

Summary
The catalytic activity of the Src family of tyrosine kinases is suppressed by phosphorylation on a tyrosine residue located near the C-terminus (Tyr 527 in c-Src), which is catalyzed by C-terminal Src Kinase (Csk). Given the promiscuity of most tyrosine kinases, it is remarkable that the C-terminal tails of the Src family kinases are the only known targets of Csk. We have determined the crystal structure of a complex between the kinase domains of Csk and c-Src at 2.9 Å resolution, revealing that interactions between these kinases position the C-terminal tail of c-Src at the edge of the active site of Csk. Csk cannot phosphorylate substrates that lack this docking mechanism because the conventional substrate binding site, used by most tyrosine kinases to recognize substrates, is destabilized in Csk by a deletion in the activation loop.
Introduction
Protein tyrosine kinases are key components of the signaling networks that regulate growth and proliferation in metazoans (Manning et al., 2002). Although kinase activity is tightly regulated by various mechanisms, these mechanisms converge on a conserved element in the active site of the kinase domain called the activation loop. The conformation of the activation loop is responsive to phosphorylation (Huse and Kuriyan, 2002), which stabilizes the active configuration of catalytic residues in most kinases, and also promotes the binding of peptide substrates (Nolen et al., 2004).
The catalytic domains of most tyrosine kinases are highly promiscuous (Miller, 2003), and specificity is achieved by controlling the localization and activity of the kinase. This is exemplified by the observation that deregulation and overexpression of tyrosine kinases results in the phosphorylation of a large and heterogeneous set of proteins (Superti-Furga et al., 1993; Takashima et al., 2003; Walkenhorst et al., 1996).
C-terminal Src Kinase (Csk) is unusual among tyrosine kinases because it readily phosphorylates only one class of substrate, a conserved tyrosine in the C-terminal tails of the Src family kinases (Tyr 527 in chicken c-Src (Takeya and Hanafusa, 1983)). This degree of specificity is retained even when Csk is highly overexpressed (Superti-Furga et al., 1993). Phosphorylation by Csk stabilizes an inactive form of the Src kinases, in which the phosphorylated tail binds to the Src homology 2 (SH2) domain (Schindler et al., 1999; Sicheri et al., 1997; Xu et al., 1999; Xu et al., 1997). Csk (like its close relative Chk) is not regulated by activation loop phosphorylation, and is instead active constitutively (Brown and Cooper, 1996), a property that is consistent with the need for Csk to inhibit the Src kinases continually (Okada et al., 1991).
Like the Src kinases, Csk possesses a Src-homology 3 (SH3) domain and an SH2 domain. The SH2 domain binds to the phosphorylated substrates of Src kinases, thereby targeting Csk to sites of Src kinase activity (Bergman et al., 1995; Howell and Cooper, 1994; Neet and Hunter, 1995; Sabe et al., 1994). These targeting mechanisms are not critical for the specificity of Csk for the Src kinases, which is apparent in vitro. Indeed, Csk displays extremely poor activity towards tyrosine-containing peptides, with the best peptides selected from a peptide library displaying KM values in the millimolar range (Sondhi et al., 1998). By comparison, the tyrosine kinases c-Src and c-Abl phosphorylate their optimal peptide substrates with KM values in the range of 5-50 μM (Songyang et al., 1995). Unlike peptide substrates, constructs of c-Src that include the folded kinase domain are efficient substrates for Csk, with KM values of ∼5 μM (Sondhi et al., 1998).
Here we address two major questions regarding the specificity of Csk for the Src kinases. Firstly, while mutagenesis experiments with Csk and c-Src have identified some of the residues involved in the interaction between the two proteins (Lee et al., 2006; Lee et al., 2003), no definitive description of the nature of the complex is as yet available. Secondly, while the catalytic mechanism, dynamics, and the roles of the SH3 and SH2 domains of Csk have been investigated extensively (Lieser et al., 2006; Lieser et al., 2005; Wong et al., 2004; Wong et al., 2005), these studies have not clarified why the Src kinases are the only significant substrates of Csk. To address the first question we have determined the crystal structure of a complex between the kinase domains of Csk and c-Src. This structure shows that Csk recognizes the C-terminal lobe of the c-Src kinase domain in a manner that positions the c-Src tail adjacent to the active site of Csk. To address the second question we present a comparative analysis of the active sites of tyrosine kinases that shows that the canonical binding site for substrates is destabilized in Csk. As a consequence, Csk is efficient at phosphorylating only the Src kinases, because they present the phosphoacceptor tyrosine directly to the active site.
Results and Discussion
Binding of Csk to c-Src and crystallization of the complex
The kinase domain of Csk is sufficient to recognize c-Src protein (Sondhi and Cole, 1999; Sun and Budde, 1999), but the dissociation constant (KD) for the Csk:c-Src interaction has not been measured directly. We used surface plasmon resonance to measure the binding of Csk to c-Src. Constructs of c-Src were immobilized on the sensor surface and the binding of different constructs of Csk to the surface was monitored as a function of Csk concentration (see Figure 1A for constructs and Figure 1B for typical binding data). The affinity of Csk for c-Src is modest and highly salt-dependent (Figure 1C, Figure S1). Constructs of Csk that include the SH2 domain (full-length Csk, CskFL, and a construct lacking the SH3 domain, CskSH2KD) bind the kinase domain of c-Src (c-SrcKD) with KD values of ∼30 μM at 150 mM NaCl, and ∼4 μM at 50 mM NaCl (Figure 1C, Figure S1B). The KD value for the kinase domain of Csk (CskKD) binding to c-SrcKD is ∼70 μM at 150 mM NaCl (Figure 1C). When the salt-dependence of the affinity is considered these values are in agreement with reports that the Michaelis constant (KM) for phosphorylation of c-Src by Csk is ∼5 μM (Lieser et al., 2005; Wang et al., 2001).
Figure 1. Binding of Csk to c-Src measured by surface plasmon resonance.

(A) Constructs of Csk and c-Src used in this paper.
(B) Representative data from the surface plasmon resonance experiments. The sensorgrams obtained with different concentrations of CskKD are shown on the left. The equilibrium response at each concentration was fitted to a single-site binding model, shown on the right, to derive the equilibrium dissociation constant.
(C) Equilibrium dissociation constants for several different constructs of Csk and c-Src at 150 mM NaCl. Values shown are the mean and standard deviation from at least two separate titrations.
Because of the relatively low affinity of Csk for c-Src we were unable to purify complexes of the two proteins. We were therefore surprised to find that CskKD and CskSH2KD both crystallized readily in complex with c-SrcKD when mixed together. It appears that the adventitious formation of a disulfide crosslink between Cys 290 in Csk and Cys 277 in c-Src prevented the otherwise facile crystallization of either protein separately. The disulfide bond links a Csk molecule from one enzyme-substrate complex to a c-Src molecule from a different complex (Figure S2A). The disulfide linkage does not appear to influence the interaction between Csk and c-Src within the biologically relevant Csk:c-Src complexes, as the interface between Csk and c-Src is essentially identical in two different crystal forms (see below), despite the fact that the disulfide-linked molecules are rotated relative to each other in the two structures (Figure S2B).
Crystals of either CskKD or CskSH2KD in complex with c-SrcKD diffracted x-rays weakly. We mutated charged residues at the surface of Csk in an effort to improve crystal quality (Derewenda, 2004), and two such mutations (Lys 361 Ala and Lys 362 Ala) improved diffraction from the CskKD:c-SrcKD crystals. While these mutations are known to decrease the activity of Csk (Lin et al., 2005), they are distant from the Csk:c-Src interface (the Cα atom of Lys 361 of Csk is more than 20 Å from the nearest Cα atom in c-Src) and are therefore unlikely to influence the manner in which c-Src binds to Csk. The CskKD:c-SrcKD crystals were originally obtained in the presence of the ATP analog AMP-PNP, but the use of the non-specific kinase inhibitor staurosporine further improved diffraction, allowing data to 2.9 Å to be measured. Staurosporine is bound to both c-Src and Csk in our structure. Structures of Csk and the Src family kinases Lck and Fyn have been determined previously in the presence of staurosporine (Kinoshita et al., 2006; Lamers et al., 1999; Zhu et al., 1999).
We have also measured x-ray data to 4.1 Å for the CskSH2KD:c-SrcKD crystals obtained in the presence of AMP-PNP. The SH2 domain of Csk is disordered in these crystals, but the kinase domains of both Csk and c-Src are positioned reliably. Although the lower resolution of this dataset provides limited information, it confirms the mode of interaction between the kinase domains of c-Src and Csk, and, in one of the molecules in the asymmetric unit, allows visualization of the c-Src tail entering the Csk active site. The discussion that follows is based on the higher resolution CskKD:c-SrcKD structure, unless otherwise noted.
