

Abstract
We identified inhibitory peptide analogs (IPAs), capable of immunomodulating experimental autoimmune uveitis (EAU), induced in B10.RIII mice by immunization with the retinal antigen interphotoreceptor-binding protein in CFA. Alanine-substituted peptides of the major pathogenic epitope, residues 161–180, were synthesized. They were tested for immunogenicity, cross-reactivity with the native 161–180 epitope, pathogenicity, and ability to prevent EAU when given in IFA before EAU challenge with native murine (m)161–180. Two peptides, 169A and 171A, were unable to elicit disease but cross-reacted with m161–180 by lymphocyte proliferation. Mice pretreated with either of the substituted peptides failed to develop EAU after challenge with the native epitope, m161–180, and had reduced cellular responses by lymphocyte proliferation and by delayed hypersensitivity. Their cytokine response profile to m161–180 showed reduced antigen-specific IFN-γ and IL-17, whereas IL-4, IL-5, IL-10, and IL-13 from IPA-protected mice were increased, and serum antibody titers to m161–180 revealed reduced IgG2a and elevated IgG1 isotypes, suggesting a Th2 shift in the response. Protection was transferable with lymphoid cells from protected donors to naïve recipients, who were subsequently immunized for EAU. Thus, IPA pretreatment prevents induction of EAU by skewing the response to a subsequent uveitogenic challenge with the native peptide to a nonpathogenic phenotype, as well as by eliciting transferable regulatory cells.
Keywords: experimental autoimmune uveitis, tolerance
INTRODUCTION
Experimental autoimmune uveitis (EAU) is an animal model for several posterior uveitic diseases in the human. EAU leads to destruction of the neural retina and related tissues. Lesions include serous retinal detachment, retinal folding, photoreceptor damage, vasculitis, retinitis, choroiditis and vitritis, and ultimately, blindness [1]. EAU is a T cell-mediated autoimmune disease that can be induced in susceptible rodents by immunization with retinal antigens or its fragments in CFA [1] or by the adoptive transfer of antigen-specific effector cells [2]. Uveitogenic antigens are typically derived from the retinal photoreceptor cells, function in the visual cycle, and are highly evolutionarily conserved. Examples are arrestin {retinal soluble antigen (S-Ag) [2]}, interphotoreceptor retinoid-binding protein (IRBP), rhodopsin, and its illuminated form opsin, recoverin, and phosducin [1]. Susceptibility to uveitis induced with these antigens varies among species and among strains. The most susceptible mouse strain is B10.RIII [3]. The most pathogenic epitope from the IRBP protein for this mouse strain is included in residues 161–180 [4].
Several immunotherapeutic approaches have been developed for the therapy of autoimmune uveitis [5, 6]. T cells recognize antigens via their TCR, bound to MHC on the cell surface. This interaction is highly specific, and minor changes in the structure of a peptide epitope can affect T cell recognition; however, there is some flexibility. Single amino acid substitutions can generate peptide analogs that are still recognized by the specific T cells, but they may generate a partial or altered response. The use of various modified peptides to treat autoimmunity in animal models and in the clinic has been reported. One example is Copaxone, a random copolymer of basic amino acids used clinically in therapy of multiple sclerosis (MS) [7, 8]. Another example is altered peptide ligands (APLs), based on epitopes derived from pathogenic proteins such as myelin, in which the contact sites between TCR and MHC have been altered. They have the ability to competitively inhibit pathogenic, autoreactive T cells from recognizing native peptide epitopes by a variety of mechanisms [9,10,11]. In the present study, we address the use of alanine-substituted analogs of the well-known uveitogenic peptide 161–180 as an antigen-specific therapy for EAU. The peptides described in this study could act similarly to APLs; however, as they were not characterized in terms of contact residues and binding affinity, the properties by which APLs are defined, we refer to them as inhibitory peptide analogs (IPAs) and not as APLs.
We were able to identify two alanine-substituted peptides, 169A and 171A, derived from the major pathogenic sequence of murine IRBP (m161–180) within the core epitope that could be used in immunomodulating EAU. They did not elicit disease when used in a uveitogenic immunization protocol and cross-reacted with the native pathogenic sequence by lymphocyte proliferation. Importantly, pretreatment with these peptides protected mice that were challenged for EAU with the native peptide 161–180. Mechanism studies showed that lymphocyte proliferation and delayed type hypersensitivity (DTH) reaction to m161–180 in the protected mice were reduced; shift in cytokine profile and antibody isotypes suggested a Th2 deviation, which may contribute to the protection. Cells from IPA-protected donors were able to transfer protection, suggesting the presence of regulatory cells as part of the mechanism. By phenotypic characterization and in vitro depletion, these regulatory cells appear to have a CD4+CD25+forkhead box P3 (FoxP3)+ phenotype.
MATERIALS AND METHODS
Mice
B10.RIII mice (H-2r), 6–8 weeks old, were supplied by The Jackson Laboratory (Bar Harbor, ME, USA). Animals were kept in a specific pathogen-free facility and given water and standard laboratory chow ad libitum. Animal care and use were in compliance with institutional guidelines and with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research.
Antigens and reagents
Heat-killed Mycobacterium tuberculosis (strain H37RA) was from Difco (Detroit, MI, USA). Bordetella pertussis toxin (PT), CFA, and IFA were purchased from Sigma-Aldrich (St. Louis, MO, USA). Overlapping 20 aa peptides representing m161–180 (sequence SGIPYVISYLHPGNTVMHVD), where each one of the original 20 aa was substituted by an alanine residue, were synthesized on a peptide synthesizer (model 432A, Applied Biosystems, Foster City, CA, USA) using fluorenylmethoxycarbonyl chemistry and subsequently, by the Center for Biologics Evaluation and Research/U.S. Food and Drug Administration (FDA) core facility (Bethesda, MD, USA). HyQ DMEM/high-glucose medium was purchased from HyClone (Logan, UT, USA). Medium supplements other than normal mouse serum (which was prepared in-house) were purchased from BioWhittaker (Gaithersburg, MD, USA).
