

Abstract
The repair of acetaldehyde/crotonaldehyde-induced guanine(N2)-guanine(N2) interstrand cross-links (ICLs), 3-(2-deoxyribos-1-yl)-5,6,7,8-(N2-deoxyguanosyl)-6(R or S)-methyl-pyrimido[1,2-α]purine-10(3H)one (Scheme 1), was studied using a shuttle plasmid bearing a site-specific ICL. Since the authentic ICLs can revert to monoadducts, a chemically stable model ICL, 1,3-bis(2′-deoxyguanos-N2-yl)butane derivative, was also employed to probe the ICL repair mechanism. Since the removal of ICL depends on the nucleotide excision repair (NER) mechanism in Escherichia coli, the plasmid bearing the model ICL failed to yield transformants in NER-deficient host cells, proving the stability of this ICL in cells. The authentic ICLs yielded transformants in the NER-deficient hosts; therefore, these transformants are produced by plasmid bearing spontaneously reverted monoadducts. In contrast, in NER-deficient human cells, the model ICL was removed by an NER-independent repair pathway, which is unique to higher eukaryotes. This repair did not associate with a transcriptional event, but with replication. The analysis of repaired molecules revealed that the authentic and model ICLs were repaired mostly (>94 %) in an error-free manner in both hosts. The major mutations observed were G→T transversions targeting the cross-linked dG located in the lagging strand template. These results support one of the current models for the mammalian NER-independent ICL repair mechanism, in which a DNA endonuclease(s) unhooks an ICL from the leading strand template at a stalled replication fork site by incising on both sides of the ICL and then translesion synthesis is conducted across the ”half-excised” ICL attached to the lagging strand template to restore DNA synthesis.
Acetaldehyde is ubiquitous in the human environment (1). It is the major metabolite of ethanol and occurs widely in the diet. It is a common environmental pollutant and one of the most prevalent carcinogens in cigarette smoke (2). Crotonaldehyde also occurs in the human environment and in cigarette smoke, and is a product of lipid peroxidation (3–5). These chemicals are genotoxic carcinogens (1, 3). DNA adduct formation is considered to be critical in their mechanisms of carcinogenesis. They react with DNA to form Interstrand cross-links (ICLs)1 in 5′CpG sequence, as illustrated in Scheme 1, and other adducts (6). Similar ICLs have been characterized in reactions with DNA of other related compounds such as acrolein and malondialdehyde (7–10).
Scheme 1.

Formation of 1, N2-propano dGuo adducts and interstrand dGuo-dGuo cross-links from acetaldehyde and crotonaldehyde. dR, 2′-deoxyribosyl; ICL-R, interstrand cross-link with R configuration at 6 and 8 positions; ICL-S, interstrand cross-link with S configuration at 6 and 8 positions; ICL-Rd, interstrand cross-link with R configuration at methyl position. G1, cross-linked guanine 1 located in the lagging strand template; G2, cross-linked guanine 2 located in the leading strand template. They appear in Figures 1B, 3 and 5.
ICLs are one of the most serious types of damage to DNA since they connect two strands of DNA covalently and thereby inhibit DNA strand separation required for various important processes such as replication, transcription and recombination. In Escherichia coli, the combined action of nucleotide excision repair (NER) and recombination or translesion synthesis (TLS) has been proposed for the repair of ICLs (reviewed in 11), in which NER incises one strand on both sides of an ICL to unhook from one strand and then the gap generated is filled by recombination or TLS. In mammalian cells, the repair mechanism appears to be more complicated. The functional NER is not an essential prerequisite for their repair since NER-defective cells except XPF and ERCC1 mutants are not particularly sensitive to ICL-inducing agents (12). Therefore, it is considered that there is an XPF-ERCC1-dependent but other NER protein-independent repair mechanism(s) in mammalian cells. Cellular and in vitro studies (e.g., 13–15) have presented several pieces of evidence supporting the idea that this repair is carried out at a replication fork: when the progression of replication is inhibited by an ICL, a structure-specific endonuclease(s) such as XPF-ERCC1 incises one strand on both sides of the ICL (13). This incision generates a gap and a double strand break at the fork (15, see Figure 5). Subsequently, these repair intermediates are repaired by TLS or homologous recombination. Thus, mammalian cells are thought to have both NER-dependent and independent ICL unhooking mechanisms: the former operates on non-replicating DNA and the latter at the replication fork. Finally, the “half-excised” ICL is removed by NER when it is available.
Figure 5.

Model for the repair of the crotonaldehyde/acetaldehyde-induced G-G interstrand cross-link in the absence of a homologous sequence. Refer to text for the explanation of pathways.
A plasmid approach with a site-specific single ICL has been employed widely to study ICL repair in E. coli, Saccaromyces cerevisiae and mammalian cells. However, in spite of the importance of their repair during replication in mammalian cells, no suitable plasmid system has been reported to date. As far as we know, all the site-specific plasmid systems thus far reported employ a plasmid that does not replicate in mammalian cells (e.g., 16, 17). Hence, the repair is confined to the replication-independent pathway. Here, we have developed a new shuttle vector system to examine how human cells repair crotonaldehyde/acetaldehyde-induced ICLs during replication. We incorporated into this plasmid the authentic and model dG-dG ICLs to study their repair and mutagenesis.
MATERIALS AND METHODS
Synthesis of interstrand cross-linked 15-mer duplex
The syntheses, purification and characterization of 15 mer-15 mer duplexes containing a crotonaldehyde/acetaldehyde-induced stereoisomeric guanine (N2)-guanine (N2) ICL, 3-(2-deoxyribos-1-yl)-5,6,7,8-(N2-deoxyguanosyl)-6(R or S)-methyl-pyrimido[1,2-α]purine-10(3H)one, or their stable 1,3-bis(2′-deoxyguanos-N2-yl)butane derivatives have been reported (18) and the same oligonucleotides were used in this study. The sequence of the duplex oligonucleotide is shown in Figure 1B. Two duplexes contained the authentic R or S stereoisomeric ICL, and a third contained the chemically stable derivative of the R isomer (Scheme 1). These ICLs were named ICL-R, ICL-S and ICL-Rd, respectively.
Figure 1.