The Csk:c-Src interface
The interaction between c-Src and Csk is restricted to the C-terminal lobes (C-lobes) of the two kinase domains, and buries ∼1200 Å2 of surface area between the proteins (Figure 2A). The interaction surface on c-Src is formed by the C-terminal α-helices I and I′ (residues 508-524; see Figure 2 for the helix notation) and the loop that precedes them (residues 504-507, part of the αH/αI loop; Figure 2B). This surface docks onto the side of the C-lobe of Csk, undergoing little adjustment in structure when compared to structures of the isolated kinase domain of c-Src (Breitenlechner et al., 2005; Seeliger et al., 2007). The interaction surface on c-Src is highly conserved amongst the Src family members (Figure S3). The catalytically important Asp-Phe-Gly (DFG) motif of c-Src adopts an inactive conformation in this structure, but the surface of c-Src that binds to Csk is distant from the active site, and the inactive conformation of the c-Src kinase domain is unlikely to be induced by the binding of Csk.
Figure 2. Structure of the complex between the kinase domains of Csk and c-Src.
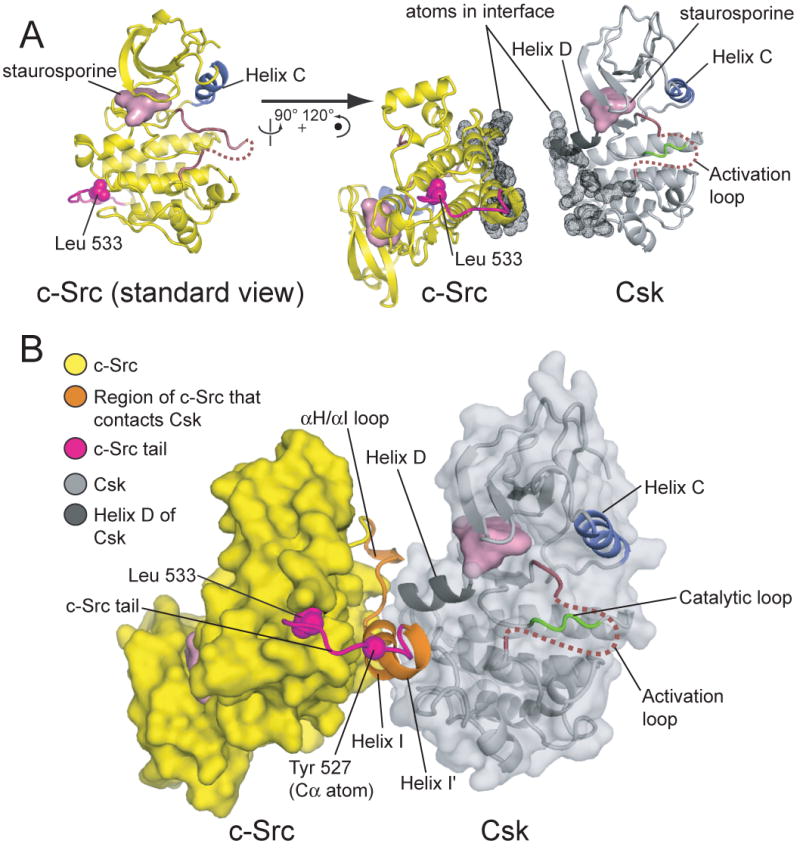
(A) The kinase domains of c-Src and Csk are depicted in their relative orientations in the structure, but separated so that the Csk:c-Src interface, highlighted as dots, is visible. The Csk:c-Src complex is oriented such that the kinase domain of Csk is shown in the standard view (active site facing the viewer). The orientation of the kinase domain of c-Src in the complex is related to the standard view (on the left) by a rotation of ∼90° about the vertical axis and ∼120° about an axis perpendicular to the page.
(B) Overview of the CskKD:c-SrcKD complex. The approximate path of the activation loop, which is disordered between residues 335 and 349, is indicated by a dotted red line. The region of the c-Src that interacts with Csk (residues 504-523), and the c-Src tail (residues 524-533), are highlighted in orange and magenta, respectively. The sidechain of the C-terminal residue of c-Src (Leu 533) is shown as spheres, as is the Cα atom of Tyr 527.
The Csk:c-Src interface is largely electrostatic, consistent with the salt-sensitive nature of the interaction: five arginine residues in Csk participate in ion-pairing networks with glutamate and aspartate residues in c-Src. c-Src residues 504-510 (from the αH/αI loop and helix I) envelop the C-terminal end of helix D of Csk (residues 274-281; Figure 3A,B). A helix capping interaction occurs between the C-terminal end of helix D of Csk and the ε-amino group of Lys 442 of c-Src (Figure 3A). Helix I, comprising residues 508 through 519, interacts with the C-lobe of Csk so as to terminate beneath the Csk active site (Figure 3B,C).
Figure 3. The Csk:c-Src interface probed by mutagenesis.
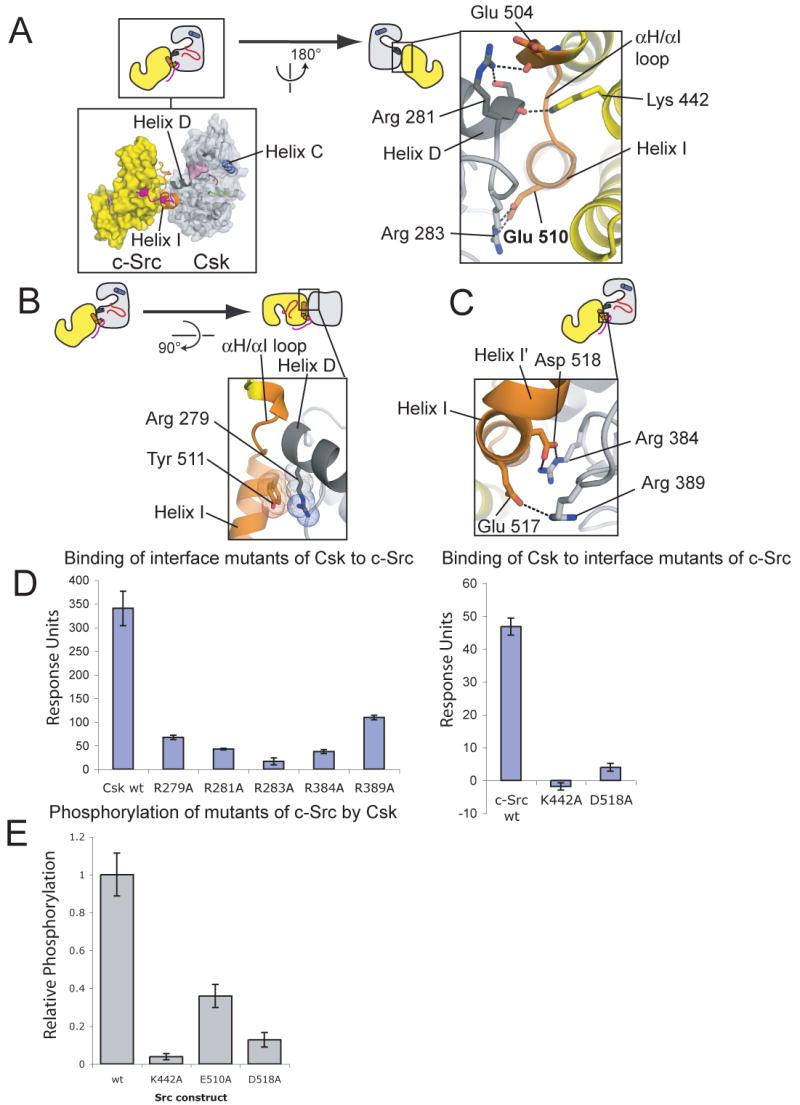
(A-C) Details of the Csk:c-Src interface are shown. The coloring scheme is the same as in Figure 2. Residues important for the binding of Csk to c-Src (see below) are indicated.
(D) The effects of mutations on the binding of Csk to c-Src as measured by surface plasmon resonance. On the left, the binding of mutants of CskFL (25 μM) to c-SrcKD is shown. On the right the binding of wt CskFL (10 μM) to mutants of c-SrcKD, immobilized on different surfaces, is shown. Values are the means and standard deviations from three series of injections.
(E) The effects of mutations in the Csk:c-Src interface on the ability of Csk to phosphorylate c-Src, measured by phosphorimaging, are shown.
