

Abstract
The characteristic properties of the blood-brain barrier (BBB) forming brain capillary endothelial cells (BCEC) are modulated by their microenvironment, but the cellular sources of the induction signals are still unclear. Apart from astrocytes, another cell type in close contact with cerebral blood vessels is the perivascular macrophages, which are known to be regularly replaced by blood-derived monocytic precursor cells. It is unknown if, and how, these cells may interact with the cerebral endothelium and modulate its BBB-specific functions. In the present study, a cell culture model of the BBB was used to investigate the effect of blood-derived human macrophages on the permeability of cultured bovine and human BCEC, determined by a transendothelial electrical resistance (TEER) measurement. We found that the TEER of postconfluent BCEC was considerably increased by a non-contact coculture with macrophages. After 24 h, we found a TEER augmentation of over 50 % compared with the control without coculture, and this effect was comparable to the response of BCEC to a C6 glioma cells coculture. Stimulation or HIV-1 infection of the macrophages did not alter their effect on BCEC monolayer permeability. Investigation of signal transduction pathways showed that TEER increase of BCEC due to macrophage coculture was cAMP-independent and involves neither phospholipase C, protein kinase C nor calmodulin. Our findings demonstrate that macrophages are able to modulate BBB-specific functions in cultured BCEC. Thus, these cells or cerebral cells of monocytic origin (e.g. perivascular macrophages), may be part of the microenvironment of BCEC that modulates their specific properties in vivo.
The term blood-brain barrier (BBB) was first used about 100 years ago to describe the failure of Prussian blue dyes to stain the brain. The proof that this was a true barrier and not a question of dye affinity was provided by Goldman working in the laboratory of Paul Ehrlich at the Georg-Speyer-Haus who showed that trypan blue injected into the blood stained virtually every tissue but the brain and that the same dye injected directly into the brain stained the brain but did not escape into the general circulation (Goldmann, 1913).
It is now recognised that brain capillary endothelial cells (BCEC) represent the structural basis of the blood-brain barrier (BBB) and that they are vastly different from the peripheral endothelium. These cells have tight circumferential junctions which effectively abolish any aqueous paracellular diffusional pathways between the cells (Brightman & Reese, 1969; Brightman & Kadota, 1992). The cerebral capillaries are thus characterised by a high transendothelial electrical resistance (TEER) of 1500–2000 Ω cm2 (Crone & Olsen, 1982; Butt et al. 1990) which effectively prevents the diffusion of polar solutes between plasma and brain extracellular fluid.
Early transplantation studies showed that the microenvironment and not the origin of the cerebral endothelium plays an essential role in induction and maintaining its specific properties (Stewart & Wiley, 1981), but even after numerous decades of BBB research the responsible factors or induction signals are still not characterised in detail. There is evidence that the differentiation of the cerebral endothelium to form tight junctions and the expression of transporters and specific markers of the BBB are induced mainly by the close association or contact between cerebral endothelial cells and the foot processes of astrocytic glial cells (Kacem et al. 1998). These glial cells apparently induce BBB differentiation on the cerebral endothelial cells at least partly by means of soluble factors (Jantzer & Raff, 1987): non-contact coculture with astrocytes or astrocyte-conditioned medium will induce many BBB features, for example polarisation of transporters and the formation of tight junctions in endothelial cells grown in culture (Dehouck et al. 1990; Rubin et al. 1991; Wolburg et al. 1994). However, it is very probable that astrocytes are not the sole part of the BBB microenvironment that modulates its specific properties, since some inconsistent observations and findings provide evidence that more complex mechanisms are involved in BBB induction and maintenance. Firstly, pial blood vessels on the surface of the brain show a high electrical resistance typical of the BBB, although they are only incompletely covered with astrocytic foot processes (Allt & Lawrenson, 1997; Cassella et al. 1997). Moreover, it was shown in developmental studies that the first BBB-specific markers of the cerebral endothelium appear at a stage where astrocytes are not present, and the complete BBB phenotype is entirely developed soon after postpartum (Qin & Sato, 1995). Thus, development and maintenance of the BBB is a prolonged and continuous process that probably involves several different factors or signals, possibly from cells other than astrocytes. For instance, nerve endings found in vicinity of cerebral capillaries could represent a possible source of these factors (Cohen et al. 1997).
Another cell type in close contact with cerebral blood vessels are the cerebral perivascular macrophages, also referred to as perivascular cells, perivascular microglia, fluorescent granular perithelial cells, or Mato cells (Williams et al. 2001). The immunophenotype, the location in the perivascular space as well as the morphology, frequency and tissue distribution clearly distinguish perivascular macrophages from pericytes and resident intraparenchymal microglia (Graeber et al. 1989, 1992). It is suggested that perivascular macrophages are a normal constituent of the human cerebral microvasculature, and studies in rodents and humans have shown that these cells are bone-marrow derived and regularly replaced by monocytic precursor cells, therefore representing tissue macrophages of the brain (Hickey & Kimura, 1988; Hickey et al. 1992; Lassmann et al. 1993; Kennedy & Abkowitz, 1997; Bechmann et al. 2001). Indeed, perivascular macrophages are recognised by markers for peripheral macrophages, confirming their origin from the monocyte lineage (Hickey et al. 1992; Lassmann et al. 1993). Circulating blood monocytes are able to interact with the cerebral endothelial cells inducing a response which allows their passage into the brain (Carlos & Harlan, 1994; Springer, 1994). It is not known if and how these cells interact with the cerebral endothelium and modulate its BBB-specific properties after they have entered the brain, reaching their final destination in close apposition to the cerebral endothelium and their differentiation into perivascular macrophages.
To obtain insight regarding the above question and to verify the contribution of human blood-derived macrophages in BBB modulation, we examined the regulation of the barrier function of cultured BCEC in a non-contact coculture system using a cell culture model of the BBB, consisting of primary bovine and human BCEC monolayers grown on porous polycarbonate membranes (Dehouck et al. 1990; Rubin et al. 1991; Miller et al. 1992). Additionally, we investigated to see if a pathological condition of the macrophages, i.e. proinflammatory stimulation or HIV-1 infection, results in an altered macrophages-BCEC interaction. Furthermore, the role of certain key signal transducers in the macrophages-modulated regulation of BCEC monolayer permeability was studied.
