

Abstract
The hereditary spastic paraplegias (HSPs) are a genetically and clinically heterogeneous group of upper-motor-neuron degenerative diseases characterized by selective axonal loss in the corticospinal tracts and dorsal columns. Although numerous mechanisms involving defective subcellular transportation, mitochondrial malfunction, and increased oxidative stress have been proposed, the pathogenic basis underlying the neuronal loss is unknown. We have performed linkage analysis to refine the extent of the SPG5 disease locus and conducted sequence analysis of the genes located within this region. This identified sequence alterations in the cytochrome P450-7B1 (CYP7B1) associated with this pure form of HSP. In the liver, CYP7B1 offers an alternative pathway for cholesterol degradation and also provides the primary metabolic route for the modification of dehydroepiandrosterone neurosteroids in the brain. These findings provide the first direct evidence of a pivotal role of altered cholesterol metabolism in the pathogenesis of motor-neuron degenerative disease and identify a potential for therapeutic intervention in this form of HSP.
Main Text
The hereditary spastic paraplegias (HSPs [MIM 182601]) encompass a clinically heterogeneous group of neurodegenerative diseases characterized by a progressive degeneration of upper motor neurons.1 The cardinal clinical feature of lower extremity weakness and spasticity may occur in isolation (“pure” HSP) or be accompanied by other symptoms including mental retardation, cerebellar ataxia, optic and peripheral neuropathy, and thin corpus callosum.2 The neuropathologic hallmark of HSP is axonal degeneration of neurons of the corticospinal tracts and the dorsal columns, with relative preservation of neuronal cell bodies.
HSP is genetically heterogeneous with >38 loci (HGNC) encompassing all modes of inheritance.3 Although autosomal-recessive forms of HSP are often associated with clinically complex phenotypes, mutations within a few genes underlie pure forms of the disease. One of the most common of these appears to be the first pure autosomal-recessive HSP locus defined, SPG5 (MIM 270800), located on chromosome 8q12.3, to which a number of families have subsequently been mapped.4–8 However, the precise nature of the causative gene has remained elusive.
Although several genes have now been identified for the various forms of HSP, the mechanisms underlying axonal degeneration and the tissue specificity of the phenotype are poorly understood. The genes identified to date appear to have diverse functions implicating a range of putative disease mechanisms including defective subcellular transportation, mitochondrial malfunction, and oxidative stress.9 Although previously not implicated in HSP, some other neurodegenerative diseases including Niemann-Pick type C (NPC [MIM 257220]) and cerebrotendinous xanthomatosis (CTX [MIM 213700]) are thought to be associated with defects in cholesterol metabolism.10–13 In the liver, primary bile acids required for normal absorption of lipids and lipid-soluble vitamins are formed from cholesterol either through the “neutral pathway” involving the 7α-hydroxylase CYP7A1, considered to be quantitatively most important in humans, or the “alternate/acidic pathway” involving the related 7α-hydroxylase CYP7B1.14 These hydroxylases are also key molecules for the metabolism of brain cholesterol; because there is little or no transfer of cholesterol across the blood-brain barrier, most cholesterol is produced in the brain locally, and there is a precise balance between the biosynthesis, storage, and catabolism of cholesterol metabolites. In the brain, CYP7B1 also provides the primary metabolic route for cholesterol derivatives dehydroepiandrosterone (DHEA) and related hydroxysteroids via 7α-hydroxylation (Figure 1).15 Here, we report mutations within CYP7B1 (MIM 603711) associated with a pure form of HSP, which defines defective cholesterol metabolism as the likely pathogenic cause and identifies a potential therapeutic strategy for this form of motor-neuron degenerative disease.
Figure 1.
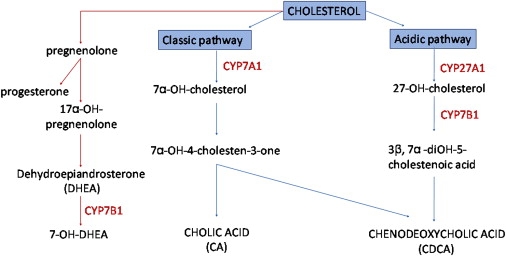
A Simplified Overview of the Major Bile-Acid and Neurosteroid Biosynthetic Pathways
The bile-acid pathway is indicated in blue, and the neurosteroid pathway is indicated in red.
