

Abstract
Activation of the renin-angiotensin system contributes to the progression of chronic kidney disease. Based on the known cellular effects of ANG II to promote inflammation, we posited that stimulation of lymphocyte responses by ANG II might contribute to the pathogenesis of hypertensive kidney injury. We therefore examined the effects of the immunosuppressive agent mycophenolate mofetil (MMF) on the course of hypertension and kidney disease induced by chronic infusion of ANG II in 129/SvEv mice. Although it had no effect on the severity of hypertension or cardiac hypertrophy, treatment with MMF significantly reduced albuminuria and ameliorated kidney injury, decreasing glomerulosclerosis and reducing lymphocyte infiltration into the renal interstitium. Attenuation of renal pathology with MMF was associated with reduced expression of mRNAs for the proinflammatory cytokines interferon-γ and tumor necrosis factor-α and the profibrotic cytokine transforming growth factor-β. As infiltration of the kidney by T lymphocytes was a prominent feature of ANG II-dependent renal injury, we carried out experiments examining the effects of ANG II on lymphocytes in vitro. We find that exposure of splenic lymphocytes to ANG II causes prominent rearrangements of the actin cytoskeleton. These actions require the activity of Rho kinase. Thus, ANG II exaggerates hypertensive kidney injury by stimulating lymphocyte responses. These proinflammatory actions of ANG II seem to have a proclivity for inducing kidney injury while having negligible actions in the pathogenesis of cardiac hypertrophy.
Keywords: inflammation, kidney diseases, T lymphocytes
the renin-angiotensin system (RAS) is a master regulator of blood pressure and fluid homeostasis. The principal effector molecule of this system, ANG II, increases blood pressure primarily through activation of type I angiotensin (AT1) receptors (13). Consistent with this notion, mice lacking the AT1A receptor, the closest functional homolog to the human AT1 receptor, have dramatically reduced blood pressures compared with wild-type controls (29). Furthermore, pharmacologic inhibition of the AT1 receptor yields impressive blood pressure reduction in human hypertensive patients (14).
In these hypertensive patients, the capacity of AT1 receptor activation to promote kidney injury is highlighted by the efficacy of AT1 receptor blockers (ARBs) in slowing the progression of chronic kidney disease (7, 36, 59). The ability of ARBs to ameliorate renal and cardiovascular disease depends at least in part on their blood pressure-lowering effects (21, 22). Clinical trials suggest that the degree of end-organ protection provided by angiotensin receptor blockade cannot be explained by blood pressure reduction alone (7, 59). One blood pressure-independent mechanism through which ARBs protect the kidney may involve direct inhibition of proinflammatory cellular actions of ANG II. For example, on a cellular level, ANG II causes lymphocyte proliferation, NF-κB activation, and generation of mononuclear cell chemokines such as MCP-1 and RANTES in the kidney (26, 43, 45, 49, 61). In models of kidney allograft rejection, blockade of AT1 receptors in the transplant recipient extends survival and reduces histologic injury independent of blood pressure control (2).
Thus far, precise characterization of proinflammatory effects of ANG II contributing to the pathogenesis of hypertensive end-organ damage remains incomplete (41, 43, 44). For example, previous studies have not clearly demonstrated that blood pressure-independent effects of lymphocyte responses induced by ANG II promote functional kidney injury as reflected by albuminuria. This issue is made even more relevant by the findings that suppression of lymphocytes (44) or the deficiency of lymphocytes (24) has the capacity to alter blood pressure responses to ANG II in some models. Moreover, whether ANG II-mediated stimulation of TGF-β, an important mediator of renal fibrosis, is due solely to activation of AT1 receptors or rather to alternative pathways linked to immune activation remains unclear. Therefore, in the present studies, we use a unique mouse model of severe hypertension to demonstrate a robust contribution of lymphocyte proliferation to the pathogenesis of ANG II-induced end-organ injury. These proinflammatory actions of ANG II have little effect on the extent of blood pressure elevation and their injurious consequences are primarily confined to the kidney. Within the kidney, these effects exaggerate not only pathologic damage but also functional injury as measured by albuminuria.
MATERIALS AND METHODS
Animals.
Wild-type 129/SvEv mice were purchased from Taconic and Agtr1a−/− mice lacking AT1A receptors for ANG II were generated as previously described (29). All mice were maintained in the animal facility of the Durham Veterans Affairs Medical Center under local and National Institutes of Health guidelines. All experiments described in this manuscript were included in an animal use protocol approved by the Durham VA Medical Center Institutional Animal Care and Use Committee. These studies used 2- to 4-mo-old male mice.
Model of ANG II-induced hypertension.
Wild-type 129/SvEv mice first underwent left nephrectomy and ANG II (1,000 ng·kg−1·day−1; Sigma) or vehicle (0.9% NaCl, n = 5) was infused continuously with subcutaneous osmotic minipumps (2004, Alzet) for 28 days as previously described (12, 35). Animals received a 6% NaCl diet (Harlan Teklad) during the ANG II infusion period to accentuate hypertension and kidney injury. To examine the contribution of the immune response to hypertensive kidney injury, the ANG II-infused mice were treated with mycophenolate mofetil (MMF; Roche; 100 mg·kg−1·day−1) or vehicle given by gavage beginning 1 day before and continuing throughout the 28-day ANG II infusion period (n ≥ 13 per group). This dose of MMF causes potent immunosuppression in mice without measurable toxicity (30, 46, 58).
Physiological assessments.
Systolic blood pressure was determined in conscious mice by the noninvasive computerized tail-cuff method as previously described (18). Animals underwent 2 wk of training before the initiation of recordings. Blood pressures were measured for 1 wk at baseline, after unilateral nephrectomy, and during 3 wk of ANG II infusion. During weeks 2 and 4 of ANG II infusion, the mice were placed in metabolic cages, and urine was collected for 24 h. Urinary concentrations of albumin were measured in individual samples using a specific ELISA for mouse albumin (Exocell, Philadelphia, PA) as previously described (18). Creatinine concentrations were measured using a picric acid-based method using a kit (Exocell). Albumin excretion is expressed as micrograms per milligram of creatinine. To measure generation of reactive oxygen species in the kidney, we quantitated urinary excretion of 8-isoprostane (32) per the manufacturer's instructions (8-Isoprostane EIA Kit, Cayman Chemical, Ann Arbor, MI). Isoprostane excretion is expressed as picograms per 24 hours.
Histopathologic analysis.
Following 28 days of ANG II infusion, hearts and kidneys were harvested, weighed, and fixed in formalin, sectioned, and stained with Masson trichrome. All of the tissues were examined by a pathologist (P.R.) without knowledge of the treatment groups. The pathological abnormalities were graded based on the presence and severity of component abnormalities including glomerulosclerosis, chronic inflammation, tubular atrophy or casts, fibrosis, and vascular injury. Grading for each component was performed using a semiquantitative scale as previously described (53, 54) where 0 was no abnormality and where 1, 2, 3, and 4 represented mild, moderate, moderately severe, and severe abnormalities, respectively. The total injury score for each kidney was a summation of these component injury scores. Percent glomerulosclerosis was defined as number of glomeruli with evidence of sclerosis divided by the total number of glomeruli in the section.
