LPF) to rank the list of breast cancer genes23. LPF represents the natural log of the product of the proportion of disease/gene double hits to disease single hits and the proportion of disease/gene double hits to gene single hits.
Using MedGene, we identified approximately 2500 cDNAs that were associated with breast cancer among all Medline abstracts. As with all automated data mining tools, MedGene has associated false positive and false negative rates. The false negative rate, the frequency of omitting cDNAs related to breast cancer in the literature, is approximately 9% and occurs with cDNAs cited only rarely23. The false positive rate (cDNAs erroneously listed as related to the disease) is dependent on the frequency of gene-disease co-citation, averaging approximately 26%23. False positives usually result because other terms such as the names of cell lines, restriction sites, plasmids, medical terms, etc., share the same names as cDNAs. The BC1000 cDNA list was assembled by favoring the highest ranked cDNAs (most related to breast cancer) where the false positive rate is lowest. When cloning attempts for some cDNAs failed, additional cDNAs were taken from the list as needed to achieve a final collection of 1000 breast cancer-related cDNA clones. All relevant information for each cDNA was obtained from NCBI and imported into the FLEXGene, our clone tracking database.
Construction of the BC1000
To ensure that the BC1000 clones would be available for the broadest range of experiments, the coding regions of the cDNAs were captured into recombinational cloning vectors. The advantage of this cloning strategy is the ability to transfer the coding sequences in frame and without mutation into any protein expression vector in a simple one-step reaction. The BC1000 gene collection was assembled into the Gateway® system (Invitrogen) (http://www.invitrogen.com/content.cfm?pageid=4072) and/or the Creator™ system (BD Biosciences)( http://www.clontech.com/clontech/products/families/creator/index.shtml). Clones were produced in two formats: one with a universal stop codon (TAG) to allow expression of native protein (closed) and one where the stop codon has been replaced with Leucine (TTG) to allow carboxyl-terminal peptide fusions (fusion). Amino-terminal peptide fusions can be added to both forms.
During clone production, we utilized an established automated clone production pipeline and laboratory information management system (LIMS), achieving a final throughput of four 96 well plates of cDNAs per week. Figure 1 illustrates a complete schematic used in creating the BC1000 collection. All processes were carried out in bar-coded containers and tracked by the Oracle™-based FLEXGene database at both sample and plate level along with any relevant process results (gel images, PCR success, colony count, etc.). The multi-step process began with selecting cDNAs for the production queue and completed with the culture and DNA preparation of 8 total candidate isolates for each cDNA (4 open isolates and 4 fusion isolates).
Figure 1.

Schematic representation of workflow utilized to create the BC1000 collection.
The clone production process occurred in two stages based on PCR template, starting with clones in the NIH Mammalian Gene Collection (MGC) and followed by first-strand cDNA. We utilized the MGC clones for all BC1000 cDNAs that were available in this collection, and all other BC1000 cDNAs were obtained from first-strand cDNA produced from human brain and placenta mRNA (1:4). MGC clones were preferred because they provided a higher concentration of well-characterized template, thereby reducing PCR cycle number and the mutation rate. The PCR products were all gel purified to avoid cloning contaminants or inappropriate splice forms and all resulting clones were single colony purified to ensure that each represents a single DNA species.
The ability to generate PCR product was the strongest predictor of cloning success rate. Extensive testing and analysis of various conditions revealed, not surprisingly, that the most important PCR parameter was template. Overall, first-strand cDNA was better than bacterially amplified library DNA. Brain, placenta and testis tissues proved to be the best sources for first-strand cDNA. PCR success rate dramatically improved from approximately 60% to 95% when utilizing the MGC clones as template. Furthermore, this method required substantially fewer PCR cycles (12 versus 40 rounds), resulting in a lower mutation rate and a reduction in cost and time for sequencing. Other factors that contributed to unsuccessful cloning were failed recombination reactions and the capture of incorrect cDNAs or cDNAs with sequences that differed from the expected wild-type sequence.
Clone validation
For each gene format (closed or fusion), one isolate was initially selected for full-length sequencing. We utilized strict criteria for inclusion in the BC1000 collection. Clones that contained any form of a truncation mutation, a reading frame mutation or that differed by more than one missense mutation from the wild-type sequence were not included in the BC1000. Silent mutations and allelic variation identified in GenBank were considered acceptable. Additional isolates were sequenced when the first isolate was deemed unacceptable.
In the NCBI GenBank database, many cDNAs have multiple mRNA sequence entries, which often differ slightly from one another presumably due to natural sequence variation. As the “expected” sequence of the clones produced from first strand cDNA was not known a priori, the reference target sequence was arbitrarily selected from among the relevant entries for that cDNA. Actual clone sequences that did not match the selected reference sequence could represent allelic variation rather than mutations. In this regard, MGC clones had the advantage that there was no ambiguity about the expected sequence. We used BLAST or our automated Polymorphism Finder25 to query all relevant clone sequence discrepancies with the GenBank database to determine whether they represented previously described variants. If these discrepancies were previously observed in GenBank, they were considered allelic variation, not mutations. We observed a cumulative estimated mutation rate of 1 base change per 1000 nucleotides. Roughly 40% of observed base pair differences between the observed and expected clone sequence were previously described in the GenBank database, such as alternate splice forms and allelic variants. Overall, the number of BC1000 clones with a potential missense mutation was fewer than 15%, and many of these will likely represent unidentified genetic variation as opposed to mutation. A complete list of cDNAs in the BC1000 collection can be viewed from the Harvard Institute of Proteomics website (http://www.hip.harvard.edu/).
Phenotypic screens
To assess the range of functional consequences of overexpressing the BC1000 cDNAs, we introduced the first 265 constructed cDNAs into a line of immortalized breast epithelial cells (MCF-10A) and subjected these cells to a single pass screen using a variety of phenotypic assays. To enhance the sensitivity of the screens, we utilized a derivative line of MCF-10A cells (10A.B2) expressing an inducibly-activated variant of ErbB2, which allowed us to analyze the functional activities of the BC1000 cDNAs in the absence or presence of a sensitized background. The 10A.B2 cells express a chimeric receptor that can be activated by dimerization mediated by the small molecule, AP151010. We initially evaluated the ability of the expressed cDNAs to promote cellular proliferation in the absence of EGF, to perturb morphogenesis of acinar structures in 3D basement membrane cultures, and to induce EGF-independent cell migration. The proliferation and morphogenesis assays were performed in the absence of dimerizer, whereas the migration assays were executed in the presence of dimerizer. These distinct screen formats were employed since dimerization of ErbB2 induces phenotypic effects in control cells in the cell proliferation and morphogenesis assays, whereas the dimerization of ErbB2 results in minimal, if any, activity in migration assays. Thus, it was feasible to activate ErbB2 in the migration assays to generate a sensitized background. Each cDNA was introduced by retroviral transduction and assayed independently using a medium-throughput parallel approach. A list of all screened cDNAs and their corresponding LPF ranking score (for those identified by MedGene) are included in Supplemental Table 1, and Supplemental Table 2 shows the genes that were identified as hits in any of the three first pass screens. LPF ranking is the log of the product frequency score generated by MedGene, for which a more negative value is indicative of a lower ranking with respect to the strength of the gene-disease relationship.