EAU induction and scoring
Induction of EAU by active immunization
B10.RIII mice were immunized with 50 μg m161–180 peptide in PBS emulsified 1:1 v/v in CFA that had been supplemented with M. tuberculosis strain H37RA to 2.5 mg/ml. A total of 200 μl emulsion was injected s.c., divided among three sites: base of tail and both thighs. The PT (in DMEM containing 1% normal mouse serum, at 0.4 μg) was given by i.p. injection concurrently with immunization. Clinical EAU was evaluated by fundoscopy under a binocular microscope after dilation of the pupil. Fundoscopies were performed on Days 10, 14, and 21 after immunization.
EAU induction by adoptive transfer of effector cells
Donor B10.RIII mice were immunized with a uveitogenic regimen of 60 μg m161–180. Splenocytes and draining lymph node (LN) cells (inguinal and iliac) were collected 14 days later, pooled, adjusted to 107 cells/ml in supplemented DMEM medium, and stimulated with 100 μg/ml m161–180 for 72 h at 37°C in 10% CO2 . Cells were transferred to new flasks every 24 h to leave behind excess of adherent (inhibitory) macrophages. After 72 h, lymphocytes were separated from debris by Lympholyte cell separation medium (Cedarlane, Burlington, NC, USA) and were counted. Recipient mice were pretreated with IPAs in IFA or with IFA alone and 14 days later, received i.v. 60 × 106 cells/mouse (ratio, two donors:one recipient) of effector cells generated by the donors. Fundoscopies for recipient mice were done every 3–4 days starting on Day 6 after the adoptive transfer of cells.
Eyes were harvested 21 days after immunization or 14 days after adoptive transfer. In all experiments, clinical appearance of disease was followed by fundus examination to confirm development and progression of disease. The final readout was by histopathology. To analyze histopathology results, globes were prefixed in 4% phosphate-buffered glutaraldehyde for 1 h (to prevent artifactual detachment of the retina) and then transferred to 10% phosphate-buffered formaldehyde until processing. Fixed and dehydrated tissue was embedded in methacrylate, and 4- to 6-μm sections were stained with standard H&E. Eye sections cut through a pupillary-optic nerve plane were scored in a masked manner. Incidence and severity of EAU are graded on a scale of 0–4 in half-point increments using the criteria described previously based on the type, number, and size of lesions present in ocular tissues [3, 12]. Incidence is shown as the number of positive animals of all animals in the group. Severity of disease is the average score of eyes from those animals in which disease developed (if disease were unilateral, both eyes are averaged).
Induction and duration of tolerance
Induction of tolerance was tested by pretreating naïve B10.RIII mice with 100 μg of the substituted peptides 169A and 171A in PBS, emulsified 1:1 v/v in IFA. A total of 200 μl emulsion was injected s.c., divided among three sites: base of tail and both thighs. As controls for protection, mice were pretreated with IFA alone. Ten to 14 days later, mice were challenged for EAU using one of the protocols mentioned above. In some experiments, mice were challenged with 300 μg of an unrelated, uveitogenic peptide in CFA, IRBP sequence 541–560 [13]. To investigate the duration of IPA protection, naïve B10.RIII mice were pretreated with the substituted peptides emulsified in IFA and were challenged with a uveitogenic regime of m161–180 peptide 2, 4, and 6 weeks later.
Determination of immunological responses
DTH
To assess DTH, 50 μg m161–180 in 10 μl PBS was injected into the ear pinna. Ear-thickness increment was measured before injection and 48 h after using a spring-loaded micrometer. The specific response was calculated as the difference between ear thickness of the m161–180-injected ear before and after injection.
Lymphocyte proliferation assay
Draining LN (inguinal and iliac) and spleens were collected 21 days after EAU induction and were pooled within the group. Triplicate 0.2 ml cultures containing 5 × 105 cells were seeded in round-bottom, 96-well microtiter plates. DMEM medium was supplemented with mouse serum 1%, 2-ME, antibiotics, glutamine, and nonessential amino acids, as described [14], and contained 30, 3, and 0.3 μg/ml m161–180 as stimulant. The cultures were incubated at 37°C in 10% CO2 for a total of 66 h. Tritiated thymidine (1 μCi/well) was added during the last 18 h.
Cytokine assay
For detection of antigen-induced cytokines in cell culture supernatants, draining LN (inguinal and iliac) and spleen cells from IPA- or IFA-immunized mice were collected after 21 days and cultured in triplicate 0.2 ml cultures containing 5 × 106 cells/well; they were seeded in flat-bottom, 96-well microtiter plates alone or with 30 μg/ml m161–180. Supernatants were collected after 48 h and were kept frozen in small aliquots at −70°C. Cytokine concentration in the supernatants was measured by multiplex ELISA using the Pierce SearchLight Technology (Pierce Boston Technology, Woburn, MA, USA; www.searchlightonline.com).
Determination of m161–180-specific IgG1 and IgG2a antibody levels in sera
Serum levels of anti-m161–180 IgG2a and IgG1 subclasses in m161–180-immunized mice pretreated with candidate IPAs or IFA were determined by ELISA, as described previously [15]. Briefly, 96-well microtiter plates (Costar, Cambridge, MA, USA) were coated with m161–180 (serial peptide dilutions were performed, using a starting concentration of 1 μg/ml for IgG1 detection and 2 μg/ml for IgG2a). After blocking the plates with BSA and overnight incubation with samples of the tested sera, the plates were developed using HRP-conjugated goat anti-mouse IgG1 or IgG2a antibodies (Southern Biotechnology Associates, Birmingham, AL, USA). The amount of each isotype bound to the m161–180-coated wells was estimated from standard curves constructed by coating wells with the same goat anti-mouse IgG1 or goat anti-mouse IgG2a antibodies and adding dilutions of Ig standards of the pertinent isotype.