To assess T lymphocyte infiltration in the kidneys, paraffin-embedded sections were stained with anti-CD3 (clone SP7) per the manufacturer's instructions (Lab Vision, Fremont, CA). For an assessment of the CD4+ and CD8+ T cell subsets, frozen sections were stained with anti-CD4 (clone RM4–5, catalog no. 550280, BD Biosciences/Pharmingen, San Diego, CA) or anti-CD8 (clone 53–6.7, catalog no. 550281, BD Biosciences/Pharmingen) as previously described (60). On each section, 20 randomly selected fields were then scored in a blinded fashion for the presence or absence of T cell infiltrates (3 or more T cells in field). To quantify the extent of vascular inflammation, vessels were identified on the sections and the severity of perivascular T cell infiltrates was scored based on a previously established method (16) by assigning vessels to quartiles: Normal, no T cells were present; Minimal, 1–4 infiltrating T cells; Moderate, infiltrates containing 5–10 T cells; and Severe, infiltrates with more than 10 T cells.
Quantification of cardiac mRNA expression.
Hearts were harvested, and total RNA was isolated by using an RNeasy Fibrous Tissue Mini Kit per the manufacturer's instructions (Qiagen, Valencia, CA). The gene expression levels of brain natriuretic peptide (BNP) and β-MHC in cardiac tissue were determined by real-time quantitative RT-PCR as previously described (33).
RNase protection assays.
Total cellular RNA was extracted from harvested kidneys using the RNeasy kit (Qiagen), according to manufacturer's instructions, and was stored in RNase-free water at −70°C. To detect cytokine mRNA, a commercially available multiprobe template set (MCK-3b; BD Biosciences, Bedford, MA) was labeled with [α-32P]UTP (PerkinElmer), according to the manufacturer's instructions, and then diluted to a concentration of 270,000 cpm/μl of hybridization buffer. All reagents used in probe synthesis were obtained from BD Biosciences (In Vitro Transcription Kit, catalog 45004K). RNase protection assays were performed using the RNase Protection Assay Kit (BD Biosciences; catalog 45014K) following the manufacturer's protocol. Gels from the assay were dried under vacuum at 80°C for 60 min and then were placed on film in a cassette with an intensifying screen and developed at −70°C. Films were scanned and bands were analyzed as a ratio of target RNA/L32 control using the Scion Image for Windows program.
Quantitation of cytoskeletal rearrangements.
To provide a quantitative assessment of actin polymerization in lymphocytes, F-actin formation was determined using flow cytometry. Mouse splenocytes were isolated as previously described (45) and were stimulated with ANG II (1 μM) or anti-CD3 (1 μg/ml) in the presence or the absence of losartan (10 μM, Merck, Whitehouse Station, NJ) or Rho kinase inhibitor Y-27632 (10 μM, Calbiochem). The cells were harvested, fixed, and permeabilized with a formaldehyde/saponin solution (Cytofix, BD Biosciences) and stained with an Alexafluor 488-conjugated phalloidin (Molecular Probes). The cells were then analyzed by flow cytometry, and the percentage of polymerized actin was determined by comparing the percent change in mean channel fluorescence in the activated vs. unstimulated cells (17). Results shown reflect the average of at least three separate experiments. In some of the actin polymerization experiments, pure populations of splenic T cells were isolated using a Pan T Cell Isolation Kit (Miltenyi Biotec, Auburn, CA). Purity of the cell population was confirmed by fluorocytometry and averaged 98%.
Statistical methods.
The values for each parameter within a group were expressed as means ± SE. For comparisons between groups with normally distributed data, statistical significance was assessed using ANOVA followed by unpaired t-test. For comparisons between groups with nonnormally distributed data, the Mann Whitney U-test was employed. For comparisons within groups, normally distributed variables were analyzed by a paired t-test, whereas nonnormally distributed variables were analyzed by the Wilcoxon signed rank test. Chi square analysis was used for analysis of the frequencies of perivascular T cell infiltrates in kidneys.
RESULTS
Immunosuppression with MMF does not alter the hypertensive response to chronic ANG II infusion and high salt feeding.
To examine the role of lymphocyte responses in ANG II-dependent hypertension, we infused uninephrectomized mice with ANG II for 4 wk while administering MMF (100 mg/kg) or its vehicle daily by gavage. The combination of ANG II infusion and high salt diet caused marked hypertension and the severity of blood pressure elevation was not significantly affected by MMF (MMF 214 ± 11 vs. vehicle 206 ± 8 mmHg; P = NS; Fig. 1A).
Fig. 1.
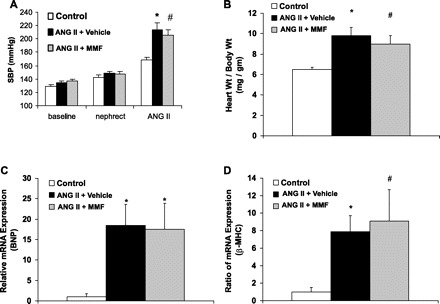
Immunosuppression with mycophenolate mofetil (MMF) during ANG II infusion does not alter blood pressure response or cardiac hypertrophy. A: blood pressures in the experimental groups at baseline, following unilateral nephrectomy (nephrect), and following 3 wk of ANG II infusion [ANG II; systolic blood pressure (SBP) by tail cuff]. *P = 0.04 vs. control. #P < 0.005 vs. control. B: mean heart-to-body wt ratios after 28 days of ANG II infusion. *P < 0.02 vs. control. #P < 0.01 vs. control. C: relative mRNA expression in the heart for brain natriuretic peptide (BNP; *P < 0.007 vs. control). D: relative mRNA expression in the heart for β-myosin heavy chain (β-MHC). *P < 0.005 vs. control. #P < 0.05 vs. control.
MMF does not attenuate cardiac hypertrophy in ANG II-dependent hypertension.
In vehicle-treated animals, ANG II and high salt feeding caused marked cardiac hypertrophy (heart wt:body wt ratio 9.8 ± 0.8 mg/g) compared with high salt diet alone (6.5 ± 0.2 mg/g; P < 0.02; Fig. 1B) or normal diet (5.4 ± 0.1 mg/g; P < 0.007). On a molecular level, ANG II induced prominent gene expression for markers of cardiac hypertrophy including BNP [18.5 ± 5.1 arbitrary units (au); Fig. 1C] and β-myosin heavy chain (β-MHC; 7.9 ± 1.8 au; Fig. 1D). Treatment with MMF during ANG II infusion had no effect on cardiac hypertrophy (heart wt:body wt ratio 9.0 ± 0.8 mg/g; P = NS; Fig. 1B) or on expression of BNP (17.5 ± 6.4 au; P = NS; Fig. 1C) or β-MHC (9.1 ± 3.6 au; P = NS; Fig. 1D). The finding of equivalent cardiac hypertrophy in vehicle- and MMF-treated groups is consistent with their similar blood pressures and suggests that immune system activation does not make a critical contribution to left ventricular hypertrophy caused by ANG II.