The N-terminal lobe of Csk is poorly ordered (Figure S4). Constructs of Csk that lack the SH2 domain have substantially reduced activity (Sondhi and Cole, 1999) and the kinase domain of Csk is therefore likely to be in an inactive conformation. The portion of Csk that interacts with c-Src is well ordered, however, and exhibits relatively low temperature factors (the average Cα temperature factor is 94 Å2 for residues of Csk within 8 Å of c-Src, compared to the average temperature factor of 129 Å2 for all Cα atoms in Csk). This region adopts a very similar conformation to that seen in the structure of full-length Csk in an active conformation (Ogawa et al., 2002), suggesting that full-length, active Csk forms a similar complex with c-Src.
This expectation is confirmed by previous mutagenesis experiments intended to investigate the determinants of the Csk:c-Src interaction (Lee et al., 2006; Lee et al., 2003). These studies predicted the importance of six residues (Arg 279, Arg 281, Arg 283 of Csk and Glu 510, Tyr 511, Asp 518 of c-Src) for the interaction between full-length Csk and c-Src, and all of these residues indeed make specific contacts in the Csk:c-Src interface. To further confirm the crystallographic interface we used surface plasmon resonance to measure the effect of mutations in the Csk:c-Src interface on the interaction between Csk and c-Src. Mutation of any of the five arginine residues of Csk that contact c-Src interferes with binding (Figure 3D). Likewise, mutation of Lys 442 or Asp 518 of c-Src to alanine also prevents binding (Figure 3D), and interferes with the phosphorylation of c-Src by Csk (Figure 3E).
The c-Src tail is disengaged from the Csk active site, but is positioned for ready entry
Kinase domains often bind substrates or inhibitory segments in a docking groove that lies directly beneath the active site cleft (Knighton et al., 1991). This docking groove is utilized by Csk to bind to c-Src (Figure 4A). In our structure, as well as in previously determined structures of c-Src, a kink occurs in helix I between Tyr 519 and Phe 520, resulting in a separate helical turn (residues 520-524), which we refer to as helix I′ (Figure 4B). The kink positions helix I′ so that it fits snugly into the docking groove of Csk.
Figure 4. The conformation of the c-Src tail.
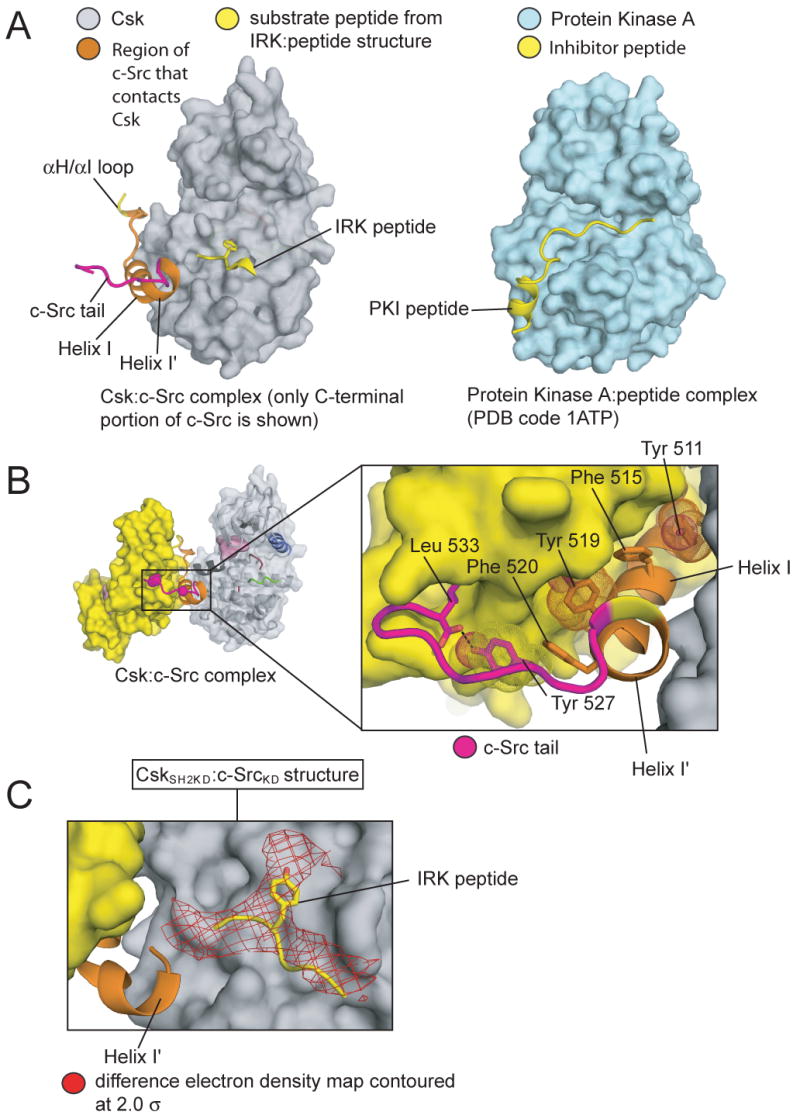
(A) On the left the kinase domain of Csk from the CskKD:c-SrcKD structure is depicted with only the C-terminal region of c-Src (the αH/αI loop, helices I and I′ and the c-Src tail) shown. The canonical binding site for peptide substrates is illustrated by the substrate peptide from the structure of the insulin receptor kinase domain (IRK), shown in yellow (Hubbard, 1997). On the right, the structure of protein kinase A, in complex with an inhibitory peptide that binds in the docking groove, is depicted for comparison (Knighton et al., 1991).
(B) An enlarged view of the C-terminal portion of c-Src in the CskKD:c-SrcKD structure. Perpendicular aromatic:aromatic interactions that stabilize this conformation of the c-Src tail are indicated with sticks and dots.
(C) In one of the complexes in the CskSH2KD:c-SrcKD structure the tail of c-Src binds in the active site of Csk. The substrate peptide from the IRK ternary complex, aligned on the kinase domain of Csk, is shown in yellow. A difference electron density map, contoured at 2 standard deviations over the mean (2σ), is shown in red. The map is calculated with coefficients (|Fo|-|Fc|)eiα(c), where |Fo| are the observed structure factor amplitudes and |Fc| and α(c) are amplitudes and phases calculated from a model lacking the c-Src tail. See Figure S5 for a more comprehensive view of the electron density map that demonstrates the significance of the features shown here.
The c-Src tail (Glu 524 - Leu 533) that follows Helix I′ does not engage the active site of Csk, but extends away from Csk to bind to the C-lobe of c-Src (Figure 4B). The sidechain of Tyr 527 packs against Phe 520, extending the perpendicular aromatic:aromatic interactions that stabilize the kink between helix I and helix I′, and the phenol hydroxyl of Tyr 527 forms a hydrogen bond to the C-terminal carboxylate group of c-Src (Figure 4B). This binding mode of the c-Src tail is also seen in structures of the isolated kinase domain of c-Src (Breitenlechner et al., 2005; Seeliger et al., 2007) and in a structure of full-length active c-Src (Cowan-Jacob et al., 2005).
That the c-Src tail can readily engage the active site of Csk is shown by aligning the crystal structure of the insulin receptor kinase (IRK), which contains a substrate peptide bound at the active site cleft (Hubbard, 1997), onto Csk in our structure. The end of helix I′ of c-Src then approaches to within 12 Å of the tyrosine residue of the IRK peptide (∼11.5 Å between the Cα atoms of Ser 522 and the tyrosine residue)(Figure 4A). The four residues that link the end of Helix I′ to Tyr 527 are sufficient to span this distance.
This fact is further confirmed by the lower resolution CskSH2KD:c-SrcKD structure, which contains two Csk:c-Src complexes in the asymmetric unit. The conformations of the c-Src and Csk kinase domains in one of the complexes is similar to the structure described above, but in the other complex the c-Src tail has disengaged from the c-Src C-lobe and instead reaches into the active site of Csk. In a difference electron density map calculated using a model lacking the c-Src tail the strongest peak (at a contour-level of 5σ above the mean value of the electron density) is located at the edge of the active site of Csk, just above helix I′ (Figure 4C, Figure S5). When contoured at a lower level (2.0σ) the electron density is consistent with the typical manner in which peptide substrates bind to tyrosine kinases. Aligning IRK onto Csk in this structure positions the IRK substrate peptide within the electron density for the c-Src tail (Figure 4C).
The fact that the c-Src tail does not engage Csk in two of the three crystallographically independent complexes reported here could merely reflect the inactive nature of Csk in our crystals. Alternatively, the Csk:c-Src interface seen in the structure may be the dominant feature that determines the specific phosphorylation of c-Src by Csk, with the binding of the c-Src tail to the active site of Csk being less important. This view is consistent with several lines of evidence. The tails of the Src kinases are poorly conserved, even though Csk readily phosphorylates all Src family members (Okada et al., 1991). Most substitutions in the tail of c-Src have little impact on the kinetics of phosphorylation by Csk (Wang et al., 2001), and a peptide comprising the sequence of the c-Src tail is an extremely poor substrate for Csk, with a value for kcat/Km that is four orders of magnitude below that for c-Src protein (Sondhi et al., 1998).