Immunogenicity, cross-reactivity, and pathogenicity of the alanine-substituted peptides
Immunogenicity of the alanine-substituted peptides was tested by immunizing B10.RIII mice with 150 μg each substituted peptide in CFA and 0.2 μg PT/mouse, as described by the EAU immunization protocol. Twelve days later, the isolated cells were stimulated with 10, 1.0, and 0.1 μM each peptide and assayed for proliferation by 3H-thymidine uptake as described above. Cross-reactivity of the alanine-substituted peptides with the native sequence was tested by immunizing B10.RIII mice with 150 μg of the native m161–180 in CFA and 0.2 μg PT/mouse and 12 days later, stimulating the cells with 10, 1, and 0.1 μM each peptide or by stimulating a m161–180-specific T cell line [16] maintained in vitro with IL-2 (20 units/ml) using 2 × 104 cells/well and splenocytes used as APC at 4 × 105 cells/well plus each one of the substituted peptides at concentrations of 10, 1.0, and 0.1 μM. Pathogenicity was tested by immunizing the mice with 50 and 200 μg each peptide in CFA and 0.5 μg PT/mouse.
Adoptive transfer of protection
Donor B10.RIII mice were pretreated with 100 μg IPAs in IFA or IFA alone as control. Ten to 14 days later, their cells were collected, suspended in a final volume of 500 μl/mouse DMEM, supplemented with 1% mouse serum, and injected i.v. into naïve, syngeneic recipients at a 1:1 donor:recipient ratio. Alternatively, 14 days after IPA pretreatment, donor mice were given a uveitogenic challenge of 50 μg m161–180 in CFA plus 0.4 μg PT, and after 7–10 days, their spleen and LN cells were collected and transferred to naïve recipients as above. EAU was induced in the recipients 3 days after adoptive transfer by immunization with 50 μg m161–180 in CFA and 0.4 μg PT/mouse. Disease was monitored 5, 7, 10, 14, and 21 days after immunization by fundoscopy, and EAU scores in recipients were confirmed on Day 21 by histopathology.
In some experiments, spleen and LN cells containing putative regulatory cells were depleted of CD25+ cells prior to transfer into recipients by using the CD25 Microbead kit from Miltenyi Biotec (Auburn, CA, USA), per the manufacturer’s instructions. Depletion was confirmed by flow cytometry staining for CD4+CD25+ cells (BD PharMingen, Franklin Lakes, NJ, USA).
Intracellular staining for FoxP3
To characterize putative regulatory cells, FoxP3 positivity was tested by flow cytometry using APC-anti-mouse/rat FoxP3 antibody and APC-rat IgG2a as isotype control (FoxP3 staining set, eBioscience, San Diego, CA, USA). In brief, splenocytes isolated from IPA-protected donors were surface-stained for CD4 and CD25, fixed, and then stained for intracytoplasmic FoxP3 following the manufacturer’s protocol.
Data presentation and statistical analysis
Experimental groups were typically composed of five mice. Experiments were repeated at least three times. Figures show combined data from repeat experiments or representative experiments as specified. Statistical significance of differences in disease scores was calculated using Snedecor and Cochran’s test [17] for linear trend in proportions, with each mouse (average of both eyes) as one statistical event. This is a nonparametric test that generates its P values by frequency analysis of the number of individuals at each possible score, thus taking into account severity and incidence of disease. Immunological responses were analyzed using the independent t-test. Probability values of P ≤ 0.05 were considered significant.
RESULTS
Characterization of substituted peptides and selection of candidate IPAs
To identify IPAs capable of immunomodulating EAU induced by the native sequence of a pathogenic epitope from IRBP, alanine-substituted peptides representing m161–180 were synthesized (Table 1). The criteria used for identifying a candidate IPA were: that the peptide would not be pathogenic when used in what would constitute a uveitogenic immunization protocol; that it would be immunogenic and thus, indicate that it can bind to MHC; that it would have detectable cross-reactivity to the native sequence; and that a pretreatment with this peptide would protect from challenge with the native epitope. From the panel of 20 alanine-substituted peptides (Table 1), sequences 169A and 171A within the core epitope of m161–180 (VISYLHPGNTVM) were selected for further characterization as candidate IPAs, as they elicited no disease when used in a uveitogenic immunization protocol at a dose of 50 μg peptide per mouse (Fig. 1A). Data were subsequently confirmed using 200 μg peptides (not shown).
TABLE 1.
Sequence of IRBP Alanine-Substituted Peptides
| Peptide | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| m161–180 | S | G | I | P | Y | V | I | S | Y | L | H | P | G | N | T | V | M | H | V | D |
| 161A | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . |
| 162A | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . |
| 163A | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . |
| 164A | . | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . |
| 165A | . | . | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . |
| 166A | . | . | . | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . | . |
| 167A | . | . | . | . | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . | . |
| 168A | . | . | . | . | . | . | . | A | . | . | . | . | . | . | . | . | . | . | . | . |
| 169A | S | G | I | P | Y | V | I | S | A | L | H | P | G | N | T | V | M | H | V | D |
| 170A | . | . | . | . | . | . | . | . | . | A | . | . | . | . | . | . | . | . | . | . |
| 171A | S | G | I | P | Y | V | I | S | Y | L | A | P | G | N | T | V | M | H | V | D |
| 172A | . | . | . | . | . | . | . | . | . | . | . | A | . | . | . | . | . | . | . | . |
| 173A | . | . | . | . | . | . | . | . | . | . | . | . | A | . | . | . | . | . | . | . |
| 174A | . | . | . | . | . | . | . | . | . | . | . | . | . | A | . | . | . | . | . | . |
| 175A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | A | . | . | . | . | . |
| 176A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | A | . | . | . | . |
| 177A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | A | . | . | . |
| 178A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | A | . | . |
| 179A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | A | . |
| 180A | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | . | A |
Fig. 1.