Given that our cloning strategy was to prioritize genes based on MedGene ranking score (LPF), the average LPF score was higher for those screened compared to the complete list of breast cancer genes obtained from MedGene (−10.07 versus −11.63, respectively; Mann-Whitney rank sum test, P-value <0.001). The overall first-pass hit rate for each initial screen ranged between 8–29% of the 239 to 265 cDNAs tested (Table 1). The relatively high hit rate likely reflects the bias of the genes that were selected for association with breast cancer. In addition, the highest hit rate in the migration assay may also be a consequence of the use of sensitization in that assay. Interestingly, the genes that scored as hits in the three first pass screens were not biased towards the well-studied genes in the literature. They had nearly the same average statistical literature ranking score (LPF) as that of the entire set of screened genes (−10.07 versus −10.15, respectively; Mann-Whitney rank sum test, P-value = 0.77), indicating that the genes identified from these screens represented a broad range of less- to well-studied breast cancer genes. The identification of less-studied breast cancer genes exemplifies the capability of using an unbiased functional proteomics approach to identify novel genes related to various aspects of disease biology. As expected, the majority of genes that scored in all three assays were proto-oncogenes, which included ARAF, BMP5, HGF, HRAS, RHOG, EDG2, RAF1, JUNB, and TGFα. The fact that these screens identified both genes that functioned only in specific assays as well as known proto-oncogenes that scored in all three assays, demonstrates the validity and power of the functional proteomics approach.
Table 1.
Percentage of single-pass hits for initial screens
| Assay | % Hit rate (no. hits/no. cDNAs screened) |
|---|---|
| Proliferation1 | 8.8% (21/239) |
| Morphogenesis2 | 22.3% (59/265) |
| Migration3 | 29.1% (75/258) |
Proliferation was assayed after plating 10A.B2 cells on 6-well dishes in Assay medium over 10 days. A cDNA was scored as a hit if it induced more than a 3 fold increase compared with control.
Morphogenesis was assessed in 3-D Matrigel cultures of 10A.B2 cells and a cDNAs was scored as a hit if any abnormalities were observed in acinar formation, morphology or lumen formation or if invasion into Matrigel was observed.
Migration was evaluated in a transwell chamber using 10A.B2 cells treated with dimerizer. A cDNA was scored as a hit if migration was more than 2 fold greater than control.
The genes that displayed strong induction of EGF-independent proliferation are predictable hits (HGF, TGFα and HRAS). In addition, ARAF induced a moderate induction of proliferation, most likely due to production of an EGFR ligand, since we have found that Raf1 and MEK2DD, an activated variant of MEK214, 26, also induce EGF-independent proliferation due to autocrine production of an EGFR ligand (Debnath et al27 (and data not shown). Several genes induced a mild, 2-to5-fold increase in cell proliferation. Further studies are required to validate these hits.
The morphogenesis and migration screens produced the greatest number of hits from the BC1000 cDNAs. A few examples of cDNAs that altered acinar morphogenesis of 10A.B2 cells are shown in Figure 2. Observed phenotypic abnormalities in the morphogenesis assays ranged from subtle differences [such as delayed lumen formation (BCL2L1 data not shown) or small spike-like projections (ORC1L data not shown)], to large, disorganized spherical structures (ARAF, GRB2), and disorganized hyperproliferative structures (JUNB), in some cases displaying protrusive branches or cords that resembled features of invasive tumor cells (EDG2, BMP5). The extent to which acinar structure was disorganized often prevented the analysis of normal acinar features [such as polarity or lumen formation (ARAF, GRB2, JUNB, BMP5, EDG2)]. Given the variety of phenotypic alterations and the subjectivity associated with scoring morphogenesis hits, further validation and analysis of hits were performed using the transwell migration assay.
Figure 2.
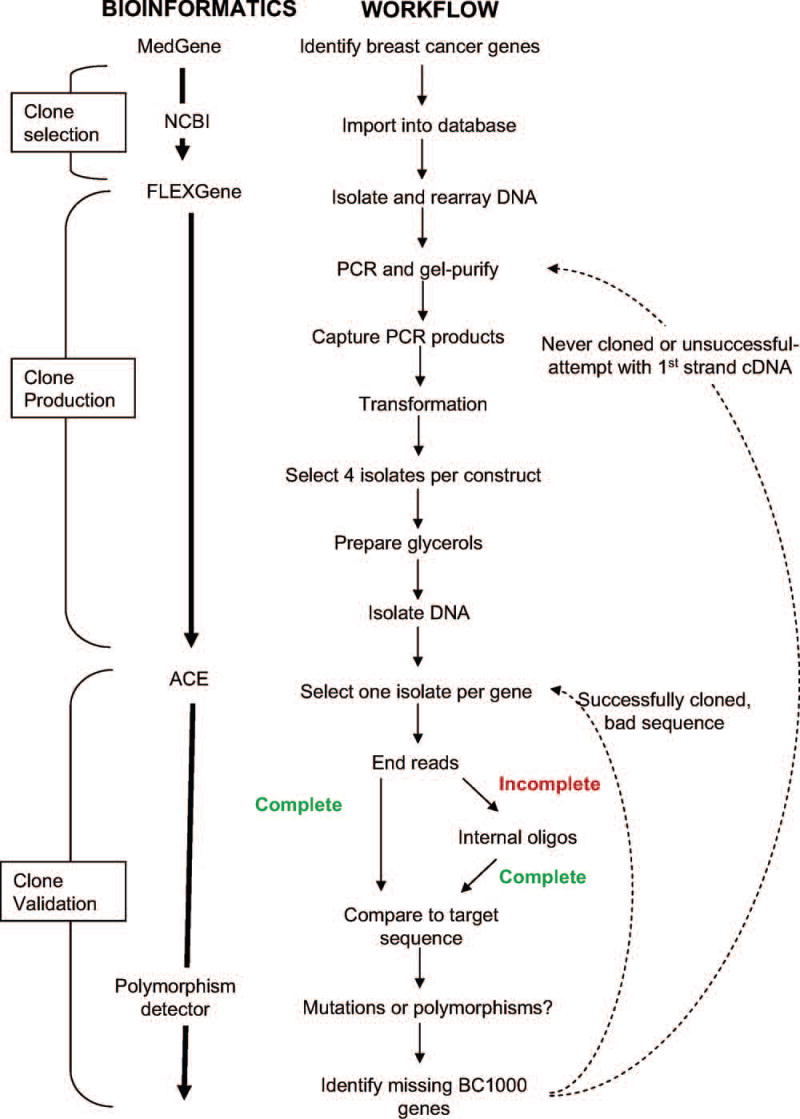
Representative 3D structures of cells expressing cDNAs that scored as hits in the morphogenesis assays. MCF-10A cells expressing the pBABE control vector (pBABE) or EDG2, ARAF1, GRB2, JUNB, or BMP5 were cultured in morphogenesis assays as described in Materials and Methods for at least 16 days. The variability of morphogenic phenotypes among structures within the same experimental well is demonstrated by the inset in the BMP5 panel.
cDNAs that induce migration of MCF-10A.B2 cells
Of the 75 cDNAs that induced cellular migration in the preliminary, single-pass screen, 66 were retested and 41 of these reproducibly scored as valid hits (Table 2 and Supplemental Table 3). These were then examined under non-sensitized conditions to evaluate the ability of the cDNA to induce migration without ErbB2 cooperation. Of these cDNAs, 14 induced migration in the absence of ErbB2 activation (shown in red). Several genes that induced migration of 10A.B2 cells in the absence of dimerizer did not induce migration of parental MCF-10A cells (e.g. TGFβ1, TGFβ3, RAD23B and EDG2)(Supplementary Table 3 and Seton Rogers et al16), suggesting that these genes may require weak signals from ligand-independent dimerization of ErbB2 to induce migration.