We have previously shown a refinement of the locus for this gene in a large English family to the location between markers D8S589 and D8S543 encompassing 23.6 cM at chromosome 8q12.3.8 In order to more precisely position the interval-defining proximal and distal recombination events in this family (family 1), we investigated additional polymorphic microsatellite markers at each site. Genotypes of microsatellite markers were determined by PCR amplification and subsequent analysis by polyacrylamide gel electrophoresis (PAGE) and visualized by silver staining as previously described.16 Genotyping and haplotype analysis more precisely defined the flanking markers as D8S1115 (proximal) and D8S1795 (distal; data not shown). Combined with very recent information which further refines the proximal boundary as marker D8S1113,5 this results in a refinement of the SPG5 critical interval from ∼24 cM to 11cM a region encompassing ∼40 transcripts. Intronic, exon-flanking primers were designed to amplify and sequence coding exons and associated splice junctions of genes located in the critical interval, with sequencing protocols as previously described.16 Systematic sequence analysis of the genes located within the interval revealed a 1088C > T (S363F) substitution in exon 5 of CYP7B1, located in the middle of this region (National Center for Biotechnology Information [accession number NM_004820]). Subsequent sequence analysis of this gene in five additional families (families 2–6) revealed nucleotide changes in all pedigrees (see Figure 2). Of these five families, families 2–4 have been previously presented and demonstrated to show linkage to the SPG5 locus.4 The phenotype of these families as well as in families 1, 5, and 6, can briefly be summarized as a pure form of HSP including posterior column sensory impairment as evidenced by diminished vibration sensation and proprioception and some degree of bladder dysfunction. The age of onset was variable in all families ranging from 1 to 40 years of age. Amyotrophy, or evidence of cerebellar or cerebral cortex dysfunction, was not present in any of the families examined. All samples were taken with LREC-approved consent.
Figure 2.
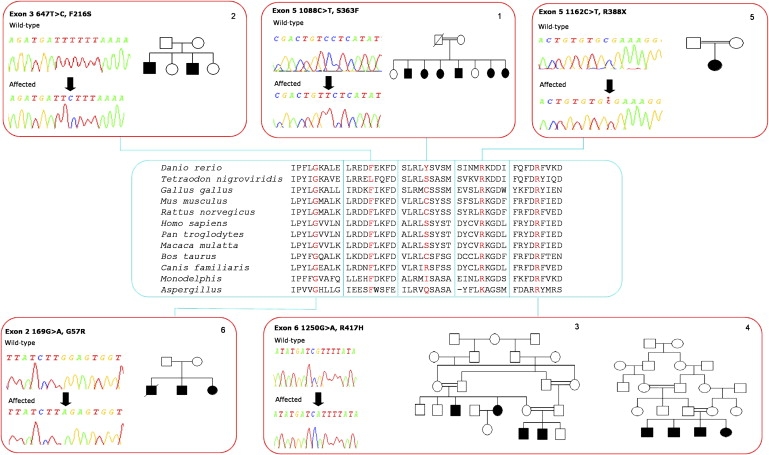
Family Trees and DNA Sequence Chromatographs Are Indicated by the Red Boxes, and Conservation of Affected Amino Acid Residues Is Shown in the Blue Box
The variants identified included four distinct nonsynonymous mutations in five of the families tested, all present in the coding sequence and in a homozygous state in affected individuals (1088C > T [S363F] in family 1, 647T > C [F216S] in family 2, 1250G > A [R417H] in families 3 and 4 and 169G > A [G57R] in family 6). Sequencing of the unaffected family members and parents of affected individuals confirmed that the variant alleles segregated with the disease. The single sporadic case (family 5) with a pure HSP phenotype was also identified and screened and was found to be homozygous for a nonsense mutation, 1162C > T (R388X). Each mutation was not found in 500 control chromosomes.