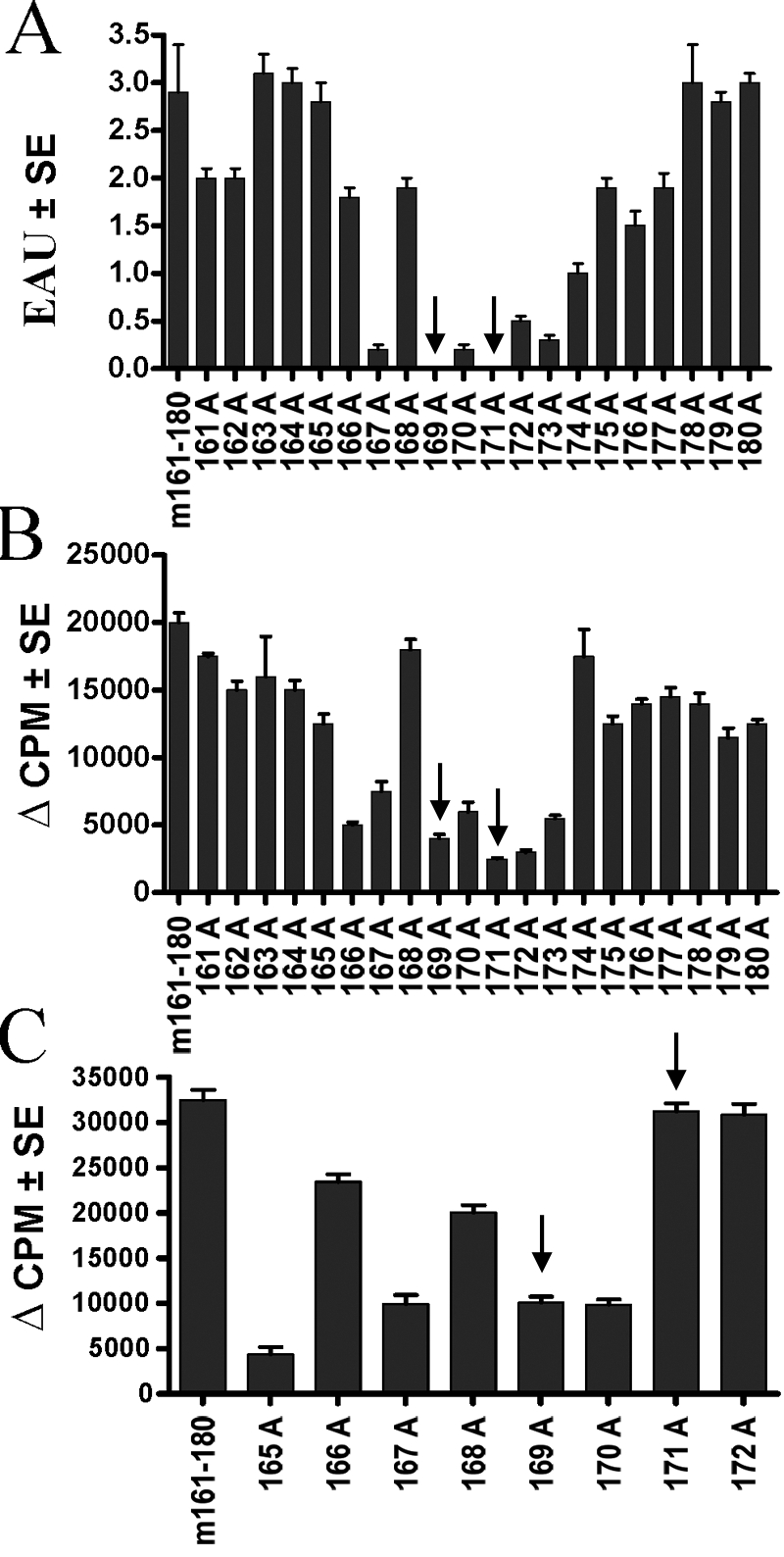
Characterization of the alanine-substituted peptides. (A) Pathogenicity. Groups of five mice were immunized with the indicated peptides in CFA and 0.4 μg PT/mouse. Shown are histopathology scores of eyes collected on Day 21 of mice immunized with 50 μg peptide. A 200-μg dose of the same peptides gave essentially the same results. The data represent an average of four combined experiments. (B) Crossreactivity. A long-term T cell line specific to m161–180 was stimulated with each of the alanine-substituted peptides (10 μM), and proliferation was determined by 3H-thymidine uptake. The graph represents proliferation from three combined experiments. (C) Immunogenicity of the alanine-substituted peptides. Mice were immunized with 150 μg of the alanine-substituted peptides in CFA + PT. Draining LN were collected 12 days after immunization and were stimulated in culture with 10 μM of the respective immunizing peptide. The data represent proliferation from three combined experiments. Arrows point to selected candidate peptides.
Cross-reactivity of the alanine-substituted peptides with the native sequence was examined by stimulating a T cell line derived against m161–180 with each of the peptides (Fig. 1B). Peptides 169A and 171A cross-reacted with the native sequence by lymphocyte proliferation, suggesting that they are indeed recognized by T cells responding to 161–180. Cross-reactivity by proliferation of splenocytes from m161–180-immunized B10.RIII mice gave similar results (data not shown). Candidate peptides were also immunogenic upon direct immunization and recall with the same peptides (Fig. 1C), indicating that they could bind to the MHC.
Candidate IPAs protect from EAU induced by the native sequence
To investigate if the selected substituted peptides could protect from EAU induced by the native epitope, susceptible B10.RIII mice were pretreated by s.c. injection of 169A or 171A in IFA. Controls received IFA alone. Two weeks later, all mice were challenged for EAU with the native epitope as described in Materials and Methods. Mice that were pretreated with peptides 169A and 171A did not develop EAU after challenge with the pathogenic epitope of m161–180, whereas controls pretreated with IFA alone developed severe disease (Fig. 2A, fundoscopy; Fig. 2B, histopathology). Protection was essentially complete in mice that were challenged for disease within 2 weeks after pretreatment with either of the IPAs (Fig. 2C).
Fig. 2.

