

Abstract
For a long time, NMR chemical shifts have been used to identify protein secondary structures. Currently, this is accomplished through comparing the observed 1Hα, 13Cα, 13Cβ, or 13C` chemical shifts with the random coil values. Here, we present a new protocol, which is based on the joint probability of each of the three secondary structural types (β-strand, α-helix, and random coil) derived from chemical-shift data, to identify the secondary structure. In combination with empirical smooth filters/functions, this protocol shows significant improvements in the accuracy and the confidence of identification. Updated chemical-shift statistics are reported, on the basis of which the reliability of using chemical shift to identify protein secondary structure is evaluated for each nucleus. The reliability varies greatly among the 20 amino acids, but, on average, is in the order of: 13Cα>13C`>1Hα>13Cβ>15N>1HN to distinguish an α-helix from a random coil; and 1Hα>13Cβ >1HN ∼13Cα∼13C`∼15N for a β-strand from a random coil. Amide 15N and 1HN chemical shifts, which are generally excluded from the application, in fact, were found to be helpful in distinguishing a β-strand from a random coil. In addition, the chemical-shift statistical data are compared with those reported previously, and the results are discussed. A JAVA User Interface program has been developed to make the entire procedure fully automated and is available via http://ccsr3150-p3.stanford.edu.
Keywords: Chemical shift, NMR, protein secondary structure, secondary structure identification
Since the late 1960's, NMR chemical shifts have been known to have a strong correlation with secondary structure (Markley et al. 1967; Nakamura and Jardetzky 1968). Although several techniques have been developed to characterize and quantify protein and peptide secondary structure using chemical-shift data (Szilagyi and Jardetzky 1989; Pastore and Saudek 1990; Wishart et al. 1991, Wishart et al. 1992; Wishart and Sykes 1994), their protocols remain basically the same — through comparing the observed chemical-shift with the random coil value. Because of the qualitative nature of the data and the simplified procedures used in these methods, the accuracy of the identification is limited. Here we describe a new approach, PSSI (Probability based Secondary Structure Identification), to identify protein secondary structure from NMR chemical-shift data. Unlike the previously reported protocols, PSSI assigns the secondary structure type (β-strand, random coil, or α-helix) to each amino acid on the basis of the joint probability, derived from the observed 1HN, 15N, 1Hα, 13Cα, 13Cβ, and 13C` chemical-shift data of each secondary structure type. PSSI shows significant improvements in both the accuracy and the confidence of identification. Testing on 36 proteins including >6100 residues, this protocol gave a global accuracy of 88%. For proteins with a well-defined secondary structure and sufficient chemical-shift data, PSSI can readily give >90% accuracy.
In recent years, the improvements in NMR instrumentation, novel multidimensional NMR experiments, higher magnetic field strengths, the wide application of protein isotopic labeling techniques, and the software for automatic assignment have greatly enhanced our ability to assign the chemical shifts for large proteins (Kay and Gardner 1997; Goto and Kay 2000). However, completion of chemical-shift assignment is just a first step on the long journey of the tertiary structure determination. Other NMR data, that is, NOE, coupling constant, etc., must be collected to allow an accurate tertiary structure determination. As the size of the protein increases, the difficulty experienced in obtaining these data grows very rapidly due to spin diffusion and resonance overlap. The method presented in this study provides a fast and reliable way to identify secondary structure before the tertiary structure can be actually determined. Not only does the accurate secondary structure identification have potential biological significance (e.g., helping to analyze putative sequences for the existence of function-related motifs and to design site-directed mutants), but it also could help in the model-building phase of experimental 3D-structure determination. In addition, the detailed statistical analysis on the chemical-shift distribution against secondary structure elements shown in this study could also provide a deeper insight into the relationship between chemical-shift and protein secondary structure.
Results and Discussion
Chemical-shift statistics
The averaged 1HN, 15N, 1Hα, 13Cα, 13Cβ, and 13C` chemical shifts, together with the standard deviations categorized according to three secondary structure types, are listed in Table 1. These statistical data are derived from a carefully prepared database containing >6100 amino acids. Since the late 1980s, several statistical studies on the chemical shifts of the 20 amino acids have been reported with the accumulation of NMR data (Wuthrich 1986; Wishart et al. 1991; Lukin et al. 1997; Schwarzinger et al. 2000). The results presented here expand upon the earlier studies in the following respects.
Table 1.