The anchoring of the activation loop of Csk is disrupted by a deletion
The question remains as to why Csk does not readily phosphorylate other substrates, particularly peptide substrates. This led us to ask whether the binding site for peptide substrates, located at the base of the active site cleft, is somehow compromised in Csk. We analyzed tyrosine kinase crystal structures in which the activation loops are well ordered and in the active conformation (Experimental Procedures). This diverse set contains six receptor tyrosine kinases and six non-receptor tyrosine kinases. Since tyrosine kinases bind substrates in a distinct manner from serine/threonine kinases (Huse et al., 1999), the conclusions of the analysis that follows may not apply to Ser/Thr kinases.
In the active conformation there are two distinct regions of the activation loop that are anchored onto the C-lobe of the kinase domain; we refer to these as anchor points 1 and 2 (Figure 5). The anchor points consist of two short antiparallel β-sheets, formed by interactions between two sections of the activation loop and two loops in the C-lobe of the kinase (the catalytic loop and the αEF/αF loop for anchor points 1 and 2, respectively). Anchor point 1 (corresponding to residues 1155-1157 in IRK) begins three residues C-terminal to the catalytically important DFG motif and stabilizes the motif in the active conformation (Figure 5A)(Nolen et al., 2004). In addition to three backbone hydrogen bonds that form the β-sheet, anchor point 1 is further characterized by the insertion of the sidechain of the last residue of the anchor (Ile 1157 in IRK) into a pocket on the surface of the C-lobe. We refer to this residue as the hydrophobic latch, because almost all tyrosine kinases have a large hydrophobic residue at this position. Anchor point 2 forms near the C-terminal end of the activation loop (corresponding to residues 1163-1165 of IRK) and includes a tyrosine phosphorylation site, when present (Figure 5A). The backbone amide and carbonyl groups of the tyrosine residue form two of the three hydrogen bonds to the kinase C-lobe.
Figure 5. Activation loop anchoring in tyrosine kinases.
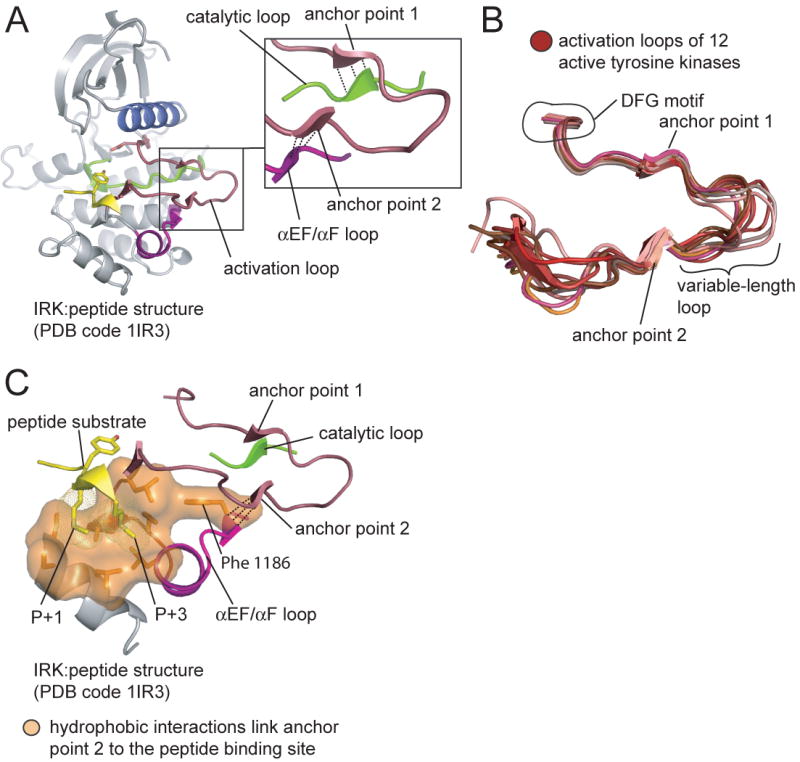
(A) The structure of the insulin receptor kinase domain (IRK) bound to a substrate peptide is shown. The panel on the right shows an enlarged view depicting the two anchor points and the loops to which they form hydrogen bonds (shown as dashed lines).
(B) The activation loops from twelve structures of active tyrosine kinases (see Experimental Procedures). The structures were aligned on the catalytic loops, but only the activation loops are shown.
(C) Hydrophobic interactions couple anchor point 2 to the peptide binding site. The structure of IRK is shown. Residues of the substrate peptide that interact with the hydrophobic residues are indicated.
The two anchor points are a structurally conserved feature of the active conformation (Figure 5B). In all twelve structures the distance between the Cα atom of the hydrophobic latch (the last residue in anchor point 1) and the Cα atom of the activation loop tyrosine (the first residue in anchor point 2) is between 6.2 and 7.2 Å. In contrast, the segment of the activation loop that connects anchor points 1 and 2 (referred to as the variable-length loop in this paper), as well as the portion of the activation loop C-terminal to anchor point 2, are relatively divergent in structure (Figure 5B).
The proximity of anchor point 2 to the C-terminal end of the activation loop, where peptide substrates bind, suggests that anchor point 2 might influence peptide binding. In particular, a conserved set of hydrophobic interactions might couple the engagement of anchor point 2 to peptide binding (Figure 5C and Figure S6). These hydrophobic residues form a crevice on the surface of the C-lobe that comprises the binding site for the sidechains located one (P+1) and three (P+3) positions C-terminal to the substrate tyrosine (Favelyukis et al., 2001; Hubbard, 1997; Levinson et al., 2006; Mol et al., 2003). Mutations at this binding site alter substrate specificity (Konkol et al., 2000; Songyang et al., 1995; Till et al., 1999; Yokoyama and Miller, 1999). Two of the hydrogen bonds formed by anchor point 2 directly stabilize a residue that is part of this network of hydrophobic interactions (Phe 1186 in IRK; Figure 5C, Figure S6). The backbone of this residue is further linked to the peptide binding site through the αEF/αF loop (Figure 5C, Figure S6). We therefore envisage that the engagement of anchor point 2 would stabilize these interactions and promote peptide binding.
A sequence alignment of the activation loops of several tyrosine kinases demonstrates that the anchoring mechanism is disrupted in Csk (Figure 6A). The variable-length loop that connects anchor points 1 and 2 is completely missing in Csk and Chk. In order for the activation loop of Csk to form both anchor points simultaneously, the ∼6.5 Å distance between the terminal Cα atoms of the anchor points would have to be bridged by a single peptide unit, which is clearly not possible without distortion of one or both anchor points (the typical distance between Cα atoms of adjacent amino acids in proteins is 3.8 Å (Creighton, 1992)). The engagement of anchor points 1 and 2 is therefore expected to be mutually exclusive in Csk. This is supported by the observation that the activation loop is at least partly disordered in all existing crystal structures of Csk. Since we expect anchor point 2 to stabilize the peptide binding site, the engagement of anchor point 1, which is required to stabilize the DFG motif in the active conformation, could interfere with peptide binding.
Figure 6. Activation loop anchoring and the activity of Csk and c-Src towards peptide substrates.