Protection from EAU by selected peptide analogs. (A) Kinetics of disease development by fundoscopic examination. Mice were pretreated with the candidate IPAs in IFA (169A and 171A) or with IFA alone as control and were challenged for EAU 10 days later as described in Materials and Methods. Eyes were evaluated for disease by fundoscopy at Days 9, 12, 19, and 21. Final scores were confirmed by histopathology performed on eyes harvested on Day 21 (not shown). The data represent EAU scores from four combined experiments. *, Statistically significant difference at P < 0.01 between IPA EAU scores and IFA control. (B) Representative histopathology. Eye at score 3 of disease (IFA alone) is shown compared with a healthy retina (171A in IFA). Note retinal folding and detachment, subretinal hemorrhage, and vitreal and choroid infiltration. (C) Duration of protection. Naïve B10.RIII mice were pretreated with the substituted peptides in IFA or with IFA alone. Pretreated mice were challenged for EAU after 2, 4, or 6 weeks with 50 μg m161–180 in CFA + PT. Shown is histopathology on Day 21 after challenge (average of three combined experiments). *, Statistically significant difference from control at P < 0.02.
Duration of protection was examined by challenging IPA-pretreated mice for EAU after increasing time intervals (Fig. 2C). After 6 weeks, EAU scores were on average still only 50% of control, but protection did not attain statistical significance at P ≤ 0.05. If each eye, rather than of each mouse, is treated as a statistical event (an approach commonly used in eye research, as disease severity often differs between the two eyes of an individual), statistical significance at P ≤ 0.05 is maintained throughout.
Polarization toward a Th2-type response by pretreatment with IPAs
To assess the effect of IPA treatment on the profile of antigen-specific lymphocyte responses that develop after m161–180 challenge in protected mice, we evaluated proliferation, DTH, cytokines, and IgG1-IgG2a antibody production of the IPA-treated and nontreated mice. Mice pretreated with IPAs or with IFA alone were challenged for disease, and their responses were evaluated after 3 weeks. DTH to m161–180 in protected mice was reduced compared with IFA controls (Fig. 3A). Similarly, lymphocytes from 169A- and 171A-pretreated mice were less responsive by proliferation compared with the lymphocyte proliferation of mice pretreated only with IFA (Fig. 3B). Antigen-specific production of IL-4, IL-5, IL-10, and IL-13 by IPA-protected mice was increased significantly (P<0.05), whereas IFN-γ and IL-17 production was reduced (P<0.02; Fig. 3C). TGF-β levels were increased in IPA-protected mice but did not attain statistical significance (data not shown). We next evaluated the humoral response profile in the protected mice. As IFN-γ and IL-4 promote antibody isotype-switching to IgG2a and IgG1, respectively, the relative titers of these antibody isotypes are a good indicator of the type of response that develops to the antigen in vivo. We therefore assayed the anti-m161–180 antibodies of each isotype in sera of IPA-protected mice (Fig. 3D). Protected mice had elevated IgG1 and reduced IgG2a serum antibodies to m161–180. Overall, IPA treatment seems to favor a switch from a Th1 to a Th2 response rather than induction of unresponsiveness. As EAU is a Th1/Th17-mediated disease, these results suggest that the mechanism of IPA protection is mediated, at least in part, by immune deviation.
Fig. 3.

Immune responses in protected mice. (A) DTH. Mice were challenged for DTH with m161–180 into the ear pinna. Ear-thickness increment was measured before and 48 h after injection, and the specific response was calculated as the difference between the two measurements. Data are from a representative experiment of three. (B) Proliferation to m161–180. Spleens and the LN draining the site of immunization were collected on Day 21, pooled within the group, and stimulated with 30, 3, or 0.3 μg/ml m161–180. The data are shown as cpm after background subtraction. Data are from a representative experiment of three. (C) Antigen-specific cytokine responses. Spleen and LN cells collected after 21 days from protected mice and controls were stimulated with m161–180. Cytokines in 48 h supernatants were assayed by multiplex ELISA. The graph represents data from two combined experiments. *, Statistically significant difference between IPA (171A) and IFA cytokine production at P < 0.05; **, P < 0.02. (D) Determination of m161–180-specific antibody isotypes in serum. Sera were collected 21 days after immunization and were assayed by ELISA for anti-161–180 antibody isotypes IgG1 and IgG2a. The graph represents data from a representative experiment of three.
Protection involves transferable CD25+ regulatory cells
To address the question of whether IPA-induced protection involves transferable regulatory cells, pooled splenocytes and draining LN cells from mice that were pretreated with IPAs and then challenged for EAU and shown to be protected were transferred into naïve B10.RIII mice. Recipients of cells from such protected donors were themselves protected from a uveitogenic challenge with m161–180 (Fig. 4A).
Fig. 4.

Protection appears to be mediated by regulatory cells. (A) Adoptive transfer of cells from protected (challenged) donors. Splenocytes and draining LN cells were collected from IPA-protected donors. Control cells came from mice pretreated with IFA alone. The donor cells (60×106) were adoptively transferred into naïve recipients who were challenged for EAU 72 h later. Fundoscopies were done at the indicated time-points. Data are an average of five mice per group from a representative experiment out of three that gave similar results. *, Statistically significant increase in disease scores. (B) Adoptive transfer of protection with CD4+CD25+ and CD4+CD25− fractions. Splenocytes and LN cells from IPA (171A)-protected mice and from IFA controls were depleted, or not, of CD25+ cells and were adoptively transferred into naïve recipients who were subsequently challenged for EAU. The graph shows normalized data combined from three experiments, and EAU scores in recipients of nondepleted cells were set to 100% (average EAU score of 0.5 in recipients of cells from IPA-pretreated and of 1.75 in recipients of cells from IFA-pretreated donors). *, Statistically significant increase in disease scores. (C) IPA-protected mice have increased numbers of CD4+CD25+FoxP3+ T cells. Spleens cells from mice treated as A were surface-stained for CD4 and CD25, fixed, and stained for intracytoplasmic FoxP3. Panels represent cells gated for the CD4+ stain. The IPA/IFA group was naïve B10.RIII mice pretreated with IPAs emulsified in IFA, and the IFA group was a control group pretreated with IFA alone. Data are from a representative experiment out of four that gave similar results.