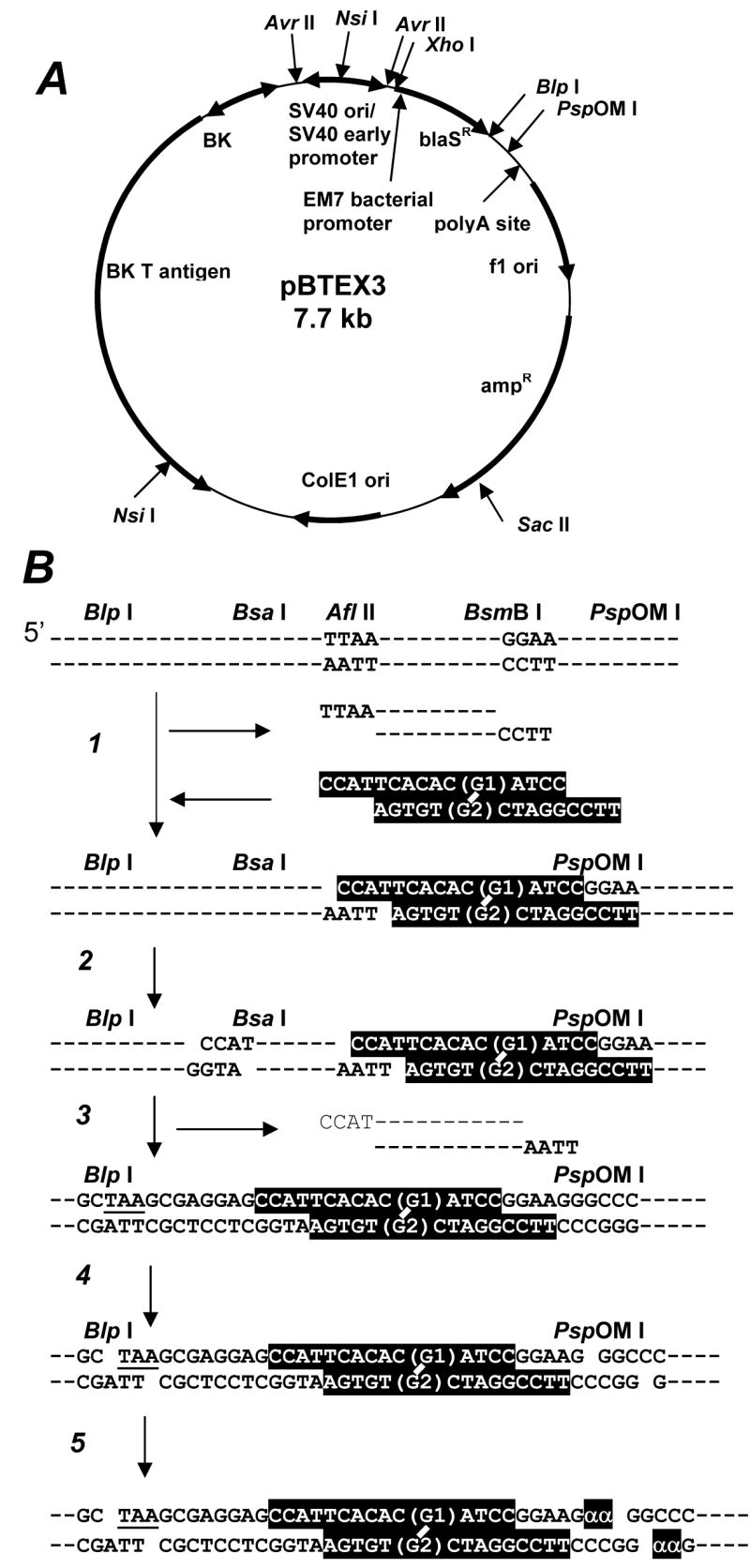
Structure of a shuttle vector (A), strategy for site-specific incorporation of a single ICL and characterization of a construct (B). A: The vector is maintained stably in human cells and confers blaR on human and E. coli cells. B: (1) Digestion with Afl II and BsmB I and ligation of ICL-oligonucleotide (highlighted) to the BsmB I-cleaved end. The duplex oligomer does not ligate to the Afl II-cleaved end. (2) Digestion with the second enzyme, Bsa I. (3) Ligation of the inserted oligonucleotide to the Bsa I-cleaved end to form closed circular DNA, which is purified by centrifugation in a cesium chloride-ethidium bromide solution (Figure 2A). (4) To confirm ligation at both sites, purified DNA is digested with Blp I and PspOM I. (5) DNA is labeled with [α-32P] dGMP (shown as α) by Klenow enzyme. The ICL-containing fragment is a labeled 65 mer (Figure 2B). The ICL is located 18/19 nucleotides downstream of the stop codon (TAA) of the blaR gene and hence within this transcriptional unit. The upper and bottom strands serve as templates for the lagging and leading strand syntheses, respectively.
Construction of plasmid
The parental plasmid, pBTE, has been described (19). This plasmid replicates and is stably maintained extrachromosomally in human cells. It confers blasticidin S resistance (blaR) on human and E. coli cells. The Blp I-PspOM I fragment (67 mer/68 mer duplex) of this plasmid was replaced with another Blp I-PspOM I fragment (94 mer/95 mer duplex) containing unique Bsa I, Afl II and BsmB I sites (Figure 1). This plasmid was named pBTEX3, and the Bsa I and BsmB I sites were used to incorporate the modified oligonucleotides. Three derivatives of pBTEX3 were also constructed: the first derivative, named pTRC, is defective in the transcription of the blaR gene in human cells. It was constructed by removing the Avr II-Avr II fragment [390 base pairs (bps)] containing the SV40 early promoter (Figure 1A). The second derivative, named pREP, is defective in the replication in human cells. It was constructed by removing the Nsi I-Nsi I fragment (2835 bp) containing the SV40 and BK virus replication origins and the BK T antigen gene (Figure 1A). The third derivative was constructed to serve as an internal control to quantify the relative ICL repair efficiency. This plasmid was constructed by replacing the Xho I-Sac II fragment (2927 bp) containing the blaR- and ampicillin-resistant (ampR) genes with a kanamycin-resistant (kmR) gene cassette (960 bp) prepared by PCR amplification of the cassette in pCR4Blunt-TOPO (Invitrogen, Carlsbad, CA). This plasmid was named pINTC.