Immunosuppression reduces ANG II-dependent kidney injury.
As a functional measure of the extent of kidney injury, we measured urinary albumin excretion in the experimental groups. ANG II infusion and high salt diet caused marked albuminuria in the vehicle-treated animals at 2 (7,705 ± 1,581 μg albumin/mg creatinine) and 4 wk (10,390 ± 2,324 μg albumin/mg Cr). Despite its lack of effect on blood pressure, administration of MMF reduced urinary albumin excretion by over 60% (Fig. 2A) after 2 (2,528 ± 449 μg albumin/mg Cr; P < 0.008 vs. vehicle) and 4 wk (4,058 ± 1,212 μg albumin/mg Cr; P < 0.04 vs. vehicle).
Fig. 2.
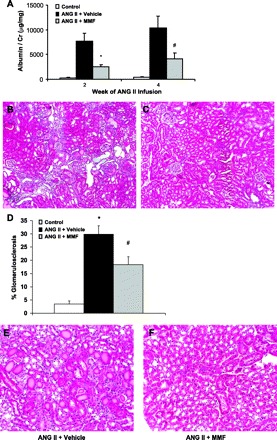
MMF therapy during ANG II infusion reduces albuminuria and kidney injury. A: urinary albumin excretion after ANG II infusion. *P < 0.008 vs. ANG II + vehicle. #P < 0.04 vs. ANG II + vehicle. B: representative kidney section from ANG II + vehicle group showing glomerulosclerosis, interstitial inflammation, and mild perivascular fibrosis. C: representative kidney section from ANG II + MMF group. D: average percent glomerulosclerosis (# glomeruli with sclerosis/total # of glomeruli in section) in experimental groups. *P < 0.0002 vs. control. #P < 0.009 vs. control, P < 0.02 vs. ANG II + vehicle. E: representative ANG II + vehicle section showing tubular cast formation. F: comparable section from representative ANG II + MMF kidney.
After 4 wk of ANG II infusion and high salt feeding, kidneys from the vehicle-treated group exhibited significant pathology including glomerulosclerosis, proteinacious casts in renal tubules, robust inflammatory cell infiltration, and interstitial fibrosis (Fig. 2B). Treatment with MMF significantly ameliorated kidney injury reducing the severity of the overall pathology score from 11.5 ± 0.9 to 7.8 ± 0.7 au (P = 0.003; Fig. 2, B–C, Table 1). This included effects of MMF therapy to reduce glomerulosclerosis (18.4 ± 3.0 vs. 29.9 ± 3.2%; P < 0.02; Fig. 2D) and cast formation (1.3 ± 0.2 vs. 2.1 ± 0.2 au; P < 0.008; Fig. 2, E–F). Within the renal interstitium, MMF significantly attenuated inflammatory cell infiltration (1.9 ± 0.2 vs. 3.2 ± 0.4 au; P = 0.006; Fig. 2, B–C) and interstitial fibrosis (0.2 ± 0.1 vs. 0.7 ± 0.2 au; P = 0.03; Fig. 2, B–C). In contrast to negligible consequences in the heart, these data suggest a significant role for immune activation in ANG II-dependent kidney injury.
Table 1.
MMF therapy during ANG II infusion reduces albuminuria and kidney injury
| Glomerulosclerosis | Vascular Injury | Interstitial Disease | Total Injury | |
|---|---|---|---|---|
| Control | 0.8±0.2 | 0.4±0.4 | 1.8±0.2 | 3.0±0.5 |
| ANG II + vehicle | 2.4±0.2 | 3.1±0.2 | 6.0±0.7 | 11.5±0.9 |
| ANG II + MMF | 1.8±0.2† | 2.6±0.3 | 3.4±0.3‡ | 7.8±0.7* |
Values are means ± SE of kidney injury scores in arbitiary units (au). MMF, mycophenolate mofetil.
P = 0.003 vs. ANG II + vehicle.
P = 0.03 vs. ANG II + vehicle.
P = 0.002 vs. ANG II + vehicle.
Activation of intrarenal lymphocyte responses promotes hypertensive kidney injury.
We used immunostaining to examine the surface phenotype of cells infiltrating kidneys from the ANG II-infused mice (Fig. 2B). As shown in Fig. 3B, we found that CD3+ cells, presumably T lymphocytes, are a prominent component of the mononuclear cell infiltrates in the kidneys from vehicle-treated mice infused with ANG II. T cells were particularly concentrated in perivascular regions (Fig. 3E) and 83% of vessels from kidneys of mice receiving ANG II infusion and vehicle had moderate to severe perivascular T cell infiltrates. Treatment with MMF caused a dramatic diminution of these T cell infiltrates (Fig. 3, C–D, F–G). This was manifested by a substantial reduction in the frequency of renal vessels with moderate to severe T cell infiltrates to only 28% (P < 0.0001 vs. vehicle; Fig. 3F).
Fig. 3.
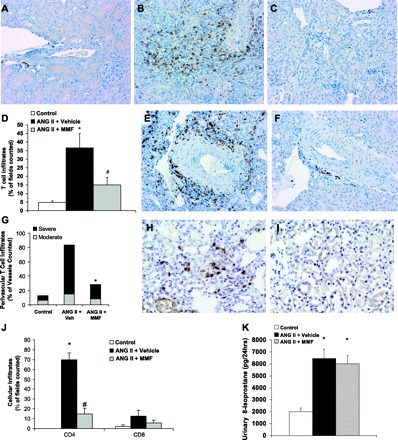
MMF treatment diminishes T lymphocyte infiltration into the kidney during ANG II infusion. T cells stain brown. Representative kidney sections from control group (A), ANG II + vehicle group (B), and ANG II + MMF (C). D: scores for interstitial T cell infiltration into kidney, defined as % of fields in section with T cell infiltration. *P < 0.02 vs. control. #P = 0.03 vs. ANG II + vehicle. E: blood vessel from representative ANG II + vehicle kidney showing perivascular T cell infiltrates. F: comparable vessel from representative ANG II + MMF kidney. G: percent of renal vessels with perivascular T cell infiltrates. Moderate and severe infiltrates are composed of 5–10 T cells and >10 T cells, respectively (vessels counted n = 16 in control group, n ≥ 50 in ANG II + vehicle and ANG II + MMF groups; *P < 0.0001 vs. ANG II + vehicle using chi square analysis). H: representative ANG II + vehicle kidney stained for CD4. I: representative ANG II + MMF kidney stained for CD4. J: scores for interstitial CD4+ and CD8+ T cell infiltration into kidney. *P < 0.00001 vs. control. #P = 0.00001 vs. ANG II + vehicle. K: renal generation of reactive oxygen species as measured by urinary 8-isoprostane excretion (*P < 0.005 vs. control).