Table 2.
| I. Growth Factors/Hormones | II. Signal Transduction | III. Transcription/Translation Factors | IV. Extracellular Matrix Proteins |
|---|---|---|---|
| BMP5 | CEBPγ | IBSP | |
| IGF1 (mild) | ARAF | EIF2AK2 | SPP1 |
| HGF | CART | ENO1 | |
| INSL61 | EDG2 | ELF3 | |
| IL4 | GRB2 | ETS1 (mild) | |
| IL11 | HRAS | FOS | |
| IL13 | RAF1 | HOXB7 | |
| PRL | RARB | JUNB | |
| TGFA | RHOG | PCGF4 | |
| TGFβ1 | SGK1 | PSIP1 (mild) | |
| TGFβ3 | TNFRSF10B | ||
|
| |||
| V. Cell Cycle/DNA repair | VI. Proteases | VII. Energy Pathways | VIII. Unknown |
| CTSD | COX6A1 (mild) | IER2 | |
| CDC5L | CTSG | COX6A2 (mild) | |
| ORC1L | |||
| RAD23B | |||
SGK and INSL6 were only tested with activated with ErbB2.
Categorization of the known functional activities of cDNAs determined to be validated hits from sensitized migration screens using 10A.B2 cells with activated ErbB2. cDNAs depicted in red confer migratory ability in both sensitized and non-sensitized conditions. cDNAs were scored as a hit if they induced at least a 3-fold increase in migration over the vector-transduced control unless they are indicated as having a mild (2 fold) effect.
The 41 validated migration hits were also reassessed in the morphogenesis assay using 10A.B2 cells, with and without dimerization of ErbB2. Of these the 41 migration hits, 20 induced alterations in the morphology of 3-D structures in absence of dimerizer (Table 3 and Supplemental Table 3). The majority of these cDNAs induced the formation of disorganized structures, many of which showed a mild protrusive behavior (examples shown in Figures 2 and 3). Twelve of these 21 cDNAs were also hits in the ErbB2-independent migration assay discussed above (GRB2, HRAS, ELF3, FOS, RHOG, EDG2, HGF, RAF1, JUNB, IBSP, IL13 and TGFα).
Table 3.
| Gene Symbols | Observed phenotype (−) ErbB2 | Observed phenotype (+) ErbB2 |
|---|---|---|
| I. Growth Factors/Hormones | ||
| BMP5 | Moderate protrusive behavior | Enhanced protrusive behavior |
| GRB2 | Larger structures, disorganized, mild protrusive behavior | Enhanced protrusive behavior |
| HGF | Larger structures, disorganized | Enhanced protrusive behavior |
| TGFα | Disorganized, mild protrusive behavior | Enhanced protrusive behavior |
| TGFβ1 | Smaller structures, disorganized | Highly Invasive16 |
| TGFβ3 | Smaller structures, disorganized | Highly Invasive16 |
| IL13 | Smaller structures, disorganized | No enhanced effect |
| INSL6 | Normal | Mild protrusive behavior |
| II. Signal transduction | ||
| ARAF | Larger structures, disorganized | No enhanced effect |
| EDG2 | Larger structures, disorganized, protrusive behavior | No enhanced effect |
| HRAS | Highly Invasive | Not screened |
| RAF1 | Very disorganized | Increased disorganization, mild protrusive behavior |
| RHOG | Disorganized, clumping together, protrusive behavior | Highly Invasive |
| III. Transcription/Translation Factors | ||
| ELF3 | Some disorganized structures, mild protrusive behavior | No enhanced effect |
| FOS | Disorganized, mild protrusive behavior | Enhanced protrusive behavior |
| JUNB | Multiacinar-like, mild protrusive behavior | Enhanced protrusive behavior |
| HOXB7 | Mild protrusive behavior | Enhanced protrusive behavior |
| IV. Extracellular Proteins | ||
| IBSP | Larger structures, disorganized | No enhanced effect |
| SPP1 | Large structures, filled, disorganized | No enhanced effect |
| V. Cell Cycle/DNA repair | ||
| ORC1L | Mild protrusive spikes | No enhanced effect |
| p16 (CDKN2A) | Motile networks | Not screened |
| p21 (CDKN1A) | Motile networks | Not screened |
Figure 3.
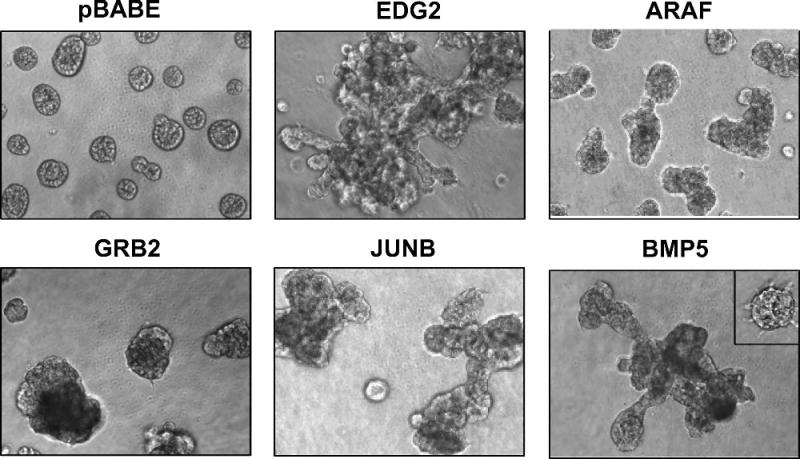
Representative 3D structures +/− ErbB2 dimerization, of 10A.B2 cells expressing cDNAs that scored in the sensitized migration assays. 10A.B2 cells were cultured with EGF in upper panels and with AP1510 in lower panels as described in Materials and Methods for at least 16 days.
Activation of the chimeric ErbB2 receptor by dimerization in acinar structures causes constitutive, EGF-independent proliferation and the formation of multi-acinar structures that display filling of the luminal space (10 and Figure 3E). Most of the 19 cDNAs that displayed migratory activity in the absence of activated ErbB2 provoked a more dramatic perturbation of structure when ErbB2 was activated, frequently involving disruption of the spheroid acinar structure and the formation of spike-like protrusions, invasive cell cords, or branch-like tubular extensions (examples in Figure. 3). This screen thus identified genes that can cooperate with ErbB2 to convert a non-invasive phenotype into one which displays features that have been associated with invasive behavior in breast carcinoma.