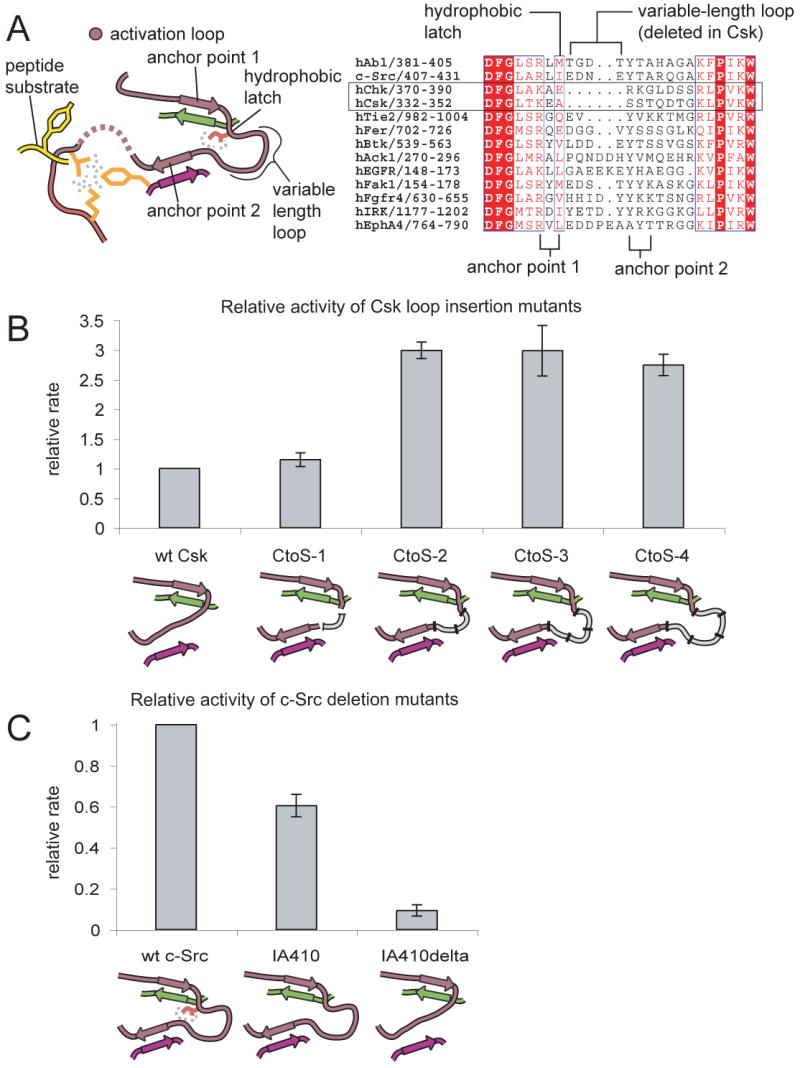
A) On the left a schematic diagram is shown that highlights the characteristic features of the anchoring mechanism. An alignment of the activation loop sequences of several tyrosine kinases is shown on the right.
(B) The kinase activity of constructs of Csk containing insertions in the activation loop is shown relative to wt Csk. The constructs are referred to as CtoS-1 (for Csk to Src), CtoS-2, CtoS-3, and CtoS-4 depending on whether 1, 2, 3, or 4 residues respectively were inserted. The mean value and standard deviation of the relative rates from three experiments are shown.
(C) The relative kinase activity of mutants of c-Src. These constructs of the kinase domain either had the hydrophobic latch mutated to alanine alone (IA) or in combination with the deletion of the variable-length loop (IA delta). The graphs show the mean and standard deviation from two experiments.
Activation loop anchoring affects the activity of Csk and c-Src towards peptides
In an attempt to restore activation loop anchoring in Csk we made insertions in the activation loop (by introducing amino acids derived from the variable-length loop of c-Src) that would bridge the distance between anchor points 1 and 2. We also mutated the hydrophobic latch of Csk from alanine to isoleucine (the hydrophobic latch residue in c-Src). A kinetic assay demonstrates that when Csk is engineered to contain two or more residues connecting the anchor points, the resulting proteins possess a three-fold greater activity than wildtype Csk towards peptide substrates (Figure 6B). Insertion of a single amino acid, in contrast, has no effect on activity (Figure 6B). This is consistent with the architecture of the anchor points. Strikingly, all tyrosine kinases other than Csk and Chk possess a minimum of 2 amino acids between the anchor points.
In a previous study, replacement of the entire activation loop of Csk with that of c-Src did not result in an increase in catalytic activity (Lin et al., 2003). It is possible that this more extensive replacement was not accommodated by the core of the kinase domain, whereas the insertions we have made are located at the edge of the kinase domain, and are therefore tolerated.
To further test the importance of activation loop anchoring we deleted the variable-length loop in c-Src and mutated the hydrophobic latch residue to alanine. This c-Src mutant was well folded, as indicated by an 1H-15N TROSY NMR spectrum (Figure S7), but displays a ten-fold reduction in activity towards peptides (Figure 6C). At least part of this decrease is due to an elevated KM for the peptide substrate (Figure S7). These results suggest that activation loop anchoring is important for the phosphorylation of peptide substrates, and that the poor activity of Csk towards peptides is at least partly a consequence of disrupted anchoring.
Csk can discriminate between tail-phosphorylated and unphosphorylated c-Src
The Src kinases are localized to membranes constitutively through N-terminal myristoylation (Brown and Cooper, 1996). The restriction of protein molecules to membrane surfaces can lead to increases in effective concentrations of as much as three orders of magnitude (Kholodenko et al., 2000). While the affinity of Csk for c-Src in solution is relatively weak (KD ∼30μM), recruitment of Csk to the membrane, which occurs when the Src kinases are activated, could potentially result in saturation of the c-Src binding sites on Csk. We therefore wondered whether Csk can distinguish between active and inactive Src kinases.
We aligned the structure of inactive assembled c-Src (Xu et al., 1997) onto the C-terminal lobe of the kinase domain of c-Src (residues 350-520) in our structure. In this alignment, residues 523-526 of c-Src, corresponding to a portion of the phosphorylated c-Src tail, clash with Csk (Figure 7A). A similar result is obtained using the structure of the inactive form of the Src kinase Hck (Sicheri et al., 1997). Hence we expect that Csk cannot bind these inactive forms of the Src kinases. Surface plasmon resonance experiments demonstrate that phosphorylation of c-Src on Tyr 527 by Csk prior to immobilization on the sensor surface prevents the binding of Csk to c-Src (Figure 7B). Clearly Csk can discriminate between active c-Src molecules and those that have already been tail-phosphorylated, preventing inactive c-Src from competing with active c-Src for the binding site on Csk. We expect that the same mechanism is at work in Chk to distinguish unphosphorylated c-Src from phosphorylated c-Src (Chong et al., 2006).
Figure 7. The inactive assembled state of c-Src is incompatible with the Csk:c-Src complex.
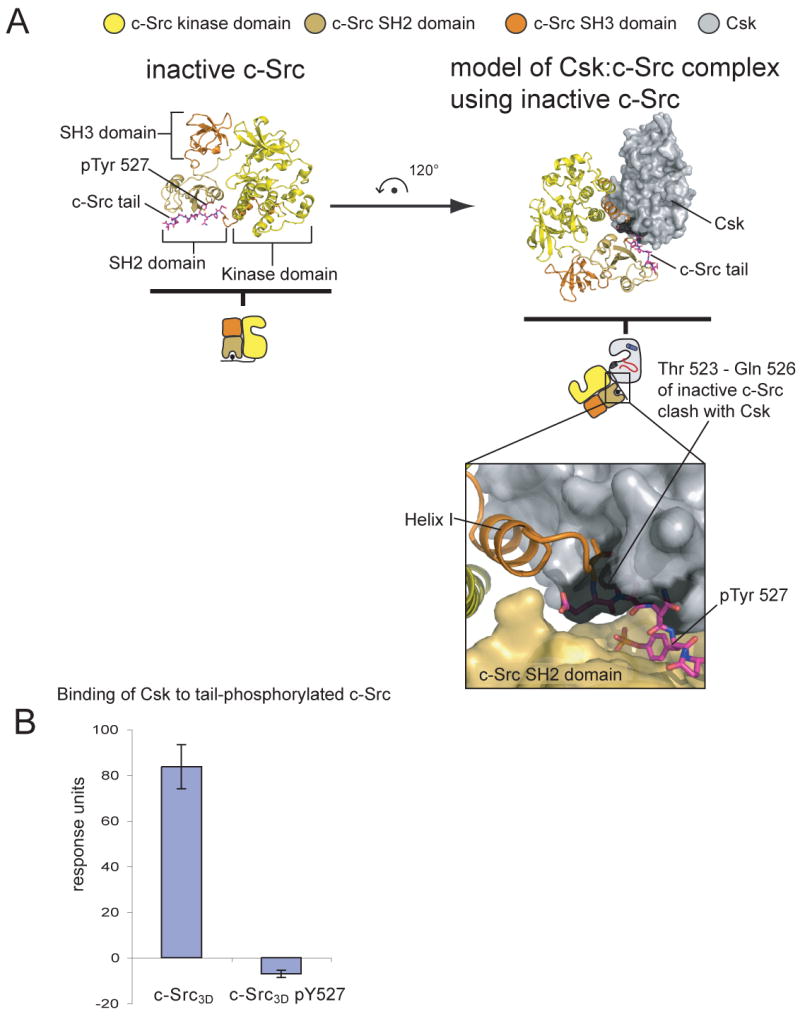
(A) The crystal structure of inactive assembled c-Src (pdb code 2SRC) is aligned on the C-terminal lobe of c-Src from our CskKD:c-SrcKD structure. Clashes between the tail of inactive c-Src and the C-lobe of Csk are highlighted in black.