The mutations all disrupt residues that are highly conserved throughout evolution; G57 and R417 are invariant in all species examined, F216 is conserved in all but pufferfish, and S363 is conserved in human, gorilla, monkey, and pufferfish (Figure 2). An evaluation of the likely outcome of the missense mutations was investigated by NetPhos 2.0. Two of the mutations are likely to affect phosphorylation of CYP7B1. The serine at position 363 is strongly predicted to be phosphorylated (NetPhos score 0.985). Also, the substitution of the phenylalanine at position 216 for a serine creates a putative phosphorylation site that is predicted to be the most likely phosphorylated residue in the mutant molecule, assuming that it is expressed (NetPhos score of 0.997).
The identification of mutations in CYP7B1 associated with HSP presented here provides the first direct evidence for abnormalities in cholesterol metabolism in the pathogenesis of motor-neuron degeneration. Levels of cholesterol in the human brain and spinal cord are particularly high, and as such, neurons may well be particularly susceptible to abnormalities in cholesterol homeostasis.17 Mutations were defined in all families displaying linkage to the SPG5 locus. The clinical phenotype of the families in whom we identified CYP7B1 mutations is that of a pure (uncomplicated) form with progressive spastic paraplegia, with variable bladder and sensory (loss of proprioception and vibration sense) involvement. Age of onset showed both interfamilial and intrafamilial variation with a range of 1 to 40 years. Those patients with long disease duration required walking aids, and a few were wheelchair dependent. Although the genetic defect remains to be clarified, a recent study reported two families linked to SPG5 in which the HSP was associated with mild cerebellar signs in individuals with long disease duration.5 Should CYP7B1 mutations subsequently be described in these patients, this would suggest that the phenotype of CYP7B1-associated HSP may be broader than the pure presentation in the cases documented here. Such phenotypic variability is not unusual, and complicated phenotypes have been reported previously for autosomal-recessive (SPG7 [MIM 607259], SPG27 [MIM 610244]) as well as autosomal-dominant (SPG4 [MIM 604277]) forms of HSP.18–20
The mutations identified here are likely to be highly detrimental to CYP7B1 functionality. Phosphorylation has previously been shown to act as an important functional regulatory switch for a number of cytochrome p450 molecules,21 and the S363F and F216S CYP7B1 mutations are predicted to abolish and create novel phosphorylation sites, respectively. Importantly, one of the mutations identified here (R388X) has been defined previously and shown to result in loss of function of CYP7B1.22 Whereas the phenotype of our patient solely involves that of motor-neuron degeneration with no other features, the previous study demonstrated homozygosity for the R388X mutation associated with a severe liver disease in a neonate who presented with severe cholestasis, cirrhosis, and liver synthetic failure and who died at the age of 6 months.22 The mechanism of liver injury in this patient was proposed to be due to the accumulation of hepatotoxic monohydroxy bile acids exacerbated by a lack of primary bile acids. A probable explanation for this phenotypic discrepancy may be provided by the serum and urine principal oxysterols analysis that revealed hugely increased levels of substrates in the affected neonate (in particular 27-hydroxycholesterol) but the complete absence of 7α-hydroxycholesterol. This latter finding is puzzling given that sequence analysis of the six coding exons of the CYP7A1 (MIM 118455) gene, which is responsible for production of 7α-hydroxycholesterol via the neutral pathway, was thought to be normal in this child. These findings were understandably interpreted to suggest that in early human development, bile-acid synthesis occurs mainly via the alternate CYP7B1 pathway and is essential for normal liver development. Other studies show that patients with a homozygous deletion mutation in CYP7A1, which results in loss of the active site and enzyme function, survive and display high levels of hepatic cholesterol content and a markedly deficient rate of bile-acid excretion. These patients also display evidence for upregulation of the alternative bile-acid pathway CYP7B1,23 a situation that is also mirrored in studies of murine Cyp7a1 knockouts.24–26 Taken together with the findings of our study, it seems likely that the loss of one 7α-hydrolase pathway may be compensated for, at least in part, by the remaining pathway. Consequently, perhaps the best explanation for the discrepancy between the phenotype of the CYP7B1 mutations presented here and that of the severe neonatal liver disease is that in the latter situation, the fatal liver disease in fact arises because of loss of function of both α-hydroxylating enzymes. This could be mediated through other sequence variants either within regions of the CYP7A1 gene not examined in this individual or at a distinct locus important for CYP7A1 function, perhaps within a gene encoding a CYP7A1 cofactor, thereby resulting in loss of function of both 7-hydroxylating enzymes. Such a result could explain the complete absence of 7α-hydroxycholesterol (it is noteworthy that this child was the offspring of a consanguineous first-cousin union). When considered together, these factors are consistent with the notion that the production of 7α-hydroxycholesterol (via the classic CYP7A1 pathway) is sufficient for normal liver development and function and that loss of function of CYP7B1 results specifically in upper-motor-neuron degeneration.