Cell cycle inhibitors p16 and p21 induce migration and abnormal morphology
In independent studies in our lab, we observed that the CDK inhibitors (CKIs) p16 and p21 (CDKN2A and CDKN1A) induced interesting migration phenotypes in MCF-10A cells. As neither of these CDKIs scored in our preliminary screens, we hypothesized that ectopic expression of these cell cycle inhibitors could inhibit cell division and be lost upon cell passage. Therefore, these genes were then reevaluated in both the migration and morphogenesis assays without passaging the MCF-10A cells post transduction. Upon infection of MCF-10As with retroviral constructs expressing p16 or p21 (referred to as 10A.p16 cells and 10A.p21 cells, respectively), the cells showed a flattened, enlarged phenotype, consistent with a growth arrested state. G1 growth arrest was confirmed in 10A.p16 and 10A.p21 cells using propidium iodide staining and FACS analysis for DNA content (data not shown). Interestingly, in 3D morphogenesis assays, 10A.p16 or 10A.p21 cells exhibited a very striking motile network phenotype (Figure 4). Within 1 hour of seeding on Matrigel, 10A.p16 and 10A.p21 cells began to move. By 6 hours post-seeding (Figure 4 B, C), both 10A.p16 and 10A.p21 cells had begun to form networks that extended throughout the well. By 24 hours these networks were extensive and consisted of many cells (Figure 4 E, F). In contrast, vector control (pBABE) cells showed neither motility nor the ability to form networks (Figure 4 A, D).
Figure 4.
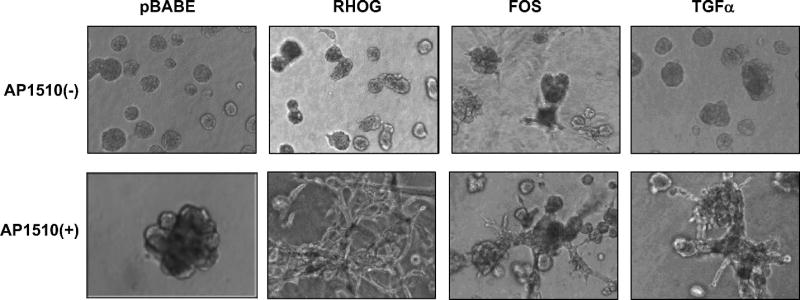
Motile network structure of p16 and p21. Images taken six (upper) or 24 (lower) hours post-seeding on Matrigel, (A,D) MCF-10As plus EGF expressing vector control, (B,E) MCF-10A cells plus EGF expressing p16, and (C,F) MCF-10A cells plus EGF expressing p21.
Discussion
In model organisms, ectopic expression screens have proved a powerful method for identifying genes that confer specific behaviors or phenotypes on cells. This approach has not been widely exploited in mammalian settings due to the lack of available expression-ready gene collections and inadequate high-throughput methodologies for introducing cDNAs into mammalian cells. Recent advances in high-throughput molecular cloning technologies present the opportunity to address some of these challenges. For example, the availability of recombinational cloning systems provides a method for readily transferring cDNAs into virtually any expression vector without interfering with the reading frame or introducing mutations. Applying this technology to create indexed clone sets containing unique, full length sequence-verified cDNAs will enable efficient, high-throughput experimentation to examine the function of each gene. Systematic screens of gene function complemented with traditional methods for validation provide a more comprehensive approach to studying human disease development, which may ultimately impart valuable insight into the underlying mechanisms.
To apply this functional proteomics approach in breast cancer research, we created the BC1000, the first sequence-validated and individually arrayed collection of 1300 cDNAs associated with a specific disease. The availability of this collection will enable pilot experimentation and accelerate the development of high-throughput methodologies for studying breast oncogenesis in a mammalian setting. There are several advantages of using defined cDNA collections like the BC1000 compared to random cDNA libraries. First, the identity of each clone is known. Thus, complex and inefficient capture strategies are not required to recover and identify any potential hits20–22. Second, each clone is known to be of good quality, i.e. full-length and lack mutations. Lastly, complex phenotypic assays are feasible as it is not necessary to sample millions of clones to compensate for redundancy and to ensure that rare cDNAs are represented. While this set of genes covers a broad spectrum of breast cancer-related genes, it is not all inclusive. The addition of new genes to this collection, including genes more recently linked to breast cancer and genes more difficult to clone, is an ongoing effort. By coupling this and other growing collections of human cDNAs with automation and data tracking, it will soon be feasible to perform such screens at the scale of the entire proteome.
To investigate the genetic events involved in the different aspects of breast carcinogenesis, we conducted three phenotypic assays with a derivative of an immortalized mammary epithelial cell line, MCF-10A, on a subset of the BC1000 gene collection. The process of carcinogenesis is complex and involves the activation of many different cellular programs including: misregulated cell proliferation, apoptosis, differentiation, motility, angiogenesis, invasion, and metastasis. Proteins that regulate these various processes act at many different levels; master regulators, which are often oncogenic, activate many programs simultaneously, whereas other proteins act downstream along specific defined pathways. In order to identify both classes of proteins, we conducted screens to find proteins whose overexpression induced premalignant phenotypes resembling events that take place during tumor initiation and progression, specifically uncontrolled cellular proliferation, disruption of acinar morphogenesis, and cell migration, in the presence or absence of activated ErbB2. All three assays yielded well-studied proto-oncogenes expected to be involved in these processes, such as RAF1, JUNB, FOS, and GRB2, validating our cell-based screening approach. All of these assays were performed in the same genetic background, highlighting the robust and pleiotropic activities of these genes. Moreover, one of the strengths of such a parallel screening approach is its ability to identify genes that could contribute to the oncogenic process in a more selective fashion by altering specific cellular processes. While many such hits were validated in the migration screens, further studies are required to validate and understand the mechanisms of these the genes that produced the unpredicted activities in the initial EGF independent proliferation (EIF2AK2 and SPARC) and 3D morphogenesis assay (TACSTD2, GNRH1 and ID2). The cDNAs used in these experiments were selected because of a known or implicated connection to breast cancer. Often, these associations are very weak and the roles played by the genes, if any, in breast cancer development have not been characterized. Systematic approaches such as this one may facilitate the annotation of any novel function/s of these proteins.
Characterization of Validated Migration Hits
Many different classes of proteins were found to induce the migration of the MCF-10A cells, including extracellular ligands, matrix proteins, and transcription factors, all of which can stimulate multiple regulatory programs. This is not unexpected because the conversion from a non-motile to a motile state would require the activation and repression of numerous cellular pathways. The best characterized motogen for MCF-10A cells is EGF. EGF family ligands generate a complex array of signals by binding to and activating ErbB receptors, specifically ErbB1 (EGFR/HER1), ErbB2 (HER2/NEU), ErbB3 and ErbB4, via both homo- and heterodimerization. In our screen, several known activators and targets of ErbB family members were identified as pro-migratory. Specifically, TGFα, GRB2, HRAS, RAF1, JUNB, FOS induced migration of MCF-10A cells independent of ErbB2 activation, supporting the important role of EGFR pathways in cell migration. Interestingly GRB2, HRAS, RAF1 all act upstream of Erk and activate transcription of the Fos transcription factor, which together with Jun forms the AP1 transcription complex28. The Erk pathway and AP1 complex has previously been implicated in migration and invasion through direct effects of Erk phosphorylation in the cytoplasm as well as AP-1 induced transcriptional changes29–35. Figure 5 highlights the involvement of some of the cDNAs identified in our screens in various cellular pathways believed to be involved in motility and migration.
Figure 5.
Genes identified in our screens in relation to various cellular pathways believed to be involved in motility, invasion and migration. Hits are in red.