To determine which T cell subsets infiltrated the kidneys of the ANG II-infused animals, we stained kidney sections for CD4 and CD8. ANG II infusion caused a dramatic accumulation of CD4+ T helper lymphocytes in the kidney (Fig. 3, H and J), an effect almost completely reversed by treatment with MMF (Fig. 3, I–J). By contrast, although ANG II caused a numerical increase in the fields containing CD8+ cells, this increase was not significant compared with the control group or the group treated with ANG II plus MMF (Fig. 3J). As CD8+ cells are capable of eliciting reactive oxygen species (ROS) (39), we quantitated renal generation of ROS in the experimental groups by measuring urinary 8-isoprostane excretion. ANG II induced a brisk generation of ROS that was not altered by treatment with MMF (Fig. 3K), indicating that induction of ROS by ANG II depends on cell lineages other than CD8+ cytotoxic T cells.
To further assess the effects of MMF on the intrarenal inflammatory response, we examined mRNA expression for key inflammatory cytokine genes. In control mice fed high salt without ANG II infusion, renal expression of inflammatory cytokines IFN-γ, TNF-α, and MIF and profibrotic cytokine TGF-β was negligible (data not shown). Chronic infusion of ANG II prominently increased intrarenal gene expression for this entire panel of cytokines. MMF therapy dramatically attenuated expression of IFN-γ and TNF-α but did not alter the enhanced expression of MIF, suggesting some specificity of the response (Fig. 4, A–B). It has been suggested that ANG II may directly stimulate intrarenal production of the profibrotic molecule TGF-β (31) which may then contribute to the development of kidney fibrosis. Indeed, expression of TGF-β was significantly upregulated in kidneys from ANG II-infused animals. However, MMF dramatically reduced TGF-β mRNA levels suggesting complex control of TGF-β expression in this setting (Fig. 4, A–B).
Fig. 4.
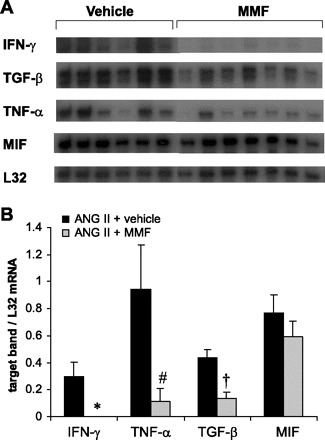
MMF treatment ameliorates renal expression of inflammatory genes induced by ANG II. A: mRNA expression for IFN-γ, TGF-β, TNF-α, MIF, and housekeeping gene L32 detected by RNAse protection assay (RPA) using kidney RNA from experimental animals. The vehicle and MMF lanes were run on the same gel but were noncontiguous. B: densitometry analysis of RPA for IFN-γ, TNF-α, TGF-β, and MIF expression. *P = 0.01 vs. ANG II + vehicle, P < 0.04 vs. ANG II + vehicle. †P = 0.002 vs. ANG II + vehicle.
ANG II stimulates cytoskeletal rearrangements in T lymphocytes.
Our findings from experiments in intact animals suggest that one of the pathways contributing to kidney injury in ANG II-dependent hypertension involves accumulation of T cells around renal vasculature and enhanced expression of cytokines that promote inflammation and fibrosis. In T cells, rearrangements of the actin-myosin cytoskeleton are crucial to the development of full activation and development of the immunological synapse (1, 8, 50). Because ANG II triggers cytoskeletal changes in vascular tissues (27, 34, 55) and in glomerular epithelial cells (52), we considered the possibility that another pathway for control of T cell activation by ANG II might involve effects on actin polymerization.
To test this hypothesis, murine splenocytes were stimulated with ANG II (1 μM) and subsequently stained with a fluorescent phalloidin that binds the polymerized or “filamentous” form of actin (F-actin). We then performed flow cytometry to measure ANG II-dependent F-actin formation as reflected by mean channel fluorescence in wild-type and AT1A receptor-deficient (Agtr1a−/−) lymphocytes. Using this assay, we found that ANG II induced marked actin polymerization in wild-type lymphocytes (27.9 ± 8.4%; P = 0.01 vs. vehicle) but did not stimulate F-actin formation in splenocytes from Agtr1a−/− mice (−16.3 ± 7.5%; P = 0.003 vs. wild-type). Consistent with the notion that cytoskeletal rearrangements facilitate T lymphocyte activation, we found that T cell receptor stimulation with anti-CD3 (1 μg/ml) caused significant actin polymerization in wild-type lymphocytes (28.5 ± 4.0%). This effect was significantly blunted in Agtr1a−/− lymphocytes (11.5 ± 3.0%; P = 0.007). Furthermore, activation of highly enriched wild-type T lymphocytes with anti-CD3 and anti-CD28 (2 μg/ml) triggered marked actin polymerization, an effect significantly attenuated by coincubation with the ARB losartan (38.9 ± 8.4 vs. 13.3 ± 7.0%; P = 0.009). Thus, ANG II stimulates cytoskeletal rearrangements in T lymphocytes through activation of the AT1 receptor.
We next explored the mechanism through which ANG II modulates the cytoskeleton in lymphocytes. In vascular smooth muscle cells, ANG II influences actin-myosin cytoskeletal rearrangements via pathways involving Rho-GTPases and their distal effectors such as Rho Kinase; these actions may promote vascular injury (34). We considered the possibility that similar pathways might be responsible for the effects of ANG II to modulate the actin cytoskeleton in lymphocytes. To test this possibility, splenic lymphocytes were exposed to 1 μM ANG II in the presence or absence of the specific Rho kinase inhibitor, Y-27632 (10 μM). As before, exposure to ANG II stimulated F-actin formation (17.1 ± 6.8%) in lymphocytes and this was completely abrogated by Y-27632 (−3.0 ± 5.8%; P < 0.05 vs. ANG II alone). Thus, by stimulating AT1 receptors on T lymphocytes, ANG II promotes cytoskeletal rearrangements through a pathway requiring Rho kinase.
DISCUSSION
The critical role that the RAS plays in hypertension is well-established (9). In hypertension, activation of the RAS may contribute independently to both elevation of blood pressure and progression of cardiovascular and renal disease (7, 15, 59). In clinical trials, blockade of the RAS appears to reduce target organ injury more than can be explained by blood pressure reduction alone. Nevertheless, determining the precise mechanisms through which ANG II promotes hypertensive organ damage has proved challenging.