Interestingly, mice with disrupted Cyp7b1 have previously been generated, and although plasma and tissue levels of 27-hydroxycholesterol were markedly elevated, these animals developed normally and were grossly indistinguishable from wild-type mice.27 A lack of a motor-neuron phenotype in these animals may reflect their age of examination. Previous studies of another mouse model of HSP (paraplegin-SPG7 [MIM 607259-MIM 602783]) have shown that the first features of neurodegeneration occur relatively late (after 8–12 months of age) and that paraplegin-deficient mice ultimately become affected by a distal axonopathy of spinal and peripheral axons, characterized by axonal swelling and degeneration.28 Consequently, because a motor-neuron phenotype associated with Cyp7b1 knockout would not necessarily be predicted, it is possible that any features may have been subtle and late in onset and have occurred after the mice evaluation that took place at 3–4 months of age.27 Evidence to show that abnormal oxysterol levels may be responsible for motor-neuron degeneration in mice is provided by studies of liver X receptor α (LXRα) null mice. LXRα is a ligand-activated receptor, which plays an important role in the regulation of Cyp7a1 and Cyp7b1 function, and null mutation of this gene results in an adult-onset motor-neuron degenerative phenotype.29 This is associated with lipid accumulation and a selective loss of lower motor neurons associated with impaired motor function in animals from 7 months of age.
Cytochrome P450s have been detected in neurons,30 and it is of note that a recent study utilizing monoclonal antibodies specific for CYP7B1 detected particularly high expression in pyramidal cells of human hippocampal sections, consistent with an important role in motor neurons that specifically degenerate in HSP.31 DHEA is converted to 7-OH-DHEA by CYP7B1, and there is growing evidence to suggest that this group of neurosteroids act as neuroprotective factors and can reduce ischemia-induced neurodegeneration in both in vitro and in vivo models.32 By using a model of hypoxia that produces an injury similar to mild ischemia in rat hippocampal slice cultures, a recent study revealed that these 7-hydroxy metabolites strongly protect against neurodegeneration of rat hippocampal CA1 pyramidal cells.32 This neuroprotection was thought to be dependent on the metabolism of the steroid so that efficacy was mediated by long-term modification of intracellular processes. Consequently, as well as possible neurotoxicity associated with aberrant oxysterol levels, the neurodegeneration associated with the CYP7B1 mutation may also be associated with a loss of neuroprotective function of DHEA-related neurosteroids.
Interestingly, the previous association of cerebrotendinous xanthomatosis with mutations in CYP27A (MIM 606530), which produces the 27-hydroxylated substrate for CYP7B1 action, raises important possibilities for treatment of this form of HSP. Cerebrotendinous xanthomatosis characteristically occurs in late childhood or early adulthood and is caused by the accumulation of cholesterol and cholestanol in various tissues.33 As well as tendon xanthomata, cataracts, and atherosclerosis, symptoms include dementia and motor dysfunction including spastic paraparesis.34 Early diagnosis and treatment with chenodeoxycholic acid, which normalizes the level of the cholesterol precursor, results in a reduction of cholestanol-containing xanthomas in the brain and contributes to a better prognosis by preventing the progression of neurological symptoms.35 Our findings suggest that the adoption of a similar strategy here may well offer a potentially important therapeutic advance for this debilitating form of motor-neuron degenerative disease.