Most of the migration hits (28) required activation of ErbB2, indicating that induction of migration of these genes is dependent on the activation of cellular pathways induced by ErbB2 activation. It is likely that the Erk pathway may represent a key ErbB2 pathway since many of the genes that induced ErbB2-independent migration in our screens are activators of the Erk pathway and since we previously found that constitutive activation of the Erk kinase through ectopic expression of an activated variant of MEK2 could substitute for ErbB2 in promoting migration induced by TGFβ1 and TGFβ316. In addition, we found that migration induced by ErbB2 and TGFβ was dependent on the secretion of an EGFR-ligand released by these cells. Indeed, conditioned medium from these cells could replace ErbB2 in inducing cell migration. These results suggest that Erk and EGFR pathways are key in mediating the role of TGFβ as a pro-migratory factor with activated ErbB216.
The cooperative ErbB2 migratory screen identified several cDNAs that had been previously implicated in migration although the mechanisms have not been fully elucidated. For example, osteopontin (OPN, also known as SPP1) has been shown to have pro-migratory effects on various mammary epithelial cell lines, specifically 21PT, 21NT, and MDA-MB-435 cell lines36. As with TGFβ family members, OPN induced migration is also believed to be mediated by the EGFR pathway. Integrin-binding sialoprotein (IBSP), another bone matrix protein that cooperatively induced migration with ErbB2, has been previously shown to enhance migration of MDA-MB-231 breast cancer cells in basement membrane37. Furthermore, IBSP-transfected clones injected into the mammary fat pad of nude mice increased tumor growth rate37, 38. Our screen supports the existing data that these bone matrix proteins are involved in migration. Interestingly, we also identified several cytokines that stimulate migratory behavior in MCF-10A cells, specifically, IL4, IL11and IL13. Certain cytokines that promote osteolysis have been previously implicated in breast cancer metastasis to the bone39. Endogenous expression of IL11, which induced migration of MCF-10A cells irrespective of ErbB2 activation, has been documented in invasive primary breast tumors and associated with a higher rate of bone metastases39–41. In addition, soluble IL11has been previously shown to have a dose-dependent effect on migration of breast carcinoma cells, MDA-MB-231 cells, in a biocoat matrigel invasion chamber assay42 The results of our screen support previous findings and implications that cytokines involved in osteoclast formation may be involved in bone metastases, and reflect the diverse functional activities of proteins that contribute to migratory behavior.
The identification of cDNAs already known to play a role in cell migration validates this approach as a format to identify relevant cDNAs. Furthermore, we also identified novel functional activities for cDNAs known to be involved in other aspects of carcinogenesis. Several unexpected cDNAs were capable of inducing migration cooperatively with activated ErbB2. For example, SGK (serum and glucocorticoid-regulated kinase-1) and TNFRSF10B (tumor necrosis factor receptor, 10B) were both identified as pro-migratory, however they were previously recognized for their involvement in survival. The finding that cDNAs known to be involved in other cellular processes may also play a role in migration suggests that this approach may help uncover unanticipated activities for previously identified proteins.
Validated Migration Hits that Induced Abnormal Morphology
All of the confirmed migration hits were retested in the 3-D Matrigel cultures of MCF-10A cells for their ability to perturb acinar morphogenesis with and without activated ErbB2. Approximately one third of the genes that scored in the transwell migration assay caused alterations in morphogenesis. In the absence of ErbB2 activation, the majority of the these cDNAs induced the formation of disorganized structures, some that showed protrusive projections that emanated from cells within disorganized masses or displayed cords of cells that invaded through the Matrigel bed. For most genes, there was a dramatic enhancement of the phenotype in the context of activated ErbB2. Almost all cDNAs displayed significant protrusive behavior when co-expressed with activated ErbB2.
Amplification or overexpression of ErbB2 (also known as HER2/Neu) occurs in up to 85% of comedo-type ductal carcinoma in situ (DCIS), an early non-invasive, yet high grade breast carcinoma, suggesting that ErbB2 plays a role in the induction of this specific histological variant of DCIS43–45. It is of interest to understand what types of genetic or epigenetic alterations can contribute to the progression towards an invasive tumor. Thus, one rationale for screening genes in combination with ErbB2 was to identify genes that could be considered as candidate “second hits” in the development of invasive breast carcinomas. The genes that were found to promote significant protrusive phenotypes in combination with ErbB2 in the 3D Matrigel cultures could be considered as candidate genes that might contribute to the conversion of non-invasive lesions that carry amplified or overexpressed ErbB2 to invasive structures.
Several of the genes that induced significant protrusive/invasive activity in these screens have been implicated in cell migration or invasion. For example, RHOG, a Rac homologue that also induced migration and disrupted morphogenesis, is known to have a central role in regulating the actin cytoskeleton during cell migration, as well as mediating several cellular morphological processes. This small GTPase has been shown to interact with Elmo and Dock180 to induce activation of Rac1 and cell spreading mediated by integrins46. EDG2, another identified pro-migratory gene that induced dramatic alterations in MCF-10As in Matrigel, has also been shown to activate Rac and induce cell spreading and cell migration47. EDG2 is a member of G-protein coupled receptors which binds lysophosphatidic acid (LPA) to activate a host of cellular responses.
Our results confirm recent data demonstrating that expression of ELF3 (also known as ESX), an epithelial-restricted member of a large family of Ets transcription factors, confers a transformed and invasive phenotype in MCF-12A cells48. Similar to the MCF-10A cell line, MCF-12A is an immortalized, but nontransformed mammary cell line that forms highly organized duct-like structures in 3D membrane cultures. MCF-12A cells expressing ESX formed solid, disorganized structures, which invaded the surrounding matrix48. It has been suggested that the ESX promoter represents a transcriptional target of ErbB2.
The CDK inhibitors (CKIs), p16 and p21 (CDKN2A and CDKN1A, respectively), are two interesting migration and morphogenesis hits that also appear to exert multiple biological activities. Both of these CKIs inhibit the G1-S transition. p16 binds to CDK4 or CDK6 to inhibit cyclin D-CDK4/6 complexes, while p21 binds and inhibits CDK2 complexes. Both CKIs prevent downstream Rb phosphorylation and S phase gene transcription in mid-G1More recently, p21 has been shown to play a growth-promoting role in early G1 by promoting cyclin D-CDK4/6 complex assembly69, 70. The identification of cell cycle inhibitors p16 and p21 as pro-migratory is consistent with previously reported roles for p21 and for another CKI, p27, in enhancing cell motility. Specifically, cytoplasmic p21 has been shown to inhibit ROCK and therefore compromise stress fiber formation by the Rho/ROCK/LIMK/cofilin pathway leading to enhanced motility70, 71. In addition, p27has been shown to bind directly to RhoA, blocking interactions between Rho A and its activators. Furthermore, p27-deficient fibroblasts display a dramatic decrease in motility, suggesting that it may act as both a tumor suppressor and an oncogene, depending on its subcellular location, 73.
Interestingly, p16 and p21 did not score as hits in our initial screens due to the design of the experimental screening protocols which incorporated passaging of the MCF-10A cells after transduction but before the assays. Other cDNAs that cause a block in cell cycle or growth retardation may have also been missed in our initial screens for this reason. Overall, our screens were successful in identifying proto-oncogenes and other cDNAs known to be involved in these specific aspects of breast carcinogenesis.
In summary, we have built an arrayed and sequence verified cDNA collection of human breast cancer- associated cDNAs, known as the BC1000. To our knowledge, the BC1000 represents the first publicly available disease-based cDNA collection. Here, we have used this resource in three complex cell-based assays to investigate the genetic events involved in different stages of breast carcinogenesis. Through this comprehensive approach, we identified potentially novel functional activities for known breast cancer-associated cDNAs, as well as provided insight into the multiple biological effects of various candidate oncogenes. Subsequent analyses of the mechanisms underlying these findings may provide important insight into the process of breast cancer development.