Averaged chemical shift (in ppm) and standard deviation values (in parentheses) categorized according to secondary structure type
| 13Cα | 13Cβ | 13C` | |||||||
| Amino acid | β-strand | Random coil | α-helix | β-strand | Random coil | α-helix | β-strand | Random coil | α-helix |
| Ala | 50.86 (1.28) | 52.67 (1.76) | 54.86 (0.94) | 21.72 (1.77) | 19.03 (1.27) | 18.27 (1.08) | 175.3 (1.61) | 177.39 (1.45) | 179.58 (1.39) |
| Arg | 54.63 (1.50) | 55.96 (1.94) | 59.05 (1.21) | 32.36 (1.82) | 30.53 (1.77) | 30.00 (0.83) | 175.04 (1.18) | 175.91 (1.27) | 178.11 (1.70) |
| Asn | 52.48 (1.18) | 52.94 (1.43) | 55.67 (0.99) | 40.43 (1.89) | 38.22 (1.47) | 38.28 (1.12) | 174.55 (1.28) | 174.98 (1.38) | 176.74 (1.66) |
| Asp | 53.41 (1.15) | 54.09 (1.59) | 57.04 (1.00) | 42.78 (1.75) | 40.76 (1.34) | 40.50 (1.12) | 175.15 (1.54) | 176.01 (1.45) | 178.07 (1.80) |
| Cys | 57.64 (1.94) | 58.8 (2.06) | 62.86 (1.85) | 29.48 (1.97) | 29.75 (1.86)b | 26.99 (0.84)b | 173.86 (1.83)a | 174.77 (1.38)b | 177.42 (1.35)b |
| 54.19 (1.64)a | 57.68 (1.43)a,b | 58.57 (1.59)a,b | 43.79 (4.04)a | 38.38 (1.39)a,b | 40.02 (1.78)a,b | 172.73 (1.05)a | 175.85 (1.58)a,b | 176.84 (0.47)a,b | |
| Gln | 54.33 (1.39) | 55.94 (1.83) | 58.61 (1.04) | 31.92 (1.74) | 28.67 (1.73) | 28.33 (0.79) | 174.58 (0.94) | 175.88 (1.53) | 178.35 (1.15) |
| Glu | 55.55 (1.45) | 56.39 (1.84) | 59.30 (1.05) | 32.45 (1.96) | 30.02 (1.62) | 29.20 (0.77) | 175.01 (1.24) | 176.11 (1.47) | 178.46 (1.34) |
| Gly | 45.08 (1.20) | 45.34 (1.17) | 47.02 (0.90) | 173.01 (2.59) | 174.30 (1.80) | 176.31 (1.50) | |||
| His | 54.8 (1.75) | 55.78 (2.02) | 59.62 (1.57) | 32.2 (2.52) | 29.62 (1.99) | 29.91 (1.67) | 173.80 (2.24) | 174.88 (1.68) | 176.83 (1.16) |
| Ile | 60.00 (1.51) | 60.64 (2.08) | 64.68 (1.66) | 40.09 (1.85) | 38.26 (2.06) | 37.59 (1.08) | 174.79 (1.41) | 175.46 (1.65) | 177.49 (1.62) |
| Leu | 53.94 (1.19) | 54.85 (1.79) | 57.54 (0.98) | 44.02 (1.99) | 41.87 (1.70) | 41.40 (1.11) | 175.16 (1.31) | 176.61 (1.77) | 178.42 (1.70) |
| Lys | 55.01 (1.00) | 56.40 (1.80) | 59.11 (1.19) | 34.86 (1.79) | 32.57 (1.30) | 32.31 (1.08) | 174.93 (1.25) | 176.15 (1.40) | 177.79 (2.22) |
| Met | 54.10 (1.46) | 55.12 (1.79) | 58.45 (1.66) | 34.34 (2.44) | 32.93 (3.05) | 31.70 (1.72) | 174.64 (1.47) | 175.93 (1.54) | 177.76 (1.77) |
| Phe | 56.33 (1.31) | 56.94 (1.98) | 60.74 (1.63) | 41.64 (1.65) | 39.43 (1.93) | 38.91 (1.49) | 174.15 (1.93) | 175.28 (1.88) | 176.42 (1.74) |
| Pro | 62.79 (1.22) | 63.53 (1.26) | 65.52 (1.01) | 32.45 (0.93) | 31.87 (0.96) | 31.08 (0.84) | 176.41 (1.50) | 176.91 (1.72) | 178.34 (1.53) |
| Ser | 57.14 (1.11) | 58.35 (1.78) | 60.86 (1.27) | 65.39 (1.48) | 63.88 (1.24) | 62.81 (0.58) | 173.52 (1.55) | 174.33 (1.22) | 176.51 (1.40) |
| Thr | 61.10 (1.71) | 61.59 (2.04) | 65.89 (1.55) | 70.82 (2.11) | 69.75 (1.29) | 68.64 (0.98) | 173.47 (1.39) | 174.62 (1.45) | 176.62 (1.24) |
| Trp | 56.28 (1.52) | 57.62 (1.25) | 60.03 (1.94) | 31.78 (1.76) | 29.27 (1.10) | 28.74 (1.15) | 175.10 (1.80) | 175.91 (1.32) | 177.81 (1.62) |
| Tyr | 56.56 (1.59) | 57.72 (2.14) | 61.07 (1.72) | 40.79 (1.77) | 38.71 (2.00) | 38.38 (0.89) | 174.65 (1.64) | 175.32 (1.54) | 177.05 (1.51) |
| Val | 60.72 (1.59) | 61.80 (2.25) | 65.96 (1.39) | 33.81 (1.79) | 32.68 (1.76) | 31.41 (0.74) | 174.66 (1.36) | 175.76 (1.63) | 177.75 (1.49) |
| 1HN | 1Cα | 15N | |||||||
| Amino acid | β-strand | Random coil | α-helix | β-strand | Random coil | α-helix | β-strand | Random coil | α-helix |
| a Cys in the oxidized form. | |||||||||
| b Number of the chemical shifts used in the statistical analysis is less than 10. | |||||||||
| c Averaged value fro Gly. | |||||||||
| Those that show significant discrepancies (15N > 1.0ppm, 1Hα > 0.2, HN > 0.3, 13C` > 0.5) when compared with earlier reported value (see Results and Discussion) are indicated in bold. | |||||||||
| Ala | 8.59 (0.76) | 8.11 (0.68) | 7.99 (0.57) | 4.87 (0.46) | 4.25 (0.35) | 4.03 (0.31) | 125.57 (4.80) | 132.52 (3.51) | 121.65 (2.52) |
| Arg | 8.57 (0.69) | 8.17 (0.77) | 8.03 (0.56) | 4.85 (0.47) | 4.33 (0.37) | 4.00 (0.33) | 122.60 (4.74) | 120.59 (4.42) | 118.99 (2.56) |
| Asn | 8.70 (0.55) | 8.33 (0.72) | 8.20 (0.66) | 5.26 (0.41) | 4.60 (0.38) | 4.45 (0.20) | 122.70 (4.18) | 118.48 (4.58) | 117.60 (2.37) |
| Asp | 8.56 (0.62) | 8.39 (0.66) | 8.05 (0.55) | 5.01 (0.36) | 4.64 (0.29) | 4.44 (0.22) | 123.82 (4.70) | 120.69 (4.45) | 119.90 (2.03) |
| Cys | 9.00 (0.45) | 7.81 (0.62) | 8.22 (0.53) | 5.18 (0.57) | 4.63 (0.37)b | 4.16 (0.25)b | 123.27 (5.69) | 117.01 (2.50)b | 117.47 (3.04)b |
| 8.68 (0.98)a | 8.53 (0.59)a,b | 8.58 (0.48)a,b | 5.21 (0.49)a | 4.44 (0.29)a,b | 4.53 (0.18)a,b | 121.81 (4.34)a | 118.62 (4.25)a,b | 119.51 (2.44)a,b | |
| Gln | 8.51 (0.83) | 8.25 (0.75) | 8.11 (0.52) | 4.97 (0.43) | 4.26 (0.39) | 4.03 (0.23) | 123.14 (4.89) | 119.73 (3.85) | 118.59 (2.59) |
| Glu | 8.66 (0.60) | 8.29 (0.53) | 8.32 (0.63) | 4.76 (0.44) | 4.28 (0.30) | 3.99 (0.21) | 123.52 (4.29) | 120.87 (3.94) | 119.89 (2.85) |
| Gly | 8.27 (1.06) | 8.34 (0.83) | 8.23 (0.78) | 4.09c (0.46) | 3.95c (0.40) | 3.84 (0.43)c | 110.19 (4.20) | 109.94 (4.09) | 107.34 (2.82) |
| His | 8.76 (0.79) | 8.09 (0.83) | 8.03 (0.68) | 5.07 (0.50) | 4.50 (0.51) | 4.06 (0.54) | 121.65 (5.16) | 118.87 (4.98) | 118.09 (3.17) |