Acknowledgments
The authors are grateful to the family members of the English, American, Australian, and Tunisian kindreds for participation in this research project and Dr. Phillipa J. Lamont of the Royal Perth Hospital of Australia. This work was supported at St George's in London by the Birth Defects Newlife Foundation (UK) and Action Research, and at Northwestern University Feinberg School of Medicine (in part) by The National Institutes of Health (NS046535, NS050641, and ES014469), The Les Turner ALS Foundation, Vena E. Schaff ALS Research Fund, Harold Post Research Professorship Fund, Herbert and Florence C. Wenske Foundation, Ralph and Marian Falk Medical Research Trust, The David C. Asselin MD Memorial Fund, and the Les Turner ALS Foundation/Herbert C. Wenske Foundation Professor . N.G.L. was supported by Australian National Health and Medical Research Council Fellowship 403904.
Web Resources
The URLs for data presented herein are as follows:
HGNC HUGO Gene Nomenclature Committee, http://www.genenames.org/
NCBI National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/
UCSC Genome Browser, http://genome.ucsc.edu/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
Accession Numbers
The GenBank accession number for CYP7B1 reported in this paper is NM_004820.
References
- 1.Fink J.K. Hereditary spastic paraplegia. Curr. Neurol. Neurosci. Rep. 2006;6:65–76. doi: 10.1007/s11910-996-0011-1. [DOI] [PubMed] [Google Scholar]
- 2.Harding A.E. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–1155. doi: 10.1016/s0140-6736(83)92879-9. [DOI] [PubMed] [Google Scholar]
- 3.Casari G., Rugarli E. Molecular basis of inherited spastic paraplegias. Curr. Opin. Genet. Dev. 2001;11:336–342. doi: 10.1016/s0959-437x(00)00199-4. [DOI] [PubMed] [Google Scholar]
- 4.Hentati A., Pericak-Vance M.A., Hung W.Y., Belal S., Laing N., Boustany R.M., Hentati F., Ben Hamida M., Siddique T. Linkage of ‘pure’ autosomal recessive familial spastic paraplegia to chromosome 8 markers and evidence of genetic locus heterogeneity. Hum. Mol. Genet. 1994;3:1263–1267. doi: 10.1093/hmg/3.8.1263. [DOI] [PubMed] [Google Scholar]
- 5.Klebe S., Durr A., Bouslam N., Grid D., Paternotte C., Depienne C., Hanein S., Bouhouche A., Elleuch N., Azzedine H. Spastic paraplegia 5: Locus refinement, candidate gene analysis and clinical description. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2007;144:854–861. doi: 10.1002/ajmg.b.30518. [DOI] [PubMed] [Google Scholar]
- 6.Muglia M., Criscuolo C., Magariello A., De Michele G., Scarano V., D'Adamo P., Ambrosio G., Gabriele A.L., Patitucci A., Mazzei R. Narrowing of the critical region in autosomal recessive spastic paraplegia linked to the SPG5 locus. Neurogenetics. 2004;5:49–54. doi: 10.1007/s10048-003-0167-7. [DOI] [PubMed] [Google Scholar]
- 7.Ouahchi K., Madrid R.E., Hentati A., Hung W.-Y., Kaplan J., Siddique T. Linkage of a family of Italian descent with recessive familial spastic paraplegia to chromosome 8 markers. Am. J. Hum. Genet. 1998;63(Suppl):A304. [Google Scholar]
- 8.Wilkinson P.A., Crosby A.H., Turner C., Patel H., Wood N.W., Schapira A.H., Warner T.T. A clinical and genetic study of SPG5A linked autosomal recessive hereditary spastic paraplegia. Neurology. 2003;61:235–238. doi: 10.1212/01.wnl.0000069920.42968.8d. [DOI] [PubMed] [Google Scholar]
- 9.Crosby A.H., Proukakis C. Is the transportation highway the right road for hereditary spastic paraplegia? Am. J. Hum. Genet. 2002;71:1009–1016. doi: 10.1086/344206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porter F.D. Human malformation syndromes due to inborn errors of cholesterol synthesis. Curr. Opin. Pediatr. 2003;15:607–613. doi: 10.1097/00008480-200312000-00011. [DOI] [PubMed] [Google Scholar]