Insertion of a modified duplex oligonucleotide into plasmid
The strategy for incorporating a cross-linked oligonucleotide is shown in Figure 1B. pBTEX3, pTRC and pREP were digested with Afl II and BsmB I, and a large fragment was purified by Qiaquick PCR purification Kit (Qiagen, Valencia, CA). 5′-Phosphorylated duplex oligonucleotide was ligated to the digested vector at the BsmB I-cleaved site at 4 °C overnight (Step 1) and then digested with Bsa I (Step 2). Following the purification of a large fragment, DNA was self-ligated to form closed circular DNA (Step 3). The ligation mixture was redigested with Afl II to remove the residual parental plasmid. The modified construct was purified by ultracentrifugation in a CsCl-ethidium bromide solution (Figure 2A). The amount of DNA was quantified by a UV spectrophotometer. Purified constructs were named pICL-R, pICL-S and pICL-Rd according to the ICL inserted. The ICL was located 18 and 19 nucleotides downstream of the stop codon (TAA in Figure 1B) of the blaR gene: the ICL site was 5′ to the polyadenylation site and therefore within the mammalian transcription unit. All experiments were conducted in the absence of the sequence homologous to the cross-linked region.
Figure 2.
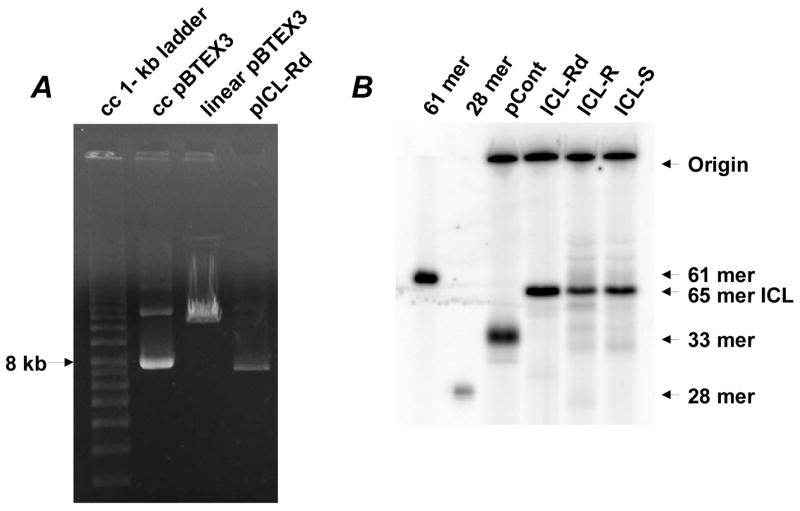
Confirmation of the closed circular (cc) nature of purified DNA construct (A) and characterization of constructs (B). Refer to Figure 1B legend for a procedure. Samples were run in denaturing 20 % polyacrylamide gel. Note that ICL-containing 65 mer (65 mer ICL) migrated faster than a single strand 61 mer.
Characterization of purified constructs
DNA constructs (100 ng) were digested with Blp I and PspOM I and then incubated with exonuclease-deficient Klenow enzyme in the presence of [α-32P] dGTP (3000 Ci/mmol, Amersham Biosciences, Piscataway, NJ) at room temperature for 20 min (Figure 1B, Steps 4 and 5). These treatments labeled DNA fragments with two [α-32P] dGMP (shown as αα) inserted into the PspOM I-cleaved ends, generating a labeled 33 mer/32 mer duplex connected by the ICL. A labeled 33 mer was generated from control DNA, pCont. Samples were run in denaturing 20 % polyacrylamide gel.
Introduction of modified plasmid into host cells and recovery of plasmid
NER-proficient (GM637) and defective (XPA, GM04429) cells were obtained from Coriell Institute (Camden, NJ) and cultured in Dulbecco’s modified Eagle’s medium supplemented with 10 % fetal bovine serum, penicillin (100 units/mL) and streptomycin (100 μg/mL) under 5 % CO2 at 37 °C. This XPA cell line shows less than 2 % of normal UV-induced unscheduled DNA synthesis (Coriell Institute). Cells were plated at 1×106 cells in a 25-cm2 flask and maintained overnight, after which they were transfected overnight with 500 ng of a DNA construct by the FuGENE6 (Roche, Nutley, NJ) method according to the manufacturer’s instruction and then detached by treating with trypsin-EDTA. A portion (0.5 – 2 %) of cells was plated in a 10-cm dish, and blasticidin S was added to the medium at 5 μg/mL on the next day. The culture was maintained until resistant cells formed visible colonies, at which time cells were stained with a Giemsa solution. Since ICL was incorporated immediately downstream of the blaR gene, plasmid repaired without the induction of a large deletion in the ICL region establishes the antibiotic resistance. The remaining cells were plated into a 150-cm2 flask and cultured in the absence of blasticidin S, allowing the collection of all types of repair events including large deletions. When cells became nearly confluent, progeny plasmid was recovered by the method of Hirt (20) and analyzed for repair events (see below).
E. coli MM1992 (as AB1157, but uvrA6, endA7::cam, mutS201::Tn5) and MM1993 (as AB1157, but endA7::cam, mutS201::Tn5) were employed. Electroporation was used to transform bacteria with 50 ng of a construct by an E. coli Pulser (Bio-Rad, Hercules, CA). Following the incubation at 37 °C for 40 min, various volumes of a transformation mixture were plated on YT (1×) agar plates (21) containing ampicillin (100 μg/mL medium) to determine the number of transformants in the mixture. Transformants were individually picked up and analyzed for repair events.
Analysis of plasmid for repair events
In the experiments with human cells, 5 ng of pVgRXR (Invitrogen), which coded for Zeocin resistance and served as an internal control for Dpn I digestion, was added to the recovered plasmid. The mixture was treated with Dpn I (1 unit) to remove nonreplicated input DNA and then used to transform E. coli DH10BMax electrocompetent cells [F−, mcrA, Δ(mrr-hsdRMS-mcrBC), φ80lacZΔM15, ΔlacX74, deoR, recA1, endA1, araΔ139, Δ(ara, leu)7697, galU, galK, λ−, rpsL, nupG, tonA)] (Invitrogen) by an E. coli Pulser. The transformation mixture was plated on YT (1×) agar plates with ampicillin (100 μg/mL medium) or with Zeocin (25 μg/mL medium). Very large deletions spanning into the ampR gene will not be detected in this analysis. E. coli transformants were picked up individually and subjected to oligonucleotide hybridization (22) using probes shown in Figure 3A to determine DNA sequence at the ICL site. Probe CG detected error-free repair events while probe CT detected G1→T transversions. Examples of hybridization are shown in Figure 3B and C. When a colony did not hybridize to either of them, DNA sequencing was conducted. Thus, this strategy detected all types of repair events including base substitutions, frameshifts, deletions and insertions without bias.