Supplementary Material
Supplemental Table 1. Complete list of breast cancer associated cDNAs screened in proliferation, morphogenesis, and migration assays. LPF ranking is the log of the product frequency score generated by MedGene, for which a more negative value is indicative of a lower ranking with respect to the strength of the gene-disease relationship.
Supplemental Table 2. cDNAs that displayed activity in the first pass screens.1 Migration was evaluated in a transwell chamber using 10A.B2 cells treated with dimerizer. A cDNA was scored as a hit if migration was more than 2 fold greater than control.2 Morphogenesis was assessed in 3-D Matrigel cultures of 10A.B2 cells and a cDNAs was scored as a hit if any abnormalities were observed in acinar formation, morphology or lumen formation or if invasion into Matrigel was observed.3 Proliferation was assayed after plating 10A.B2 cells on 6-well dishes in Assay medium over 10 days. A cDNA was scored as a hit if it induced more than a 3 fold increase compared with control. “X” indicates activity observed for cDNA in a specific assay. LPF ranking is the log of the product frequency score generated by MedGene, for which a more negative value is indicative of a lower ranking with respect to the strength of the gene-disease relationship.
Supplemental Table 3. Confirmed hits from screens in proliferation, morphogenesis and migration assays. The symbol “o” indicates normal cellular behavior, “x” indicates aberrant activity of the transduced cDNA in the assay. The descriptions of the relative effects of the cDNAs reflect the following fold increases relative to control cells without transduced cDNA. Migration assay [none (no migration), mild (2−5X), moderate (6−15X), High (16−50+X)]. NS indicates cDNAs “not screened”. NV (not validated) indicates cDNA hits that were observed but were not repeated for validation. Proliferation assay [none (no proliferation), mild (3−5X), moderate (6−15X), High (16+X)]. NS indicates cDNAs “not screened”. Relative effects of Matrigel invasion are in comparison to controls[none (typical acinar structures), mild (mild spike protrusions), moderate (branching, protrusive structures), high (completely invasive behavior). NS indicates cDNAs “not screened”. NV (not validated) indicates cDNA hits that were observed but were not repeated for validation. The morphogenesis description column provides additional detail about disrupted morphogenesis in ErbB2-independent morphogenesis assays. In addition this Table includes data from secondary screens of the migration ‘hits’ from the first screen.
References
- 1.Nielsen KV, Niebuhr E, Ejlertsen B, Holstebroe S, Madsen MW, Briand P, Mouridsen HT, Bolund L. Molecular cytogenetic analysis of a nontumorigenic human breast epithelial cell line that eventually turns tumorigenic: validation of an analytical approach combining karyotyping, comparative genomic hybridization, chromosome painting, and single-locus fluorescence in situ hybridization. Genes Chromosomes Cancer. 1997;20:30–37. [PubMed] [Google Scholar]
- 2.Nishizaki T, DeVries S, Chew K, Goodson WH, 3rd, Ljung BM, Thor A, Waldman FM. Genetic alterations in primary breast cancers and their metastases: direct comparison using modified comparative genomic hybridization. Genes Chromosomes Cancer. 1997;19:267–272. doi: 10.1002/(sici)1098-2264(199708)19:4<267::aid-gcc9>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 3.Perou CM, Jeffrey SS, van de Rijn M, Rees CA, Eisen MB, Ross DT, Pergamenschikov A, Williams CF, Zhu SX, Lee JC, Lashkari D, Shalon D, Brown PO, Botstein D. Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc Natl Acad Sci U S A. 1999;96:9212–9217. doi: 10.1073/pnas.96.16.9212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porter DA, Krop IE, Nasser S, Sgroi D, Kaelin CM, Marks JR, Riggins G, Polyak K. A SAGE (serial analysis of gene expression) view of breast tumor progression. Cancer Res. 2001;61:5697–5702. [PubMed] [Google Scholar]
- 5.Ma XJ, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P, Payette T, Pistone M, Stecker K, Zhang BM, Zhou YX, Varnholt H, Smith B, Gadd M, Chatfield E, Kessler J, Baer TM, Erlander MG, Sgroi DC. Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci U S A. 2003;100:5974–5979. doi: 10.1073/pnas.0931261100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacquemier J, Ginestier C, Rougemont J, Bardou VJ, Charafe-Jauffret E, Geneix J, Adelaide J, Koki A, Houvenaeghel G, Hassoun J, Maraninchi D, Viens P, Birnbaum D, Bertucci F. Protein expression profiling identifies subclasses of breast cancer and predicts prognosis. Cancer Res. 2005;65:767–779. [PubMed] [Google Scholar]
- 7.Somiari RI, Somiari S, Russell S, Shriver CD. Proteomics of breast carcinoma. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;815:215–225. doi: 10.1016/j.jchromb.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 8.Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–6086. [PubMed] [Google Scholar]
- 9.Petersen OW, Ronnov-Jessen L, Howlett AR, Bissell MJ. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc Natl Acad Sci U S A. 1992;89:9064–9068. doi: 10.1073/pnas.89.19.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001;3:785–792. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 12.Shaw KR, Wrobel CN, Brugge JS. Use of three-dimensional basement membrane cultures to model oncogene-induced changes in mammary epithelial morphogenesis. J Mammary Gland Biol Neoplasia. 2004;9:297–310. doi: 10.1007/s10911-004-1402-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer. 2005;5:675–688. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- 14.Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 2002;111:29–40. doi: 10.1016/s0092-8674(02)01001-2. [DOI] [PubMed] [Google Scholar]
- 15.Wrobel CN, Debnath J, Lin E, Beausoleil S, Roussel MF, Brugge JS. Autocrine CSF-1R activation promotes Src-dependent disruption of mammary epithelial architecture. J Cell Biol. 2004;165:263–273. doi: 10.1083/jcb.200309102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seton-Rogers SE, Lu Y, Hines LM, Koundinya M, LaBaer J, Muthuswamy SK, Brugge JS. Cooperation of the ErbB2 receptor and transforming growth factor beta in induction of migration and invasion in mammary epithelial cells. Proc Natl Acad Sci U S A. 2004;101:1257–1262. doi: 10.1073/pnas.0308090100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang YA, Dukhanina O, Tang B, Mamura M, Letterio JJ, MacGregor J, Patel SC, Khozin S, Liu ZY, Green J, Anver MR, Merlino G, Wakefield LM. Lifetime exposure to a soluble TGF-beta antagonist protects mice against metastasis without adverse side effects. J Clin Invest. 2002;109:1607–1615. doi: 10.1172/JCI15333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muraoka RS, Koh Y, Roebuck LR, Sanders ME, Brantley-Sieders D, Gorska AE, Moses HL, Arteaga CL. Increased malignancy of Neu-induced mammary tumors overexpressing active transforming growth factor beta1. Mol Cell Biol. 2003;23:8691–8703. doi: 10.1128/MCB.23.23.8691-8703.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siegel PM, Shu W, Cardiff RD, Muller WJ, Massague J. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci U S A. 2003;100:8430–8435. doi: 10.1073/pnas.0932636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hannon GJ, Sun P, Carnero A, Xie LY, Maestro R, Conklin DS, Beach D. MaRX: an approach to genetics in mammalian cells. Science. 1999;283:1129–1130. doi: 10.1126/science.283.5405.1129. [DOI] [PubMed] [Google Scholar]