- 11.Yau J.L., Rasmuson S., Andrew R., Graham M., Noble J., Olsson T., Fuchs E., Lathe R., Seckl J.R. Dehydroepiandrosterone 7-hydroxylase CYP7B: Predominant expression in primate hippocampus and reduced expression in Alzheimer's disease. Neuroscience. 2003;121:307–314. doi: 10.1016/s0306-4522(03)00438-x. [DOI] [PubMed] [Google Scholar]
- 12.Cali J.J., Hsieh C.L., Francke U., Russell D.W. Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J. Biol. Chem. 1991;266:7779–7783. [PMC free article] [PubMed] [Google Scholar]
- 13.Boadu E., Choi H.Y., Lee D.W., Waddington E.I., Chan T., Asztalos B., Vance J.E., Chan A., Castro G., Francis G.A. Correction of apolipoprotein A-I-mediated lipid efflux and high density lipoprotein particle formation in human Niemann-Pick type C disease fibroblasts. J. Biol. Chem. 2006;281:37081–37090. doi: 10.1074/jbc.M606890200. [DOI] [PubMed] [Google Scholar]
- 14.Chiang J.Y. Regulation of bile acid synthesis. Front. Biosci. 1998;3:d176–d193. doi: 10.2741/a273. [DOI] [PubMed] [Google Scholar]
- 15.Rose K.A., Stapleton G., Dott K., Kieny M.P., Best R., Schwarz M., Russell D.W., Bjorkhem I., Seckl J., Lathe R. Cyp7b, a novel brain cytochrome P450, catalyzes the synthesis of neurosteroids 7alpha-hydroxy dehydroepiandrosterone and 7alpha-hydroxy pregnenolone. Proc. Natl. Acad. Sci. USA. 1997;94:4925–4930. doi: 10.1073/pnas.94.10.4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simpson M.A., Cross H., Proukakis C., Pryde A., Hershberger R., Chatonnet A., Patton M.A., Crosby A.H. Maspardin is mutated in mast syndrome, a complicated form of hereditary spastic paraplegia associated with dementia. Am. J. Hum. Genet. 2003;73:1147–1156. doi: 10.1086/379522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lathe R., Seckl J.R. Genetics of Steroid Biosynthesis and Function. Volume 6. Harwood Academic; Amsterdam: 2002. Neurosteroids and brain sterols; pp. 405–472. [Google Scholar]
- 18.Ribai P., Stevanin G., Bouslam N., Pontier B., Nelson I., Fontaine B., Dussert C., Charon C., Durr A., Brice A. A new phenotype linked to SPG27 and refinement of the critical region on chromosome. J. Neurol. 2006;253:714–719. doi: 10.1007/s00415-006-0094-2. [DOI] [PubMed] [Google Scholar]
- 19.De Michele G., De Fusco M., Cavalcanti F., Filla A., Marconi R., Volpe G., Monticelli A., Ballabio A., Casari G., Cocozza S. A new locus for autosomal recessive hereditary spastic paraplegia maps to chromosome 16q24.3. Am. J. Hum. Genet. 1998;63:135–139. doi: 10.1086/301930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heinzlef O., Paternotte C., Mahieux F., Prud'homme J.F., Dien J., Madigand M., Pouget J., Weissenbach J., Roullet E., Hazan J. Mapping of a complicated familial spastic paraplegia to locus SPG4 on chromosome 2p. J. Med. Genet. 1998;35:89–93. doi: 10.1136/jmg.35.2.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oesch-Bartlomowicz B., Oesch F. Cytochrome-P450 phosphorylation as a functional switch. Arch. Biochem. Biophys. 2003;409:228–234. doi: 10.1016/s0003-9861(02)00558-1. [DOI] [PubMed] [Google Scholar]