Figure 3.
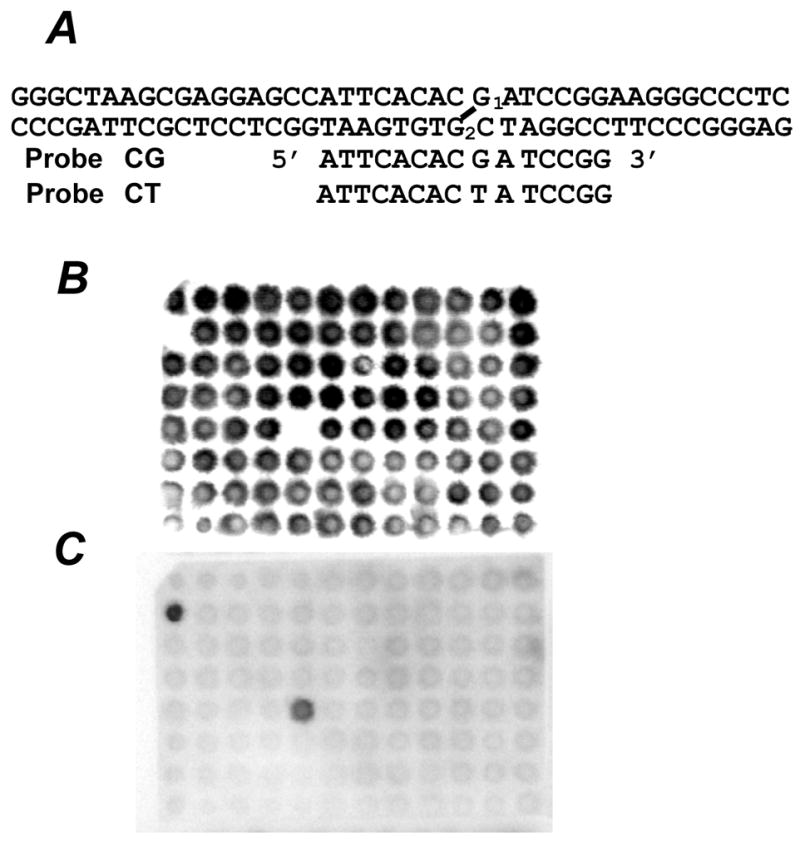
Sequences of two oligonucleotide hybridization probes, CG and CT (A) and filter hybridized with probe CG that detects accurate repair (B) or with probe CT that detects G1→T transversion (C). Two colonies contained G1→T transversions (C) and all the rest showed correct repair (B).
RESULTS
Construction of modified plasmid
Since crotonaldehyde/acetaldehyde-induced ICLs are reversible to monoadducts (Scheme 1), the results obtained with the authentic ICLs (ICL-R and ICL-S) may represent those for both ICLs and monoadducts. Therefore, we included an irreversible model ICL (ICL-Rd) of the R isomer and compared with the results for authentic ICLs. The gel analysis of labeled products revealed two bands (Figure 2B): one corresponded to the cross-linked 65 mer, which migrated faster than single-strand 61 mer, and the other was the large fragment of the vector, which did not migrate in the gel. The control construct yielded a 33 mer as expected. When the authentic ICLs revert to their monoadducts, the modified 33 mer will be observed. The analysis did not reveal a clear 33-mer band in any lanes of modified DNAs, indicating that the authentic ICLs were quite stable during the construction (Figure 2B). However, this does not necessarily mean that they are stable in a cellular environment. The ICL-modified 15 mer/15 mer duplex also migrated faster than a single strand 30 mer (data not shown),
Role for NER in ICL repair in E. coli
In the initial experiment, we examined the effects of NER on the repair of these ICLs. When the modified constructs were introduced into the uvrA strain (MM1992), pICL-R and pICL-S yielded reduced but substantial numbers of ampR transformants (Table 1). In contrast, pICL-Rd failed to yield transformants. When the NER-proficient strain (MM1993) was used, all constructs yielded transformants. These results imply that portions of the two authentic ICLs reverted to their monoadducts in bacteria or during storage while the model ICL-Rd did not. This result confirms the essential role for NER in ICL repair in E. coli (23) and the stability of ICL-Rd.
Table 1.
E. coli transformation efficiency of ICL-bearing constructs
| Construct | No. of ampR transformants per ng construct | |
|---|---|---|
| MM1993 (uvr+) | MM1992 (uvrA6) | |
| Pcont | 426 (100)a | 1012 (100)a |
| pICL-S | 227 (53) | 203 ( 20) |
| pICL-R | 161 (38) | 197 ( 19) |
| pICL-Rd | 184 (43) | 3 ( 0.3) |
% to the value for pCont.