METHODS
Isolation of cerebral blood capillaries from bovine and human brain tissue
Fresh bovine brain tissue was obtained from the local slaughterhouse and kept in cold phosphate buffered saline (PBS, Biochrom, Berlin, Germany) until preparation. Human brain material was obtained during forensic autopsies with permission of the Ethical Commission of the Medical Faculty, 6 h post mortem in accordance with national ethical standards. After removal of the meninges the grey matter was collected and minced into small pieces. The minced brain tissue was digested with 10 U ml−1 dispase (Sigma-Aldrich, Taufkirchen, Germany) for 70 min at 37 °C. Then, the released capillaries were separated from the tissue debris by diluting the homogenised tissue in a 1:2 ratio with a 40 % (v/v in PBS) Percoll solution (Biochrom) and density centrifugation was made at 2685 g for 20 min at 4 °C (Minifuge RF, Heraeus Instruments, Hanau, Germany). The pelleted blood vessels were further digested with 500 U ml−1 collagenase (Sigma) for 20 min at 37 °C. This step was followed by a digestion step with 0.1 U ml−1 DNase (Boehringer, Mannheim, Germany) at 37 °C until dispersion of visible clumps of capillaries occurred. Large macrovascular blood vessels and incomplete digested capillary fragments were then removed by filtration through a 40 μm cell strainer (BD Biosciences, San Jose, CA, USA). Finally, the capillaries were separated from other contaminating cells by centrifugation on a continuous 57 % (v/v in PBS) Percoll gradient at 1135 g for 10 min at 10 °C.
Culture of brain capillary endothelial cells
Brain capillary fragments were plated directly after isolation onto porous polycarbonate membranes of cell culture plate inserts with a diameter of 12 mm, a pore size of 3 μm and a pore density of 2 × 106 pores cm−2 (Transwell 3402, Corning Life Sciences, Wiesbaden, Germany). Before use, the Transwells were precoated with type IV collagen (15 μg cm−2, Sigma) in accordance with the manufacturers instructions.
After one day, brain capillary endothelial cells (BCEC) had fully grown out from the dispersed capillary fragments. For the first two days, they were cultured in supplemented Dulbecco's modified Eagle's medium (DMEM, BioWhittaker, Verviers, Belgium) with 10 % fetal calf serum (Life Technologies, Eggenstein, Germany) and 2 % retinal extract as a crude source of fibroblast growth factors (Wolburg et al. 1994). On day 2 after isolation, the BCEC cultures were washed to remove debris from detached cells and dissolved capillary fragments, and for conditions of coculture the medium was switched to supplemented medium 199 (M199, Sigma) with 10 % human AB serum (Sigma) and 2 % retinal extract. For light microscopy and immunocytochemistry, BCEC were cultured in 24-well cell culture plates (Corning Life Sciences) under the same conditions as the Transwell cultures. All BCEC cultures were kept in an incubator (Cytoperm, Heraeus Instruments, Hanau, Germany) at 37 °C with 7 % CO2 and 90 % humidity.
The bovine BCEC cultures were generally confluent on days 2–3 in vitro and developed BBB-specific low permeability cell monolayers, which were checked regularly by transendothelial electrical resistance (TEER, see below) measurement, and occasionally by determination of their paracellular permeability for radiolabelled sucrose as an additional control.
Cultured postconfluent BCEC were routinely characterised by their typical spindle-shaped morphology, their expression of von Willebrand's factor and the characteristic staining pattern of the tight junction-associated protein zonula occludens-(ZO-)1 (not shown). Only an occasional occurrence of smooth muscle cell actin-positive pericytes in the BCEC cultures was found (> 98 % purity).
Transendothelial electrical resistance (TEER)
Transendothelial electrical resistance of BCEC monolayers was determined using a combination of an Millicell-ERS apparatus (Millipore, Eschborn, Germany) together with chamber electrodes (Endohm, World Precision Instruments, Berlin, Germany) that ensured accurate measurements. The Transwell inserts with the BCEC cultures were placed into the chamber electrode filled with Hepes-buffered (25 mM, Life Technologies) serum-free DMEM under sterile conditions, so that sequential daily measurements could be performed. Resistance values of multiple Transwell inserts of an experimental group were measured sequentially and the mean was expressed in the common unit (Ω cm2) after subtraction of the value of a blank cell-free filter. Normally, TEER of all bovine BCEC monolayers reached its maximum between days 6 and 8 in vitro and was typically above 1000 Ω cm2 for most preparations. For the sole preparation of human BCEC that was performed, a TEER maximum of about 200 Ω cm2 was achieved on day 9 in vitro.
Isolation and culture of human monocytes and/or macrophages from peripheral blood
Human monocytes and/or macrophages (MO/MAC) were isolated from peripheral blood with the Ficoll-Hypaque method. Informed written consent was obtained from the donors. Briefly, peripheral blood mononuclear cells (PBMC) were separated from erythrocytes by density centrifugation on a Ficoll (Biochrom) gradient, washed and resuspended in culture media (5 × 106 cells ml−1) consisting of supplemented RPMI 1640 (Biochrom) with 4 % human AB serum (Sigma); the PBMC cultures consisted of about 90 % lymphocytes and 10 % MO/MAC. The cell suspension was transferred into teflon bags to prevent firm attachment of the MO/MAC, allowing plating of these cells in cell culture dishes at any time (Andreesen et al. 1983).
Non-contact coculture system of BCEC with human macrophages
Transendothelial electrical resistance of bovine BCEC monolayers reached its maximum usually around day 7 in vitro, so all experiments were performed on day 7 after isolation of the BCEC, except the sole coculture experiment with human BCEC which was performed on day 9 after isolation.
After 7 or 14 days of maturation in the teflon bag, the macrophages were plated into 12-well cell culture plates (Corning Life Science) at a density of 2.5 × 105 (7-day-old macrophages) or 2.0 × 105 (14-day-old macrophages) cells cm−2. After 1 h, the non-adherent lymphocytes were removed by repeated thorough washing steps. After day 1, the coculture system was initiated by transferring the Transwell inserts with the BCEC into the cell culture plates with the macrophages cultures. For control, inserts were set into empty wells without cells. Change of BCEC monolayer permeability was monitored by daily TEER measurements.
For coculture of BCEC with proinflammatory stimulated macrophages, the macrophages were cultured before start of the coculture experiment for 4 h in the presence of 100 ng ml−1 lipopolysaccharide (LPS, kindly provided by Dr Galanos, MPI Freiburg, Germany).
For coculture of BCEC with HIV-1-infected macrophages, the macrophages were also plated on day 7 in vitro and the lymphocytes removed by washing. After one day, the macrophages were infected with two different macrophagotropic HIV-1 isolates (strain D117III and ADA-M) by incubating with virus supernatant (von Briesen et al. 1990). After 24 h, the supernatant was washed out and replaced by fresh medium. After day 6, the HIV-1-infected macrophages were used for the coculture experiments as described above. A productive virus infection of the macrophages was monitored by measuring reverse transcriptase activity in the culture supernatant (Rübsamen-Waigmann et al. 1986).
Non-contact coculture system of BCEC with C6 glioma cells
Additional cocultures with C6 glioma cells (European Collection of Cell Cultures, Salisbury, UK) instead of macrophages were used and performed similarly in parallel experiments. C6 cultures were subcultured one day before start of coculture experiments and plated into cell culture plates (2.5 × 105 cells cm−2), to form a confluent monolayer when coculture started on day 7 after isolation of the BCEC.