| Ile | 8.74 (0.66) | 7.94 (0.66) | 8.06 (0.56) | 4.72 (0.42) | 4.13 (0.36) | 3.66 (0.30) | 124.12 (4.93) | 121.07 (5.17) | 120.22 (2.75) |
| Leu | 8.63 (0.67) | 8.12 (0.72) | 8.02 (0.56) | 4.85 (0.43) | 4.35 (0.36) | 4.00 (0.27) | 125.69 (4.14) | 121.53 (4.30) | 120.18 (2.46) |
| Lys | 8.54 (0.63) | 8.13 (0.66) | 8.04 (0.61) | 4.96 (0.46) | 4.28 (0.31) | 3.98 (0.26) | 123.29 (4.76) | 121.44 (4.19) | 119.90 (2.93) |
| Met | 8.43 (0.65) | 8.37 (0.51) | 8.05 (0.48) | 4.94 (0.48) | 4.55 (0.38) | 4.03 (0.35) | 121.67 (4.12) | 120.19 (3.46) | 118.69 (2.36) |
| Phe | 8.80 (0.70) | 7.95 (0.90) | 8.21 (0.66) | 5.17 (0.47) | 4.62 (0.42) | 4.11 (0.40) | 121.95 (4.38) | 119.41 (4.75) | 119.12 (4.05) |
| Pro | 4.72 (0.45) | 4.41 (0.30) | 4.13 (0.39) | ||||||
| Ser | 8.57 (0.65) | 8.26 (0.74) | 8.11 (0.50) | 5.08 (0.48) | 4.48 (0.35) | 4.20 (0.19) | 117.44 (4.19) | 115.94 (4.13) | 114.78 (2.39) |
| Thr | 8.50 (0.58) | 8.22 (0.74) | 8.10 (0.55) | 4.81 (0.46) | 4.33 (0.38) | 4.02 (0.27) | 118.09 (4.86) | 114.41 (5.70) | 115.30 (3.72) |
| Trp | 8.83 (0.73) | 7.59 (0.84) | 8.24 (0.82) | 5.24 (0.41) | 4.54 (0.24) | 4.35 (0.40) | 124.04 (5.43) | 120.57 (3.58) | 120.48 (2.89) |
| Tyr | 8.69 (0.73) | 7.90 (0.79) | 8.10 (0.70) | 5.00 (0.51) | 4.55 (0.45) | 4.14 (0.36) | 122.55 (4.70) | 120.05 (4.23) | 119.67 (3.19) |
| Val | 8.73 (0.61) | 7.88 (0.75) | 7.99 (0.63) | 4.66 (0.42) | 4.14 (0.40) | 3.57 (0.34) | 123.27 (5.05) | 119.66 (5.62) | 119.53 (3.19) |
First, the most recently published chemical-shift assignments and structure data are included in the statistical analysis. All of the proteins selected in this study are large in size (>80 residues), double or triple isotope labeled, and their chemical-shifts are assigned by use of multidimensional NMR techniques. The larger chemical-shift and protein coordinate database allows us to specify and take into account other conditions such as chemical-shift reference, pH, and temperature to avoid their possible effects on chemical shift. Table 2 provides a detailed list of the proteins together with BMRB and PDB entries selected for this study. Although most of the BMRB files contain one or more corresponding PDB access numbers, of the 36 proteins used in this study, only 2 can be found in the list of BMRB entries manually matched to PDB NMR entries. Manually matching chemical-shift data (BMRB entry) with the known protein coordinates (PDB entry) is essential for this study. As shown in this table, sometimes sequence renumbering is necessary to obtain a correct match.
Table 2.
Listing of proteins used in chemical shift statistical analysis
| Protein (no. of residues) | BMRB entries (conditions)a | PDB entries (Exp Method) | PDB entries Manually Matched in BMRB |
| β2-GP1 domain V (86) | 4981 (pH6.0, 298K) | 1G4F (NMR) | N/A |
| Vam3p N-terminal (123) | 4945 (pH6.0, 302K)b | 1HS7 (NMR)b | N/A |
| Mouse doppel (132) | 4938 (pH5.2, 299K)b | 1I17 (NMR)b | N/A |
| DLC8 (89) | 4931 (pH7.0, 303K) | 1F95 (NMR) | N/A |
| ERp29 C-domain (120) | 4920 (pH4.9, 308K)b | 1G7D (NMR)b | N/A |
| ERp29 N-domain (137) | 4919 (pH4.9, 308K)b | 1G7E (NMR)b | N/A |
| Dynein light chain 8 (89) | 4911 (pH7.0, 303K) | 1F96 (NMR) | N/A |
| Human galectiin-3, C-terminal (143) | 4909 (pH7.4, 303K) | 1A3K (X-ray) | N/A |
| CDC4P (141) | 4851 (pH6.5, 303K) | 1GGW (NMR) | N/A |
| Hepatitis A 3C protease (217) | 4836 (pH5.4, 298K) | 1QA7 (X-ray) | N/A |
| EPPIb (164) | 4765 (pH6.2, 308K) | 2NUL (X-ray) | N/A |
| Xylanase (185) | 4705 (pH7.4, 298K) | 1C5I (X-ray) | N/A |
| Apo CRBP II (134) | 4681 (pH7.4, 298K) | 1OPB (X-ray) | N/A |
| Pathogenesis-related protein (159) | 4671 (pH7.0, 298K) | 1E09 (NMR) | N/A |
| HTLV-I capsid protein (134) | 4649 (pH6.0, 302K) | 1G03 (NMR) | N/A |
| Phosphoglycerate mutase (211) | 4648 (pH6.4, 310K) | 1FZT (NMR) | N/A |
| Human prion protein (146) | 4641 (pH4.6, 299K) | 1F07 (NMR) | N/A |
| Anti-dansyl Fv fragment (237) | 4580 (pH6.0, 310K)b | 2DLF (X-ray)b | N/A |
| Bet v 1-L (159) | 4417 (pH7.0, 298K) | 1B6F (NMR) | 1B6F |
| HPrP (210) | 4402 (pH4.5, 293K) | 1QLX (NMR) | N/A |
| Cyclic AMP receptor protein (209) | 4388 (pH6.0, 313K) | 1RUN (X-ray) | N/A |
| CA RSV (262) | 4384 (pH6.0, 303K) | 1D1D (NMR) | N/A |
| Rabphilin_3_C2B (140) | 4360 (pH6.1, 304K) | 3RPB (NMR) | N/A |
| FvNPN43C9 (230) | 4349 (pH6.8, 298K)b | 43C9 (X-ray)b | N/A |
| α-β T cell receptor (249) | 4330 (pH5.0, 298K) | 1D9K (X-ray) | N/A |
| p55 (166) | 4321 (pH6.5, 300K) | 5GCN (NMR) | N/A |
| HTLV-I capsid (214) | 4311 (pH6.0, 303K) | 1QRJ (NMR) | N/A |
| CaM:SEF2-1 (148) | 4310 (pH6.0, 306K) | 1CDL (X-ray) | N/A |
| P14a (135) | 4301 (pH5.5, 303K) | 1CFE (NMR) | N/A |
| XRCC1 N-terminal (183) | 4282 (pH6.8, 298K) | 1XNA (NMR)c | N/A |
| Superoxide Dismutase (153) | 4202 (pH5.0, 298K) | 1MFM (X-ray) | 1BA9 (NMR) |
| Intimin cell adhesion domain (280) | 4111 (pH5.0, 310K)b | 1FOO (X-ray)b | N/A |
| metallo-β-lactamase (232) | 4102 (pH7.0, 295K) | 2BMI (X-ray) | N/A |
| Core binding factor (143) | 4092 (pH6.6, 293K) | 2JHB (NMR) | N/A |
| Protoporphyrin IX (147) | 4038 (pH5.0, 297K) | 1VRF (NMR) | N/A |
| Human carbonic anhydrase I (260) | 4022 (pH6.2, 303K) | 1HUG (X-ray) | N/A |