Role for NER in ICL repair in human cells
In experiments with human cells, a portion of transfected cells were plated in a 10-cm plate and cultured in the presence of blasticidin S (5 μg/mL medium) until cells formed visible resistant colonies (7–10 days). Since the ICL was located immediately downstream of the blaR gene (Figure 1A), only repair events that are not associated with large deletions give rise to resistant colonies. pICL-R and pICL-S established numerous resistant cells in both NER-proficient and defective cells as in E. coli (picture not shown). In contrast to the result in E. coli, a good number of resistant cells were obtained even in NER-defective XPA cells when pICL-Rd was used (Figure 4B). A repeated experiment showed that the numbers of resistant colonies were 422 for pCont and 193 (46 %) for pICL-Rd when 0.5 % of cells were plated. These results indicate that the human NER-independent ICL repair mechanism operates without losing a large segment of DNA. In this experiment, the repair of ICL was estimated by determining the number of blaR colonies. However, this estimation is not accurate and the repair efficiency is overestimated since host cells likely absorb more than one molecule of the construct during transfection and the repair of one of them renders the cell resistant to the antibiotic, thus leading to the overestimation. It is technically difficult to control the number of molecules absorbed to each cell during transfection. To overcome this problem, an internal control vector (pINTC) was designed, which replicates in human cells and codes for only kmR. In this strategy, the repair efficiency is determined by the ratio of the number of ampR colonies and kmR E. coli colonies. This approach is useful when comparing results obtained in different cell lines since a transfection efficiency can be very different from one cell line to another. A mixture (500 ng:500 ng) of pINTC and pICL-Rd was transfected into GM637 and XPA cells overnight in a 25-cm2 flask, cells were replated in a 150-cm2 flask and cultured in the absence of blasticidin S for 4 days before collecting progeny plasmid. Following Dpn I digestion, DH10BMax was transformed with progeny plasmid and various volumes of a transformation mixture were plated on amp (for pICL-Rd) and km (for pINTC) plates. On the next day, transformants were counted and the ratio (No. of ampR/No. of kmR) was determined. They were 0.29 (204 ampR/714 kmR) for GM637 and 0.06 (46 ampR/800 kmR) for XPA. Thus, the value for GM637 is almost 5 times higher than that for XPA, implying the repair efficiency in XPA cells was 20 % of that in the GM cells. In another experiment, the value was 16 %. Thus, although NER-independent repair mechanism significantly contributes to the repair, NER still plays a major role.
Figure 4.
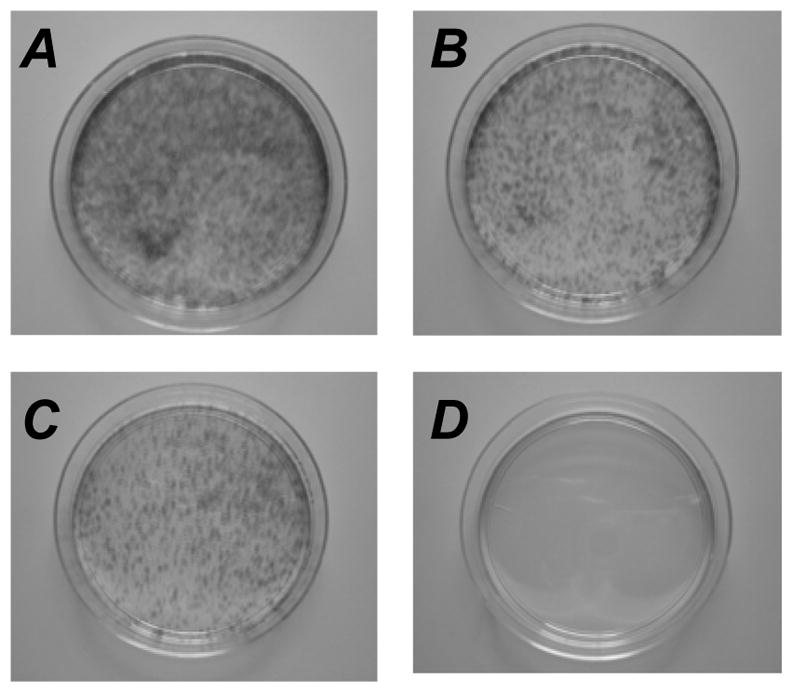
Establishment of blaR XPA cells by various DNA constructs. (A): pCont, replication-competent plasmid with unmodified insert; (B): pICL-Rd, replication-competent plasmid with ICL-Rd; (C): pREP-Cont, replication-incompetent plasmid with unmodified insert; (D) pREP-Rd, replication-incompetent plasmid with ICL-Rd. Two (A and B) and ten (C and D) percent of cells were plated following overnight transfection treatment and selected with 5 μg/mL blasticidin S.
Fidelity of ICL repair in E. coli
To determine a DNA sequence at the cross-linked region, we employed oligonucleotide hybridization, which could detect even one-base mismatches (Figure 3) and hence allowed us to detect any sequence changes including base substitutions and large/small deletions and insertions. To reduce the number of oligonucleotide probes, we first employed a probe which detected correct repair (probe CG). A preliminary analysis revealed that the majority of plasmid hybridized to the CG probe and the sequencing of hybridization-negative plasmid identified G1→T transversions as major targeted mutations. Therefore, to determine the fidelity of the repair, we employed the hybridization with these two probes and DNA sequencing.
The analysis of plasmid revealed that the vast majority (>97%) of plasmid had the correct sequence of 5′ CG at the site of ICL for all ICLs (Table 2). There were no significant differences in the fidelity of repair or the specificity of miscoding events among the three ICLs. The frequencies of targeted point mutations were <3% and they were almost exclusively G1→T transversions. No mutations were observed at the G2 site.
Table 2.
Nucleotide sequence at ICL sites in progeny plasmid
| Host and DNA | No. of NMa | No. of targeted mutations at
|
Targeted point MFb (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| G1 | G2 | ||||||||||
| G | T | A | C | Δd | T | A | C | Δ | Othersc | ||
| E. coli MM1993 | |||||||||||
| pCont | 192 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | < 0.5 | 0 |
| pICL-R exp I | 184 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2.1 | 0 |
| exp II | 182 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2.7 | 1 |
| pICL-S exp I | 190 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1.0 | 0 |
| exp II | 188 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2.1 | 0 |
| pICL-Rd | 184 | 3 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 2.1 | 0 |
| E. coli MM1992 (uvrA) | |||||||||||
| pICL-R | 190 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | < 0.6 | 1 |
| pICL-S | 188 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1.6 | 0 |
| Human XPA cells | |||||||||||
| pCont | 288 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | < 0.3 | 0 |
| pICL-R | 281 | 4 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1.7 | 1 |
| pICL-S | 274 | 7 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 2.8 | 1 |
| pICL-Rd | 284 | 2 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 1.4 | 0 |
| pTRC-Rd | 250 | 5 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 2.3 | 1 |
| Human GM637 cells | |||||||||||
| pCont | 288 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | < 0.5 | 0 |
| pICL-R | 280 | 4 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 2.1 | 1 |
| pICL-S | 271 | 9 | 4 | 2 | 0 | 0 | 1 | 0 | 0 | 5.6 | 1 |
| pICL-Rd | 275 | 5 | 0 | 1 | 0 | 0 | 2 | 0 | 0 | 2.8 | 4 |
No. of nonmutagenic event, “G” represents “G1/G2→G”.