Signal transduction involvement in tight junction regulation
To study involvement of certain signal transduction pathways in the interaction of macrophages (or C6 cells) with the cocultured BCEC and the regulation of tight junctions, BCEC were treated with various pharmacological agents known to influence key signal transducers (see Table 1). These agents were added to the BCEC culture medium in the upper compartment of the BBB model system directly after the start of the coculture experiments. All agents were purchased from Biomol (Hamburg, Germany).
Table 1.
Agents and concentrations used to study involvement of certain signal transduction mediators in the interaction of macrophages (or C6 cells) with the cocultured BCEC and the regulation of tight junctions
| Agent | Description | Concentration (JOA) |
|---|---|---|
| Forskolin | Adenylate cyclase activator | 50 |
| RO 20–1724 | Selective inhibitor of cAMP-specific type IV-phosphodiesterases | 50 |
| SQ22536 | Adenylate cyclase inhibitor | 100 |
| H-7 | Inhibits protein kinases C, A, G and myosin light chain kinase | 50 |
| H-8 | Inhibits protein kinases A and G selectively over protein kinase C and myosin light chain kinase | 50 |
| U-73122 | Inhibits agonist-induced phospholipase C (PLC) activation | 10 |
| W-7 | Binds to calmodulin, inhibiting Ca2+-calmodulin-regulated enzyme activity | 50 |
Data presentation and statistical analysis
All results are expressed as the mean ± S.E.M. Differences between experimental groups with respect to TEER value were analysed using the non-parametric Wilcoxon-Mann-Whitney U test (Siegel & Castellan, 1988; Sachs, 1992). Statistical significance was defined at P < 0.02 values.
RESULTS
Human macrophages enhance barrier function of bovine and human BCEC comparable with C6 glioma cells
Non-contact coculture of postconfluent primary bovine or human brain capillary endothelial cells (BCEC) with human blood-derived macrophages substantially decreased their paracellular permeability determined by transendothelial electrical resistance (TEER) measurement. As a representative experiment shown in Fig. 1, the coculture of bovine BCEC with macrophages resulted in a considerable increase of the TEER from a baseline of about 1400 Ω cm2 before coculture to about 2000 Ω cm2 after 24 h (P = 0.001), whereas TEER of control BCEC cultures without coculture decreased. This represented a TEER augmentation of over 50 % of the control without coculture at that time, comparable to the coculture of BCEC with C6 glioma cells (see also Fig. 5 and Fig. 6). The macrophage-induced increase in the TEER of BCEC was always reproducible: similar results were found in a number of other coculture trials performed (see also Figs 3–6).
Figure 1. TEER of bovine BCEC increases due to coculture with human macrophages and is comparable with the coculture with C6 glioma cells.
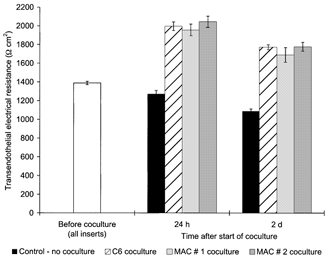
Transendothelial electrical resistance (TEER) across postconfluent bovine brain capillary endothelial cell (BCEC) monolayers after 24 h and 2 days coculture with either C6 glioma cells or human blood-derived macrophages (MAC); in the control group, the BCEC remained without coculture (as described in Methods). Data shown are the combined results of two independent BCEC preparations with approximately similar TEER baseline levels before coculture, and each BCEC population was cocultured with macrophages from two different donors (MAC 1 and 2) and the same C6 cell line (n = 6 for all groups). The mean TEER value before start of the coculture of all inserts used was 1388 ± 17 Ω cm2. The TEER of all coculture groups was found to be statistically different from those of the control group without coculture (P = 0.001).
Figure 5. Treatment of BCEC cocultured with macrophages or C6 cells with adenylate cyclase activator or phosphodiesterase inhibitor resulted in an additional TEER increase.
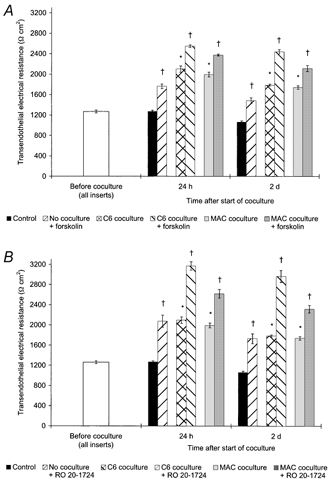
TEER across postconfluent bovine BCEC monolayers after 24 h and 2 days of coculture with C6 glioma cells or human blood-derived macrophages (MAC) and/or treatment with either 50 μM adenylate cyclase activator forskolin (A) or 50 μM phosphodiesterase inhibitor RO 20–1724 (B). In the control group, the BCEC remained untreated and without coculture. Data shown are the combined results of two independent BCEC preparations with approximately similar TEER baseline levels before coculture, and each BCEC population was cocultured with macrophages from two different donors and the same C6 cell line (n = 6 for all groups). The mean TEER value before start of the coculture of all inserts used was 1265 ± 28 Ω cm2. The TEER of all untreated coculture groups was found to be statistically different from those of the control group without coculture (*P = 0.001), and the TEER of each forskolin- or RO 20–1724-treated group was found to be statistically different from those of the corresponding group without †P≤ 0.012).
Figure 6. No effect of adenylate cyclase inhibitor on the TEER of BCEC cocultured with macrophages or C6 cells.
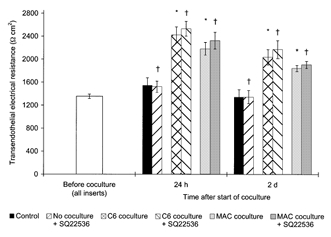
TEER across postconfluent bovine BCEC monolayers after 24 h and 2 days of coculture with C6 glioma cells or human blood-derived macrophages (MAC) and/or treatment with 100 mm adenylate cyclase inhibitor SQ22536. In the control group, the BCEC remained untreated and without coculture. Data shown are the combined results of two independent BCEC preparations with approximately similar TEER baseline levels before coculture, and each BCEC population was cocultured with macrophages from two different donors and the same C6 cell line (n = 6 for all groups). The mean TEER value before start of the coculture of all inserts used was 1353 ± 39 Ω cm2. The TEER of all untreated coculture groups was found to be statistically different from those of the control group without coculture (*P = 0.001). No statistical differences were found between the TEER of each SQ22536-treated group from those of the corresponding group without †P≥ 0.1).
Figure 3. Proinflammatory stimulation of macrophages did not interfere with their effect on the TEER of the cocultured BCEC.