In the present experiments, we wished to assess the extent to which lymphocyte stimulation contributes to the pathogenesis of ANG II-dependent hypertension. To this end, we utilized a simple model wherein ANG II is infused chronically into mice in sufficient quantities to cause marked increases in blood pressure, primarily driven by activation of AT1 angiotensin receptors (12). To augment the severity of target organ damage, animals were uninephrectomized and fed a high salt diet. Moreover, we used 129/SvEv mice, a strain with enhanced susceptibility to renal damage (25, 28, 38). This targeted approach resulted in impressive kidney injury allowing us to characterize not only renal pathologic changes but also functional damage as measured by albuminuria. To determine the contribution of lymphocyte responses, one group of animals was given MMF. MMF is an immunosuppressant that acts primarily by inhibiting lymphocyte proliferation.
In this model, treatment with MMF had no effect on the magnitude of the hypertensive response; blood pressures were nearly identical in the MMF- and vehicle-treated groups. As heart enlargement correlates tightly with blood pressure elevation during hypertension (12), we also measured heart weights in the animals. Accordingly, the similar degree of cardiac hypertrophy in the vehicle- and MMF-treated groups suggests that the blood pressures in the two groups were truly similar. Thus, in this model of ANG II-dependent hypertension, AT1 receptor actions to promote lymphocyte activation do not play a significant role to increase blood pressure.
Our finding that actions of lymphocytes do not contribute to ANG II-dependent hypertension in the 129/SvEv mouse strain contrasts sharply with the studies of Guzik et al. (24) using the C57BL/6 mouse strain in which the absence of lymphocytes dramatically attenuated the chronic hypertensive response to ANG II. There are several differences between the two models that could explain this discrepancy. First, in our model, the absence of blood pressure effects with MMF treatment may relate to our use of the 129/SvEv mouse strain. Using the (C57BL/6 × 129/SvEv) F1 strain, we previously showed that AT1 receptors on lymphocytes and in all other extrarenal compartments do not contribute to blood pressure elevation during ANG II infusion (12). The feature common to both the studies from our laboratory in which lymphocytes do not influence blood pressure elevation is the partial or complete presence of the 129/SvEv strain, which is more prone to kidney injury than the C57BL/6 strain. Thus, the ANG II-induced kidney injury in the 129/SvEv strain may confer a degree of salt sensitivity and, in turn, hypertension that cannot be rectified by suppression of lymphocytes. Second, the lower dose of ANG II employed in the Harrison study (∼500 mg·kg−1·day−1) may have caused less kidney injury and/or allowed more sensitive detection of the blood pressure effects of lymphocytes. Third, the Harrison model used ANG II infusion in mice with two kidneys receiving a normal salt diet. In contrast, we infused ANG II into uninephrectomized mice receiving a high salt diet. The severe kidney injury in our model may therefore have precluded an effect of MMF on blood pressure. Nonetheless, this attribute of the model allowed us to examine how lymphocytes can influence kidney injury in the setting of hypertension independently of effects on blood pressure elevation.
Studies addressing the effects of lymphocytes on blood pressure elevation in rats have shown inconsistent results. In the double transgenic rat (dTGR) model of RAS activation (44) and in the Dahl S salt-sensitive rat model (40), lymphocyte suppression lowered blood pressure. By contrast, in hypertension induced by chronic inhibition of nitric oxide synthase (l-NAME model) or by 5/6 nephrectomy (19, 48), treatment with MMF did not influence blood pressure even though kidney injury was ameliorated. As the RAS is suppressed in the Dahl S rat (40) but activated in the l-NAME model (10, 37), different levels of RAS activation in these models cannot reconcile the divergent effects of lymphocytes on blood pressure. Rather, the differential effects of lymphocytes on blood pressure elevation in these models may relate to the mechanisms through which hypertension was induced.
In our model, the combination of ANG II infusion, uninephrectomy, and high salt diet caused marked cardiac hypertrophy, ≈25% greater than we observed in previous studies with ANG II infusion alone (12). While pressure load from elevated blood pressure clearly contributes to the development of cardiac hypertrophy, it has also been suggested that direct cellular actions of ANG II via AT1 receptors may also contribute to cardiac pathology in this setting. In the present study, we find that suppressing the cellular immune response with MMF had no appreciable effect to mitigate the cardiac hypertrophy induced by chronic ANG II infusion (Fig. 1B). Thus, activation of the immune system by ANG II does not contribute to cardiac damage in this setting. This finding is consistent with our previous work indicating that ANG II activation of AT1 receptors in extrarenal tissues, including the immune system, is not sufficient to induce cardiac hypertrophy in the absence of hypertension (12). Instead, the extent of LVH depends directly on the magnitude of blood pressure elevation.
In contrast, our experiments indicate that stimulation of lymphocyte responses by ANG II plays a major role in promoting hypertensive kidney injury. Treatment with MMF reduced the level of albuminuria by more than 60%, indicating that in the setting of hypertension ANG II-dependent lymphocyte responses exaggerate proteinuria. Along with its anti-proteinuric effects, we find that MMF also provides striking protection against renal pathological injury and these protective actions are seen across the glomerular, interstitial, and tubular compartments. Although others showed that ANG II-induced lymphocyte responses may promote kidney injury (44, 47), the present studies are the first to our knowledge to link these lymphocyte responses directly to glomerulosclerosis, consistent with the effects seen on albuminuria (Fig. 2A). However, MMF did not attenuate renal arteriosclerosis. As vascular hypertrophy and sclerosis are typical pathological features associated with hypertensive nephrosclerosis, the absence of vascular protection is likely a direct reflection of the lack of blood pressure lowering by MMF. On the other hand, the striking attenuation of glomerular and tubulointerstitial injury by MMF occurs in the setting of persistent, severe blood pressure elevation, consistent with those studies showing that lymphocyte suppression can protect from kidney injury without altering blood pressure (19, 48).
In view of previously described actions of ANG II to promote T cell activation and well-characterized immunosuppressive properties of MMF, it is likely that T lymphocytes are a critical cell population mediating kidney injury in our studies. In the present studies we find impressive T cell infiltration of the renal interstitium, especially around the renal vasculature, in animals infused with ANG II. These infiltrates are dramatically attenuated by immunosuppression with MMF. Previous studies demonstrated that infusion of ANG II in rats causes a shift in T lymphocyte populations toward the Th1 phenotype characterized by increased IFN-γ production (51). Consistent with this notion, ANG II in our model causes accumulation in the kidney of CD4+ but not CD8+ lymphocytes with increased expression of IFN-γ and TNF-α. In turn, actions of both IFN-γ and TNF-α have been linked to disease progression in diverse models of kidney injury (3, 42, 44, 57). Moreover, these cytokines have direct effects on glomerular cell lineages to augment proteinuria. For example, IFN-γ induces expression of MHC class II antigens in glomerular endothelial cells (23) and podocytes (4, 11), which may sensitize these cells to further immunologic injury. TNF-α causes glomerular endothelial cell damage (5, 20) and downregulates nephrin in podocytes (62), a cardinal feature of proteinuric renal disease in which the glomerular slit diaphragm is compromised. In the present experiments, MMF therapy essentially abolished ANG II-induced expression of both IFN-γ and TNF-α in the kidney, suggesting that ANG II may promote hypertensive kidney injury in part by inducing expression of proinflammatory cytokines. Thus, blocking these proinflammatory mediators may represent an important avenue for protecting the kidney in ANG II-dependent hypertension.