(B) Csk cannot bind tail-phosphorylated c-Src as measured by surface plasmon resonance. A construct of c-Src that contains the SH2, SH3 and kinase domains (Src3D) was phosphorylated by Csk on Tyr 527 (Src3D pY527) prior to immobilization on the sensor surface. The binding of CskFL (10 μM) to the surface was then measured. The graphs show the mean and standard deviation of two experiments.
A surprising feature of the alignment of inactive c-Src onto the c-Src kinase domain in our structure is that the SH2/SH3 unit of c-Src is brought into close proximity to the C-lobe of Csk without clashing with it. The SH3 and SH2 domains of c-Src may dock onto the c-Src kinase domain to form the assembled structure even in the absence of tail phosphorylation (Faraldo-Gomez and Roux, 2007), and so the Csk:c-Src interaction may have evolved to permit binding of assembled but unphosphorylated c-Src, while discriminating against tail phosphorylated c-Src.
The mechanism by which Csk discriminates between tail-phosphorylated and unphosphorylated Src kinases would be circumvented when the Src kinases are activated by SH2 and SH3 ligands. With the SH2 domain displaced from the tail, Csk could bind to c-Src even though the tail is phosphorylated. This could potentially sequester Csk molecules and delay the down-regulation of Src kinase signaling.
Conclusion
There are two distinct factors that explain the unusual specificity of Csk for the Src kinases, one that is intrinsic to Csk itself, and another that depends on features that are unique to the Src kinases. The unusually short activation loop of Csk impedes the simultaneous engagement of two anchor points in the loop, one important for catalysis and one for peptide substrate binding. As a result, Csk does not readily phosphorylate most proteins. The Src kinases have evolved to circumvent the destabilized peptide binding site by binding to Csk through a specific protein:protein interface that orients the Src kinase tail towards the active site of Csk.
Most tyrosine kinases are tightly regulated, and the disruption of their inhibitory mechanisms is frequently associated with cancer. In contrast, Csk is constitutively active under normal conditions. Csk recognizes c-Src through a protein-protein interaction that relies primarily on electrostatic complementarity and hydrogen bonding. Such interactions have high intrinsic specificity, allowing Csk to select the Src kinases as unique targets. The inability of Csk to bind and phosphorylate substrates other than the Src kinases explains why a conventional regulatory mechanism like those of other tyrosine kinases is not critical for Csk.
Experimental Procedures
Protein purification
The purification of the kinase domain of wildtype chicken c-Src (residues 251-533, c-SrcKD) and a construct of c-Src that contains the SH2, SH3 and kinase domains (residues 83-533, c-Src3D) from bacteria is described in detail elsewhere (Seeliger et al., 2005). All constructs of Csk (CskFL, residues 1-450, CskSH2KD, residues 68-450, and CskKD, residues 187-450) were expressed as fusions with glutathione S-transferase (GST). The insertions in the activation loop of Csk (CskCtoS-1 etc) were made in the CskFL background by site-directed mutagenesis, with the insertions derived from the c-Src variable-length loop (encoding Glu-Asp-Asn-Glu). Csk constructs were expressed in E.coli BL21 overnight at 18°C. Cell pellets were resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 10% glycerol, 1 mM DTT) and lysed using a cell disrupter. Cleared lysate was loaded on glutathione sepharose resin (GE Healthcare). Protein was eluted with elution buffer (50 mM Bis-Tris, pH 6.0, 10% glycerol, 10 mM reduced glutathione) and cleaved with Precision Protease (Amersham) to release GST. After digestion, protein was loaded onto a HiTrap SP cation exchange column (GE Healthcare). The eluted sample was buffer-exchanged by gel filtration chromatography (50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 5% glycerol).
Crystallization of the Csk:c-Src complex
Protein concentrations were determined by absorbance at 280 nm, and Csk and c-Src proteins were combined in a 1:1 molar ratio prior to crystallization. In the case of the CskKD:c-SrcKD crystals, the protein mixture was concentrated in the presence of a 2-fold molar excess of staurosporine (Sigma) prior to crystallization. The nucleotide analog AMPPNP (Sigma) was added directly to crystallization samples in the case of the CskSH2KD:c-SrcKD crystals. The crystallization samples did not include reducing agents, which in retrospect was critical to obtaining the crystals of the complex (see main text).
Crystals of the CskSH2KD:c-SrcKD complex grew in 0.2 M sodium malonate pH 7.0, 20% PEG 3350 while the CskKD:c-SrcKD crystals were obtained in 1.1 M sodium tartrate pH 7.0. All crystals were cryoprotected in 25% glycerol prior to being frozen in liquid nitrogen. X-ray diffraction data were collected at the Advanced Light Source beamlines 8.2.1, 8.2.2 and 8.3.1.
Structure Determination and Refinement
Structures were solved by molecular replacement using Phaser (McCoy et al., 2005), with models of the kinase domains of Csk and c-Src (1K9A chain A residues 188-450 for Csk, 1YOM residues 260-520 for c-Src). Refinement was performed with CNS (Brunger et al., 1998) and PHENIX (Adams et al., 2002), and model building with Coot (Emsley and Cowtan, 2004). For the CskSH2KD:c-SrcKD structure, rigid body refinement was performed exclusively. The SH2 domain of Csk could not be located and is not included in the model. For the CskKD:c-SrcKD structure individual atomic refinement was performed. The model displays relatively low B-factors for the c-Src kinase domain, the Csk:c-Src interface and the portion of Csk in the vicinity of the interface (Figure S4). For the rest of the Csk model there is a strong correlation between the B-factor of an individual atom and the distance of that atom from the Csk:c-Src interface. While the electron density for the c-Src kinase domain was strong, there was clear evidence of structural heterogeneity in the conformation of the activation loop of c-Src and the N-terminal lobe of Csk. We chose to model only one conformation of these segments.
Surface Plasmon Resonance Experiments
Surface plasmon resonance experiments were performed with a BIACORE 2000 instrument. c-Src protein (c-SrcKD or c-Src3D) in 10 mM sodium acetate pH 5.0 was coupled to the chip surface in one of the flowcells using standard amine coupling chemistry, to a level of 2000 response units (RU), and a flow cell that was activated and blocked without exposure to protein was used as a reference in all experiments. A flowcell to which the kinase domains of CamKII or EGFR had been coupled was used to confirm that the binding of Csk to the surface was specific for c-Src.
Csk constructs were diluted from concentrated stocks into HPS buffer (10 mM Hepes pH 7.5, 150 mM NaCl, 3 mM EDTA, 0.005% P20 surfactant and 2 mM TCEP, a reducing agent that precludes disulfide bond formation between Csk and c-Src). Because of rapid binding kinetics, 60-second injections were sufficient to reach equilibrium. The equilibrium response values obtained from injections of Csk at different concentrations were fit to a “one site-specific binding” model using GraphPad Prism version 5.0a (www.graphpad.com) to derive the dissociation constant of the interaction.
Kinetic Assays
The phosphorylation of peptides by constructs of Csk and c-Src was measured using a coupled kinase assay (Seeliger et al., 2005), in reaction buffer (100 mM Tris-HCl pH 8.0, 10 mM MgCl2, 1 mM TCEP) with 1 mM ATP, and either 500 nM Csk or 16 nM c-Src. The peptide sequences used were the optimal peptide substrate for c-Src (AEEEIYGEFEAKKKK) at 1 mM and the optimal peptide substrate for Csk (KKKKEEIYFFF) at 250 μM. Peptides were prepared by Dr. David King (University of California, Berkeley).
The phosphorylation of c-Src mutants by Csk was assayed by measuring the incorporation of 32P phosphate into c-Src. To avoid complications due to c-Src autophosphosphorylation, an inactive mutant form of c-Src was used (Lys 295 Met). Reactions were performed in 60 mM Tris-HCl pH 8.0, 30 mM NaCl, 50 μM unlabelled ATP, 0.5 μCi of γ32P ATP, 2 mM MnCl2, 10 mM DTT, 200 μg/ml BSA with 5 nM Csk and 5 μM c-Src, for 2 minutes at 30 °C. Radioactivity was quantified using phosphorimaging with a typhoon imager (GE Healthcare). Under these conditions the phosphorylation of c-Src is linear with time (Wang et al., 2001).
NMR spectroscopy
15N-labeled c-SrcKD constructs were expressed in standard M9 media with 15N ammonium chloride as the sole nitrogen source and purified as described above. Proteins were concentrated to 250 μM and exchanged into 100 mM MES pH 5.8, 200 mM NaCl, 1 mM DTT, 250 μM Dasatinib and 10 % D2O. {1H-15N} Trosy NMR spectra (Pervushin et al., 1997) were recorded at 303 K on a Bruker Avance 800 MHz (18.8T) spectrometer, processed with NMRPipe and visualized in NMRdraw (Delaglio et al., 1995).