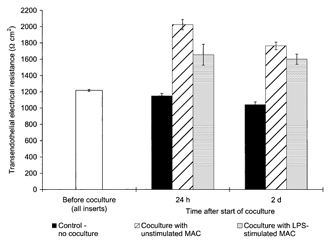
TEER across postconfluent bovine BCEC monolayers after 24 h and 2 days coculture with human blood-derived macrophages (MAC) which were stimulated with lipopolysaccharide (LPS, 100 ng ml−1) or unstimulated. In the control group, the BCEC remained without coculture. In each coculture group, data shown are the combined results for BCEC cocultured with macrophages derived from two different donors (n = 6 for all coculture groups, n = 3 for control group). The mean TEER value before start of the coculture of all inserts used was 1216 ± 12 Ω cm2. TEER of the group in coculture with unstimulated macrophages was found to be statistically different from those of the control group without coculture (P = 0.011). No statistical differences were found between the TEER of the group cocultured with stimulated macrophages with those of the corresponding group cocultured with unstimulated macrophages (P≥ 0.032).
Coculture of BCEC of human origin with macrophages resulted in a delayed TEER increase. The increase, however, was more sustained and continued for several days, whereas TEER of the control continuously diminished. A considerable and significant TEER augmentation of about 60 % was first established after 2 days of macrophage coculture (P < 0.02), and after 6 days the TEER of BCEC cocultured with macrophages was about 9-fold higher than that of the controls without coculture (see Fig. 2).
Figure 2. Delayed increase of the TEER of human BCEC due to coculture with human macrophages.
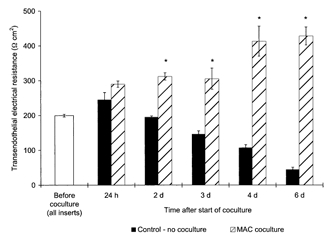
TEER across postconfluent human BCEC monolayers during prolonged coculture with human blood-derived macrophages (MAC); in the control group, the BCEC remained without coculture. In the macrophage group, the data shown are the combined results of two BCEC populations that were cocultured with diversely differentiated macrophages (8 days and 15 days in culture) from two different donors (n = 6 for coculture group, n = 3 for control group). The mean TEER value before start of the coculture of all inserts used was 200 ± 4 Ω cm2. After 2 days of coculture, the TEER of the macrophages coculture group was found to be statistically different from those of the control group without coculture (*P < 0.02).
Stimulation or HIV-1 infection of macrophages did not interfere with their effect on BCEC monolayer permeability
Treatment of the macrophages with lipopolysaccharide (LPS, 100 ng ml−1) showed no significant influence of this proinflammatory stimulation on their effect on BCEC monolayer permeability (see Fig. 3). After 24 h, the TEER of BCEC cocultured with LPS-stimulated macrophages was slightly reduced, but the difference to the group with unstimulated macrophages was found to be not significant (P≥ 0.032).
Similarly, coculture of BCEC with macrophages infected with two different HIV-1 strains (D117III and ADA-M) showed that HIV-1 infection had no influence on the effect of macrophages on BCEC monolayer permeability. As a representative experiment shown in Fig. 4, coculture of bovine BCEC with macrophages resulted in an increase of the TEER, irrespective of whether cells were infected or not. After 24 h, a TEER augmentation of over 70 % was found for both HIV-1 infected- and uninfected-macrophage populations. After a long-term coculture period of 9 days, the TEER of all coculture groups was about 5-fold higher than that of the control without coculture. No differences could be found between the infected and corresponding uninfected coculture groups for all points of time (P≥ 0.032).
Figure 4. HIV-1 infection of macrophages did not interfere with their effect on the TEER of the cocultured BCEC.
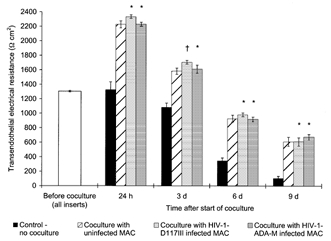
TEER across postconfluent bovine BCEC monolayers during prolonged coculture with human blood-derived macrophages (MAC) which were infected with two different macrophagotropic HIV-1 isolates (strain D117III and ADA-M) or uninfected; in the control group, the BCEC remained without coculture. In each coculture group, data shown are the combined results for BCEC cocultured with macrophages from two different donors (n = 6 for all coculture groups, n = 3 for control group). The mean TEER value before start of the coculture of all inserts used was 1304 ± 11 Ω cm2. The TEER of all coculture groups was found to be statistically different from those of the control group without coculture (P < 0.013). No statistical differences were found between the TEER of the groups cocultured with HIV-1-infected macrophages to those of the corresponding group cocultured with uninfected macrophages (*P > 0.1, †P = 0.032).
A productive HIV-1 infection of the macrophages during coculture was verified and confirmed by measurement of the HIV-1 specific enzyme reverse transcriptase in the culture medium of the macrophage populations (data not shown).
Signal transduction involvement in macrophages: BCEC interaction and tight junction regulation
The investigation of certain signal transduction pathways showed that a rise of the intracellular cAMP level of the BCEC cocultured with macrophages or C6 glioma cells by treatment with either the adenylate cyclase activator forskolin or the phosphodiesterase inhibitor RO 20–1724 resulted in an additional increase of TEER. As a representative experiment shown in Fig. 5, TEER of untreated BCEC increased about 60 % after 24 h in all coculture groups, compared with control without coculture (P = 0.001). Treatment with forskolin resulted in an additional and significant (P≤ 0.012) TEER increase of about 30 % in the coculture groups. Total TEER increase was 86 % in the macrophages coculture group and 100 % in the C6 coculture group, whereas TEER increase of the corresponding forskolin-treated group without coculture was only 38 %. A similar result was found for the parallel RO 20–1724 treatments: after 24 h, an additional TEER augmentation of 43 % was found in the RO 20–1724-treated BCEC group cocultured with macrophages (in total 106 %) and 86 % in the treated C6 coculture group (in total 149 %), whereas TEER of the corresponding treated group without coculture increased only about 63 % in total (P≤ 0.012). Two days after the start of the experiment, the TEER of all groups had slightly diminished, compared with the corresponding 24 h values, but a similar relationship between the groups still existed.
In contrast, the TEER augmentation induced by macrophages or C6 coculture could not be influenced by treatment of the BCEC with the adenylate cyclase inhibitor SQ22536 (see Fig. 6). Both 24 h and 2 days after the start of the experiment, TEER of all coculture groups was about 50 to 80 % higher than those of the control group without coculture (P = 0.001), and no difference was found between each SQ22536-treated group and the corresponding untreated group (P≥ 0.1).
A similar result was found when the macrophages or C6 cocultured BCEC were treated with other specific inhibitors of various signal transduction pathways: protein kinase inhibitors H-7 and H-8, phospholipase C inhibitor U-73122 and Ca2+-calmodulin inhibitor W-7 also failed to change the effect of macrophages and C6 on BCEC monolayer permeability, since no differences were found between the treated and the corresponding untreated coculture groups (P≥ 0.1, data not shown).