- 21.Randow F, Seed B. Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat Cell Biol. 2001;3:891–896. doi: 10.1038/ncb1001-891. [DOI] [PubMed] [Google Scholar]
- 22.Peeper DS, Shvarts A, Brummelkamp T, Douma S, Koh EY, Daley GQ, Bernards R. A functional screen identifies hDRIL1 as an oncogene that rescues RAS-induced senescence. Nat Cell Biol. 2002;4:148–153. doi: 10.1038/ncb742. [DOI] [PubMed] [Google Scholar]
- 23.Hu Y, Hines LM, Weng H, Zuo D, Rivera M, Richardson A, LaBaer J. Analysis of genomic and proteomic data using advanced literature mining. J Proteome Res. 2003;2:405–412. doi: 10.1021/pr0340227. [DOI] [PubMed] [Google Scholar]
- 24.Marsischky G, LaBaer J. Many paths to many clones: a comparative look at high-throughput cloning methods. Genome Res. 2004;14:2020–2028. doi: 10.1101/gr.2528804. [DOI] [PubMed] [Google Scholar]
- 25.Chang CY, LaBaer J. DNA polymorphism detector: an automated tool that searches for allelic matches in public databases for discrepancies found in clone or cDNA sequences. Bioinformatics. 2005;21:2133–2135. doi: 10.1093/bioinformatics/bti298. [DOI] [PubMed] [Google Scholar]
- 26.Gopalbhai K, Jansen G, Beauregard G, Whiteway M, Dumas F, Wu C, Meloche S. Negative regulation of MAPKK by phosphorylation of a conserved serine residue equivalent to Ser212 of MEK1. J Biol Chem. 2003;278:8118–8125. doi: 10.1074/jbc.M211870200. [DOI] [PubMed] [Google Scholar]
- 27.Debnath J, Walker SJ, Brugge JS. Akt activation disrupts mammary acinar architecture and enhances proliferation in an mTOR-dependent manner. J Cell Biol. 2003;163:315–326. doi: 10.1083/jcb.200304159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- 29.Stupack DG, Cho SY, Klemke RL. Molecular signaling mechanisms of cell migration and invasion. Immunol Res. 2000;21:83–88. doi: 10.1385/IR:21:2-3:83. [DOI] [PubMed] [Google Scholar]
- 30.Mulder KM. Role of Ras and Mapks in TGFbeta signaling. Cytokine Growth Factor Rev. 2000;11:23–35. doi: 10.1016/s1359-6101(99)00026-x. [DOI] [PubMed] [Google Scholar]
- 31.Reddy KB, Nabha SM, Atanaskova N. Role of MAP kinase in tumor progression and invasion. Cancer Metastasis Rev. 2003;22:395–403. doi: 10.1023/a:1023781114568. [DOI] [PubMed] [Google Scholar]
- 32.Xia Y, Karin M. The control of cell motility and epithelial morphogenesis by Jun kinases. Trends Cell Biol. 2004;14:94–101. doi: 10.1016/j.tcb.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 33.Hennigan RF, Hawker KL, Ozanne BW. Fos-transformation activates genes associated with invasion. Oncogene. 1994;9:3591–3600. [PubMed] [Google Scholar]
- 34.Milde-Langosch K, Roder H, Andritzky B, Aslan B, Hemminger G, Brinkmann A, Bamberger CM, Loning T, Bamberger AM. The role of the AP-1 transcription factors c-Fos, FosB, Fra-1 and Fra-2 in the invasion process of mammary carcinomas. Breast Cancer Res Treat. 2004;86:139–152. doi: 10.1023/B:BREA.0000032982.49024.71. [DOI] [PubMed] [Google Scholar]
- 35.Johnston IM, Spence HJ, Winnie JN, McGarry L, Vass JK, Meagher L, Stapleton G, Ozanne BW. Regulation of a multigenic invasion programme by the transcription factor, AP-1: re-expression of a down-regulated gene, TSC-36, inhibits invasion. Oncogene. 2000;19:5348–5358. doi: 10.1038/sj.onc.1203927. [DOI] [PubMed] [Google Scholar]
- 36.Tuck AB, Hota C, Wilson SM, Chambers AF. Osteopontin-induced migration of human mammary epithelial cells involves activation of EGF receptor and multiple signal transduction pathways. Oncogene. 2003;22:1198–1205. doi: 10.1038/sj.onc.1206209. [DOI] [PubMed] [Google Scholar]
- 37.Chen J, Rodriguez JA, Barnett B, Hashimoto N, Tang J, Yoneda T. Bone sialoprotein promotes tumor cell migration in both in vitro and in vivo models. Connect Tissue Res. 2003;44(Suppl 1):279–284. [PubMed] [Google Scholar]
- 38.Sharp JA, Waltham M, Williams ED, Henderson MA, Thompson EW. Transfection of MDA-MB-231 human breast carcinoma cells with bone sialoprotein (BSP) stimulates migration and invasion in vitro and growth of primary and secondary tumors in nude mice. Clin Exp Metastasis. 2004;21:19–29. doi: 10.1023/b:clin.0000017167.17065.61. [DOI] [PubMed] [Google Scholar]
- 39.Sloan EK, Anderson RL. Genes involved in breast cancer metastasis to bone. Cell Mol Life Sci. 2002;59:1491–1502. doi: 10.1007/s00018-002-8524-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sotiriou C, Lacroix M, Lespagnard L, Larsimont D, Paesmans M, Body JJ. Interleukins-6 and -11 expression in primary breast cancer and subsequent development of bone metastases. Cancer Lett. 2001;169:87–95. doi: 10.1016/s0304-3835(01)00524-9. [DOI] [PubMed] [Google Scholar]
- 41.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- 42.Arihiro K, Oda H, Kaneko M, Inai K. Cytokines facilitate chemotactic motility of breast carcinoma cells. Breast Cancer. 2000;7:221–230. doi: 10.1007/BF02967464. [DOI] [PubMed] [Google Scholar]
- 43.Witton CJ, Reeves JR, Going JJ, Cooke TG, Bartlett JM. Expression of the HER1-4 family of receptor tyrosine kinases in breast cancer. J Pathol. 2003;200:290–297. doi: 10.1002/path.1370. [DOI] [PubMed] [Google Scholar]
- 44.Weeks BH, He W, Olson KL, Wang XJ. Inducible expression of transforming growth factor beta1 in papillomas causes rapid metastasis. Cancer Res. 2001;61:7435–7443. [PubMed] [Google Scholar]
- 45.Harari D, Yarden Y. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene. 2000;19:6102–6114. doi: 10.1038/sj.onc.1203973. [DOI] [PubMed] [Google Scholar]
- 46.Katoh H, Negishi M. RhoG activates Rac1 by direct interaction with the Dock180-binding protein Elmo. Nature. 2003;424:461–464. doi: 10.1038/nature01817. [DOI] [PubMed] [Google Scholar]
- 47.Van Leeuwen FN, Olivo C, Grivell S, Giepmans BN, Collard JG, Moolenaar WH. Rac activation by lysophosphatidic acid LPA1 receptors through the guanine nucleotide exchange factor Tiam1. J Biol Chem. 2003;278:400–406. doi: 10.1074/jbc.M210151200. [DOI] [PubMed] [Google Scholar]
- 48.Schedin PJ, Eckel-Mahan KL, McDaniel SM, Prescott JD, Brodsky KS, Tentler JJ, Gutierrez-Hartmann A. ESX induces transformation and functional epithelial to mesenchymal transition in MCF-12A mammary epithelial cells. Oncogene. 2004;23:1766–1779. doi: 10.1038/sj.onc.1207391. [DOI] [PubMed] [Google Scholar]