Structural Alignments of Active Kinases
The twelve tyrosine kinases (PDB codes in parentheses) used in the comparative analysis are: Lck (3LCK)(Yamaguchi and Hendrickson, 1996), Fyn (2DQ7)(Kinoshita et al., 2006), c-Abl (2F4J)(Young et al., 2006), IRK (1IR3)(Hubbard, 1997), Insulin-like growth factor 1 receptor (1K3A)(Favelyukis et al., 2001), epidermal growth factor receptor (2GS6)(Zhang et al., 2006), c-Kit (1PKG)(Mol et al., 2003), fibroblast growth factor receptor (2PVF)(Chen et al., 2007), Ret (2IVV)(Knowles et al., 2006), Syk (1XBA)(Atwell et al., 2004), Janus kinase 3 (1YVJ)(Boggon et al., 2005) and Fes (3BKB).
Supplementary Material
Supplemental Data: Supplemental Data include figures and tables and can be found online at http://www.cell.com/cgi/content/full/134/1/124/DC1/
Acknowledgments
We thank David King for the synthesis of the two substrate peptides, Caleb Cassidy-Amstutz for help with cloning and mutagenesis and Luke Chao and Natalia Jura for providing CamKII and EGFR kinase domain used as controls in the surface plasmon resonance experiments. We are grateful to David Wemmer and Jeff Pelton for support and discussion regarding the NMR experiments. We also thank Xuewu Zhang, Tanya Freedman, Brian Kelch and Meindert Lamers for helpful discussions and Tanya Freedman, Natalia Jura and Jonathon Winger for critical reading of the manuscript. Supported in part by grants from the NIH (PC). M.A.S. is supported by NIH grant K99 GM080097.
Footnotes
Accession Numbers: The structures described here have been deposited in the Protein Data Bank with ID codes 3D7T for CskKD:c-SrcKD and 3D7U for CskSH2KD:c-SrcKD.
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Atwell S, Adams JM, Badger J, Buchanan MD, Feil IK, Froning KJ, Gao X, Hendle J, Keegan K, Leon BC, et al. A novel mode of Gleevec binding is revealed by the structure of spleen tyrosine kinase. J Biol Chem. 2004;279:55827–55832. doi: 10.1074/jbc.M409792200. [DOI] [PubMed] [Google Scholar]
- Bergman M, Joukov V, Virtanen I, Alitalo K. Overexpressed Csk tyrosine kinase is localized in focal adhesions, causes reorganization of alpha v beta 5 integrin, and interferes with HeLa cell spreading. Mol Cell Biol. 1995;15:711–722. doi: 10.1128/mcb.15.2.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggon TJ, Li Y, Manley PW, Eck MJ. Crystal structure of the Jak3 kinase domain in complex with a staurosporine analog. Blood. 2005;106:996–1002. doi: 10.1182/blood-2005-02-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitenlechner CB, Kairies NA, Honold K, Scheiblich S, Koll H, Greiter E, Koch S, Schafer W, Huber R, Engh RA. Crystal structures of active SRC kinase domain complexes. J Mol Biol. 2005;353:222–231. doi: 10.1016/j.jmb.2005.08.023. [DOI] [PubMed] [Google Scholar]
- Brown MT, Cooper JA. Regulation, substrates and functions of src. Biochim Biophys Acta. 1996;1287:121–149. doi: 10.1016/0304-419x(96)00003-0. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Chen H, Ma J, Li W, Eliseenkova AV, Xu C, Neubert TA, Miller WT, Mohammadi M. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol Cell. 2007;27:717–730. doi: 10.1016/j.molcel.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong YP, Chan AS, Chan KC, Williamson NA, Lerner EC, Smithgall TE, Bjorge JD, Fujita DJ, Purcell AW, Scholz G, et al. C-terminal Src kinase-homologous kinase (CHK), a unique inhibitor inactivating multiple active conformations of Src family tyrosine kinases. J Biol Chem. 2006;281:32988–32999. doi: 10.1074/jbc.M602951200. [DOI] [PubMed] [Google Scholar]
- Cowan-Jacob SW, Fendrich G, Manley PW, Jahnke W, Fabbro D, Liebetanz J, Meyer T. The crystal structure of a c-Src complex in an active conformation suggests possible steps in c-Src activation. Structure. 2005;13:861–871. doi: 10.1016/j.str.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Creighton TE. Proteins: Structures and Molecular Properties. W.H. Freeman & Company; 1992. [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Derewenda ZS. The use of recombinant methods and molecular engineering in protein crystallization. Methods. 2004;34:354–363. doi: 10.1016/j.ymeth.2004.03.024. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Faraldo-Gomez JD, Roux B. On the importance of a funneled energy landscape for the assembly and regulation of multidomain Src tyrosine kinases. Proc Natl Acad Sci U S A. 2007;104:13643–13648. doi: 10.1073/pnas.0704041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favelyukis S, Till JH, Hubbard SR, Miller WT. Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat Struct Biol. 2001;8:1058–1063. doi: 10.1038/nsb721. [DOI] [PubMed] [Google Scholar]
- Howell BW, Cooper JA. Csk suppression of Src involves movement of Csk to sites of Src activity. Mol Cell Biol. 1994;14:5402–5411. doi: 10.1128/mcb.14.8.5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard SR. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. Embo J. 1997;16:5572–5581. doi: 10.1093/emboj/16.18.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse M, Chen YG, Massague J, Kuriyan J. Crystal structure of the cytoplasmic domain of the type I TGF beta receptor in complex with FKBP12. Cell. 1999;96:425–436. doi: 10.1016/s0092-8674(00)80555-3. [DOI] [PubMed] [Google Scholar]
- Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- Kholodenko BN, Hoek JB, Westerhoff HV. Why cytoplasmic signalling proteins should be recruited to cell membranes. Trends Cell Biol. 2000;10:173–178. doi: 10.1016/s0962-8924(00)01741-4. [DOI] [PubMed] [Google Scholar]
- Kinoshita T, Matsubara M, Ishiguro H, Okita K, Tada T. Structure of human Fyn kinase domain complexed with staurosporine. Biochem Biophys Res Commun. 2006;346:840–844. doi: 10.1016/j.bbrc.2006.05.212. [DOI] [PubMed] [Google Scholar]
- Knighton DR, Zheng JH, Ten Eyck LF, Xuong NH, Taylor SS, Sowadski JM. Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:414–420. doi: 10.1126/science.1862343. [DOI] [PubMed] [Google Scholar]
- Knowles PP, Murray-Rust J, Kjaer S, Scott RP, Hanrahan S, Santoro M, Ibanez CF, McDonald NQ. Structure and chemical inhibition of the RET tyrosine kinase domain. J Biol Chem. 2006;281:33577–33587. doi: 10.1074/jbc.M605604200. [DOI] [PubMed] [Google Scholar]
- Konkol L, Hirai TJ, Adams JA. Substrate specificity of the oncoprotein v-Fps: site-specific mutagenesis of the putative P+1 pocket. Biochemistry. 2000;39:255–262. doi: 10.1021/bi992096j. [DOI] [PubMed] [Google Scholar]
- Lamers MB, Antson AA, Hubbard RE, Scott RK, Williams DH. Structure of the protein tyrosine kinase domain of C-terminal Src kinase (CSK) in complex with staurosporine. J Mol Biol. 1999;285:713–725. doi: 10.1006/jmbi.1998.2369. [DOI] [PubMed] [Google Scholar]
- Lee S, Ayrapetov MK, Kemble DJ, Parang K, Sun G. Docking-based substrate recognition by the catalytic domain of a protein tyrosine kinase, C-terminal Src kinase (Csk) J Biol Chem. 2006;281:8183–8189. doi: 10.1074/jbc.M508120200. [DOI] [PubMed] [Google Scholar]
- Lee S, Lin X, Nam NH, Parang K, Sun G. Determination of the substrate-docking site of protein tyrosine kinase C-terminal Src kinase. Proc Natl Acad Sci U S A. 2003;100:14707–14712. doi: 10.1073/pnas.2534493100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson NM, Kuchment O, Shen K, Young MA, Koldobskiy M, Karplus M, Cole PA, Kuriyan J. A Src-like inactive conformation in the abl tyrosine kinase domain. PLoS Biol. 2006;4:e144. doi: 10.1371/journal.pbio.0040144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieser SA, Shaffer J, Adams JA. SRC tail phosphorylation is limited by structural changes in the regulatory tyrosine kinase Csk. J Biol Chem. 2006;281:38004–38012. doi: 10.1074/jbc.M607824200. [DOI] [PubMed] [Google Scholar]