DISCUSSION
In this study, it was found that a non-contact coculture of postconfluent primary bovine or human brain capillary endothelial cells (BCEC) with human blood-derived macrophages substantially increased their TEER and consequently decreased their paracellular permeability. This TEER increase induced by macrophage factors was comparable with the response of BCEC to coculture with C6 glioma cells; these C6 cells are often used to reduce the permeability of cultured primary BCEC and to improve the barrier function of in vitro BBB model systems (Rubin et al. 1991; Raub et al. 1992; Wolburg et al. 1994).
In the heterologous system using BCEC of bovine origin, both the absolute and relative TEER increase was maximal 24 h after the start of coculture with human macrophages. The peak TEER of bovine BCEC cocultured with macrophages or C6 glioma cells without further influence of both cell populations was always about 2000 Ω cm2 and in some cases substantially above that; therefore in the range or even higher than the TEER of pial blood capillaries in vivo (Crone & Olsen, 1982; Butt et al. 1990). This TEER value is clearly superior to other BBB cell culture systems and is produced by a highly optimised in vitro BBB model system which is characterised by a high baseline TEER per se. The high intrinsic TEER level thus allowed very sensitive measurements of additional signals modulating the tight junctions and barrier function of the cultured BCEC.
In contrast, in the homologous system using a BBB cell culture model consisting of human BCEC, the TEER increase as a consequence of the macrophage coculture was delayed. The absolute peak TEER was lower and was only reached after prolonged coculture over 6 days, most probably due to the much lower TEER baseline of the human BCEC monolayers before the start of coculture. This result is probably due to the lower quality of the human brain autopsy material, compared with fresh bovine brain tissue obtained from the slaughterhouse under controlled conditions.
The barrier enhancing effect of macrophages on the BCEC was found not only with normal cells, but also when the macrophages were in a pathological condition. Neither the proinflammatory stimulation of the macrophages with LPS nor the highly productive HIV-1 infection altered the TEER enhancing effect of the macrophages on the cocultured BCEC.
The finding that infection of macrophages with HIV-1 did not interfere with their barrier enhancing effect on the cocultured BCEC seems to be inconsistent with the observation that the permeability of the BBB is increased in patients with AIDS, and that the integrity of the tight junctions of the brain capillaries are partially disturbed in patients with HIV-1 encephalitis (HIVE) and AIDS dementia complex (Petito & Cash, 1992; Power et al. 1993; Dallasta et al. 1999; Berger et al. 2000). These alterations in the BBB are often associated with perivascular accumulation of HIV-1 infected and activated macrophages and microglial cells (Gelman et al. 1992; Fischer-Smith et al. 2001), and both cell types have been identified as the primary source of various cytokines which are known to impair the barrier function of BCEC in vitro (Achim & Wiley, 1996; de Vries et al. 1996; Giulian et al. 1996; Blum et al. 1997; Mark & Miller, 1999). Thus, it has often been supposed that these cells are mainly responsible for the BBB impairment observed in AIDS patients. However, relevant evidence suggests that this simple conclusion is insufficient. First, HIV-1-infection per se does not transform macrophages into an activated state with increased secretory and cytotoxic activity; this activation is not achieved unless an additional secondary immune stimulation of the cells occurs (Genis et al. 1992; Nottet & Gendelman, 1995; Esser et al. 1996). Second, the alterations in the BBB were only observed in patients with symptomatic AIDS and HIVE, but were never found in HIV-1-infected individuals without any neuropathological symptoms (Petito & Cash, 1992; Power et al. 1993; Dallasta et al. 1999; Berger et al. 2000), although HIV-1 enters the brain during acute HIV-1 infection in a very early stage soon after seroconversion and remains present (Ho et al. 1985; Chiodi et al. 1986; Goudsmit et al. 1986; Petito et al. 1986; von Briesen et al. 1987). However, a detectable disturbance in BBB function occurs only after a prolonged latent period and entrance into the symptomatic AIDS phase. Most probably, HIV-1 enters the brain via infected blood-derived monocytes/ macrophages, which represent a primary target of HIV-1 in the blood (the Trojan horse theory, Peluso et al. 1985). Following brain entry, the infiltrating HIV-1-infected monocytes/macrophages probably remain in the vicinity of the cerebral microvasculature and initially spread the virus over the whole localised perivascular macrophage population representing the major target of HIV-1 in the brain (Smith et al. 1990; Peudenier et al. 1991; Jones et al. 2000; Fischer-Smith et al. 2001). Thus, the cerebral endothelium is already exposed to the influence of HIV-1-infected blood-derived monocytes/macrophages and perivascular macrophages at an early stage of infection, but obviously without any impact on BBB function. This observation is consistent with our results and confirmed by our findings that there was no influence of a productive HIV-1 infection on the barrier-enhancing effect of macrophages on the BCEC.
The investigation of certain signal transduction pathways showed that increasing the intracellular cAMP level (by incubation with either the adenylate cyclase activator forskolin or the phosphodiesterase inhibitor RO 20–1724) resulted in a further TEER increase of the BCEC cocultured with macrophages (or C6 glioma cells, respectively), thus both effects are additive. In contrast, the TEER of the BCEC was not affected by inhibition of the adenylate cyclase with SQ22536, irrespective of whether the treated BCEC were cocultured with macrophages or C6 cells or no cells. Thus, it is unlikely that the TEER enhancing effect of the macrophages or C6 cells on the cocultured BCEC is mediated via a signal transduction pathway involving adenylate cyclase or cAMP. This result is in accordance with other studies that showed both an additive TEER increasing effect of raised cAMP levels in combination with astrocyte or C6 coculture and that the intracellular cAMP level of BCEC was not altered by astrocyte or C6 coculture (Rubin et al. 1991; Wolburg et al. 1994; Raub, 1996).
Inhibition of protein kinases A, C, G and myosin light chain kinase (MLCK) by incubation of the BCEC with the potent inhibitors H-7 and H-8 could not prevent the macrophage coculture-induced TEER increase, indicating that the effect of macrophages on the cocultured BCEC was not mediated via these signal transduction mechanisms. The same result was found in parallel experiments for the effect of C6 glioma cells on the BCEC barrier function. This finding seems to be inconsistent with the results of a study showing a TEER increase in C6-cocultured BCEC by activation of the protein kinase C and a TEER reduction by inhibition with H-7 (Raub, 1996). However, the BBB cell culture model used in that study was characterised by a low TEER baseline level, so it is very likely that the C6 coculture-induced TEER augmentation was primarily based on induction of de novo synthesis or fundamental reorganisation processes of the tight junctions. It was shown that processes involving tight junction synthesis, assembly and reorganisation require protein phosphorylation events, thus they are supported by protein kinase activation or prevented by their inhibition (Citi, 1992; Balda et al. 1993; Stuart & Nigam, 1995; Klingler et al. 2000).