Previous studies suggested that direct stimulation of TGF-β expression by activation of AT1 receptors is an important pathway promoting renal fibrosis and structural injury in a variety of renal diseases (31). Conversely, blockade of this pathway by ARBs and ACE inhibitors has been correlated with their actions to prevent progression of chronic kidney disease leading some to propose that TGF-β might be a useful biomarker for assessing the extent of RAS blockade in these diseases (6). Consistent with this hypothesis, expression of TGF-β was markedly upregulated in kidneys of control mice infused with ANG II. Yet, surprisingly MMF caused a striking reduction in TGF-β expression indicating critical involvement of the immune system in this response and thereby suggesting that regulation of TGF-β by ANG II in the diseased kidney may depend not only on AT1 receptor activation but also on stimulation of lymphocyte responses.
In the present studies, we explored the mechanism through which ANG II modulates T cell activation. Cytoskeletal rearrangements are critical to T cell activation as they facilitate formation of the immunological synapse between the T cell and an antigen-presenting cell (1, 50), but whether ANG II modulates these processes has not been previously demonstrated. Through both direct visualization and flow cytometry, we find that ANG II stimulates cytoskeletal rearrangements in T lymphocytes via an AT1 receptor-dependent pathway. As it has been suggested that ANG II stimulates vascular remodeling through the interaction of small GTP-binding protein Rho with Rho kinase (34), which also plays a critical role in the adaptive immune response (1, 56), we considered the possibility that ANG II promotes cytoskeletal rearrangements in T cells through a Rho kinase-dependent mechanism. Indeed, we showed that inhibition of Rho kinase completely abrogated F-actin formation induced by ANG II. In sum, our data suggest that activation of AT1 receptors directly stimulates cytoskeletal rearrangements, promoting T cell activation and proliferation.
In summary, although blood pressure elevation is clearly responsible for a major portion of kidney damage in the setting of hypertension (22), some clinical trials suggest that ACE inhibitors and ARBs protect the kidney more than can be explained solely by blood pressure reduction (7, 59). In our studies, suppression of the lymphocyte responses during ANG II-dependent hypertension reduces proteinuria by over 60% and attenuates kidney injury including glomerulosclerosis and interstitial inflammation by ∼40% without affecting blood pressure. Our studies further suggest that the cellular mechanism of this effect involves direct effects of ANG II to facilitate T lymphocyte activation. Thus, ANG II causes renal injury through the activation of the cellular immune system, and the kidney seems to be much more susceptible to this immune-mediated mechanism of injury than the heart. Since therapy with ARBs and ACEIs slows but does not arrest the progression of chronic kidney disease, interventions to suppress cellular immune responses and T cell activation may represent useful therapeutic options to further diminish kidney injury in hypertension.
GRANTS
This work was supported by National Institutes of Health Grants DK-38108 and HL-56122, by funding from the Medical Research Service of the Veterans Administration, and by the Edna and Fred L. Mandel Center for Hypertension and Atherosclerosis Research.
Acknowledgments
The authors acknowledge outstanding administrative support from N. Barrow.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Acuto O, Cantrell D. T cell activation and the cytoskeleton. Annu Rev Immunol 18: 165–184, 2000 [DOI] [PubMed] [Google Scholar]
- 2.Amuchastegui SC, Azzollini N, Mister M, Pezzotta A, Perico N, Remuzzi G. Chronic allograft nephropathy in the rat is improved by angiotensin II receptor blockade but not by calcium channel antagonism. J Am Soc Nephrol 9: 1948–1955, 1998 [DOI] [PubMed] [Google Scholar]
- 3.Balomenos D, Rumold R, Theofilopoulos AN. Interferon-gamma is required for lupus-like disease and lymphoaccumulation in MRL-lpr mice. J Clin Invest 101: 364–371, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baudeau C, Delarue F, He CJ, Nguyen G, Adida C, Peraldi MN, Sraer JD, Rondeau E. Induction of MHC class II molecules HLA-DR, -DP and -DQ and ICAM 1 in human podocytes by gamma-interferon. Exp Nephrol 2: 306–312, 1994 [PubMed] [Google Scholar]
- 5.Bertani T, Abbate M, Zoja C, Corna D, Perico N, Ghezzi P, Remuzzi G. Tumor necrosis factor induces glomerular damage in the rabbit. Am J Pathol 134: 419–430, 1989 [PMC free article] [PubMed] [Google Scholar]
- 6.Border WA, Noble NA. Interactions of transforming growth factor-beta and angiotensin II in renal fibrosis. Hypertension 31: 181–188, 1998 [DOI] [PubMed] [Google Scholar]
- 7.Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 345: 861–869, 2001 [DOI] [PubMed] [Google Scholar]
- 8.Bretscher MS. Getting membrane flow and the cytoskeleton to cooperate in moving cells. Cell 87: 601–606, 1996 [DOI] [PubMed] [Google Scholar]
- 9.Brunner HR. Experimental and clinical evidence that angiotensin II is an independent risk factor for cardiovascular disease. Am J Cardiol 87: 3C–9C, 2001 [DOI] [PubMed] [Google Scholar]
- 10.Casellas D, Benahmed S, Artuso A, Jover B. Candesartan and progression of preglomerular lesions in NG-nitro-l-arginine methyl ester hypertensive rats. J Am Soc Nephrol 10, Suppl 11: S230–S233, 1999 [PubMed] [Google Scholar]
- 11.Coers W, Vos JT, Huitema S, Dijk F, Weening JJ. Biological alterations of rat podocytes cultured under basolateral hydrostatic pressure. Pathobiology 64: 222–232, 1996 [DOI] [PubMed] [Google Scholar]
- 12.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci USA 103: 17985–17990, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crowley SD, Tharaux PL, Audoly LP, Coffman TM. Exploring type I angiotensin (AT1) receptor functions through gene targeting. Acta Physiol Scand 181: 561–570, 2004 [DOI] [PubMed] [Google Scholar]