- Lieser SA, Shindler C, Aubol BE, Lee S, Sun G, Adams JA. Phosphoryl transfer step in the C-terminal Src kinase controls Src recognition. J Biol Chem. 2005;280:7769–7776. doi: 10.1074/jbc.M411736200. [DOI] [PubMed] [Google Scholar]
- Lin X, Ayrapetov MK, Lee S, Parang K, Sun G. Probing the communication between the regulatory and catalytic domains of a protein tyrosine kinase, Csk. Biochemistry. 2005;44:1561–1567. doi: 10.1021/bi048142j. [DOI] [PubMed] [Google Scholar]
- Lin X, Lee S, Sun G. Functions of the activation loop in Csk protein-tyrosine kinase. J Biol Chem. 2003;278:24072–24077. doi: 10.1074/jbc.M210596200. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- Miller WT. Determinants of substrate recognition in nonreceptor tyrosine kinases. Acc Chem Res. 2003;36:393–400. doi: 10.1021/ar020116v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mol CD, Lim KB, Sridhar V, Zou H, Chien EY, Sang BC, Nowakowski J, Kassel DB, Cronin CN, McRee DE. Structure of a c-kit product complex reveals the basis for kinase transactivation. J Biol Chem. 2003;278:31461–31464. doi: 10.1074/jbc.C300186200. [DOI] [PubMed] [Google Scholar]
- Neet K, Hunter T. The nonreceptor protein-tyrosine kinase CSK complexes directly with the GTPase-activating protein-associated p62 protein in cells expressing v-Src or activated c-Src. Mol Cell Biol. 1995;15:4908–4920. doi: 10.1128/mcb.15.9.4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell. 2004;15:661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Ogawa A, Takayama Y, Sakai H, Chong KT, Takeuchi S, Nakagawa A, Nada S, Okada M, Tsukihara T. Structure of the carboxyl-terminal Src kinase, Csk. J Biol Chem. 2002;277:14351–14354. doi: 10.1074/jbc.C200086200. [DOI] [PubMed] [Google Scholar]
- Okada M, Nada S, Yamanashi Y, Yamamoto T, Nakagawa H. CSK: a protein-tyrosine kinase involved in regulation of src family kinases. J Biol Chem. 1991;266:24249–24252. [PubMed] [Google Scholar]
- Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci U S A. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabe H, Hata A, Okada M, Nakagawa H, Hanafusa H. Analysis of the binding of the Src homology 2 domain of Csk to tyrosine-phosphorylated proteins in the suppression and mitotic activation of c-Src. Proc Natl Acad Sci U S A. 1994;91:3984–3988. doi: 10.1073/pnas.91.9.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler T, Sicheri F, Pico A, Gazit A, Levitzki A, Kuriyan J. Crystal structure of Hck in complex with a Src family-selective tyrosine kinase inhibitor. Mol Cell. 1999;3:639–648. doi: 10.1016/s1097-2765(00)80357-3. [DOI] [PubMed] [Google Scholar]
- Seeliger MA, Nagar B, Frank F, Cao X, Henderson MN, Kuriyan J. c-Src binds to the cancer drug imatinib with an inactive Abl/c-Kit conformation and a distributed thermodynamic penalty. Structure. 2007;15:299–311. doi: 10.1016/j.str.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Seeliger MA, Young M, Henderson MN, Pellicena P, King DS, Falick AM, Kuriyan J. High yield bacterial expression of active c-Abl and c-Src tyrosine kinases. Protein Sci. 2005;14:3135–3139. doi: 10.1110/ps.051750905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicheri F, Moarefi I, Kuriyan J. Crystal structure of the Src family tyrosine kinase Hck. Nature. 1997;385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- Sondhi D, Cole PA. Domain interactions in protein tyrosine kinase Csk. Biochemistry. 1999;38:11147–11155. doi: 10.1021/bi990827+. [DOI] [PubMed] [Google Scholar]
- Sondhi D, Xu W, Songyang Z, Eck MJ, Cole PA. Peptide and protein phosphorylation by protein tyrosine kinase Csk: insights into specificity and mechanism. Biochemistry. 1998;37:165–172. doi: 10.1021/bi9722960. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Carraway KL, 3rd, Eck MJ, Harrison SC, Feldman RA, Mohammadi M, Schlessinger J, Hubbard SR, Smith DP, Eng C, et al. Catalytic specificity of protein-tyrosine kinases is critical for selective signalling. Nature. 1995;373:536–539. doi: 10.1038/373536a0. [DOI] [PubMed] [Google Scholar]
- Sun G, Budde RJ. Mutations in the N-terminal regulatory region reduce the catalytic activity of Csk, but do not affect its recognition of Src. Arch Biochem Biophys. 1999;367:167–172. doi: 10.1006/abbi.1999.1253. [DOI] [PubMed] [Google Scholar]
- Superti-Furga G, Fumagalli S, Koegl M, Courtneidge SA, Draetta G. Csk inhibition of c-Src activity requires both the SH2 and SH3 domains of Src. Embo J. 1993;12:2625–2634. doi: 10.1002/j.1460-2075.1993.tb05923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima Y, Delfino FJ, Engen JR, Superti-Furga G, Smithgall TE. Regulation of c-Fes tyrosine kinase activity by coiled-coil and SH2 domains: analysis with Saccharomyces cerevisiae. Biochemistry. 2003;42:3567–3574. doi: 10.1021/bi0272499. [DOI] [PubMed] [Google Scholar]
- Takeya T, Hanafusa H. Structure and sequence of the cellular gene homologous to the RSV src gene and the mechanism for generating the transforming virus. Cell. 1983;32:881–890. doi: 10.1016/0092-8674(83)90073-9. [DOI] [PubMed] [Google Scholar]
- Till JH, Chan PM, Miller WT. Engineering the substrate specificity of the Abl tyrosine kinase. J Biol Chem. 1999;274:4995–5003. doi: 10.1074/jbc.274.8.4995. [DOI] [PubMed] [Google Scholar]
- Walkenhorst J, Goga A, Witte ON, Superti-Furga G. Analysis of human c-Abl tyrosine kinase activity and regulation in S. pombe. Oncogene. 1996;12:1513–1520. [PubMed] [Google Scholar]
- Wang D, Huang XY, Cole PA. Molecular determinants for Csk-catalyzed tyrosine phosphorylation of the Src tail. Biochemistry. 2001;40:2004–2010. doi: 10.1021/bi002342n. [DOI] [PubMed] [Google Scholar]
- Wong L, Lieser S, Chie-Leon B, Miyashita O, Aubol B, Shaffer J, Onuchic JN, Jennings PA, Woods VL, Jr, Adams JA. Dynamic coupling between the SH2 domain and active site of the COOH terminal Src kinase, Csk. J Mol Biol. 2004;341:93–106. doi: 10.1016/j.jmb.2004.05.060. [DOI] [PubMed] [Google Scholar]
- Wong L, Lieser SA, Miyashita O, Miller M, Tasken K, Onuchic JN, Adams JA, Woods VL, Jr, Jennings PA. Coupled motions in the SH2 and kinase domains of Csk control Src phosphorylation. J Mol Biol. 2005;351:131–143. doi: 10.1016/j.jmb.2005.05.042. [DOI] [PubMed] [Google Scholar]
- Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell. 1999;3:629–638. doi: 10.1016/s1097-2765(00)80356-1. [DOI] [PubMed] [Google Scholar]
- Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Hendrickson WA. Structural basis for activation of human lymphocyte kinase Lck upon tyrosine phosphorylation. Nature. 1996;384:484–489. doi: 10.1038/384484a0. [DOI] [PubMed] [Google Scholar]
- Yokoyama N, Miller WT. Identification of residues involved in v-Src substrate recognition by site-directed mutagenesis. FEBS Lett. 1999;456:403–408. doi: 10.1016/s0014-5793(99)00992-8. [DOI] [PubMed] [Google Scholar]
- Young MA, Shah NP, Chao LH, Seeliger M, Milanov ZV, Biggs WH, 3rd, Treiber DK, Patel HK, Zarrinkar PP, Lockhart DJ, et al. Structure of the kinase domain of an imatinib-resistant Abl mutant in complex with the Aurora kinase inhibitor VX-680. Cancer Res. 2006;66:1007–1014. doi: 10.1158/0008-5472.CAN-05-2788. [DOI] [PubMed] [Google Scholar]
- Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Zhu X, Kim JL, Newcomb JR, Rose PE, Stover DR, Toledo LM, Zhao H, Morgenstern KA. Structural analysis of the lymphocyte-specific kinase Lck in complex with non-selective and Src family selective kinase inhibitors. Structure. 1999;7:651–661. doi: 10.1016/s0969-2126(99)80086-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data: Supplemental Data include figures and tables and can be found online at http://www.cell.com/cgi/content/full/134/1/124/DC1/