In contrast, the BBB model system used in our studies is initially characterised by an high TEER baseline per se, so that any de novo synthesis or fundamental assembly and reorganisation steps are probably not required for additional reduction of the BCEC tight junction permeability (e.g. induced by C6 or macrophages coculture), but are rather accomplished by direct regulation of the permeability of completely established tight junctions. Obviously, this process is mediated via distinct mechanisms which do not involve any protein phosphorylation events, and therefore it is not affected by protein kinase activity. This finding is in agreement with findings made in epithelial cell systems which showed that the inhibition of protein kinases prevented tight junctions synthesis and organisation, whereas completely established tight junctions and a high electrical resistance of low permeable cell monolayers were not affected (Stuart & Nigam, 1995).
Similar results were found when the macrophage- or C6- cocultured BCEC were treated with the phospholipase C inhibitor U-73122, showing that TEER was not affected. Our finding is in agreement with other results showing that phospholipase C is not involved in the TEER increase of BCEC induced by coculture with C6 glioma cells (Raub, 1996).
In contrast, intracellular Ca2+ has an essential role in tight junction biogenesis, since accumulation and organisation of the tight junction-associated proteins and their assembly into tight junctions is heavily dependent on the Ca2+-calmodulin pathway (Stuart et al. 1994). Nevertheless, treatment of the BCEC with the Ca2+-calmodulin pathway inhibitor W-7 showed no effect on the increase of the TEER resulting from macrophage or C6 coculture, indicating that intracellular Ca2+ and Ca2+-dependent signal pathways are also not involved in the mechanisms regulating permeability of fully developed tight junctions of BCEC.
Taken together, our findings show that none of the signal transduction pathways investigated is involved in the enhancement of the barrier function of cultured BCEC in response to macrophages coculture. The same result was found for the equivalent effect of C6 glioma cells on BCEC, suggesting that the effects of both cell types are mediated via the same or at least related mechanisms. Certainly, these mechanisms allow the direct regulation of the tight junction permeability and modulation of the barrier function of the BCEC via signal pathways that are distinct from those involved in tight junction biogenesis and organisation. Despite many years of research, these regulatory pathways have still not been characterised in detail, and it remains uncertain whether the principal effect of primary astrocytes or C6 glioma cells on BCEC tight junction formation is mediated via soluble factors or direct cell-cell contact, or both acting together. Our findings suggest that the interaction between macrophages or C6 glioma cells with the BCEC is mediated via soluble factors, since there was no direct cell-cell contact between the two cell types in our coculture system.
In conclusion, we have demonstrated that blood-derived macrophages which do not originate from brain parenchyma are - in addition to cells of neuroectodermal origin (e.g. astrocytes) - able to induce and/or maintain blood-brain barrier specific functions in cultured BCEC. This interaction is most probably mediated via soluble factors. This indicates that macrophages or related brain-parenchymal cells of blood-monocytic lineage, such as the perivascular macrophages located in close proximity to the cerebral endothelium, might be a part of the microenvironment of the BCEC that modulates their specific blood-brain barrier functions. Certainly, peripheral macrophages do not really represent differentiated perivascular macrophages of the brain, but they seem to be the best available cell model for perivascular macrophages, since the latter cannot be isolated from the brain due to their low frequency and special location, and both cell types are nearly indistinguishable in their phenotype.
However, it was remarkable that stimulation of the macrophages or even HIV-1-infection itself did not affect their ability to increase the barrier function, in contrast to previous assumptions that pathological leakiness of the blood-brain barrier could have been caused by e.g. HIV-1 infection of these cells.
Acknowledgments
We are grateful to Karin Becker-Peters, Silke Deckert and Elisabeth Kurunci for their skilful technical assistance. Furthermore, we thank Uwe Alex for his support in statistical analysis. The Georg-Speyer-Haus is supported by the Bundesministerium für Gesundheit and the Hessisches Ministerium für Wissenschaft und Kunst.
REFERENCES
- Achim CL, Wiley CA. Inflammation in AIDS and the role of the macrophage in brain pathology. Curr Opin Neurol. 1996;9:221–225. doi: 10.1097/00019052-199606000-00013. [DOI] [PubMed] [Google Scholar]
- Allt G, Lawrenson JG. Is the pial microvessel a good model for blood-brain barrier studies. Brain Res Brain Res Rev. 1997;24:67–76. doi: 10.1016/s0165-0173(97)00011-8. [DOI] [PubMed] [Google Scholar]
- Andreesen R, Picht J, Löhr GW. Primary cultures of human blood-born macrophages grown on hydrophobic Teflon membrances. J Immunol Methods. 1983;56:295–304. doi: 10.1016/s0022-1759(83)80019-2. [DOI] [PubMed] [Google Scholar]
- Balda MS, Gonzales-Mariscal L, Matter K, Cereijido M, Anderson JM. Assembly of tight junctions: the role of diacylglycerol. J Cell Biol. 1993;123:293–302. doi: 10.1083/jcb.123.2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechmann I, Priller J, Kovac A, Bontert M, Wehner T, Klett FF, Bohsung J, Stuschke M, Dirnagl U, Nitsch R. Immune surveillance of mouse brain perivascular spaces by blood-borne macrophages. Eur J Neurosci. 2001;14:1651–1658. doi: 10.1046/j.0953-816x.2001.01793.x. [DOI] [PubMed] [Google Scholar]
- Berger JR, Nath A, Greenberg RN, Andersen AH, Greene RA, Bognar A, Avison MJ. Cerebrovascular changes in the basal ganglia with HIV dementia. Neurology. 2000;54:921–926. doi: 10.1212/wnl.54.4.921. [DOI] [PubMed] [Google Scholar]
- Blum MS, Toninelli E, Anderson JM, Balda MS, Zhou J, O'Donnell L, Pardi R, Bender JR. Cytoskeletal rearrangement mediates human microvascular endothelial tight junction modulation by cytokines. Am J Physiol. 1997;273:H286–294. doi: 10.1152/ajpheart.1997.273.1.H286. [DOI] [PubMed] [Google Scholar]