- 14.Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, Lindholm LH, Nieminen MS, Omvik P, Oparil S, Wedel H. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 359: 995–1003, 2002 [DOI] [PubMed] [Google Scholar]
- 15.Devereux RB, Dahlof B, Gerdts E, Boman K, Nieminen MS, Papademetriou V, Rokkedal J, Harris KE, Edelman JM, Wachtell K. Regression of hypertensive left ventricular hypertrophy by losartan compared with atenolol: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) trial. Circulation 110: 1456–1462, 2004 [DOI] [PubMed] [Google Scholar]
- 16.Dragun D, Tullius SG, Park JK, Maasch C, Lukitsch I, Lippoldt A, Gross V, Luft FC, Haller H. ICAM-1 antisense oligodesoxynucleotides prevent reperfusion injury and enhance immediate graft function in renal transplantation. Kidney Int 54: 590–602, 1998 [DOI] [PubMed] [Google Scholar]
- 17.Flaishon L, Lantner F, Hershkoviz R, Levo Y, Shachar I. Low levels of IFN-gamma downregulate the integrin-dependent adhesion of B cells by activating a pathway that interferes with cytoskeleton rearrangement. J Biol Chem 276: 46701–46706, 2001 [DOI] [PubMed] [Google Scholar]
- 18.Francois H, Athirakul K, Mao L, Rockman H, Coffman TM. Role for thromboxane receptors in angiotensin-II-induced hypertension. Hypertension 43: 364–369, 2004 [DOI] [PubMed] [Google Scholar]
- 19.Fujihara CK, Avancini Costa Malheiros DM, de Lourdes Noronha I, De Nucci G, Zatz R. Mycophenolate mofetil reduces renal injury in the chronic nitric oxide synthase inhibition model. Hypertension 37: 170–175, 2001 [DOI] [PubMed] [Google Scholar]
- 20.Gomez-Chiarri M, Ortiz A, Lerma JL, Lopez-Armada MJ, Mampaso F, Gonzalez E, Egido J. Involvement of tumor necrosis factor and platelet-activating factor in the pathogenesis of experimental nephrosis in rats. Lab Invest 70: 449–459, 1994 [PubMed] [Google Scholar]
- 21.Griffin KA, Abu-Amarah I, Picken M, Bidani AK. Renoprotection by ACE inhibition or aldosterone blockade is blood pressure dependent. Hypertension 41: 201–206, 2003 [DOI] [PubMed] [Google Scholar]
- 22.Griffin KA, Bidani AK. Progression of renal disease: renoprotective specificity of renin-angiotensin system blockade. Clin J Am Soc Nephrol 1: 1054–1065, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Groenewegen G, Buurman WA, van der Linden CJ. Lymphokine dependence of in vivo expression of MHC class II antigens by endothelium. Nature 316: 361–363, 1985 [DOI] [PubMed] [Google Scholar]
- 24.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hartner A, Cordasic N, Klanke B, Veelken R, Hilgers KF. Strain differences in the development of hypertension and glomerular lesions induced by deoxycorticosterone acetate salt in mice. Nephrol Dial Transplant 18: 1999–2004, 2003 [DOI] [PubMed] [Google Scholar]
- 26.Hernandez-Presa M, Bustos C, Ortego M, Tunon J, Renedo G, Ruiz-Ortega M, Egido J. Angiotensin-converting enzyme inhibition prevents arterial nuclear factor-kappa B activation, monocyte chemoattractant protein-1 expression, and macrophage infiltration in a rabbit model of early accelerated atherosclerosis. Circulation 95: 1532–1541, 1997 [DOI] [PubMed] [Google Scholar]
- 27.Ishida T, Ishida M, Suero J, Takahashi M, Berk BC. Agonist-stimulated cytoskeletal reorganization and signal transduction at focal adhesions in vascular smooth muscle cells require c-Src. J Clin Invest 103: 789–797, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishola DA Jr, van der Giezen DM, Hahnel B, Goldschmeding R, Kriz W, Koomans HA, Joles JA. In mice, proteinuria and renal inflammatory responses to albumin overload are strain dependent. Nephrol Dial Transplant 21: 591–597, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Ito M, Oliverio MI, Mannon PJ, Best CF, Maeda N, Smithies O, Coffman TM. Regulation of blood pressure by the type 1A angiotensin II receptor gene. Proc Natl Acad Sci USA 92: 3521–3525, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jonsson CA, Svensson L, Carlsten H. Beneficial effect of the inosine monophosphate dehydrogenase inhibitor mycophenolate mofetil on survival and severity of glomerulonephritis in systemic lupus erythematosus (SLE)-prone MRLlpr/lpr mice. Clin Exp Immunol 116: 534–541, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kagami S, Border WA, Miller DE, Noble NA. Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-beta expression in rat glomerular mesangial cells. J Clin Invest 93: 2431–2437, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawada N, Dennehy K, Solis G, Modlinger P, Hamel R, Kawada JT, Aslam S, Moriyama T, Imai E, Welch WJ, Wilcox CS. TP receptors regulate renal hemodynamics during angiotensin II slow pressor response. Am J Physiol Renal Physiol 287: F753–F759, 2004 [DOI] [PubMed] [Google Scholar]
- 33.Kim HS, Lee G, John SWM, Maeda N, Smithies O. Molecular phenotyping for analyzing subtle genetic effects in mice: application to an angiotensinogen gene titration. Proc Natl Acad Sci USA 99: 4602–4607, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kobayashi N, Nakano S, Mita S, Kobayashi T, Honda T, Tsubokou Y, Matsuoka H. Involvement of Rho-kinase pathway for angiotensin II-induced plasminogen activator inhibitor-1 gene expression and cardiovascular remodeling in hypertensive rats. J Pharmacol Exp Ther 301: 459–466, 2002 [DOI] [PubMed] [Google Scholar]
- 35.Lautrette A, Li S, Alili R, Sunnarborg SW, Burtin M, Lee DC, Friedlander G, Terzi F. Angiotensin II and EGF receptor cross-talk in chronic kidney diseases: a new therapeutic approach. Nat Med 11: 867–874, 2005 [DOI] [PubMed] [Google Scholar]
- 36.Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I. Renoprotective effect of the angiotensin receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 345: 851–860, 2001 [DOI] [PubMed] [Google Scholar]
- 37.Luvara G, Pueyo ME, Philippe M, Mandet C, Savoie F, Henrion D, Michel JB. Chronic blockade of NO synthase activity induces a proinflammatory phenotype in the arterial wall: prevention by angiotensin II antagonism. Arterioscler Thromb Vasc Biol 18: 1408–1416, 1998 [DOI] [PubMed] [Google Scholar]