a DSS was used directly or indirectly as zero chemical shift reference of 1H, 15N, and 13C for all of the proteins.
b Manually renumbering of the sequence is needed to match the BMRB entry to PDB entry.
c Coordinate is not available for the random coil region (resides 152–183) in the PDB file.
Second, an explicit and exact protocol is applied to define a protein's secondary structure from its 3D coordinates. Accurate assignment of secondary structures in proteins with known 3D structures is crucial for the determination of the relationship between protein secondary structure and the observed chemical shift. However, the correlation always remains subjected to some inaccuracy due to the differences in the concept of secondary structure, as well as errors and inconsistencies in experimental structural data. This was shown in the comparison (Colloch et al. 1993) of three methods for structure determination, DSSP (Kabsch and Sander 1983), DEFINE (Richards and Kundrot 1988), and P-curve (Sklenar et al. 1989) on a set of 154 proteins in which all 3 methods agree only 63% of the time. Another comparison (Cuff and Barton 1999) of DSSP, DEFINE, and STRIDE (Frishman and Argos 1995) on a set of 126 proteins showed an overall agreement of only 71%. Among these various protocols/programs, we tried to choose one by which the secondary structures defined could be in a better agreement with the observed chemical-shift data. For this purpose, three automatic programs, VADAR (Wishart et al. 1995a), DSSP, and STRIDE, were used to define the secondary structure during our statistical study. All three programs agree well with each other in defining both the location and the length of long (≥ 4 residues) and well-formed (in terms of dihedral angle, hydrogen bond, etc.) β-strand/α-helix, but not the short (< 4 residues) β-strand/α-helix and poorly-formed β-strand/α-helix. In most cases, a separated β-bridge (one residue) defined by DSSP/STRIDE is assigned as a three-residue β-strand by VADAR. A separated 310-helix (only three residues) defined by DSSP/STRIDE is normally assigned to random coil by VADAR. For these two situations, we found that the results from VADAR agree with the observed chemical-shift data. For a long β-strand/α-helix with poorly formed boundary, VADAR defines more β-strand/α-helix. Sometimes, modifications had to be made on one or two residues at the end of β-strand or α-helix defined by VADAR, taking into account the backbone dihedral angle, to which the chemical shift is sensitive (Williamson et al. 1992; Wishart and Nip 1997), as well as the results from DSSP and STRIDE.
Furthermore, the 3D coordinates, from which the secondary structures were extracted and used as reference, were carefully selected and priority was given to high-resolution X-ray structures, which have always been the gold standard for the determination of NMR parameters such as 3JHNHα coupling constants, etc. When NMR structures were used, at least three individual structures were selected to generate a secondary structure reference.
The averaged chemical shifts in this study are compared with the earlier reported values and those showing significant discrepancies are indicated in bold in Table 1. More specifically, 1HN and 1Hα were compared with Wishart's result (Wishart et al. 1991); 15N, 13Cα, 13Cβ, and 13C` with that of Lukin's (Lukin et al. 1997). The statistics of the chemical-shift difference, Δδ (averaged chemical-shift value in this study minus previously reported value), are also listed in Table 3. As shown in Tables 1 and 3, despite significant differences for certain amino acids, on average, the 1HN, 1Hα, and 13Cα chemical shifts reported in this study are in good agreement with the earlier results, whereas for 15N, 13Cβ, and 13C` chemical shifts, the discrepancies are relatively larger for both the overall averaged chemical shift and certain individual amino acids. Because the random coil chemical shift is usually used as a reference in the identification of secondary structure, a number of random coil data sets have been published. A comparison of the random coil chemical shift obtained in this study with those most recently obtained by Schwarzinger et al. (2000) on Ac-GGXGG-NH2 hexapeptides in pH 2.3, 8 M urea is listed in Table 4. As shown in Table 4, despite the difference in the methods applied, on average, the random coil chemical shifts of 1HN, 1Hα, 13Cβ, and 13Cα reported in this study are in good agreement with those of Schwarzinger et al. (2000). However, as is apparent from this table, systematic deviation of nearly 1.0 ppm is observed between the two sets of data for 13C` for most of the amino acids except Asp and Cys. As the data of Schwarzinger et al. (2000) were obtained at very low pH (2.3), the CO may be partly protonated and, hence, affect the observed the chemical shifts. It is of great interest to find that significant deviations also exist for 13Cβ chemical shift on Asp and Cys. One possible reason for this could be the formation of an intra-residue hydrogen bond, OH-CO for Asp and SH-CO for Cys. The other few significant deviations could be attributed to the different method used.