- Brightman MW, Kadota Y. Nonpermeable and permeable vessels of the brain. NIDA Res Monogr. 1992;120:87–107. [PubMed] [Google Scholar]
- Brightman MW, Reese TJ. Junctions between intimately apposed cell membranes in the vertebrate brain. J Cell Biol. 1969;40:648–677. doi: 10.1083/jcb.40.3.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt AM, Jones HC, Abbott NJ. Electrical resistance across the blood-brain barrier in anaesthetized rats: a developmental study. J Physiol. 1990;429:47–62. doi: 10.1113/jphysiol.1990.sp018243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlos RM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. 1994;84:2068–2101. [PubMed] [Google Scholar]
- Cassella JP, Lawrenson JG, Firth JA. Development of endothelial paracellular clefts and their tight junctions in the pial microvessels of the rat. J Neurocytol. 1997;26:567–575. doi: 10.1023/a:1015438624949. [DOI] [PubMed] [Google Scholar]
- Chiodi F, Asjo B, Fenyo EM, Norkrans G, Hagberg L, Albert J. Isolation of human immunodeficiency virus from cerebrospinal fluid of antibody-positive virus carrier without neurological symptoms. Lancet. 1986;ii:1276–1277. doi: 10.1016/s0140-6736(86)92699-1. [DOI] [PubMed] [Google Scholar]
- Citi S. Protein kinase inhibitors prevent dissociation induced by low extracellular calcium in MDCK epithelial cells. J Cell Biol. 1992;117:169–178. doi: 10.1083/jcb.117.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen Z, Molinatti G, Hamel E. Astroglial and vascular interactions of noradrenaline terminals in the rat cerebral cortex. J Cereb Blood Flow Metab. 1997;17:894–904. doi: 10.1097/00004647-199708000-00008. [DOI] [PubMed] [Google Scholar]
- Crone C, Olsen S-P. Electrical resistance of brain microvascular endothelium. Brain Res. 1982;241:49–55. doi: 10.1016/0006-8993(82)91227-6. [DOI] [PubMed] [Google Scholar]
- Dallasta LM, Pisarov LA, Esplen JE, Werley JV, Moses AV, Nelson JA, Achim CL. Blood-brain barrier tight junction disruption in human immunodeficiency virus-1 encephalitis. Am J Pathol. 1999;155:1915–1927. doi: 10.1016/S0002-9440(10)65511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehouck MP, Méresse S, Delorme P, Fruchart JC, Cecchelli R. An easier, reproducible, and mass-production method to study the blood-brain barrier in vitro. J Neurochem. 1990;54:1798–1801. doi: 10.1111/j.1471-4159.1990.tb01236.x. [DOI] [PubMed] [Google Scholar]
- de Vries HE, Brom-Roosemalen MCM, De Boer AG, van Berkel TJC, Breimer DD, Kuiper J. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J Neuroimmunol. 1996;64:37–43. doi: 10.1016/0165-5728(95)00148-4. [DOI] [PubMed] [Google Scholar]
- Esser R, Glienke W, Von Briesen H, Rübsamen-Waigmann H, Andreesen R. Differential regulation of proinflammatory and hematopoietic cytokines in human macrophages after infection with human immunodeficiency virus. Blood. 1996;88:3474–3481. [PubMed] [Google Scholar]
- Fischer-Smith T, Croul S, Sverstiuk AE, Capini C, L'Heureux D, Regulier EG, Richardson MW, Amini S, Morgello S, Khalili K, Rappaport J. CNS invasion by CD14+/CD16+ peripheral blood-derived monocytes in HIV dementia: perivascular accumulation and reservoir of HIV infection. J Neurovirol. 2001;7:528–541. doi: 10.1080/135502801753248114. [DOI] [PubMed] [Google Scholar]
- Gelman BB, Rodriguez-Wolf MG, Wen J, Kumar S, Campbell GR, Herzog N. Siderotic cerebral macrophages in the acquired immunodeficiency syndrome. Arch Pathol Lab Med. 1992;116:509–516. [PubMed] [Google Scholar]
- Genis P, Jett M, Berton EW, Gelbard HA, Dzenko K, Keane R, Resnick L, Volsky DJ, Epstein LG, Gendelman HE. Cytokines and arachidonic acid metabolites produced during HIV-1 infected macrophage-astroglial interactions: implications for the neuropathogenesis of HIV disease. J Exp Med. 1992;176:1703–1718. doi: 10.1084/jem.176.6.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Yu J, Li X, Tom D, Li J, Wendt E, Lin SN, Schwarcz R, Noonan C. Study of receptor-mediated neurotoxins released by HIV-1-infected mononuclear phagocytes found in human brain. J Neurosci. 1996;16:3139–3153. doi: 10.1523/JNEUROSCI.16-10-03139.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann EE. Vitalfärbung am Zentralnervensystem. Abhandlungen der Preusslschen Akademie der Wissenschaften. Physikalisch-Mathematische Klasse. 1913;1:1–60. [Google Scholar]
- Goudsmit J, Dewolf F, Paul DA, Epstein LG, Lange JMA, Krone WJA, Speelman H, Wolters EC, van der Noordaa J, Oleske JM, van der Helm HJ, Coutinho RA. Expression of human immunodeficiency virus antigen (HIV-Ag). in serum and cerebrospinal fluid during acute and chronic infection. Lancet. 1986;ii:177–180. doi: 10.1016/s0140-6736(86)92485-2. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Streit WJ, Buringer D, Sparks DL, Kreutzberg GW. Ultrastructural location of major histocompatibility complex (MHC) class II positive perivascular cells in histologically normal human brain. J Neuropathol Exp Neurol. 1992;51:303–311. doi: 10.1097/00005072-199205000-00009. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Streit WJ, Kreutzberg GW. Identity of ED2-positive perivascular cells in rat brain. J Neurosci Res. 1989;22:103–106. doi: 10.1002/jnr.490220114. [DOI] [PubMed] [Google Scholar]
- Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 1988;239:290–292. doi: 10.1126/science.3276004. [DOI] [PubMed] [Google Scholar]
- Hickey WF, Vass K, Lassmann H. Bone-marrow derived elements in the central nervous system: an immunohistochemical and ultrastructural survey of rat chimeras. J Neuropathol Exp Neurol. 1992;51:246–256. doi: 10.1097/00005072-199205000-00002. [DOI] [PubMed] [Google Scholar]
- Ho DD, Rota TR, Schooly RT, Kaplan JC, Allan JD, Groopman JE, Resnick L, Felsenstein D, Andrews CA, Hirsch MS. Isolation of HTLV-III from cerebrospinal fluid and neural tissue of patients with neurological syndromes related to the acquired immunodeficiency syndrome. N Engl J Med. 1985;313:1493–1497. doi: 10.1056/NEJM198512123132401. [DOI] [PubMed] [Google Scholar]
- Jantzer RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature. 1987;325:253–257. doi: 10.1038/325253a0. [DOI] [PubMed] [Google Scholar]
- Jones MV, Bell JE, Nath A. Immunolocalization of HIV envelope gp120 in HIV encephalitis with dementia. AIDS. 2000;14:2709–2713. doi: 10.1097/00002030-200012010-00010. [DOI] [PubMed] [Google Scholar]