To test if transfer of protection was a result of cells with CD25+ [T regulatory cell (Treg)] phenotype, splenocytes isolated from IPA-protected mice and from IFA controls were immunomagnetically depleted (or not) of CD25+ cells. The proportion of CD25+ cells in the CD4+ population, as analyzed by flow cytometry, was reduced from 32.17% to 0.94% in IPA-pretreated donors (97% depletion) and from 33.56% to 0.67% in IFA controls (98% depletion). The depleted and nondepleted populations were then transferred into naive recipients who were challenged 3 days later for EAU by immunization with m161–180. CD25-depleted cells from IPA-protected donors induced EAU, which was twice as severe as that induced by the nondepleted cells. In contrast, recipients of CD25-depleted cells from IFA-only controls developed minimally up-regulated disease scores over recipients of the nondepleted population (Fig. 4B). These results support the notion that the Tregs have a CD25+ phenotype.
We next examined whether IPA pretreatment expands FoxP3+ cells in the cell population that transfers protection. To test this, we performed FACS analysis on whole splenocytes from IPA-protected mice and IFA control. Results showed that after gating for CD4+ cells, splenocytes of IPA-protected mice contained double the number of CD4+CD25+FoxP3+ cells compared with IFA-only, pretreated mice (Fig. 4C). This is compatible with the notion that at least some of the Tregs induced by the IPA pretreatment are FoxP3+.
IPA-induced protection is effective in a reversal protocol against already-primed effector cells
Regulatory cells can act in an afferent manner, preventing priming of T cells into effector cells, or they can also inhibit the function of already-primed effector T cells. The latter activity is particularly relevant to therapy of chronic disease during relapsing or recurrent episodes. To test whether the substituted peptides could protect from a challenge with effector cells, we used an established adoptive transfer system, where EAU was induced with activated effector cells from 161–180-immunized donor mice. Cells were adoptively transferred into IPA- or IFA-pretreated mice. Recipient mice that received pretreatment with IPA prior to transfer developed low and transient disease, whereas control mice that were pretreated only with IFA developed progressively higher disease scores (Fig. 5).
Fig. 5.
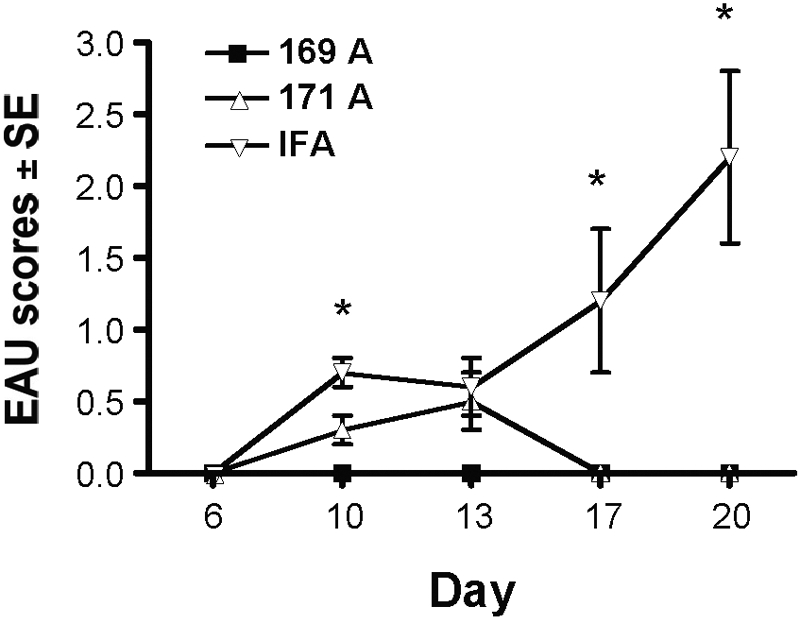
IPA-induced regulation affects already-primed effector cells. Donor mice were immunized with a uveitogenic protocol of m161–180. In parallel, recipients were pretreated with IPAs in IFA. Twelve days later, the donor spleen and LN cells were collected and cultured for 3 days with m161–180, and 60 × 106 cells were infused into the IPA-pretreated recipients (ratio, two donors:one recipient), which were examined by fundoscopy starting on Day 6 after the adoptive transfer. Data show an average of three experiments. *, Statistically significant difference at P < 0.05 between IPAs and IFA.
IPAs based on m161–180 partially protect from challenge with an unrelated IRBP epitope
We next examined whether IPA pretreatment could protect from a challenge with an unrelated H-2r haplotype-restricted pathogenic epitope of the IRBP molecule (position 541–560, sequence SLGWATLVGEITAGNLLHTR). Mice pretreated with IPAs and challenged with the native 161–180 epitope were completely protected. Mice challenged with peptide 541–560 were partly protected (Fig. 6). This indicates the presence of linked suppression, compatible with a mechanism mediated by regulatory cells and cytokines.
Fig. 6.

IPAs of m161–180 protect from challenge with an unrelated IRBP epitope. Naïve B10.RIII mice were pretreated with peptide 171A in IFA or IFA alone. Two weeks later, mice were challenged with a uveitogenic protocol of m161–180 or with another pathogenic IRBP peptide, sequence 541–560. The graph represents an average of two experiments. *, Statistically significant difference at P < 0.005; **, P < 0.05.
DISCUSSION
EAU is a well-characterized, robust, and reproducible model for autoimmune uveitis in humans as well as a model for autoimmunity in general. Several approaches have been attempted for the therapy of autoimmune uveitis [5, 6]. In the present study, we examined the use of IPAs as a potential candidate therapy. To be selected as IPAs, peptides were required to be nonpathogenic but immunogenic and cross-reactive with the native epitope and be able to protect from a uveitogenic challenge with the native epitope. Modified autoantigens were used to treat autoimmunity in animal models [18] and have also made their way to the clinic. One example is the Copolymer-1 (Copaxone), which is now a FDA-approved treatment for MS [7, 8]. However, another approach to altered peptide therapy of MS was not as successful [19, 20].