Table 3.
Statistic result of Δδ (averaged chemical shift in this study—previously reported valuea)
| 1HN | 1Hα | 15N | |||||||
| Amino acid | β-strand | Random coil | α-helix | β-strand | Random coil | α-helix | β-strand | Random coil | α-helix |
| Average | 0.07 | −0.07 | 0.03 | 0.11 | 0.02 | 0.00 | 0.14 | −0.57 | −0.58 |
| Std | 0.16 | 0.16 | 0.11 | 0.10 | 0.11 | 0.07 | 0.88 | 0.74 | 0.66 |
| Max | 0.32 | 0.22 | 0.26 | 0.3 | 0.32 | 0.15 | 1.56 | 1.21 | 0.58 |
| Min | −0.27 | −0.44 | −0.11 | −0.04 | −0.16 | −0.16 | −1.18 | −1.68 | −2.0 |
| 13Cα | 13Cβ | 13C` | |||||||
| Amino acid | β-strand | Random coil | α-helix | β-strand | Random coil | α-helix | β-strand | Random coil | α-helix |
| a 1HN and 1Hα are from Wishart et al. (1991); 15N, 13Cα, 13Cβ, and 13C` are from Lukin et al. (1997). | |||||||||
| Average | −0.10 | −0.09 | −0.07 | 0.16 | −0.32 | −0.25 | −0.09 | −0.11 | −0.31 |
| Std | 0.25 | 0.46 | 0.34 | 0.46 | 0.38 | 0.33 | 0.50 | 0.32 | 0.52 |
| Max | 0.5 | 0.89 | 0.69 | 0.95 | 0.19 | 0.13 | 1.23 | 0.37 | 1.16 |
| Min | −0.5 | −1.21 | −0.98 | −0.9 | −1.16 | −1.45 | −0.9 | −0.73 | −1.03 |
Table 4.
The difference of the random coil chemical shifts (previously reported valuea minus that from this study)
| Amino acid | 13C` | 13Cβ | 13Cα | 1HN | 1Hα | 15N |
| Ala | 1.11 | 0.23 | 0.15 | 0.24 | 0.1 | 1.48 |
| Arg | 1.19 | 0.40 | 0.52 | 0.22 | 0.05 | 0.61 |
| Asn | 1.12 | 0.87 | 0.39 | 0.18 | 0.19 | 0.52 |
| Asp | −0.11b | −2.43b | −1.1 | 0.17 | 0.18 | −1.59 |
| Cys | 0.17b | −4.10b | 0.56 | 0.6 | −0.04 | 1.22 |
| Gln | 0.92 | 0.86 | 0.28 | 0.19 | 0.12 | 0.77 |
| Glu | 0.69 | −1.14 | −0.3 | 0.11 | 0.14 | −0.67 |
| Gly | 0.60 | 0.05 | 0.07 | 0.02 | −2.44 | |
| His | 0.22 | −0.5 | −0.39 | 0.47 | 0.29 | −0.77 |
| Ile | 1.64 | 0.65 | 0.98 | 0.23 | 0.08 | −0.67 |
| Leu | 1.59 | 0.59 | 0.62 | 0.16 | 0.03 | 0.87 |
| Lys | 1.25 | 0.64 | 0.31 | 0.23 | 0.08 | 0.16 |
| Met | 1.17 | 0.01 | 0.65 | 0.05 | −0.03 | 0.11 |
| Phe | 1.32 | 0.32 | 1.15 | 0.36 | 0.03 | 1.29 |
| Pro | 0.89 | 0.35 | 0.17 | 0.04 | ||
| Ser | 1.07 | 0.18 | 0.32 | 0.17 | 0.03 | −0.44 |
| Thr | 0.98 | 0.26 | 0.42 | 0.03 | 0.1 | −2.41 |
| Trp | 1.19 | 0.48 | −0.02 | 0.63 | 0.16 | 1.53 |
| Tyr | 1.38 | 0.23 | 0.56 | 0.36 | 0.03 | 0.85 |
| Val | 1.24 | 0.14 | 0.81 | 0.28 | 0.02 | −0.36 |
| Average | 0.98 | 0.27 | 0.31 | 0.24 | 0.08 | 0.00 |
| SD | 0.46 | 0.49 | 0.51 | 0.17 | 0.08 | 1.18 |
a Schwarzinger et al. (2000).
b Data is not included in the calculation of average and S.D.
Most important is that the standard deviation of the chemical-shift distribution, which is ignored in earlier reports, is reported for each individual amino acid in this study. As we show later, the standard deviation plays an important role in evaluating the reliability of the identification. As shown in Table 1, the value of standard deviation varies greatly with the amino acid as well as the secondary structure type. In earlier studies, the standard deviation value was either not considered (Wishart et al. 1992; Wishart and Sykes 1994) or used as an averaged value (Lukin et al. 1997). As shown in Figure 1 ▶, the chemical-shift distribution varies from one amino acid to another. Using the standard deviation data specified for each of the individual amino acid and secondary structure type certainly increases the accuracy of the secondary structure identification. The statistics of the chemical-shift data in Table 1, we believe, will have other potential applications, such as automatic chemical-shift assignment.
Fig. 1.

Simulated 13Cα chemical-shift distribution of (a) Ala and (b) Met. (•) Strand; (♦) coil; (▴) helix.
Reliability of identification
When using observed chemical shifts to identify secondary structure, we would like to know the reliability of the result. In this study, the reliability of using an observed chemical shift to identify secondary structure is investigated. This is accomplished by evaluating R, the resolution of the chemical-shift distribution between the three secondary structure types. The calculated Rstrand vs. coil and Rhelix vs. coil values (see Materials and Methods for definitions) for the 20 amino acids are listed in Table 5. The higher the Rstrand vs. coil or Rhelix vs. coil value, the higher the reliability of distinguishing a β-strand or an α-helix from a random-coil. A High-Low-Average chart of Rstrand vs. coil and Rhelix vs. coil is shown in Figure 2 ▶. Both Rstrand vs. coil and Rhelix vs. coil vary greatly with the nucleus and the amino acid. On the basis of the calculated values of Rstrand vs. coil and Rhelix vs. coil, on average, the reliability is in the order of: 13Cα>13C`>1Hα>13Cβ>15N>1HN to distinguish an α-helix from a random coil; and 1Hα>13Cβ >1HN ∼13Cα∼13C`∼15N to distinguish a β-strand from a random coil. In this study, we also found that the amide 15N and 1HN chemical shifts, which were generally excluded in prior work, have almost the same ability/resolution as those of 13Cα and 13C` to distinguish a β-strand from random coil. It is of interest that the Rstrand vs. coil values of amide 1HN are relatively higher for amino acids Cys, Phe, Ile, Val, Trp, Tyr, and His. Most of these amino acids are of high β-strand propensity (Kim and Berg 1993).