Miscoding frequency
Include untargeted mutations and small deletions/insertions
Single nucleotide deletion
Fidelity of ICL repair in human cells
Similar to the results in E. coli, the majority (>94%) of plasmid was repaired correctly. Targeted mutation frequencies were <3 % except pICL-S in GM637, where it was a little higher (5.6 %). The major targeted mutations were G1→T transversions, and limited numbers of other types of targeted mutations such as G1→A and G1→C were also observed. The three ICLs showed similar miscoding specificity in human cells. Interestingly, targeted mutations were limited to G1 in XPA cells while a limited number of mutations (G2→A and G2→C) were also observed at G2 in GM637 cells. In addition to these targeted point mutations, limited numbers of deletions and insertions, especially the former, were also detected. These deletions were ranging from 8 to 125 nucleotides, and most of them had two - four nucleotides microhomology at the junction of deletions. There were also a few plasmid in each group, whose sequencings failed. Based on their sizes, we assume that these plasmids had large deletions, which were not determined here.
Effects of transcription and plasmid replication on ICL repair
As revealed in the bacterial experiments, certain fractions of the authentic ICLs revert to their monoadducts, and this reversion confounds the study of ICL processing in cells. The model ICL, ICL-Rd, appears to be stable in cells and its genotoxic properties also appear to be similar to those of the authentic ICLs. Therefore, further studies were conducted using pICL-Rd. In our construct, the ICL-Rd was incorporated between the blaR gene and its polyadenylation site (Figure 1). Hence, it is possible that transcription rather than replication is important to the repair in NER-deficient XPA cells. To test this possibility, the transcription-defective derivative, pTRC, was constructed (see Materials and Methods). Unmodified pTRC (1 μg) was introduced into XPA cells and cells were selected for blaR (5 μg/mL). When 10 % of transfected cells were plated, no resistant colonies were established unlike the parental pBTEX3 that yielded an uncountable number of resistant colonies. This proved the lack of expression of the gene in pTRC. The ICL-Rd was then inserted into this plasmid and the modified plasmid, pTRC-Rd, was introduced into XPA cells. Transfected XPA cells did not give rise to blaR colonies as expected. However, Dpn I-resistant (replicated) progeny plasmid was recovered efficiently from the transfected cells. This suggests that the transcription event is not critical to the NER-independent repair mechanism. The ampR colomies were analyzed for the fidelity of repair. As reported in Table 2, the repair fidelity for pTRC-Rd was the same as that observed with the transcription-competent pICL-Rd. Thus, the inactivation of transcription did not influence the ICL repair. In conclusion, the transcription does not play a significant role in the NER-independent ICL repair.
In the next experiment, the replication-defective derivative, pREP, was used to examine the role for replication. Control pREP or modified pREP-Rd (1 μg) was transfected overnight into XPA cells. All transfected cells were plated with different portions, ranging from 5 to 30% per dish, in 10-cm dishes and selected for blaR. Replication-defective control plasmid, pREP, established resistant cells (Figure 4C), but the modified construct did not yield even a single colony (Figure 4D). The total numbers of resistant colonies were more than 4000 for pREP and <20 for pREP-Rd, based on the number of resistant colonies per plate seeded with 5 % of cells. This result is in contrast to that with the replication-competent modified construct, pICL-Rd, (Figure 4B) and suggests the importance of the replication ability for the repair events. The replication-incompetent modified plasmid may be lost before or during the incorporation into chromosomes. We cannot exclude another possibility that this is due to the sensitivity difference in the detection of repair between the two types of plasmids.
DISCUSSION
ICL repair mechanism
Here we have studied how the acetaldehyde/crotonaldehyde-induced ICLs are repaired in the absence of a homologous sequence and induce mutations in cells, using a shuttle plasmid model system. To distinguish the events caused by monoadducts and ICLs, we have included a chemically stable model ICL-Rd. In E. coli, the repair of ICL-Rd absolutely required the NER function, indicating the critical role for the NER function and at the same time the stability of ICL-Rd in E. coli. Therefore, E. coli transformants obtained with the authentic ICLs in MM1992 are likely due to the reverted monoadducts. The NER function is also important in human cells as revealed by the quantitative assay, by which the repair efficiency in XPA cells was shown to be ~20 % of that in the NER-proficient cells. This is in contrast to the results obtained by another group (16, 17): the repair of a psoralen-induced dT-dT ICL (16) and a mitomycin C-induced dG-dG ICL (17), which were inserted site-specifically into plasmid, required functional NER as in E. coli. This discrepancy is explained by the difference in the ability of the plasmids to replicate in host cells: our plasmid replicates in human cells while theirs does not. These different results also support the idea that the NER-independent repair couples with replication.
Our results are consistent with one of the current models for the initial stages of ICL repair (Figure 5), in which an NER-independent repair mechanism is active on replicating DNA at the replication fork (pathway II) while NER is active on nonreplicating DNA (pathway I). When a replication fork approaches an ICL, a DNA endonuclease such as XPF-ERCC1 incises one strand 3′ and 5′ to the ICL (13). The strand incised is probably the G2-containing leading strand template since targeted mutations were limited to the G1 site in XPA cells. This implies that TLS was conducted across G1 bearing G2. This scenario also fits the enzymatic character of XPF/ERCC1. XPF/ERCC1 mutants are very sensitive to cross-linking agents while the other NER complementation group mutants are not very sensitive (12). This heterodimer incises a strand having a 3′ unpaired tail of Y-structure DNA (13, 24). This incision specificity is consistent with our results: the replication fork progresses as shown in Figure 5 in our plasmid and the G2-containing leading strand template has a 3′ unpairing tail. The incision of this strand generates a gap opposite the modified G1, across which TLS is conducted and hence targeted mutations are limited to G1 (Figure 5). In addition, the independence of this repair mechanism from transcription implies that the Y structure of DNA is not sufficient for this repair to be initiated. In the NER-dependent pathway I, the incision appears to be made on either strand since, though a limited number, targeted mutations were also observed at the G2 site in GM637 (Table 2). However, this idea requires further research since, in the NER-proficient E. coli MM1993, all targeted mutations were limited to the G1 site. Since no homologous sequence was initially available in this model system, the repair of ICL depends on TLS as previously reported (16, 17, 25).