- 49.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 50.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 51.Gu Y, Turck CW, Morgan DO. Inhibition of CDK2 activity in vivo by an associated 20K regulatory subunit. Nature. 1993;366:707–710. doi: 10.1038/366707a0. [DOI] [PubMed] [Google Scholar]
- 52.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 53.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 54.Dulic V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW, Elledge SJ, Reed SI. p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell. 1994;76:1013–1023. doi: 10.1016/0092-8674(94)90379-4. [DOI] [PubMed] [Google Scholar]
- 55.Noda A, Ning Y, Venable SF, Pereira-Smith OM, Smith JR. Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res. 1994;211:90–98. doi: 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- 56.Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato JY. D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol. 1994;14:2066–2076. doi: 10.1128/mcb.14.3.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meyerson M, Harlow E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol Cell Biol. 1994;14:2077–2086. doi: 10.1128/mcb.14.3.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell. 1992;70:993–1006. doi: 10.1016/0092-8674(92)90249-c. [DOI] [PubMed] [Google Scholar]
- 59.Hatakeyama M, Brill JA, Fink GR, Weinberg RA. Collaboration of G1 cyclins in the functional inactivation of the retinoblastoma protein. Genes Dev. 1994;8:1759–1771. doi: 10.1101/gad.8.15.1759. [DOI] [PubMed] [Google Scholar]
- 60.Mittnacht S, Lees JA, Desai D, Harlow E, Morgan DO, Weinberg RA. Distinct sub-populations of the retinoblastoma protein show a distinct pattern of phosphorylation. Embo J. 1994;13:118–127. doi: 10.1002/j.1460-2075.1994.tb06241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kitagawa M, Higashi H, Jung HK, Suzuki-Takahashi I, Ikeda M, Tamai K, Kato J, Segawa K, Yoshida E, Nishimura S, Taya Y. The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. Embo J. 1996;15:7060–7069. [PMC free article] [PubMed] [Google Scholar]
- 62.Ezhevsky SA, Nagahara H, Vocero-Akbani AM, Gius DR, Wei MC, Dowdy SF. Hypo-phosphorylation of the retinoblastoma protein (pRb) by cyclin D:Cdk4/6 complexes results in active pRb. Proc Natl Acad Sci U S A. 1997;94:10699–10704. doi: 10.1073/pnas.94.20.10699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol. 1998;18:753–761. doi: 10.1128/mcb.18.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen J, Saha P, Kornbluth S, Dynlacht BD, Dutta A. Cyclin-binding motifs are essential for the function of p21CIP1. Mol Cell Biol. 1996;16:4673–4682. doi: 10.1128/mcb.16.9.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen J, Jackson PK, Kirschner MW, Dutta A. Separate domains of p21 involved in the inhibition of Cdk kinase and PCNA. Nature. 1995;374:386–388. doi: 10.1038/374386a0. [DOI] [PubMed] [Google Scholar]
- 66.Nakanishi M, Robetorye RS, Pereira-Smith OM, Smith JR. The C-terminal region of p21SDI1/WAF1/CIP1 is involved in proliferating cell nuclear antigen binding but does not appear to be required for growth inhibition. J Biol Chem. 1995;270:17060–17063. doi: 10.1074/jbc.270.29.17060. [DOI] [PubMed] [Google Scholar]
- 67.Warbrick E, Lane DP, Glover DM, Cox LS. A small peptide inhibitor of DNA replication defines the site of interaction between the cyclin-dependent kinase inhibitor p21WAF1 and proliferating cell nuclear antigen. Curr Biol. 1995;5:275–282. doi: 10.1016/s0960-9822(95)00058-3. [DOI] [PubMed] [Google Scholar]
- 68.Lin J, Reichner C, Wu X, Levine AJ. Analysis of wild-type and mutant p21WAF-1 gene activities. Mol Cell Biol. 1996;16:1786–1793. doi: 10.1128/mcb.16.4.1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- 70.Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. Embo J. 1999;18:1571–1583. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee S, Helfman DM. Cytoplasmic p21Cip1 is involved in Ras-induced inhibition of the ROCK/LIMK/cofilin pathway. J Biol Chem. 2004;279:1885–1891. doi: 10.1074/jbc.M306968200. [DOI] [PubMed] [Google Scholar]
- 72.McAllister SS, Becker-Hapak M, Pintucci G, Pagano M, Dowdy SF. Novel p27(kip1) C-terminal scatter domain mediates Rac-dependent cell migration independent of cell cycle arrest functions. Mol Cell Biol. 2003;23:216–228. doi: 10.1128/MCB.23.1.216-228.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004;18:862–876. doi: 10.1101/gad.1185504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Complete list of breast cancer associated cDNAs screened in proliferation, morphogenesis, and migration assays. LPF ranking is the log of the product frequency score generated by MedGene, for which a more negative value is indicative of a lower ranking with respect to the strength of the gene-disease relationship.
Supplemental Table 2. cDNAs that displayed activity in the first pass screens.1 Migration was evaluated in a transwell chamber using 10A.B2 cells treated with dimerizer. A cDNA was scored as a hit if migration was more than 2 fold greater than control.2 Morphogenesis was assessed in 3-D Matrigel cultures of 10A.B2 cells and a cDNAs was scored as a hit if any abnormalities were observed in acinar formation, morphology or lumen formation or if invasion into Matrigel was observed.3 Proliferation was assayed after plating 10A.B2 cells on 6-well dishes in Assay medium over 10 days. A cDNA was scored as a hit if it induced more than a 3 fold increase compared with control. “X” indicates activity observed for cDNA in a specific assay. LPF ranking is the log of the product frequency score generated by MedGene, for which a more negative value is indicative of a lower ranking with respect to the strength of the gene-disease relationship.
Supplemental Table 3. Confirmed hits from screens in proliferation, morphogenesis and migration assays. The symbol “o” indicates normal cellular behavior, “x” indicates aberrant activity of the transduced cDNA in the assay. The descriptions of the relative effects of the cDNAs reflect the following fold increases relative to control cells without transduced cDNA. Migration assay [none (no migration), mild (2−5X), moderate (6−15X), High (16−50+X)]. NS indicates cDNAs “not screened”. NV (not validated) indicates cDNA hits that were observed but were not repeated for validation. Proliferation assay [none (no proliferation), mild (3−5X), moderate (6−15X), High (16+X)]. NS indicates cDNAs “not screened”. Relative effects of Matrigel invasion are in comparison to controls[none (typical acinar structures), mild (mild spike protrusions), moderate (branching, protrusive structures), high (completely invasive behavior). NS indicates cDNAs “not screened”. NV (not validated) indicates cDNA hits that were observed but were not repeated for validation. The morphogenesis description column provides additional detail about disrupted morphogenesis in ErbB2-independent morphogenesis assays. In addition this Table includes data from secondary screens of the migration ‘hits’ from the first screen.