Table 5.
Calculated Rstrand vs. coil and Rhelix vs. coil for the 20 amino acids
| Rstrand vs coil | Rhelix vs coil | |||||||||||
| Amino acid | 1Hα | 13Cβ | 1HN | 13Cα | 13C` | 15N | 13Cα | 13C` | 1Hα | 13Cβ | 15N | 1HN |
| Ala | 0.85 | 0.92 | 0.3 | 0.79 | 0.66 | 0.2 | 0.66 | 0.61 | 0.28 | 0.3 | 0.29 | 0.12 |
| Cys | 0.46 | 0.22 | 0.6 | 0.53 | 0.38 | 0.6 | 0.34 | 0.8 | 0.45 | 0.12 | 0.08 | 0.19 |
| Asp | 0.65 | 0.7 | 0.07 | 0.25 | 0.28 | 0.36 | 1.02 | 0.88 | 0.36 | 0.11 | 0.13 | 0.27 |
| Glu | 0.74 | 0.81 | 0.27 | 0.32 | 0.35 | 0.37 | 1.03 | 0.83 | 0.52 | 0.29 | 0.12 | 0.05 |
| Phe | 0.55 | 0.65 | 0.56 | 0.15 | 0.24 | 0.29 | 1.08 | 0.33 | 0.65 | 0.09 | 0.08 | 0.16 |
| Gly | 0.18 | 0.09 | 0.11 | 0.24 | 0.03 | 0.83 | 0.65 | 0.13 | 0.37 | 0.01 | ||
| His | 0.57 | 0.61 | 0.5 | 0.23 | 0.27 | 0.35 | 1.08 | 0.6 | 0.4 | 0.04 | 0.02 | 0.03 |
| Ile | 0.77 | 0.43 | 0.58 | 0.18 | 0.23 | 0.33 | 1.02 | 0.39 | 0.62 | 0.35 | 0.03 | 0.02 |
| Lys | 0.88 | 0.83 | 0.34 | 0.47 | 0.43 | 0.17 | 0.95 | 0.39 | 0.57 | 0.23 | 0.18 | 0.11 |
| Leu | 0.58 | 0.57 | 0.37 | 0.33 | 0.44 | 0.46 | 0.81 | 0.49 | 0.5 | 0.15 | 0.27 | 0.08 |
| Met | 0.38 | 0.32 | 0.09 | 0.16 | 0.36 | 0.17 | 1.24 | 0.56 | 0.9 | 0.25 | 0.25 | 0.25 |
| Asn | 0.86 | 0.68 | 0.21 | 0.22 | 0.18 | 0.5 | 1.1 | 0.41 | 0.19 | 0.08 | 0.2 | 0.18 |
| Pro | 0.51 | 0.37 | 0.52 | 0.15 | 0.79 | 0.7 | 0.37 | 0.42 | ||||
| Gln | 0.77 | 0.84 | 0.18 | 0.42 | 0.41 | 0.43 | 0.91 | 1.15 | 0.38 | 0.19 | 0.24 | 0.1 |
| Arg | 0.65 | 0.6 | 0.26 | 0.32 | 0.3 | 0.17 | 0.96 | 0.69 | 0.35 | 0.18 | 0.23 | 0.08 |
| Ser | 0.69 | 0.62 | 0.22 | 0.43 | 0.28 | 0.18 | 0.79 | 0.76 | 0.48 | 0.48 | 0.13 | 0.07 |
| Thr | 0.6 | 0.37 | 0.19 | 0.16 | 0.41 | 0.37 | 1.21 | 0.61 | 0.43 | 0.54 | 0.13 | 0.09 |
| Val | 0.65 | 0.31 | 0.58 | 0.28 | 0.36 | 0.37 | 1.06 | 0.53 | 0.74 | 0.52 | 0.02 | 0.02 |
| Trp | 1.12 | 0.9 | 0.69 | 0.57 | 0.01 | 0.25 | 0.53 | 0.79 | 0.14 | 0.2 | 0.17 | 0.31 |
| Tyr | 0.44 | 0.6 | 0.54 | 0.32 | 0.2 | 0.26 | 0.83 | 0.48 | 0.53 | 0.1 | 0.03 | 0.19 |
Fig. 2.

High-Low-Average charts displaying the distribution of (a) Rstand vs. coil and (b) Rhelix vs. coil among the 20 amino acids.
Probability-based secondary structure identification
In this study, the secondary structure is identified by comparing the joint probabilities derived from the observed 13Cα, 13C`, 1Hα, 13Cβ, 15N, and 1HN chemical shifts. After the secondary structure type of each residue is assigned, empirical pattern filters/functions are used to smooth poorly defined secondary structure fragments. Testing this probability-based method on a set of 30 proteins gave an accuracy (on a residue per residue basis) of 89% for β-strand, 84% for random-coil, and 89% for α-helix. For all of the residues (>6100), the global accuracy Q3 (see Materials and Methods for definition) is 88%. Testing on the same set of proteins, the current CSI methods (Wishart et al. 1992; Wishart and Sykes 1994) gave a global accuracy of 81%. This improvement in accuracy, we believe, is due to the oversimplified protocol used by the CSI method, which assigns the secondary structure type to each residue by simply comparing the observed chemical-shift with the random coil value. More specifically, if the measured chemical-shift value is within the range of the random coil (± 0.1 ppm for 1H, ± 0.5 ∼0.7 ppm for 13C), then the residue is assigned as random coil, otherwise, it is assigned as either β-strand or α-helical. The over-simplified procedure used in CSI methods certainly causes a loss of accuracy. In addition, formerly ignored amide 15N and 1HN chemical shifts are included in this study, and they improve the accuracy and the confidence of identification.
For proteins with well-defined secondary structure and sufficient 1H, 13C, and 15N chemical-shift assignments, PSSI can give an accuracy of identification as high as 95% (i.e., HTLV-I Capsid Protein). We notice that almost all of the misidentifications occur between β-strand/random coil or α-helix/random coil. Among the misidentifications, those between β-strand/α-helix are <1%. Amide 15N and 1HN chemical shifts, which were easily obtained but excluded from application, were found actually useful, specifically for distinguishing β-strand from random coil. Including these two nuclei into the calculation allows a 1.6% increase of Q3 value for the proteins we tested.