- Kacem K, Lacombe P, Seylaz J, Bonvento G. Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter: a confocal microscopy study. Glia. 1998;23:1–10. [PubMed] [Google Scholar]
- Kennedy DW, Abkowitz JL. Kinetics of central nervous system microglial and macrophage engraftment: analysis using a transgenic bone marrow transplantation model. Blood. 1997;90:986–993. [PubMed] [Google Scholar]
- Klingler C, Kniesel U, Bamforth SD, Wolburg H, Engelhardt B, Risau W. Disruption of epithelial tight junctions is prevented by cyclic nucleotide-dependent protein kinase inhibitors. Histochem Cell Biol. 2000;113:349–361. doi: 10.1007/s004180000143. [DOI] [PubMed] [Google Scholar]
- Lassmann H, Schmied M, Vass K, Hickey WF. Bone-marrow derived elements and resident microglia in brain inflammation. Glia. 1993;7:19–24. doi: 10.1002/glia.440070106. [DOI] [PubMed] [Google Scholar]
- Mark KS, Miller DW. Increased permeability of primary cultured brain microvessel endothelial cell monolayers following TNF-alpha exposure. Life Sci. 1999;64:1941–1953. doi: 10.1016/s0024-3205(99)00139-3. [DOI] [PubMed] [Google Scholar]
- Miller DW, Audus KL, Borchardt RT. Application of cultured endothelial cells of the brain microvasculature in the study of the blood-brain barrier. J Tissue Cult Methods. 1992;14:217–224. [Google Scholar]
- Nottet HSLM, Gendelman HE. Unraveling the neuroimmune mechanisms for the HIV-1-associated cognitive/motor complex. Immunol Today. 1995;16:441–448. doi: 10.1016/0167-5699(95)80022-0. [DOI] [PubMed] [Google Scholar]
- Peluso R, Haase A, Stowring L, Edwards M, Ventura P. A Trojan Horse mechanism for the spread of visna virus in monocytes. Virology. 1985;147:231–236. doi: 10.1016/0042-6822(85)90246-6. [DOI] [PubMed] [Google Scholar]
- Petito CK, Cash KS. Blood-brain barrier abnormalities in the acquired immunodeficiency syndrome: immunohistochemical localization of serum proteins in post mortem brains. Ann Neurol. 1992;32:658–666. doi: 10.1002/ana.410320509. [DOI] [PubMed] [Google Scholar]
- Petito CK, Cho E-S, Lemann W, Navia BA, Price RW. Neuropathology of acquired immunodeficiency syndrome (AIDS): an autopsy review. J Neuropathol Exp Neurol. 1986;45:635–646. doi: 10.1097/00005072-198611000-00003. [DOI] [PubMed] [Google Scholar]
- Peudenier S, Hery C, Montagnier L, Tardieu M. Human microglial cells: characterization in cerebral tissue and in primary culture, and study of their susceptibility to HIV-1 infection. Ann Neurol. 1991;29:152–161. doi: 10.1002/ana.410290207. [DOI] [PubMed] [Google Scholar]
- Power C, Kong PA, Crawford TO, Wesselingh S, Glass JD, McArthur JC, Trapp BD. Cerebral white matter changes in acquired immunodeficiency syndrome dementia: Alterations of the blood-brain barrier. Ann Neurol. 1993;34:339–350. doi: 10.1002/ana.410340307. [DOI] [PubMed] [Google Scholar]
- Qin Y, Sato TN. Mouse multidrug resistance 1a/3 gene is the earliest known endothelial cell differentiation marker during blood-brain barrier development. Dev Dyn. 1995;202:172–180. doi: 10.1002/aja.1002020209. [DOI] [PubMed] [Google Scholar]
- Raub TJ. Signal transduction and glial cell modulation of cultured brain microvessel endothelial cell tight junctions. Am J Physiol. 1996;271:C495–503. doi: 10.1152/ajpcell.1996.271.2.C495. [DOI] [PubMed] [Google Scholar]
- Raub TJ, Kuentzel SL, Sawada GA. Permeability of bovine brain microvessel endothelial cells in vitro: barrier tightening by a factor released from astroglioma cells. Exp Cell Res. 1992;199:330–340. doi: 10.1016/0014-4827(92)90442-b. [DOI] [PubMed] [Google Scholar]
- Rubin LL, Hall DE, Porter S, Barbu K, Cannon C, Horner HC, Janatpour M, Liaw CW, Manning K, Morales J, Tanner LI, Tomaselli KJ, Bard F. A cell culture model of the blood-brain barrier. J Cell Biol. 1991;115:1725–1735. doi: 10.1083/jcb.115.6.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rübsamen-Waigmann H, Becker WB, Helm EB, Brodt R, Fischer H, Henco K, Brede HD. Isolation of variants of lymphocytopathic retroviruses from the peripheral blood and cerebrospinal fluid of patients with ARC or AIDS. J Med Virol. 1986;19:335–344. doi: 10.1002/jmv.1890190406. [DOI] [PubMed] [Google Scholar]
- Sachs L. Angewandte Statistik: Anwendung Statistischer Methoden. Springer-Verlag, Berlin, Heidelberg, New York: 1992. [Google Scholar]
- Siegel S, Castellan NJ. Nonparametric Statistics for the Behavioral Sciences. New York: Blackwell Science Inc; 1988. [Google Scholar]
- Smith TW, Degirolami U, Hénin D, Bolgert F, Hauw JJ. Human immunodeficiency virus (HIV) leukoencephalopathy and the microcirculation. J Neuropathol Exp Neurol. 1990;49:357–370. doi: 10.1097/00005072-199007000-00001. [DOI] [PubMed] [Google Scholar]
- Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- Stewart PA, Wiley MJ. Developing nervous tissue induces formation of blood-brain barrier characteristics in invading endothelial cells: a study using quail-chick transplantation chimeras. Dev Biol. 1981;84:183–192. doi: 10.1016/0012-1606(81)90382-1. [DOI] [PubMed] [Google Scholar]
- Stuart RO, Nigam SK. Regulated assembly of tight junctions by protein kinase C. Proc Natl Acad Sci U S A. 1995;92:6072–6076. doi: 10.1073/pnas.92.13.6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart RO, Sun A, Panichas M, Hebert SC, Brenner BM, Nigam SK. Critical role for intracellular calcium in tight junction biogenesis. J Cell Physiol. 1994;159:423–433. doi: 10.1002/jcp.1041590306. [DOI] [PubMed] [Google Scholar]
- von Briesen H, Andreesen R, Esser R, Brugger W, Meichsner C, Becker K, Rübsamen-Waigmann H. Infection of monocytes/macophages by HIV in vitro. Res Virol. 1990;141:225–231. doi: 10.1016/0923-2516(90)90025-e. [DOI] [PubMed] [Google Scholar]
- von Briesen H, Becker WB, Henco K, Helm EB, Gelderblom HR, Brede HD, Rüebsamen-Waigmann H. Isolation frequency and growth properties of HIV-variants: multiple simultaneous variants in a patient demonstrated by molecular cloning. J Med Virol. 1987;23:51–66. doi: 10.1002/jmv.1890230107. [DOI] [PubMed] [Google Scholar]
- Williams K, Alvarez X, Lackner AA. Central nervous system perivascular cells are immunoregulatory cells that connect the CNS with the peripheral immune system. Glia. 2001;36:156–164. doi: 10.1002/glia.1105. [DOI] [PubMed] [Google Scholar]
- Wolburg H, Neuhaus J, Kniesel U, Krauβ B, Schmid E-M, Öcalan M, Farrell C, Risau W. Modulation of tight junction structure in blood-brain barrier endothelial cells. J Cell Sci. 1994;107:1347–1357. doi: 10.1242/jcs.107.5.1347. [DOI] [PubMed] [Google Scholar]