It is important to underscore that the peptides identified here, as well as IRBP itself, constitute proof of concept but are not themselves candidates for use in therapy of human uveitis. Studies in double-knockout mice, deficient in the autoimmune regulator molecule and in IRBP, led to the conclusion that IRBP is a naturally dominant self-antigen for uveitis in the mouse species [21]. In contrast, retinal arrestin, also known as retinal S-Ag is the most frequent antigen responded to by human uveitis patients but does not elicit EAU in wild-type (WT) mice [22]. This species-specific dominance may be in part determined by MHC, as “humanized” HLA transgenic mice not only develop EAU with S-Ag but also respond to the same dominant epitope known as “peptide N,” recognized by human patients. Although humans and WT mice dominantly recognize different retinal antigens, cellular mechanisms that lead to pathology appear similar. This notion is upheld by the findings that experimental approaches that modulate the experimental disease are often found to be efficacious in the clinic [23].
The two IPAs identified and characterized in this study elicited no pathology at a dose as high as 200 μg/mouse, which is at least five times higher than the quantity needed to elicit pathology with the native epitope [4; and P. B. Silver and Rajeev K. Agarwal, unpublished data]. Substituted peptides 167A and 170A exhibited some trace pathogenicity, and for that reason, they were not selected for further study. There appeared to be a correlation between cross-reactivity and pathogenicity from the tested peptides, and the more cross-reactive ones (at least by proliferation) were more pathogenic. It is not known which residues of m161–180 are TCR contacts and which are MHC contacts, but this issue, although certainly of interest, does not affect data interpretation in the current study.
The conditions under which T cells are primed, including the strength of TCR signaling and the innate signals accompanying it, play an important role in determining whether effector cells or suppressor cells are generated. Our data indicated that pretreatment with IPA skewed the subsequent immune response to m161–180 in protected mice toward noninflammatory, type 2 cytokines, including IL-4, IL-5, IL-10, and IL-13, and decreased production of IFN-γ and IL-17. This is in line with other studies that demonstrated immune deviation as a result of altered peptide treatment [8, 18]. There is also evidence that some protective, modified peptides might act by generation of regulatory cells [8, 9]. Protection induced with our two IPAs was associated with an increase in the number of CD4+CD25+FoxP3+ T cells and with generation of transferable CD25+ Tregs. Although a Th2 switch and Treg generation are nonmutually exclusive mechanisms, our data do not negate the possibility that at least some of the CD4+CD25+Tregs, whose depletion prevents transfer of suppression, might be of the Th2 phenotype. However, Th2 cells are not as a rule FoxP3+ so that existence of two separate populations that contribute to the observed protection is likely. Recently, we have examined another type of “vaccination for tolerance,” based on hydrodynamic delivery of plasmid DNA encoding the first homologous repeat of IRBP that contains the p161–180 epitope. Although no Th2 skewing was observed in the hydrodynamic DNA vaccination model, the protection involved participation of antigen-specific CD4+CD25+FoxP3+ Tregs and lasted for at least 10 weeks [24].
In our hands, the protective effect of IPA pretreatment persisted for up to 4 weeks but started to wane after that, suggesting that as a possible therapeutic approach in the clinic, repeated treatment might be needed to maintain the protective effect. The relatively shorter duration of protection compared with DNA vaccination [24] could be a result of the fact that the tolerogen is a partially cross-reactive peptide analog rather than the native epitope, but that property also underlies its advantage, as the analog is nonpathogenic. A need for repeated dosing with antigen was seen in an open-label study, where uveitis patients received retinal arrestin (S-Ag) orally for induction of protective tolerance [25]. Similarly, repeated treatment with Copaxone is needed to maintain its therapeutic efficacy in MS. Importantly, IPA therapy was able to afford significant (P<0.05) protection from a challenge with already-primed effector cells specific to the native epitope, suggesting that such an approach could be useful in established disease. No less importantly, there was evidence for linked or “bystander” suppression, such that protection was afforded against challenge with an unrelated epitope from the IRBP molecule. The mechanism of this is likely to involve Tregs and anti-inflammatory cytokines triggered by recognition of the native 161–180 epitope released from damaged tissue. In human disease, the antigenic specificity(ies) are often unknown, and more than one epitope may be involved in driving disease. As well, still unknown in human disease is the issue of epitope spreading that was defined in animal models [26]. Such linked, or bystander, suppression could go toward addressing these issues. Overall, these results point to IPA therapy as a potentially useful approach to therapy of ocular autoimmune disease.
Acknowledgments
The authors thank Ms. Mary Alice Crawford, Mr. Joseph Hackett, Ms. Iris Wise, and Mr. Julius Boykin from the National Eye Institute Histology Core Facility for their technical assistance in preparation of the histological specimens. We thank Drs. Rajeev Agarwal, Shao-Bo Su, and Wei Zhu for assistance in specific experimental procedures.