The ICL-Rd repair observed in XPA cells provides direct cellular evidence for NER-independent ICL repair in human cells. This mechanism can not be base excision repair initiated by a DNA glycosylase. If so, the enzyme would generate an apurinic site at G1 or G2 and this lesion has to be bypassed by TLS with a preferential insertion of dCMP opposite the apurinic site to restore the original G:C pair. However, it is generally agreed that other nucleotides, especially dATP, are preferably inserted opposite the lesion in mammalian cells (26–28). Thus, the base excision repair mechanism can not explain the repair with such a high fidelity. Furthermore, we have obtained the evidence for the involvement of ERCC1 in this repair (unpublished data).
Translesion events across authentic and model ICLs
The results of the E. coli transformation experiments indicate that the authentic ICLs revert to their monoadduct forms (Scheme 1, Table 2). Therefore, the results in MM1992 (uvrA) should represent the fidelity of TLS across the G1 monoadducts. This TLS is quite accurate with the insertion of the correct dC opposite the monoadducts, and targeted G1→T transversions were observed at a frequency of <2 %. In MM1993, the results may reflect TLS across both the G1 monoadducts and “half-excised” ICL. The results in this strain were similar to those in MM1992, suggesting that the TLS across the “half-excised” ICL adducts is also quite accurate as observed for pICL-Rd, and dA was occasionally inserted opposite these lesions.
In human cells, the results obtained in XPA cells represent the TLS across monoadducts and the replication-coupled repair intermediate for the authentic ICLs. TLS across the monoadducts has been characterized in human cells (22) and simian kidney cells (29). The study with human cells (22) reported that the S monoadduct was more miscoding than was the R monoadduct (10% vs 5%). Interestingly, pICL-S is somewhat more miscoding than is pICL-R in the two human cell lines. This may reflect the contribution of those stereoisomeric monoadducts to the miscoding events here. The TLS across the repair intermediate (pICL-Rd under XPA in Table 2) is also mostly accurate with occasional misinsertion of dA opposite the intermediate. Therefore, G1→T transversions observed with pICL-R and S in XPA cells may be induced by both types of lesions. In GM637 cells, in addition to the TLS across these lesions, TLS across NER intermediates may also occur, totaling three different TLS. As shown in Table 2, the results in GM637 are not very different from those in XPA cells. Therefore, all of these results suggest that the fidelity of TLS across these three types of lesions is similar in human cells though the TLS mechanisms may be very different from each other: correct dC is preferentially inserted opposite these lesions with occasional insertions of incorrect nucleotides, mainly dA. The DNA polymerase(s) catalyzing these TLS is not known. However, since the “half-excised” G1-G2 ICL adduct is very bulky, it is very likely that the responsible polymerase is a Y family DNA polymerase (30) and/or pol ζ. Indeed, the involvements of pol η (17) and pol ζ (31, 32) have been recently reported.
Acknowledgments
We thank Dr. Orlando Schärer for critical reading of a manuscript.
Footnotes
This study was supported by U.S. Public Health Service Grants ES11297, CA76163 and CA47995.
Abbreviations: ampR, ampicillin resistant; blaR, blasticidin S resistant; bp, base pair; ICL, interstrand cross-link; kmR, kanamycin resistant; NER, nucleotide excision repair; TLS, translesion synthesis/syntheses.
References
- 1.U.S. Department of Health and Human Services. Report on Carcinogens. 11. Research Triangle Park, NC: 2004. pp. III-1–III-3. [Google Scholar]
- 2.International Agency for Research on Cancer. IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans. Tobacco Smoke and Involuntary Smoking; Lyon, France. 2004. pp. 53–94. [Google Scholar]
- 3.International Agency for Research on Cancer. IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans. Dry Cleaning, Some Chlorinated Solvents and Other Industrial Chemicals; Lyon, France. 1995. pp. 272–391. [Google Scholar]
- 4.Chung FL, Chen HJC, Nath RG. Lipid peroxidation as a potential source for the formation of exocyclic DNA adducts. Carcinogenesis. 1996;17:2105–2111. doi: 10.1093/carcin/17.10.2105. [DOI] [PubMed] [Google Scholar]
- 5.Chung FL, Nath RG, Nagao M, Nishikawa A, Zhou GD, Randerath K. Endogenous formation and significance of 1, N2-propanodeoxyguanosine adducts. Mutat Res. 1999;424:71–81. doi: 10.1016/s0027-5107(99)00009-3. [DOI] [PubMed] [Google Scholar]
- 6.Wang M, McIntee EJ, Cheng G, Shi Y, Villalta PW, Hecht SS. Identification of DNA adducts and acetaldehyde. Chemical Res Toxicol. 2000;13:1149–1157. doi: 10.1021/tx000118t. [DOI] [PubMed] [Google Scholar]
- 7.Kozekov ID, Nechev LV, Moseley MS, Harris CM, Rizzo CJ, Stone MP, Harris TM. DNA interchain cross-links formed by acrolein and crotonaldehyde. J Am Chem Soc. 2003;125:50–61. doi: 10.1021/ja020778f. [DOI] [PubMed] [Google Scholar]
- 8.Kozekov ID, Nechev LV, Sanchez A, Harris CM, Lloyd RS, Harris TM. Interchain cross-linking of DNA mediated by the principal adduct of acrolein. Chem Res Toxicol. 2001;14:1482–1485. doi: 10.1021/tx010127h. [DOI] [PubMed] [Google Scholar]