If a residue is in a well-defined secondary fragment, in most cases it has a probability of that secondary structure type above 0.9 (the sum of the probabilities of all the secondary structure types are normalized to 1) and is readily distinguished from the other two types. Plotted in Figure 3 ▶, are the normalized joint probabilities calculated from 1H, 15N, and 13C chemical-shifts for a fragment of pathogenesis-related protein P14a as well as the secondary structures extracted from its 3D coordinates. The probability distributions shown in this figure provide high-confidence identification of the β-strand, random coil, and α-helix. Those with a poorly defined secondary structure type, for example, the isolated short β-bridge and 310-helix, are clearly indicated by the ambiguity of the probability between the three secondary structure types.
Fig. 3.

Normalized joint probabilities of the three secondary structure types for a fragment from Pathogenesis-related Protein P14a. The secondary structures derived from its 3D coordinates are marked at top. (▴) Strand; (▪) coil; (•) helix.
A JAVA user interface graphic program, PSSI, has been developed to make the process automatic. The chemical-shift data can be read from a file in the NMR-str format (essential for the deposition of the chemical-shift data in the BioMagResBank) or input by the user directly. Its output includes: (1) The normalized joint probability of each secondary structure type, the basis of which the secondary structure is automatically assigned. As we discussed earlier, in this study, empirical filters/functions are used to smooth the poorly defined secondary structure fragments. However, users can also make judgements by themselves on the basis of the probabilities provided by PSSI. (2) The secondary structure, which is editable, derived from chemical-shift data. (3) A graphic output displaying the identified secondary structure. A graphic output from PSSI showing the secondary structure derived from NMR chemical-shift data is shown in Figure 4 ▶. Because chemical-shift references, other than DSS, could be used, a function to calibrate the chemical-shift reference is also provided. The whole package is available at http://ccsr3150-p3.stanford.edu.
Fig. 4.
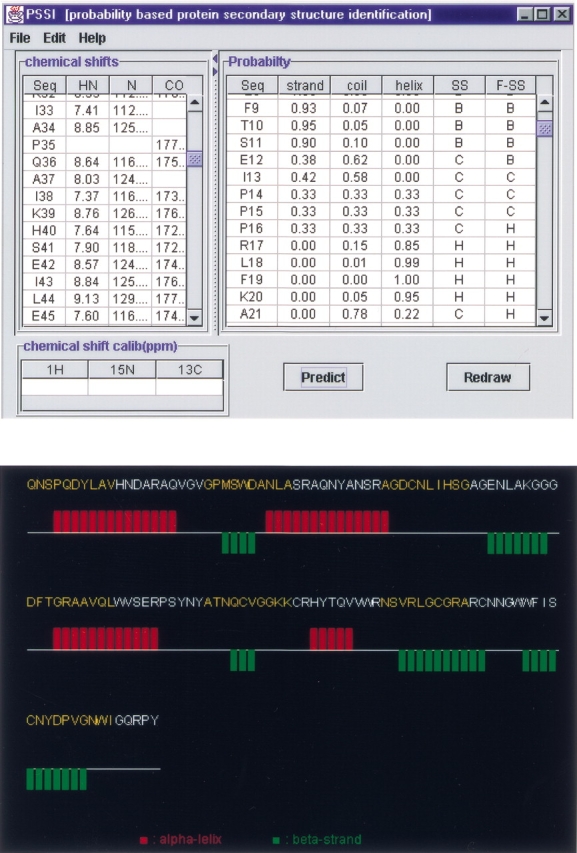
PSSI (top), and its graphic output (bottom) showing the secondary structure derived from NMR chemical-shift data.
Materials and methods
A data-set containing chemical shifts and secondary structures was established as follows: the whole BioMagResBank database was downloaded and entries meeting the following criteria were selected first. (1) 1H, 15N, and 13C chemical shifts were referred to DSS (Wishart et al. 1995b); (2) the temperature at which the NMR data were collected was 293–313 K; (3) pH of the sample is 4.5–8.5; and (4) the sequence length is >80. For each initial chemical-shift entry obtained, the corresponding coordinate file was carefully identified either on the basis of the information provided in chemical-shift file and/or by a key words search from Protein Data Bank (PDB). The second selection was based on availability of the 3D coordinates, number of assignments, and quality of the structures. In cases in which several PDB files were available for one protein, the priority of selection was given to the high-resolution X-ray structure. If only NMR structures were available, the consensus result from at least three structures was used. For some proteins, modification of the sequence numbering had to be made to precisely match the chemical-shift data with the coordinate data. Finally, a data-set of 36 distinct, nonparamagnetic proteins including >6100 amino acid residues, was set up for the statistical analysis.
Three programs, VADAR, DSSP, and STRIDE were compared and used to define the reference secondary structure from the 3D coordinate file. Consensus results from the three programs, taking into account the backbone φ and ξ dihedral angles of each residue, were used as the secondary structure reference.
During the statistical analysis of the chemical-shift distribution against secondary structure (β-strand, random coil, and α-helix), extremely large or small values (>4.5 standard deviation) were automatically removed. The chemical-shift data of the very first N-terminal and last C-terminal residues of each protein were also excluded to avoid possible terminal effects. The averaged chemical shift, standard deviation, and the distribution resolutions of each nucleus were calculated by use of either the JAVA program or MS Excel for all of the 20 amino acids. The resolution is defined as:
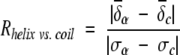 |
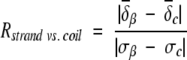 |
in which the averaged chemical shift and standard deviations are categorized according to the three secondary structure types (β-strand, random-coil, and α-helix).
Secondary structure identification
Two steps were used to identify the secondary structure elements from the observed NMR chemical-shift data.
Step l: Joint probability
Given a { δn} (n = δN, δCA, δCO, δCB, δHA, δHN) are the observed chemical shifts for amino acid i; the secondary structure type of this amino acid is adjudged by comparing the joint probability of the three secondary structure types, Ps,i.
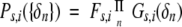 |
Fs,i represents the probability for amino acid i at the secondary structure type s (s = β-strand, random coil, or α-helix); Gs,i is the Gaussian distribution.