References
- Caspi R R. Immune mechanisms in uveitis. Springer Semin Immunopathol. 1999;21:113–124. doi: 10.1007/BF00810244. [DOI] [PubMed] [Google Scholar]
- Caspi R R, Roberge F G, McAllister C G, el-Saied M, Kuwabara T, Gery I, Hanna E, Nussenblatt R B. T cell lines mediating experimental autoimmune uveoretinitis (EAU) in the rat. J Immunol. 1986;136:928–933. [PubMed] [Google Scholar]
- Agarwal R K, Caspi R R. Rodent models of experimental autoimmune uveitis. Methods Mol Med. 2004;102:395–419. doi: 10.1385/1-59259-805-6:395. [DOI] [PubMed] [Google Scholar]
- Silver P B, Rizzo L V, Chan C C, Donoso L A, Wiggert B, Caspi R R. Identification of a major pathogenic epitope in the human IRBP molecule recognized by mice of the H-2r haplotype. Invest Ophthalmol Vis Sci. 1995;36:946–954. [PubMed] [Google Scholar]
- Whitcup S M, Nussenblatt R B. Immunologic mechanisms of uveitis. New targets for immunomodulation. Arch Ophthalmol. 1997;115:520–525. doi: 10.1001/archopht.1997.01100150522013. [DOI] [PubMed] [Google Scholar]
- Caspi R R. Regulation, counter-regulation, and immunotherapy of autoimmune responses to immunologically privileged retinal antigens. Immunol Res. 2003;27:149–160. doi: 10.1385/IR:27:2-3:149. [DOI] [PubMed] [Google Scholar]
- Arnon R, Aharoni R. Mechanism of action of glatiramer acetate in multiple sclerosis and its potential for the development of new applications. Proc Natl Acad Sci USA. 2004;101:14593–14598. doi: 10.1073/pnas.0404887101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela M, Mozes E. Therapeutic vaccines in autoimmunity. Proc Natl Acad Sci USA. 2004;101:14586–14592. doi: 10.1073/pnas.0404826101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontoura P, Garren H, Steinman L. Antigen-specific therapies in multiple sclerosis: going beyond proteins and peptides. Int Rev Immunol. 2005;24:415–446. doi: 10.1080/08830180500379655. [DOI] [PubMed] [Google Scholar]
- Dayan M, Sthoeger Z, Neiman A, Abarbanel J, Sela M, Mozes E. Immunomodulation by a dual altered peptide ligand of autoreactive responses to the acetylcholine receptor of peripheral blood lymphocytes of patients with myasthenia gravis. Hum Immunol. 2004;65:571–577. doi: 10.1016/j.humimm.2004.02.030. [DOI] [PubMed] [Google Scholar]
- Illes Z, Waldner H, Reddy J, Bettelli E, Nicholson L B, Kuchroo V K. T cell tolerance induced by cross-reactive TCR ligands can be broken by superagonist resulting in anti-inflammatory T cell cytokine production. J Immunol. 2005;175:1491–1497. doi: 10.4049/jimmunol.175.3.1491. [DOI] [PubMed] [Google Scholar]
- Caspi R R. Experimental autoimmune uveoretinitis (EAU)—mouse and rat. Coligan J E, Kruisbeek A M, Margulies D H, Shevech E M, Strober W, editors. New York, NY; San Diego, CA, USA.: 2003 [Google Scholar]
- Cortes L M, Mattapallil M J, Silver P B, Donoso L A, Liou G I, Zhu W, Chan C-C, Caspi R R. Repertoire analysis and new pathogenic epitopes of IRBP in C57BL/6 (H-2b) and B10.RIII (H-2r) mice. Invest Ophthalmol Vis Sci. 2008;49:1946–1956. doi: 10.1167/iovs.07-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R R, Roberge F G, Chan C C, Wiggert B, Chader G J, Rozenszajn L A, Lando Z, Nussenblatt R B. A new model of autoimmune disease. Experimental autoimmune uveoretinitis induced in mice with two different retinal antigens. J Immunol. 1988;140:1490–1495. [PubMed] [Google Scholar]
- Rizzo L V, DeKruyff R H, Umetsu D T, Caspi R R. Regulation of the interaction between Th1 and Th2 T cell clones to provide help for antibody production in vivo. Eur J Immunol. 1995;25:708–716. doi: 10.1002/eji.1830250312. [DOI] [PubMed] [Google Scholar]
- Rizzo L V, Silver P, Wiggert B, Hakim F, Gazzinelli R T, Chan C C, Caspi R R. Establishment and characterization of a murine CD4+ T cell line and clone that induce experimental autoimmune uveoretinitis in B10.A mice. J Immunol. 1996;156:1654–1660. [PubMed] [Google Scholar]
- Snedecor G W, Cochran W G. Ames, IA, USA: Iowa State University Press; Statistical Methods. 1967 [Google Scholar]
- Young D A, Lowe L D, Booth S S, Whitters M J, Nicholson L, Kuchroo V K, Collins M. IL-4, IL-10, IL-13, and TGF-β from an altered peptide ligand-specific Th2 cell clone down-regulate adoptive transfer of experimental autoimmune encephalomyelitis. J Immunol. 2000;164:3563–3572. doi: 10.4049/jimmunol.164.7.3563. [DOI] [PubMed] [Google Scholar]
- Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, Gran B, Eaton J, Antel J, Frank J A, McFarland H F, Martin R. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6:1167–1175. doi: 10.1038/80516. [DOI] [PubMed] [Google Scholar]
- Kappos L, Comi G, Panitch H, Oger J, Antel J, Conlon P, Steinman L. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. The Altered Peptide Ligand in Relapsing MS Study Group. Nat Med. 2000;6:1176–1182. doi: 10.1038/80525. [DOI] [PubMed] [Google Scholar]
- DeVoss J, Hou Y, Johannes K, Lu W, Liou G I, Rinn J, Chang H, Caspi R R, Fong L, Anderson M S. Spontaneous autoimmunity prevented by thymic expression of a single self-antigen. J Exp Med. 2006;203:2727–2735. doi: 10.1084/jem.20061864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennesi G, Mattapallil M J, Sun S H, Avichezer D, Silver P B, Karabekian Z, David C S, Hargrave P A, McDowell J H, Smith W C, Wiggert B, Donoso L A, Chan C C, Caspi R R. A humanized model of experimental autoimmune uveitis in HLA class II transgenic mice. J Clin Invest. 2003;111:1171–1180. doi: 10.1172/JCI15155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenblatt R B, Whitcup S M. Philadelphia, PA, USA: Mosby (Elsevier); UveitisFundamentals and Clinical Practice. 2004 [Google Scholar]
- Silver P B, Agarwal R K, Su S B, Suffia I, Grajewski R S, Luger D, Chan C C, Mahdi R M, Nickerson J M, Caspi R R. Hydrodynamic vaccination with DNA encoding an immunologically privileged retinal antigen protects from autoimmunity through induction of regulatory T cells. J Immunol. 2007;179:5146–5158. doi: 10.4049/jimmunol.179.8.5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenblatt R B. Bench to bedside: new approaches to the immunotherapy of uveitic disease. Int Rev Immunol. 2002;21:273–289. doi: 10.1080/08830180212067. [DOI] [PubMed] [Google Scholar]
- Vanderlugt C L, Miller S D. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. 2002;2:85–95. doi: 10.1038/nri724. [DOI] [PubMed] [Google Scholar]