- 9.Cho Y-J, Wang H, Kozekov ID, Kurtz AJ, Jacob J, Voehler M, Smith J, Harris TM, Lloyd RS, Rizzo CJ, Stone MP. Stereospecific formation of interstrand carbinolamine DNA cross-links by crotonaldehyde- and acetaldehyde-derived α-CH3-γ-OH-1, N2-propano-2′-deoxyguanosine adducts in the 5′-CpG-3′ sequence. Chem Res Toxicol. 2006;19:195–208. doi: 10.1021/tx050239z. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dooley PA, Tsarouhtsis D, Korbel GA, Nechev KL, Shearer J, Zegar IS, Harris CM, Stone MP, Harris TM. Structural studies of an oligonucleotide containing a trimethylene interstrand cross-link in a 5′-(CpG) motif: model of a malondialdehyde cross-link. J Am Chem Soc. 2001;123:1730–1739. doi: 10.1021/ja003163w. [DOI] [PubMed] [Google Scholar]
- 11.Dronkert MLG, Kanaar R. Repair of DNA interstrand cross-links. Mutat Res. 2001;486:217–247. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- 12.de Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol. 2000;20:7980–7990. doi: 10.1128/mcb.20.21.7980-7990.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuraoka I, Kobertz WR, Ariza RR, Biggerstaff M, Essigmann JM, Wood RD. Repair of an interstrand DNA cross-link initiated by ERCC1-XPF repair/recombinational nuclease. J Biol Chem. 2000;275:26632–26636. doi: 10.1074/jbc.C000337200. [DOI] [PubMed] [Google Scholar]
- 14.Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NGJ, Beverloo HB, Hoeijmakers JHJ, Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol. 2004;24:5776–5787. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bessho T. Induction of DNA replication-mediated double strand breaks by psoralen DNA interstrand cross-links. J Biol Chem. 2003;278:5250–5254. doi: 10.1074/jbc.M212323200. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Peterson CA, Zheng H, Nairn RS, Legerski RJ, Li L. Involvement of nucleotide excision repair in a recombination-independent and error-prone pathway of DNA interstrand cross-link repair. Mol Cell Biol. 2001;21:713–720. doi: 10.1128/MCB.21.3.713-720.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng H, Wang X, Warren AJ, Legerski RJ, Nairn RS, Hamilton JW, Li L. Nucleotide excision repair- and polymerase η-mediated error-prone removal of mitomycin C interstrand cross-links. Mol Cell Biol. 2003;23:754–761. doi: 10.1128/MCB.23.2.754-761.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lao Y, Hecht SS. Synthesis and properties of an acetaldehyde-derived oligonucleotide interstrand cross-link. Chem Res Toxicol. 2005;18:711–721. doi: 10.1021/tx0497292. [DOI] [PubMed] [Google Scholar]
- 19.Yang IY, Johnson F, Grollman AP, Moriya M. Genotoxic mechanism for the major acrolein-derived deoxyguanosine adduct in human cells. Chemical Res Toxicol. 2002;15:160–164. doi: 10.1021/tx010123c. [DOI] [PubMed] [Google Scholar]
- 20.Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 21.Yang I-Y, Hossain M, Miller H, Khullar S, Johnson F, Grollman AP, Moriya M. Responses to the major acrolein-derived deoxyguanosine adduct in Escherichia coli. J Biol Chem. 2001;276:9071–9076. doi: 10.1074/jbc.M008918200. 2001. [DOI] [PubMed] [Google Scholar]
- 22.Scott S, Lao Y, Yang I-Y, Hecht SS, Moriya M. Genotoxicity of acetaldehyde- and crotonaldehyde-induced 1, N2-propanodeoxyguanosine DNA adducts in human cells. Mutat Res. 2005 doi: 10.1016/j.mrgentox.2006.01.009. in press. [DOI] [PubMed] [Google Scholar]
- 23.Cole RS. Repair of DNA containing interstrand crosslinks in Escherichia coli: sequential excision and recombination. Proc Natl Acad Sci USA. 1973;70:1064–1068. doi: 10.1073/pnas.70.4.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Laat WL, Appeldoorn E, Jaspers NGJ, Hoeijmakers JHJ. DNA structural elements required for ERCC1-XPF endonuclease activity. J Biol Chem. 1998;273:7835–7842. doi: 10.1074/jbc.273.14.7835. [DOI] [PubMed] [Google Scholar]
- 25.Berardini M, Mackay W, Loechler EL. Evidence for a recombination-independent pathway for the repair of DNA interstrand corss-links based on a site-specific study with nitrogen mustard. Biochemistry. 1997;36:3506–3513. doi: 10.1021/bi962778w. [DOI] [PubMed] [Google Scholar]
- 26.Gentil A, Cabral-Neto JB, Mariage-Samson R, Margot A, Imbach JL, Rayner B, Sarasin A. Mutagenicity of a unique apurinic/apyrimidinic site in mammalian cells. J Mol Biol. 1992;227:981–984. doi: 10.1016/0022-2836(92)90513-j. [DOI] [PubMed] [Google Scholar]
- 27.Takeshita M, Eisenberg W. Mechanism of mutation on DNA templates containing synthetic abasic sites: study with a double strand vector. Nucleic Acids Res. 1994;22:1897–1902. doi: 10.1093/nar/22.10.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cabral Neto JB, Gentil A, Caseira Cabral RE, Sarasin A. Mutation spectrum of eat-induced abasic sites on a single-stranded shuttle vector replicated in mammalian cells. J Biol Chem. 1992;267:19718–19723. [PubMed] [Google Scholar]
- 29.Fernandes PH, Kanuri M, Nechev LV, Harris TM, Lloyd RS. Mammalian cell mutagenesis of the DNA adducts of vinyl chloride and crotonaldehyde. Environ Mol Mutagen. 2005;45:455–459. doi: 10.1002/em.20117. [DOI] [PubMed] [Google Scholar]
- 30.Ohmori H, Friedberg EC, Fuchs RPP, Goodman MF, Hanaoka F, Hinkle D, Kunkel TA, Lawrence CW, Livneh Z, Nohmi T, Prakash L, Prakash S, Todo T, Walker GC, Wang Z, Woodgate R. The Y-family of DNA polymerases. Mol Cell. 2001;8:7–8. doi: 10.1016/s1097-2765(01)00278-7. [DOI] [PubMed] [Google Scholar]
- 31.Sarkar S, Davies AA, Ulrich HD, McHugh PJ. DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase ζ. EMBO J. 2006;25:1285–1294. doi: 10.1038/sj.emboj.7600993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen X, Jun S, O’Neal LE, Sonoda E, Bemark M, Sale JE, Li L. REV3 and REV1 play major roles in recombination-independent repair of DNA interstrand cross-links mediated by monoubiquitinated PCNA. J Biol Chem. 2006;281:13869–13872. doi: 10.1074/jbc.C600071200. [DOI] [PubMed] [Google Scholar]