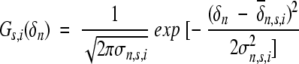 |
During the calculation, Gs,i(δn) is set to 1 if δn is not available. G of three secondary structure types is normalized so that its sum is equal to 1.0, as is the joint probability, Ps, for each residue. The secondary structure type is initially assigned to B, C, or H (representing β-strand, random coil, and α-helix, respectively) based on joint probability of each type, that is, B if Pβ,i>Pc,i and Pβ,i>Pα,i.
Step 2: Smoothing/filtering
This step is optional. Users can make their own adjustments on the basis of the calculated probabilities. The result from step 1 can be further smoothed/filtered by use of empirical patterns and smoothing functions. For example, (1) if a local density of either B or C exceeds one-half for a five-residue window, its secondary structure type is adjusted on the basis of Ps values, that is, a BBCBC segment will be adjusted to BBBBB (if Pβ of the last residue is >0.35) or BBBBC (if Pβ of the last residue = <0.35); (2) if a C-type residue is not covered by the smoothing/filtering in (1) and is located either at the end or in the middle of B or H segment, its Ps is recalculated and its secondary structure type is readjudged accordingly. For example, for a CHHH segment, the Ps,i of the first residue is recalculated as P s,i = 0.5*Ps,1 + 0.3*P s,2 + 0.2*Ps,3 for the second N-terminal residue; (3) separated short (less than two residues) B or H segments are smoothed to C.
The global accuracy of identification Q3 (Schulz and Schimer 1979) is defined as:
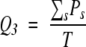 |
in which Ps is the number of residues identified in the s (β-strand, random coil, and α-helix) state, effectively observed in the s state; T is the total number of residues.
The JAVA user interface program (PSSI), which makes the entire procedure fully automated, was developed by one of the authors (Y.J. Wang)
Acknowledgments
NIH grant 1R01 Gm33385 is acknowledged.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.3180101.
References
- Colloch, N., Etchebest, C., Thoreau, E., Henrissat, B, and Mornon, J.P. 1993. Comparison of three algorithms for the assignment of secondary structure in proteins: The advantages of a consensus assignment. Protein Eng. 377–382. [DOI] [PubMed]
- Cuff, J.A and Barton, G.J. 1999. Evaluation and improvement of multiple sequence methods for protein secondary structure prediction. Proteins: Struct. Funct. Genet. 34 508–519. [DOI] [PubMed] [Google Scholar]
- Frishman, D and Argos, P. 1995. Knowledge-based protein secondary structure assignment. Proteins 23 566–579. [DOI] [PubMed] [Google Scholar]
- Goto, N.K and Kay, L.E. 2000. New developments in isotope labeling strategies for protein solution NMR spectroscopy. Curr. Opin. Struct. Biol. 10 585–592. [DOI] [PubMed] [Google Scholar]
- Kabsch, W, and Sander, C. 1983. A dictionary of protein secondary structure. Biopolymers 22 2577–2637. [DOI] [PubMed] [Google Scholar]
- Kay, L.E. and Gardner, K.H. 1997. Solution NMR spectroscopy beyond 25 kDa. Curr. Opin. Struct. Biol. 7 722–731. [DOI] [PubMed] [Google Scholar]
- Kim, C.A. and Berg, J.M. 1993. Thermodynamic β-sheet propensities measured using a zinc-finger host peptide. Nature 362 267–270. [DOI] [PubMed] [Google Scholar]
- Lukin, J.A., Gove, A.P., Talukdar, S.N., and Ho, C. 1997. Automated probabilistic method for assigning backbone resonance of 13C, 15N-labeled proteins. J. Biomol. NMR. 9 151–166. [DOI] [PubMed] [Google Scholar]
- Markley, J.L., Meadows, D.H., and Jardetzky, O. 1967. Nuclear magnetic resonance studies of helix-core transitions in polyamino acids. J. Mol. Biol. 27 25–35. [DOI] [PubMed] [Google Scholar]
- Nakamura, A. and Jardetzky, O. 1968. Systematic analysis of chemical shifts in the nuclear magnetic resonance spectra of peptide chains. II. Oligoglycines. Biochemistry 7 1226–1230. [DOI] [PubMed] [Google Scholar]
- Pastore, A. and Saudek, V. 1990. The relationship between chemical shifts and secondary structure in proteins. J. Magn. Reson. 90 165–176. [Google Scholar]
- Richards, F.M. and Kundrot, C.E. 1988. Identification of structural motifs from protein coordinate data: secondary structure and first-level supersecondary structure. Proteins. 3 71–84. [DOI] [PubMed] [Google Scholar]
- Schulz, G.E. and Schimer, R.H. 1979. Principles of proteins structure. Springer-Verlag, New York.
- Schwarzinger, S., Kroon, G.J.A., Foss, T.R., Wright, P.E., and Dyson, H.J. 2000. Random coil chemical shifts in acidic 8 M urea: Implementation of random coil shift data in NMRView. J. Biomol. NMR 18 43–48. [DOI] [PubMed] [Google Scholar]
- Sklenar, H., Etchebest, C., and Lavery, R. 1989. Describing protein structure: A general algorithm yielding complete helicoidal parameters and a unique overall axis. Proteins. 6 46–60. [DOI] [PubMed] [Google Scholar]
- Szilagyi, L, and Jardetzky, O. 1989. α-Proton chemical-shifts and secondary structure in proteins. J. Magn. Reson. 83 441–449. [Google Scholar]
- Williamson, M.P., Asakura, T., Nakamura, E., and Demura, M. 1992. A method for the calculation of protein α-CH chemical-shifts. J. Biomol. NMR. 2 93–98. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S. and Sykes, B.D. 1994. The 13C chemical-shift index: A simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR. 4 171–180. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S. and Nip, A.M. 1998. Protein chemical shift analysis: A practical guide. Biochem. Cell Biol. 76 1–10. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S., Sykes, B.D., and Richards, F.M. 1991. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 222 311–333. [DOI] [PubMed] [Google Scholar]
- ———. 1992. The chemical shift index: A fast and simple method for the assignment of protein secondary structure through NMR spectroscopy Biochemistry 31 1647–1651. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S., Willard, L., and Sykes, B.D. 1995a. VADAR 1.1– University of Alberta (http://redpoll.pharmacy.ualberta.ca).
- Wishart, D.S., Bigam, C.G., Yao, J., Abildgaard, F., Dyson, H.J., Oldfield, E., Markley, J.L, and Sykes, B.D. 1995b. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J. Biomol. NMR. 6 135–140. [DOI] [PubMed] [Google Scholar]
- Wuthrich, K. 1986. NMR of proteins and nuclei acids. Wiley, New York.