- 22.Setchell K.D., Schwarz M., O'Connell N.C., Lund E.G., Davis D.L., Lathe R., Thompson H.R., Weslie T.R., Sokol R.J., Russell D.W. Identification of a new inborn error in bile acid synthesis: Mutation of the oxysterol 7alpha-hydroxylase gene causes severe neonatal liver disease. J. Clin. Invest. 1998;102:1690–1703. doi: 10.1172/JCI2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pullinger C.R., Eng C., Salen G., Shefer S., Batta A.K., Erickson S.K., Verhagen A., Rivera C.R., Mulvihill S.J., Malloy M.J. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J. Clin. Invest. 2002;110:109–117. doi: 10.1172/JCI15387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erickson S.K., Lear S.R., Deane S., Dubrac S., Huling S.L., Nguyen L., Bollineni J.S., Shefer S., Hyogo H., Cohen D.E. Hypercholesterolemia and changes in lipid and bile acid metabolism in male and female cyp7A1-deficient mice. J. Lipid Res. 2003;44:1001–1009. doi: 10.1194/jlr.M200489-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Ishibashi S., Schwarz M., Frykman P.K., Herz J., Russell D.W. Disruption of cholesterol 7alpha-hydroxylase gene in mice. I. Postnatal lethality reversed by bile acid and vitamin supplementation. J. Biol. Chem. 1996;271:18017–18023. doi: 10.1074/jbc.271.30.18017. [DOI] [PubMed] [Google Scholar]
- 26.Schwarz M., Lund E.G., Setchell K.D., Kayden H.J., Zerwekh J.E., Bjorkhem I., Herz J., Russell D.W. Disruption of cholesterol 7alpha-hydroxylase gene in mice. II. Bile acid deficiency is overcome by induction of oxysterol 7alpha-hydroxylase. J. Biol. Chem. 1996;271:18024–18031. doi: 10.1074/jbc.271.30.18024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li-Hawkins J., Lund E.G., Turley S.D., Russell D.W. Disruption of the oxysterol 7alpha-hydroxylase gene in mice. J. Biol. Chem. 2000;275:16536–16542. doi: 10.1074/jbc.M001811200. [DOI] [PubMed] [Google Scholar]
- 28.Ferreirinha F., Quattrini A., Pirozzi M., Valsecchi V., Dina G., Broccoli V., Auricchio A., Piemonte F., Tozzi G., Gaeta L. Axonal degeneration in paraplegin-deficient mice is associated with abnormal mitochondria and impairment of axonal transport. J. Clin. Invest. 2004;113:231–242. doi: 10.1172/JCI20138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andersson S., Gustafsson N., Warner M., Gustafsson J.A. Inactivation of liver X receptor beta leads to adult-onset motor neuron degeneration in male mice. Proc. Natl. Acad. Sci. USA. 2005;102:3857–3862. doi: 10.1073/pnas.0500634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ravindranath V., Bhamre S., Bhagwat S.V., Anandatheerthavarada H.K., Shankar S.K., Tirumalai P.S. Xenobiotic metabolism in brain. Toxicol. Lett. 1995;82–83:633–638. doi: 10.1016/0378-4274(95)03508-7. [DOI] [PubMed] [Google Scholar]
- 31.Trap C., Nato F., Chalbot S., Kim S.B., Lafaye P., Morfin R. Immunohistochemical detection of the human cytochrome P4507B1: Production of a monoclonal antibody after cDNA immunization. J. Neuroimmunol. 2005;159:41–47. doi: 10.1016/j.jneuroim.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 32.Pringle A.K., Schmidt W., Deans J.K., Wulfert E., Reymann K.G., Sundstrom L.E. 7-Hydroxylated epiandrosterone (7-OH-EPIA) reduces ischaemia-induced neuronal damage both in vivo and in vitro. Eur. J. Neurosci. 2003;18:117–124. doi: 10.1046/j.1460-9568.2003.02734.x. [DOI] [PubMed] [Google Scholar]
- 33.Clayton P.T., Verrips A., Sistermans E., Mann A., Mieli-Vergani G., Wevers R. Mutations in the sterol 27-hydroxylase gene (CYP27A) cause hepatitis of infancy as well as cerebrotendinous xanthomatosis. J. Inherit. Metab. Dis. 2002;25:501–513. doi: 10.1023/a:1021211520034. [DOI] [PubMed] [Google Scholar]
- 34.Sugama S., Kimura A., Chen W., Kubota S., Seyama Y., Taira N., Eto Y. Frontal lobe dementia with abnormal cholesterol metabolism and heterozygous mutation in sterol 27-hydroxylase gene (CYP27) J. Inherit. Metab. Dis. 2001;24:379–392. doi: 10.1023/a:1010564920930. [DOI] [PubMed] [Google Scholar]
- 35.Moghadasian M.H. Cerebrotendinous xanthomatosis: Clinical course, genotypes and metabolic backgrounds. Clin. Invest. Med. 2004;27:42–50. [PubMed] [Google Scholar]