- 38.Ma LJ, Fogo AB. Model of robust induction of glomerulosclerosis in mice: importance of genetic background. Kidney Int 64: 350–355, 2003 [DOI] [PubMed] [Google Scholar]
- 39.Martinvalet D, Zhu P, Lieberman J. Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity 22: 355–370, 2005 [DOI] [PubMed] [Google Scholar]
- 40.Mattson DL, James L, Berdan EA, Meister CJ. Immune suppression attenuates hypertension and renal disease in the Dahl salt-sensitive rat. Hypertension 48: 149–156, 2006 [DOI] [PubMed] [Google Scholar]
- 41.Mervaala EM, Muller DN, Park JK, Schmidt F, Lohn M, Breu V, Dragun D, Ganten D, Haller H, Luft FC. Monocyte infiltration and adhesion molecules in a rat model of high human renin hypertension. Hypertension 33: 389–395, 1999 [DOI] [PubMed] [Google Scholar]
- 42.Mu W, Ouyang X, Agarwal A, Zhang L, Long DA, Cruz PE, Roncal CA, Glushakova OY, Chiodo VA, Atkinson MA, Hauswirth WW, Flotte TR, Rodriguez-Iturbe B, Johnson RJ. IL-10 suppresses chemokines, inflammation, and fibrosis in a model of chronic renal disease. J Am Soc Nephrol 16: 3651–3660, 2005 [DOI] [PubMed] [Google Scholar]
- 43.Muller DN, Dechend R, Mervaala EMA, Park JK, Schmidt F, Fiebeler A, Theuer J, Breu V, Ganten D, Haller H, Luft FC. NF-κB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension 35: 193–201, 2000 [DOI] [PubMed] [Google Scholar]
- 44.Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, Hampich F, Fiebeler A, Ju X, Finckenberg P, Theuer J, Viedt C, Kreuzer J, Heidecke H, Haller H, Zenke M, Luft FC. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol 161: 1679–1693, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nataraj C, Oliverio MI, Mannon RB, Mannon PJ, Audoly LP, Amuchastegui CS, Ruiz P, Smithies O, Coffman TM. Angiotensin II regulates cellular immune responses through a calcineurin-dependent pathway. J Clin Invest 104: 1693–1701, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramos MA, Pinera C, Setien MA, Buelta L, de Cos MA, de Francisco AL, Merino R, Arias M. Modulation of autoantibody production by mycophenolate mofetil: effects on the development of SLE in (NZB × NZW)F1 mice. Nephrol Dial Transplant 18: 878–883, 2003 [DOI] [PubMed] [Google Scholar]
- 47.Rodriguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincon J, Chavez M, Parra G, Herrera-Acosta J, Gomez-Garre D, Largo R, Egido J, Johnson RJ. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int 59: 2222–2232, 2001 [DOI] [PubMed] [Google Scholar]
- 48.Romero F, Rodriguez-Iturbe B, Parra G, Gonzalez L, Herrera-Acosta J, Tapia E. Mycophenolate mofetil prevents the progressive renal failure induced by 5/6 renal ablation in rats. Kidney Int 55: 945–955, 1999 [DOI] [PubMed] [Google Scholar]
- 49.Ruiz-Ortega M, Lorenzo O, Egido J. Angiotensin III increases MCP-1 and activates NF-kappaB and AP-1 in cultured mesangial and mononuclear cells. Kidney Int 57: 2285–2298, 2000 [DOI] [PubMed] [Google Scholar]
- 50.Serrador JM, Nieto M, Sanchez-Madrid F. Cytoskeletal rearrangement during migration and activation of T lymphocytes. Trends Cell Biol 9: 228–233, 1999 [DOI] [PubMed] [Google Scholar]
- 51.Shao J, Nangaku M, Miyata T, Inagi R, Yamada K, Kurokawa K, Fujita T. Imbalance of T-cell subsets in angiotensin II-infused hypertensive rats with kidney injury. Hypertension 42: 31–38, 2003 [DOI] [PubMed] [Google Scholar]
- 52.Sharma R, Lovell HB, Wiegmann TB, Savin VJ. Vasoactive substances induce cytoskeletal changes in cultured rat glomerular epithelial cells. J Am Soc Nephrol 3: 1131–1138, 1992 [DOI] [PubMed] [Google Scholar]
- 53.Spurney RF, Ibrahim S, Butterly D, Klotman PE, Sanfilippo F, Coffman TM. Leukotrienes in renal transplant rejection in rats. Distinct roles for leukotriene B4 and peptidoleukotrienes in the pathogenesis of allograft injury. J Immunol 152: 867–876, 1994 [PubMed] [Google Scholar]
- 54.Spurney RF, Ruiz P, Pisetsky DS, Coffman TM. Enhanced renal leukotriene production in murine lupus: role of lipoxygenase metabolites. Kidney Int 39: 95–102, 1991 [DOI] [PubMed] [Google Scholar]
- 55.Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol 35: 881–900, 2003 [DOI] [PubMed] [Google Scholar]
- 56.Tharaux PL, Bukoski RC, Rocha PN, Crowley SD, Ruiz P, Nataraj C, Howell DN, Kaibuchi K, Spurney RF, Coffman TM. Rho kinase promotes alloimmune responses by regulating the proliferation and structure of T cells. J Immunol 171: 96–105, 2003 [DOI] [PubMed] [Google Scholar]
- 57.Togel F, Hu Z, Weiss K, Isaac J, Lange C, Westenfelder C. Administered mesenchymal stem cells protect against ischemic acute renal failure through differentiation-independent mechanisms. Am J Physiol Renal Physiol 289: F31–F42, 2005 [DOI] [PubMed] [Google Scholar]
- 58.Van Bruggen MC, Walgreen B, Rijke TP, Berden JH. Attenuation of murine lupus nephritis by mycophenolate mofetil. J Am Soc Nephrol 9: 1407–1415, 1998 [DOI] [PubMed] [Google Scholar]
- 59.Viberti G, Wheeldon NM. Microalbuminuria reduction with valsartan in patients with type 2 diabetes mellitus: a blood pressure-independent effect. Circulation 106: 672–678, 2002 [DOI] [PubMed] [Google Scholar]
- 60.Whiteland JL, Nicholls SM, Shimeld C, Easty DL, Williams NA, Hill TJ. Immunohistochemical detection of T-cell subsets and other leukocytes in paraffin-embedded rat and mouse tissues with monoclonal antibodies. J Histochem Cytochem 43: 313–320, 1995 [DOI] [PubMed] [Google Scholar]
- 61.Wolf G, Ziyadeh FN, Thaiss F, Tomaszewski J, Caron RJ, Wenzel U, Zahner G, Helmchen U, Stahl RA. Angiotensin II stimulates expression of the chemokine RANTES in rat glomerular endothelial cells. Role of the angiotensin type 2 receptor. J Clin Invest 100: 1047–1058, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamauchi K, Takano Y, Kasai A, Hayakawa K, Hiramatsu N, Enomoto N, Yao J, Kitamura M. Screening and identification of substances that regulate nephrin gene expression using engineered reporter podocytes. Kidney Int 70: 892–900, 2006 [DOI] [PubMed] [Google Scholar]